

Abstract
Expansion of human stem cells before cell therapy is typically performed at 20% O2. Growth in these pro-oxidative conditions can lead to oxidative stress and genetic instability. Here, we demonstrate that culture of human mesenchymal stem cells at lower, physiological O2 concentrations significantly increases lifespan, limiting oxidative stress, DNA damage, telomere shortening and chromosomal aberrations. Our gene expression and bioenergetic data strongly suggest that growth at reduced oxygen tensions favors a natural metabolic state of increased glycolysis and reduced oxidative phosphorylation. We propose that this balance is disturbed at 20% O2, resulting in abnormally increased levels of oxidative stress. These observations indicate that bioenergetic pathways are intertwined with the control of lifespan and decisively influence the genetic stability of human primary stem cells. We conclude that stem cells for human therapy should be grown under low oxygen conditions to increase biosafety.
Keywords: stem cells, MSC, oxidative stress, hypoxia, glycolysis, aneuploidy
Human mesenchymal stem cells (hMSC) are being evaluated for the treatment of a large variety of pathologies, including traumatic lesions and cardiovascular and autoimmune diseases.1, 2 Although hMSC can be obtained from several tissues, they are scarce and their quantity and quality depends on a patient's clinical history, age, gender and genetic background. Most cell therapy protocols use 10–50 million hMSC per treatment, requiring expansion of extracted stem cells ex vivo for about 8 weeks before implantation. This expansion is typically performed under ‘standard' non-physiological culture conditions, which among other factors expose cells to 20% O2, roughly 10 times the oxygen concentration encountered in their natural niches.3, 4
Previous studies have shown that exposure of mammalian cells to 20% O2 concentrations induces DNA damage, thereby contributing decisively to cell senescence and loss of viability.5, 6, 7 Conversely, culture of human stem cells over a physiological range of oxygen tensions (1–5%) improves cell growth, alters differentiation processes and extends lifespan.8 Low oxygen tensions have also been shown to reduce the levels of double-strand breaks (DSB) and chromosomal abnormalities in several types of stem cells.9, 10 This evidence suggests that the poorly defined ‘cell culture stress' can be a cause of genetic instability and therefore constitute a biological risk for cell therapy protocols. In agreement with this notion, we have found that short-term growth of hMSC at 20% O2 significantly increases oxidative stress and DNA damage markers, DSB, chromosomal aberrations, aneuploidy and telomere shortening rates compared with cells grown at 3% O2. Despite these clear correlations, the mechanisms underlying the generation of genetic instability at high O2 tension are mostly unknown.
Mammalian cells have developed oxygen-sensing mechanisms to maintain cell and tissue homeostasis (reviewed in Giaccia et al.11). When oxygen levels fall below a certain threshold, a gene expression program is initiated by the transcription factor hypoxia-inducible factor-1 (HIF-1). HIF-1 signaling is a pivotal link between oxygen availability in the cell and key processes such as energy metabolism, angiogenesis, cell proliferation and viability (reviewed in Schofield and Ratcliffe12). Activation of HIF-1a increases the levels of glycolytic enzymes when oxygen is too limiting to support oxidative phosphorylation (OXPHOS); thereby ensuring energy demands are met under hypoxic conditions.13, 14
To elucidate the mechanisms by which oxygen levels, and exogeneous oxidative stress affect the genetic stability of hMSC, we carried out detailed cytogenetic analysis, together with gene expression and bioenergetics studies, in sister cultures grown at ‘physiological' 3% O2 and ‘standard' 20% O2 conditions. Our data show that growth at physiological O2 levels is associated with a clear transcriptional activation in HIF-1a target genes that results of increased glycolytic function and inhibition of oxygen consumption. Our results suggest that cells exposed to non-physiological oxygen levels activate a cellular response in which oxygen consumption by OXPHOS is increased and glycolysis decreased. We propose that this adaptive metabolic switch in 20% O2 contributes to the generation of genetic instability by increasing the production of ROS, possibly compromising the biosafety and viability of long-term cultures of stem cells.
Results
Growth of hMSC at 20% O2 reduces lifespan, increases oxidative stress and the rate of telomere shortening
A set of four hMSC lines was acquired from a commercial source. To confirm the cell type and purity of these lines, we detected the expression of the following specific surface markers by flow cytometry: CD29, CD44, CD90, CD105, CD11b, HLA DR, CD19 and CD45 (Supplementary Figure S1a). Differentiation capacity was confirmed in osteogenic and adipogenic differentiation assays (Supplementary Figure S1b). DNA fingerprinting with nine single-nucleotide polymorphisms at early (p2) and late (p15) passages showed no evidence of cross-contamination at either oxygen concentration (data not shown).
To determine if hMSC growth dynamics are influenced by oxygen tension, we established individual adipose tissue-derived cultures from four adult female donors (aged 35–41 years) and compared their growth rates at 20% O2 versus 3% O2 over 25 passages. hMSC were sourced from donors of the same gender and similar age in order limit sources of variability such as hormonal changes or telomere length status. Cell lines grown at 3% O2 completed 35.48±0.96 population doublings (PDs), whereas the corresponding cultures grown at 20% O2 completed only 28.68±1.3 PD (Figure 1a). This difference is statistically significant (P=0.002) and was apparent even at early passages. For example, by passage 5 hMSC grown at 3% O2 completed 16.73±1.22 PD, compared with 10.98±0.86 PD for cells grown at 20% O2 (P=0.010). These results indicate that at 3% O2 we can theoretically obtain >1 × 109 cells in five passages, whereas at 20% O2 we only obtain 2 × 107 cells. Both cell yields arose from an estimated starting cell population of 1 × 104 hMSC. Despite the higher growth rate at 3% O2, our results indicate that hMSC did not overcome senescence at either oxygen concentration (Figure 1a).
Figure 1.

Effects of physiological oxygen concentration on growth and oxidative stress levels in hMSCs. (a) Growth curves of four independent hMSC primary cell lines (isolates 34, 36, 40 and 43) cultured at 3% O2 (black lines) and 20% O2 (gray lines). Growth of hMSC at 3% O2 was significantly higher from passage 2 onward; at both O2 concentrations, senescence was reached at about passage 15. (b) Flow cytometry detection of superoxide (DHE-derived fluorescence) in hMSC cultured at 3% O2 (hatched black histogram) and 20% O2 (gray histogram) at early (passage <5), middle (passages 5–10) and late passage (passage >10). For a given time in culture, superoxide levels were higher in cells grown under ‘standard' (20% O2) conditions, and ROS appear to increase with passage at both oxygen concentrations. Representative histograms from one cell line are shown (n=4). (c) Levels of protein carbonyls and MDA in hMSC lines grown at 20% O2 (white bars) and at 3% O2 (black bars). Culture at 3% O2 results in significantly lower levels of these oxidative stress-derived products (*P<0.05). Experiments were performed in triplicate with the four independent lines. Data are means±S.E.M.
To test whether the improved growth rate at low oxygen tension correlated with lower levels of reactive oxygen species (ROS), we stained early, middle and late passage hMSC with di-hydroethidine (DHE), a fluorescent marker for superoxide anion (O2−). hMSC grown at 20% O2 accumulated more superoxide anions than cells grown at 3% O2 (Figure 1b). In addition, these differences in ROS levels increased with passage number. The increased production of ROS at 20% O2 was confirmed by analysis of two surrogate markers: the levels of protein oxidation (carbonyl groups) and the mutagenic lipid peroxidation product malondialdehyde (MDA). Levels of carbonyls and MDA in hMSC grown at 20% O2 were 29% (P=0.016) and 107% (P=0.036) higher, respectively, than in parallel cultures grown at 3% O2 (Figure 1c).
To determine the contribution of telomere shortening to hMSC senescence, we analyzed interphase hMSC by quantitative fluorescence in situ hybridization (Q-FISH), using peptide-nucleic acid (PNA) telomeric probes15 (Figure 2a). In cells grown at 20% O2, the average telomere length at passage 2 was 8.43±1.28 kb, decreasing to 3.76±0.7 kb at passage 15; in contrast, cells grown over the same passages at 3% O2 showed a smaller decline in telomere length, from 8.21±1.1 kb to 5.04±0.95 kb, despite having completed 4–5 more PDs. Thus, cells grown at 20% O2 appear to exhaust their telomeric DNA faster than cells grown at 3% O2. The rates of telomere shortening were 378±46 bp per cell division at 3% O2 versus 470±61 bp at 20% O2 (P=0.016) (Figure 2b).
Figure 2.

Supra-physiological oxygen tension increases DSBs and chromosomal aberrations in hMSCs. (a) Plot of telomere length against PD in interphase hMSC. Telomere length was measured by Q-FISH at passages 2 and 15. Thin dotted and solid lines represent telomere shortening in hMSC grown at 20% O2 and 3% O2, respectively. Color insets show representative images of cells at passage 2 and passage 15 hybridized with the telomeric probe (red). Scale bars, 10 μm. (b) Quantification of telomere length reduction as kbp per PD. The quantification shows a significant reduction of telomere shortening at 3% O2 (*P=0.016). Telomere fluorescence was converted to kbp by comparison with fibroblast lines expressed known amounts of TIN2 and TIN2–13. (c) Quantification of 53BP1-positive cells grown at 3% O2 and 20% O2. A significant reduction was appreciated at 3% O2 (*P=0.013). (d) Quantification of 53BP1 foci per nucleus, showing the trend in the reduction of DSB per cell in cultures grown at 3% O2; in all cases the cells were grown for 18±2 PD (e) Examples of cells grown at 3% O2 and 20% O2 stained with 53BP1 (yellow) to mark DSB. Irradiated cells (10 Gy) are shown as a positive control; negative controls show no signal at this same time exposure. Nuclei are counterstained with DAPI (blue). The white arrows mark individual foci. Insets in the upper left corner show a magnification of representative nuclei from each condition. Scale bars, 10 μm. (f) Quantification of structural chromosomal aberrations in metaphase cells from hMSC grown at 3% O2 (black bars) and 20% O2 (white bars); 3% O2 reduced the incidence of chromosomal abnormalities, including a significant reduction in dicentric chromosomes (*P=0.049). (g) Examples of chromosomal aberrations in cells processed for FISH with centromeric (green) and telomeric (red) PNA probes. Scale bars, 1 μm. Experiments were performed in triplicate with the four independent lines. Data are means±S.E.M.
As telomere length is strongly influenced by telomerase activity, we performed Q-PCR-based telomere repeat amplification protocol (TRAP) assays.16 Telomerase activity in four hMSC cultures grown at 20% O2 was similar to that of human primary fibroblast cultures (Supplementary Figures S2a and b), consistent with their limited proliferative capacity and their senescence. Telomerase activity in hMSC was not affected by the O2 concentration (Supplementary Figures S2c and d), therefore indicating that the increased rate of telomere shortening at high oxygen tension is likely to result from increased oxidative damage and DSB under these conditions.17, 18
Growth of hMSC at 20% O2 increases DSB generation and chromosomal instability
Oxidative stress has been previously shown to cause DNA damage in mammalian fibroblasts.6 To determine if culture-derived oxidative stress is sufficient to induce DNA damage in hMSC, we performed immunofluorescence against 53BP1, a well-characterized marker of DSB.19 Of hMSC grown at 3% O2, 44.68±6.9% contained DSB foci, compared with 63.31±7.3% of cells grown at 20% O2 (Figures 2c and e). Moreover, hMSC grown at 3% O2 contained 0.69±0.11 foci per cell, whereas cells grown at 20% O2 had 1.37±0.4 foci per cell (Figures 2d and e). The number of cells with at least one DSB was significantly higher at the 20% O2 condition (P=0.013), indicating that culture at 20% O2 causes an excess of DNA breaks in human stem cells (Figure 2c).
To confirm these results, we carried out detailed cytogenetic analysis of chromosomal breaks and fusions and dicentric chromosomes, all indicative of DSB. For this, we stained metaphases with telomeric and centromeric probes to clearly define the aberrations. Cells grown at 3% O2 had 2-fold fewer chromosomal fusions, 1.5-fold fewer chromatid breaks and a significantly 2.6-fold lower level of dicentric chromosomes (P=0.049) (Figures 2f and g). Together these results indicate that hMSC grown at 20% O2 have an excess of DNA damage and of unstable chromosomal aberrations, compared with cells grown at a more physiological oxygen tension (Figures 2c–g).
Oxidative stress increases aneuploidy in hMSC
Our cytogenetic analysis suggested that culture at 20% O2 induces aneuploidy. Aneuploidy is a central characteristic of tumor cells and is associated with the partial loss of homeostasis and control of gene expression. The association between aging and an increased number of aneuploid cells has been known for decades;20 however, previous studies did not examine the association between culture conditions (passage and oxygen tension) and aneuploidy in mesenchymal stem cells.
To examine the effect of O2 concentration on aneuploidy, we analyzed interphase hMSC between passages 2 and 15 by FISH, using centromeric probes for chromosomes 8, 11 and 17 (Figures 3a and b). These chromosomes harbor important oncogenes and tumor suppressors (for example, c-myc in chr 8, CCND1 or ATM in chr 11, and p53 in chr 17), and aneuploidy in any chromosome of this set is considered an early event in primary breast cancer.21, 22 Analysis of aneuploidy in these chromosomes might therefore provide a tool for evaluating genetic instability in hMSC, thus ensuring culture biosafety. More than 100 cells per cell line and O2 condition were analyzed at interphase, because at any given time most cells are at this cell cycle stage and it is therefore more representative of the general condition of the cell population than the limited number of metaphase cells usually examined in aneuploidy studies.23, 24 Surprisingly, even at early passages (p2–p5), about 15–24% of cells cultured at 20% O2 showed aneuploidy for any of these three chromosomes. Two trends were immediately apparent. First, at both oxygen concentrations there is a clear tendency toward increased aneuploidy with passage, reaching as high as 35% by passage 15 (Figure 3a). Second, the incidence of aneuploidy was significantly lower in cells grown at 3% O2 from passages 2–10; for example, at passage 5 aneuploidy affected 16.43±0.37% of cells grown at 3% O2 compared with 24.25±0.42% of cells grown at 20% O2 (P=0.016). This trend is revealed by plotting the aneuploidy level against PD (Figure 3b). No significant differences in aneuploidy were detected between the analyzed chromosomes (data not shown).
Figure 3.

Long-term exposure to supra-physiological oxygen tension promotes aneuploidy in telomerase-negative hMSCs. (a) Mean aneuploidy (%) scored in 100–200 nuclei per hMSC cell line at 3% O2 (black bars) and 20% O2 (white bars). Aneuploidy was detected by FISH with centromeric probes for chromosomes 8, 11 and 17. Growth at 3% O2 significantly reduced the incidence of aneuploidy during passages 2–10 (*P<0.049). (b) Aneuploidy levels plotted against PD; hMSC grown at low oxygen are significantly more stable per cell division. Experiments were performed in triplicate with the four independent lines. Data are means±S.E.M. (c) Frequency histogram of chromosome counts from hMSC expanded for 18±2 PD (n=106–120). White bars represent metaphases from cells grown at 20% O2; black bars represent metaphases from cells grown at 3% O2. Chromosome numbers are as indicated. (d) Average levels of aneuploidy of two human fibroblast cell lines (passages 10 and 20) and two human BM-MSC lines (passages 10 and 14) at 20% and 3% O2. Aneuploidy was assessed by interphase FISH using Centromere Evaluation Probe (CEP) probes for chromosomes 8, 11 and 17
To confirm these results, we evaluated aneuploidy by metaphase chromosomal analysis of four cell lines cultured at both O2 concentrations at the same PD (18±2), corresponding to passages (p5–p8). These experiments indicated that 54.2% of metaphases from the hMSC grown at 3% O2 (n=120) had 46 chromosomes. In contrast, only 30% of cells grown at 20% O2 were diploid (n=106) (Figure 3c). The difference in aneuploidy between conditions was highly statistically significant (P<0.001). In general, our results suggest that long-term exposure to oxidative stress induced by ‘standard' cell culture conditions clearly promotes aneuploidy.
Owing to the relatively high levels of aneuploidy observed in hMSC, we set out to determine if this was a more general phenomenon. Human fibroblasts and bone marrow hMSC grown at 20% O2 (passage 10) presented levels of aneuploidy ranging from 12 to 18%, in line with our previous results.23, 25 Analysis of aneuploidy in cultures of these cells grown for an additional 4–10 passages at 3% O2 and 20% O2 confirmed that growth at 3% O2 reduces the levels of aneuploidy in other cell types of mesenchymal origin (Figure 3d). These results strongly suggest that growth of mesenchymal cells at 20% O2 for 5–10 passages promotes aneuploidy, and we therefore propose that ROS are a principal cause of aneuploidy in hMSC.
To validate this hypothesis, we treated hMSC grown at 3% O2 either twice a week with the superoxide generator paraquat (PQ; 40 μM) or with a single treatment of the peroxide generator H2O2 (100 μM). Treatment with PQ produced a 2.7-fold reduction in cell lifespan and a 3-fold increase in aneuploidy (Figures 4a, b and e). Treatment with H2O2 similarly led to a pronounced decrease in cell growth and to a threefold increase in aneuploidy (Figures 4c–e). These results suggest that both chemical and culture-induced sources of exogenous oxidative stress can cause senescence and aneuploidy in stem cells. At 20% O2, cells exposed to PQ (40 μM) were able to progress for only three passages, and after two passages had reached the same level of aneuploidy seen in 3% O2 cultures after five passages, indicating that higher O2 concentration enhances the toxicity of exogenous ROS generators (Supplementary Figures 3a and b).
Figure 4.

Exogenous sources of ROS cause growth defects and genetic instability in hMSCs. (a) Growth dynamics of an hMSC line cultured at 3% O2 with (black) and without (gray) treatment with 40 μM PQ as a source of ROS, showing a decrease in growth after PQ treatment. (b) Aneuploidy levels in control and PQ-treated cells. PQ treatment significantly increased aneuploidy at passages 5 (*P<0.050) and 10 (*P<0.050). (c) Growth dynamics of an hMSC line grown at 3% O2 with (black) or without (gray) treatment with hydrogen peroxide (H2O2, 100 μM) as a source of ROS, showing decreased growth and cell death after H2O2 treatment. (d) Aneuploidy levels in control and H2O2-treated cells. H2O2 treatment significantly increased aneuploidy at passage 3 (*P<0.050). (e) Representative FISH images of cells grown at 3% O2: left, controls; middle, PQ treated; right. H2O2 treated. Scale bars, 10 μm. Cells were hybridized with Centromere Evaluation Probe (CEP) probes for chromosomes 8 (red signal), 11 (green) and 17 (light blue). In the aneuploidy experiments, the fraction of aneuploid cells was calculated for each condition, and data were analyzed with Fisher's exact test for two binomials
Culture of hMSC at 20% O2 significantly increases oxygen consumption and decreases glycolytic metabolism
To further investigate the underlying mechanisms by which culture of hMSC at 20% O2 reduces lifespan and increases genetic instability, we profiled gene expression at 3% O2 and 20% O2 in four hMSC primary lines. Microarray analysis identified 822 genes whose expression was significantly different between the two conditions at a statistical significance q-value<0.050 (Supplementary Table S1). Assessment of biological function by gene ontology analysis indicated that 42 gene ontology (GO) biological processes were significantly altered (P<0.050) (Tables 1a and b). Within these processes, only 3 genes were downregulated and 12 were upregulated in cells grown at 3% O2, indicating a shift toward glycolytic metabolism (Supplementary Table S2 and Table 2). The most significant alterations (P<0.005) are linked to glucose metabolism, glycolysis, hexose, alcohol and carbohydrate catabolism (Tables 1a and b). Surprisingly, of the 42 altered GO-terms only one is related to cell survival, cell cycle or cell maintenance (GO:0008634; ‘negative regulation of survival gene product expression'). This GO category includes the proapoptotic genes BNIP3, HRK and BNIP3L (Supplementary Table S2), which are significantly altered in the gene expression pattern. BNIP3 and BNIP3L are upregulated 2.3-fold (q-value<0.001) and 1.6-fold (q-value=0.006) at 3% O2, respectively. The expression of HRK is downregulated 0.5-fold (q-value=0.010) (Supplementary Table S1).
Table 1. Simplified gene ontology analysis.

Table 2. Expression changes in genes related to carbohydrate metabolic processes and HIF-1 targets.
| Symbol | Name | Fold change | FDR BH (t) | FDR q-value (t) |
|---|---|---|---|---|
| Carbohydrate metabolism | ||||
| PDK1 | Pyruvate dehydrogenase kinase, isozyme 1 | 3.441 | 0.001 | 0.001 |
| PFKFB4 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 | 2.786 | 0.001 | 0.001 |
| PGK1 | Phosphoglycerate kinase 1 | 2.178 | 0.002 | 0.001 |
| ALDOC | Aldolase C, fructose-bisphosphate | 2.359 | 0.004 | 0.002 |
| PFKFB3 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | 2.286 | 0.004 | 0.002 |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | 2.764 | 0.004 | 0.002 |
| ENO2 | Enolase 2 (gamma, neuronal) | 2.186 | 0.009 | 0.004 |
| TPI1 | Triosephosphate isomerase 1 | 1.689 | 0.011 | 0.005 |
| ALDH5A1 | Aldehyde dehydrogenase 5 family, member A1 | 0.627 | 0.013 | 0.006 |
| LDHA | Lactate dehydrogenase A | 1.562 | 0.019 | 0.009 |
| IGF2 | Insulin-like growth factor 2 (somatomedin A) | 3.396 | 0.025 | 0.012 |
| NUDT5 | Nudix (nucleoside diphosphate linked moiety X)-type motif 5 | 0.671 | 0.026 | 0.012 |
| ENO3 | Enolase 3 (beta, muscle) | 1.412 | 0.027 | 0.012 |
| PGD | Phosphogluconate dehydrogenase | 0.749 | 0.047 | 0.021 |
| GAPDHL7 | Glyceraldehyde-3-phosphate dehydrogenase-like 7 | 1.328 | 0.062 | 0.028 |
| HIF targets | ||||
| PDK1 | Pyruvate dehydrogenase kinase, isozyme 1 | 3.441 | 0.001 | 0.001 |
| BNIP3 | BCL2/adenovirus E1B 19 kDa interacting protein 3 | 2.297 | 0.001 | 0.001 |
| PGK1 | Phosphoglycerate kinase 1 | 2.178 | 0.002 | 0.001 |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | 2.764 | 0.004 | 0.002 |
| EGLN1 | Egl nine homolog 1 (C. elegans) | 1.837 | 0.006 | 0.003 |
| PLOD1 | Procollagen-lysine 1, 2-oxoglutarate 5-dioxygenase 1 | 1.849 | 0.009 | 0.004 |
| BCL2 | B-cell CLL/lymphoma 2 | 0.566 | 0.011 | 0.005 |
| PLOD2 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 | 1.974 | 0.019 | 0.009 |
| LDHA | Lactate dehydrogenase A | 1.562 | 0.019 | 0.009 |
| VEGFA | Vascular endothelial growth factor A | 1.656 | 0.021 | 0.010 |
| ITPR1 | Inositol 1,4,5-triphosphate receptor, type 1 | 0.696 | 0.023 | 0.010 |
| ADRA2C | Adrenergic, alpha-2C-, receptor | 0.562 | 0.027 | 0.012 |
| TFR2 | Transferrin receptor 2 | 1.290 | 0.029 | 0.013 |
| CA9 | Carbonic anhydrase IX | 2.169 | 0.040 | 0.018 |
| SERPINE1 | Serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 | 1.753 | 0.053 | 0.024 |
Gene expression data for hMSC (n=4) grown at 3% O2 versus 20% O2, showing differentially expressed genes involved in carbohydrate metabolism and in HIF-1 induction. These genes are included in GO categories with P-value <0.050, and are listed in order of statistical significance (q-value <0.050)
One of the best-characterized mechanisms of glycolysis upregulation is the hypoxia response through activation of HIF-1.26 Our gene expression data showed that 12 HIF-1 target genes12 are significantly upregulated in the hMSC cultures at 3% O2, by 1.2–2.3-fold (Table 2), suggesting robust HIF-1 activation. To verify the gene expression results, we performed Taqman RT-PCR analysis and detected a significant (P<0.050) 1.5–8-fold upregulation of pyruvate dehydrogenase kinase, isozyme 1 (pdk1); HIF-3, alpha subunit; phosphoglycerate kinase 1 (pgk1); 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (pfkfb3); 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 (pfkfb4); aldolase C; fructose-bisphosphate (aldoc); triosephosphate isomerase 1 (tpi1); solute carrier family 2 (facilitated glucose transporter), member 1 (Scl2a1); lactate dehydrogenase A (ldha); and insulin-like growth factor 2 (igf2) (Figure 5a). In summary, the gene expression, gene ontology and qRT-PCR analyses indicate that in cultures at 3% O2 there is a strong upregulation of carbohydrate metabolism, particularly of glycolytic genes, that is, probably mediated through HIF-1 activation.
Figure 5.

Culture of hMSC at physiological oxygen tension increases glycolytic activity and reduces oxygen consumption. (a) RT-PCR analysis (Taqman) of pyruvate dehydrogenase kinase, isozyme 1 (pdk1); hif-3a; phosphoglycerate kinase 1 (pgk1); 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (pfkfb3); 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4, (pfkfb4); aldolase C, fructose-bisphosphate (aldoc); triosephosphate isomerase 1 (tpi1); solute carrier family 2 (facilitated glucose transporter), member 1 (Scl2a1); lactate dehydrogenase A (ldha); and insulin-like growth factor 2 (igf2). Expression is presented relative to the level in cells grown at 20% O2. Experiments were performed in triplicate with the four independent lines. Data are means±S.E.M. In all cases, the data were statistically significant (*P<0.050). (b) OCR/ECAR curves for three hMSC lines exposed to 3% O2 and 20% O2. Curves were generated using a FX96 Flux Analyzer and Seahorse technology (see Materials and Methods section). The first four points on the graphs indicate the basal OCR/ECAR ratio (expressed in pmoles/mpH); the green line indicates background correction of the blank. Oligomicin was added at time point A and the mitochondrial uncoupler at time point B. (c) Average OCR/ECAR ratios (pmoles/mpH) for three hMSC lines grown at 3% O2 and 20% O2 (*P<0.050). Bars represent means±S.E.M; experiments were performed with eight replicates for each time point. (d) Intracellular lactate concentration in hMSC grown at 3% O2 and 20% O2. Cells cultured at 3% O2 contained more lactate than cells cultured at 20% O2 (*P<0.050). Experiments were performed in triplicate with the four independent lines. Data are means±S.E.M. The color reproduction of this figure is available at the Cell Death and Differentiation journal online
To evaluate how the transcriptional upregulation of glycolysis genes at 3% O2 impacts cellular metabolism, we monitored mitochondrial oxygen consumption rate (OCR) and extra cellular acidification rate (ECAR) in three hMSC lines in both oxygen conditions, using Seahorse non-invasive technology. The basal OCR/ECAR ratios were significantly higher in hMSC cells grown at 20% O2 (2200 pMoles/mpH) than at 3% O2 (1000 pMoles/mpH) (P<0.050) (Figures 5b and c). Addition of DNP, a mitochondrial uncoupling agent, showed that maximum respiratory capacity was also significantly higher in hMCS grown at 20% O2 (Figure 5b). Addition of oligomycin to inhibit F0/F1 ATPase (complex V) revealed similar differences between culture at 20% O2 and 3% O2 (Figure 5b). These data indicate that growth at higher O2 tension promotes increased oxygen consumption and an associated drop in medium acidification. To verify that growth in 20% O2 decreases glycolysis, we analyzed lactic acid concentration. These experiments showed that hMSC grown at 3% O2 contained a twofold higher concentration of lactate (P<0.050) (Figure 5d).
Discussion
Many ongoing clinical trials use hMSC after culture expansion at 20% O2. This growth protocol is an inheritance from traditional cell culture methods established decades ago for transformed cells with very different bioenergetic characteristics.27 Accumulating evidence suggests that culture of mammalian cells at 20% O2 promotes DNA damage and senescence,5, 6, 7 and recent data suggests that long-term culture under these conditions leads to the appearance of chromosomal aberrations.28 Given the paramount importance of biosafety in cell therapy, we set out to investigate the relationships between oxygen tension, metabolism and genetic stability in hMSC expanded in vitro. In this study, we show that expansion of adipose tissue-derived hMSC at 3% O2 significantly increases their growth rate, in line with previous findings.8 Despite this faster growth, we observed no evidence of spontaneous immortalization, suggesting that, at least during 25 passages, hMSC cultured at 3% O2 do not activate mechanisms that can overcome senescence.
In the absence of compensatory mechanisms, human cell senescence is intimately related to critical telomere shortening. Previous reports indicated that telomere shortening can be accelerated by growth under hyperoxic conditions, which generates oxidative stress.17, 18, 29 In our experiments, culture at low O2 reduced the rate of telomere shortening per cell division by 24%, indicating that the increased lifespan might reflect better maintenance of telomere length. This increased lifespan is probably independent of telomerase activity because in hMSC this activity was low and insensitive to changing O2 concentration. These observations suggest that growth at 3% O2 leads to decreased telomere erosion by protecting cells from oxidative stress.
The enhanced growth of hMSC observed at 3% O2 correlated with decreased levels of mutagenic oxidative stress, with an accompanying decrease in 53BP1 foci and structural chromosomal aberrations. The fact that the levels of aneuploidy continuously accumulated with increasing passage, reaching a maximum of 35% in passage 15, and were lower in cells cultured at ‘physiological' oxygen tension strongly suggests that oxidative stress can induce genetic instability. In addition, we demonstrate that several cell types (hMSC from adipose tissue and bone marrow stroma, and skin fibroblasts) present higher levels of aneuploidy when cultured at 20% O2 rather than 3% O2, indicating that induction of aneuploidy by high O2 concentration might be a common phenomenon. Notably, significant differences in aneuploidy were not detected between the chromosomes analyzed, suggesting that aneuploidy at early-mid passages is a random process. A relationship between oxidative stress and aneuploidy is supported by the finding that acute exposure to PQ or H2O2, known inducers of ROS, significantly reduced cell growth and increased aneuploidy about threefold. The difference in the effect of PQ treatment on aneuploidy and growth reduction in cultures at 20% O2 and 3% O2 indicates that the effects of ROS on genetic instability might be a cumulative process.
Gene expression profiling and gene ontology analysis in cells grown at the two oxygen tensions showed that growth at 3% O2 causes significant transcriptional upregulation of 12 glycolytic and carbohydrate metabolism genes. The fact that many of these genes are also HIF-1 targets strongly suggests that culture at 3% O2 maintains relatively high levels of HIF-1 compared with ‘standard' growth conditions. Our analysis of respiration and glycolysis confirms that these gene expression patterns translate into bioenergetic changes; at 3% O2 the OCR/ECAR ratio is significantly lower than at 20% O2, and lactate levels are higher, providing compelling evidence that glycolysis is upregulated under these conditions. Surprisingly, higher growth at 3% O2 was reflected in only one GO category related to cell growth, survival and apoptosis. This included the genes BNIP3 and BNIP3L, whose expression has also been associated with hypoxia and HIF-1α expression.30 Our results thus strongly suggest that the main response to 3% O2 is the transcriptional and biochemical activation of bioenergetic genes. However, we cannot exclude the possibility that growth at 3% O2 might affect the profile of secreted chemokines and cytokines, influencing cell growth and senescence through autocrine or paracrine effects.31
These metabolic changes may provide an additional mechanism for lifespan extension, because increased expression of glycolytic enzymes (PGM, PGK1 and GPI) has been shown to favor immortal growth of mouse embryonic fibroblasts.32 The gene expression data, the lower OCR/ECAR ratio and the diminished ROS levels suggest that hMSC grown at 3% O2 have a bioenergetic balance that is less dependent on OXPHOS. This notion is consistent with previous studies that showed that HIF-1 decreases OXPHOS system activity (the principal endogenous inductor of ROS) by downregulating mitochondrial ATP production and oxygen consumption.13
Our findings indicate that growth of hMSC at physiological oxygen tension helps maintain the bioenergetic balance between OXPHOS and glycolysis that is disturbed at 20% O2. We propose that the reduction of mitochondrial respiration at 3% O2 decreases ROS levels, thereby decreasing the incidence of DSB and limiting telomere erosion and aneuploidy. These results indicate a plausible link between bioenergetic pathways, the control of genomic stability and the lifespan of human primary stem cells in culture. Measures to reduce oxidative stress in clinical stem cell expansion procedures, either by culture at lower O2 concentrations or other interventions that decrease ROS levels, will enhance their growth, viability and biosafety.
Materials and Methods
Cell and culture conditions
Four independent lines of hMSCs (listed 34, 36, 40 and 43), isolated from adipose tissue of female donors aged 35–41 years, were acquired from Inbiobank Stem Cell Bank (www.inbiobank.org). hMSC were cultured (1–2 × 103 cells/cm2) in high glucose Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Sigma), glutamine (2 mM), penicillin 100 U/ml and streptomycin 1000 U/ml. Media were changed twice a week and cells were passaged once a week. For acute exposures to chemical oxidative stress, subconfluent cultures (70%) were exposed to 40 μM PQ (Sigma-Aldrich, St. Louis, MO, USA) every 3 days for 10 weeks, or to a single dose of 100 μM H2O2 (Foret SA, Barcelona, Spain) and were monitored during 4 weeks. Long-term cell growth was monitored by counting cell number with a hemocytometer. Cumulative PD was calculated with the formula PD=(log (Nn/Nn−1))/log 2 (n: passage; N: cell number).
Cell differentiation assays
hMSC were seeded at 2 × 104 cells/cm2 in expansion medium, and after 24-h medium was replaced with the corresponding induction medium. Osteogenic medium contained expansion medium supplemented with 10 mM β-glycerolphosphate, 0.1 μM dexamethasone and 0.2 mM ascorbic acid. Adipogenic medium contained expansion medium supplemented with 0.01 μM dexamethasone, 0.5 mM IBMX (3-isobutyl-1-methyl xanthine) and 60 μM indomethacin. In all cases, induction medium was replaced every 3–4 days and on day 21 cells were processed for staining analysis. For osteogenic assays, cells were fixed with 70% ethanol (1 h, 4 °C) and stained with 40 mM Alizarin Red, pH 4.1 (15 min, RT). Cells cultured in adipogenic medium were fixed with 10% formalin (30 min, RT) and stained with 2% Oil Red (15 min, RT).
Cell characterization by surface marker staining
hMSC at early passage were collected by trypsin treatment and resuspended in PBS at 1.0 × 106 cells/ml. The cells were blocked for 20 min with 1% (v/v) human serum (Thermo Scientific, Waltham, MA, USA). Subsequently, the cells were incubated with surface marker-specific antibodies and isotype-matched controls in the dark at 4 °C for 30 min. Surface marker antibodies used were CD29 mIgG2a-PE, CD90 mIgG1-FITC, CD19 mIgG1-FITC, CD45 mIgG1-FITC, CD44 rIgG2b-PE (BD Biosciences, San Jose, CA, USA), CD105 mIgG1-FITC, CD11b mIgG1-PE, HLADR mIgG3-FITC (AbD Serotec, Oxford, UK) and their specific isotype-matched controls mIgG2a-PE, rIgG2b-PE, mIgG1-FITC, mIgG1-PE, mIgG3-FITC (BD Biosciences). The cells were washed twice with PBS and analyzed by flow cytometry in a BDFACS Canto flow cytometer (BD Biosciences), and data were analyzed using Summit v4.3 software (Dako Inc., Carpinteria, CA, USA).
Aneuploidy analysis
Cells were incubated with 10 μg/ml colcemid for 4 h at 37 °C, and then treated with 0.56% KCl for 15 min at 37 °C and fixed in metanol:acetic acid (3 : 1). Cell suspensions were dropped onto clean slides and air-dried for 24 h. Centromere Evaluation Probe for chromosomes 8, 11 and 17, from the Breast Aneusomy Multi-Color Probe kit (Abbott Laboratories, Libertyville, IL, USA), were applied (in 3 μl) to the cells according to the manufacturer's instructions. At least 100 cells per line were analyzed for each time-point and oxygen concentration. For chromosome analysis, metaphase cells were dropped onto clean wet slides, aged overnight and stained with DAPI (4,6 diamidino-2-phenylindole) in Vectashield H-1200 mounting medium (Vector Laboratories, Burlingame, CA, USA). Chromosome counts were performed on at least 100 metaphases per oxygen concentration. Fluorescence images were acquired with a Nikon 90i microscope (Nikon Instruments, Melville, NY, USA) fitted with a 100 × planfluor 1.3 N/A objective, appropriate filters, and an Hg Intensilight fluorescence unit. Digital images were acquired with Cytovision Genus software (Genetix, Boston, MA, USA) coupled to a JAI monochrome CCD cooled camera (Kushima City, Miyazaki, Japan).
Telomere length quantification by Q-FISH
Q-FISH was carried out using a Cy3-labeled LL(CCCTAA)3 PNA telomeric probe (Eurogentec, Liège, Belgium) as described33 with the following modifications. After hybridization, slides were washed three times with PBS-0.1% Tween for 10 min at 60 °C and dehydrated through an ethanol series (70, 90 and 100% 5 min each). Slides were then counterstained and mounted in Vectashield H-1200 mounting medium. Digital images were acquired as described above. Telomere signals were captured with the same exposure time in all samples. Telomere length (in kb) was extrapolated from the fluorescence of hTert-immortalized 82-6 fibroblasts expressing either TIN2 or TIN2-13 proteins with known and stable telomere lengths (3.4 and 8.4 kb, respectively; see Rubio et al.34), kindly provided by Dr. Judith Campisi. Telomere signals from at least 20–30 nuclei per group were quantified using TFL-Telo (version 2) (Vancouver, BC, Canada), kindly provided by Dr. Peter Lansdorp (British Columbia Cancer Centre, Vancouver). All images were captured and analyzed in parallel on the same day by an experimenter blinded to the treatment groups.
Chromosomal aberrations
FISH was carried out as described above, using a Cy3-labeled LL(CCCTAA)3 PNA telomeric probe and a FITC-labeled LL(ATTCGTTGGAAACGGGA) PNA alpha satellite probe (Eurogentec). Structural chromosomal aberrations were detected by superimposing telomere and centromere images on the DAPI stained chromosomes in imageJ program supported by National Institutes of Health Image (Bethesda, MD, USA). At least 20 metaphases of each cell line and condition were analyzed. Chromosomal aberrations were identified as follows: chromatid or chromosomal breakages: gaps in one or two chromatids whose corresponding centromere was identified; chromosomal fusions: two chromosomes joined without telomere signals at the fusion point with one centromeric signal; dicentrics: chromosomal fusions between two chromosomes joined without telomere signals at the fusion point with two centromeric signals. The percentage of each type of aberration in each cell line and condition was used for statistical analysis.
TRAP assay
Telomerase assays were performed on 5000 hMSC as described,16 with the following modifications. In all, 2 μl of the extension reaction was added to 23 μl PCR reaction mix containing 1 × PCR Master Mix power SYBR Green, 5 mM EGTA, 4 ng/μl Oligo ACX and 2 ng/μl Oligo TS. PCR was carried out over 40 cycles of 94 °C for 10 min; 94 °C for 15 s and 60 °C for 1 min. PCR products were quantified with an ABI PRISM 7900 quantitative PCR apparatus (Applied Biosystems, Foster City, CA, USA) and analyzed with SDS v2.3 software (Applied Biosystems).
Transduction with hTert lentiviral vector
Primary hMSC at passage 5 in 20% O2 were infected with lentivirus (pRRL.SIN18) encoding the human telomerase reverse transcriptase catalytic subunit (hTert), as described.34 The hTert lentivirus was a generous gift from Dr. Judith Campisi (Buck Institute for Age Research, Novato, CA, USA).
Immunohistochemistry
DNA damage was evaluated by immunofluorescence staining for 53BP1. For this, sham-treated or -irradiated cells (10 Gy Gamma irradiation) were fixed in acetone at 4 °C for 10 min, permeabilized in 0.5% Tx-100, blocked in 10% horse serum, and stained overnight with a 1 : 500 dilution of IgM-mouse antibody against 53BP1 (clone 05725, Cell Signaling Technology, Danvers, MA, USA) in 1% horse serum in PBS-1%Tween; 1% horse serum in PBS-1%Tween was used as a negative control. Stain was developed with an Alexa 546-conjugated goat antibody, and nuclei were counterstained with DAPI. Quantification of positive cells for 53BP1 foci was performed by fluorescence microscopy at × 40 magnification. At least 100 nuclei were imaged for each cell line and culture condition in randomly selected fields using Cytovision Genus software (Newcastle Upon Tyne, UK).
Flow cytometry detection of ROS
Early passage (p5) hMSC cultures were washed in PBS, trypsinized and incubated in phenol-red-free, reduced-serum OptiMEM medium containing 5 μM DHE (Molecular Probes, Carlsbad, CA, USA) for 30 min in the dark. ROS were detected by flow cytometry in a BDFACS Canto flow cytometer (BD Biosciences), and data were analyzed using Summit v4.3 software (Dako Inc.).
Microarray gene expression profiling
Sample labeling and microarray hybridization
The One-Color Microarray-Based Gene Expression Analysis Protocol (Agilent Technologies, Palo Alto, CA, USA) was used to amplify and label RNA. Briefly, 400 ng of total RNA was reverse transcribed using T7 promoter primer and MMLV-RT. cDNA was then converted to aRNA using T7 RNA polymerase, which simultaneously amplifies target material and incorporates cyanine 3-labeled CTP. Cy3-labeled aRNA (1.65 μg) was hybridized to a Whole Human Genome Microarray 4 × 44K (G4112F, Agilent Technologies) for 17 h at 65 °C in 1 × GEx Hybridization Buffer HI-RPM in a hybridization oven (G2545A, Agilent Technologies) set to 10 r.p.m. Arrays were washed according to the manufacturer's instructions, dried by centrifugation, and scanned at 5 mm resolution on an Agilent DNA Microarray Scanner (G2565BA, Agilent Technologies), with the default settings for 4 × 44 K format one-color arrays. Scanned images were analyzed with Feature Extraction software (Agilent Technologies).
Data processing
Data were read into R and processed using the Agi4x44PreProcess Bioconductor package as follows. Agi4x44PreProcess options were set to use the MeanSignal and the BGMedianSignal as foreground and background signals, respectively. Data were then background corrected by the half, which produces a positive background-corrected signal by subtracting the background signal from the foreground signal, assigning intensities below 0.5 to 0.5 to produce positive corrected intensities. Data were normalized between arrays by the quantile method.35 A constant value of 50 was added to the intensities before the log transformation in order to reduce the signal variability for genes expressed at low intensity. The AFE image analysis software attaches to each feature a set of flags that identify different quantification properties of the signal. Agi4x44PreProcess uses these flags to filter out features that (1) are controls (2) are out of the dynamic range of the scanner and (3) are outliers. To keep features within the dynamic range three independent levels of filtering can be performed to ensure that (1) the signal is distinguishable from the background, (2) the signal is found and (3) that the signal is not saturated. In each filtering step, we required that more than the 75% of the replicates of each feature in at least one experimental condition had a quantification flag denoting that the signal was within the dynamic range. In addition, for each replicated feature across the whole set of samples, we filtered out those probes for which >25% of the replicates in at least one experimental condition were flagged to indicate the presence of outliers. After the completion of all pre-processing steps, there were 26 670 (T20 versus T3 data set) features available for the statistical analysis. Finally, Agi4x44PreProcess maps each Agilent probe identifier to its corresponding accession number, gene symbol, gene description and GO identifiers (The Gene Ontology Consortium) using the Bioconductor annotation hgug4112a.db.
Statistical analysis
Differential expression analysis was done using the linear modeling features implemented in the Bioconductor limma package, which incorporates empirical Bayes methods36 to obtain moderated statistics. To estimate differential expression between the different experimental conditions in the two data sets (T20 versus T3 and P21 versus P2), the following linear model was fitted to each gene:
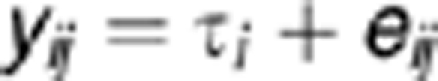 |
where yij is the observation of the ith treatment for individual j, τij is the effect of the ith treatment and eij is the experimental error, assumed to be normally distributed with 0 mean and variance σe2. Genes differentially expressed in the two oxygen concentrations were identified by testing the null hypothesis (no differences in signal) for each gene τi.
To reduce the number of genes used for multiple testing correction without missing relevant information, genes that showed either constant expression between samples (IQR<0.50) or a low expression signal (log2 expression <5 in all samples) were removed using a nonspecific filter in the Bioconductor genefilter function. As the eBayes function estimates the average variability of genes on the microarray, this nonspecific filtering was done after the eBayes correction. Multiple comparisons of genes were then taken into account by controlling for the false discovery rate, which was estimated in terms of the q-value statistic37 using the Bioconductor qvalue package.
To integrate significant expression profiles into functional categories we performed a GO-based statistical analysis using the hyperGTest function of the GOstats package.38 hyperGTest computes hypergeometric P-values to test for over-representation and under-representation of each GO term in a given subset of genes compared with its distribution in a defined universe of genes. The universe included all genes with an above-background signal and that had a known GO term in the corresponding database. Duplicated genes were removed before GO analysis.
TaqMan assays
hMSC RNA was extracted with TRIzol by standard methods. The RT reaction was carried out with the SuperScript III Reverse Transcriptase kit (Invitrogen, Carlsbad, CA, USA). In all, 10 ng of cDNA per reaction was added to 10 μl of 2 × PCR Master Mix (Applied Biosystems). TaqMan Gene Expression Assays (Applied Biosystems) were used for all mRNA quantifications.
Detection of protein carbonyls and MDA
Carbonyl derivatives in hMSC were measured by an adaptation of the method of Levine.39 Cell suspensions were mixed with 30% (v/v) trichloroacetic acid. Protein precipitates were resuspended in 10 mM 2,4-dinitrophenylhydrazine (DNPH) and incubated for 60 min at 37 °C. Samples were then precipitated with 20% trichloroacetic acid, and centrifuged for 10 min at 1000 × g at 4 °C. The precipitate was washed twice with ethanol-ethyl acetate (50% v/v) to remove free DNPH, and resuspended in 6 M guanidine, 2 mM phosphate buffer, and pH 2.3. Samples were incubated for 40 min at 37 °C and centrifuged for 5 min at 3000 × g at 4 °C. The levels of protein carbonyls, expressed as μmols/103 cells, were calculated from the absorbance of supernatants at 360 nm. MDA was detected by HPLC as described by Wong et al.,40 and results expressed as nmols/103 cells.
OCR and ECAR measurement
The XF96 Flux analyzer and Prep Station (Seahorse Bioscience XF96 Instrument, North Billerica, MA, USA) were used according to the manufacturer's instructions. Briefly, hMSC were seeded in XF96 cell culture plates at 2500 cells per well at 20% O2 or 3% O2 and cultured for 48 h. The XF96 sensor cartridges were hydrated overnight with 200 μl of Seahorse Bioscience XF96 Calibrant at ph 7.4 and stored at 37 °C without CO2 for 24 h. The culture medium was replaced with serum-free high glucose DMEM supplemented with glutamine and pen/strep and lacking bicarbonate (pH 7.4). Cells were then incubated for 15 min at 37 °C without CO2 and measurements were performed. Drugs (1 μM oligomicin and 130 μM 2,4 dinitrophenol) were added according to the supplier's technical specifications.
Lactate measurement
Cellular lactate was measured with the Lactate Colorimetric Assay Kit (Abcam, Cambridge, MA, USA). Lactate concentration was expressed relative to cell number.
Acknowledgments
We are indebted to Dr. Judith Campisi (Buck Institute for Age Research) for providing the telomere length control cells and the hTert lentiviral vector, to Jose Manuel Ligos and the Cytometry Unit (CNIC) for advice on FACS analysis, Antonio Diez-Juan and Kenneth McCreath for critical reading of the paper, Marta Ramón (CNIC) for secretarial assistance and Simon Bartlett (CNIC) for editorial support. This work was supported by grants to AB from the Ministry of Science and Innovation (SAF 2008-02099; PLE2009-0147 and PSE-010000-2009-3), the Comunidad Autónoma de Madrid (P-BIO-0306-2006) and the Red de Terapia Celular del Instituto de Salud Carlos III (TerCel); to ES from the Fundación Mutua Madrileña, the Ministry of Education (Ramon y Cajal program), and the Ministry of Health (FIS PI071023) to JAE from the Ministry of Science and Innovation (SAF 2009-08007 and CSD2007-00020). JCE is currently a predoctoral fellow funded by TerCel. The CNIC is supported by the Spanish Ministry of Science and Innovation and the Pro-CNIC Foundation.
Glossary
- ECAR
extra cellular acidification rate
- FDR
false discovery rate
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- TRAP
telomere repeat amplification protocol
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by R De Maria
Supplementary Material
References
- Siegel G, Schafer R, Dazzi F. The immunosuppressive properties of mesenchymal stem cells. Transplantation. 2009;87 (9 Suppl:S45–S49. doi: 10.1097/TP.0b013e3181a285b0. [DOI] [PubMed] [Google Scholar]
- Miyahara Y, Nagaya N, Kataoka M, Yanagawa B, Tanaka K, Hao H, et al. Monolayered mesenchymal stem cells repair scarred myocardium after myocardial infarction. Nat Med. 2006;12:459–465. doi: 10.1038/nm1391. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Okunieff P, Neuringer LJ. Blood flow, tissue oxygenation, pH distribution, and energy metabolism of murine mammary adenocarcinomas during growth. Adv Exp Med Biol. 1989;248:835–845. doi: 10.1007/978-1-4684-5643-1_95. [DOI] [PubMed] [Google Scholar]
- Fischer B, Bavister BD. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil. 1993;99:673–679. doi: 10.1530/jrf.0.0990673. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci USA. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busuttil RA, Rubio M, Dolle ME, Campisi J, Vijg J. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2003;2:287–294. doi: 10.1046/j.1474-9728.2003.00066.x. [DOI] [PubMed] [Google Scholar]
- Fehrer C, Brunauer R, Laschober G, Unterluggauer H, Reitinger S, Kloss F, et al. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell. 2007;6:745–757. doi: 10.1111/j.1474-9726.2007.00336.x. [DOI] [PubMed] [Google Scholar]
- Li TS, Marban E. Physiological levels of reactive oxygen species are required to maintain genomic stability in stem cells. Stem Cells. 2010;28:1178–1185. doi: 10.1002/stem.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth NR, Musio A, Vezzoni P, Simpson AH, Noble BS, McWhir J. Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells. 2006;8:16–23. doi: 10.1089/clo.2006.8.16. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004;18:2183–2194. doi: 10.1101/gad.1243304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, et al. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685–691. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- Herbert BS, Hochreiter AE, Wright WE, Shay JW. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat Protoc. 2006;1:1583–1590. doi: 10.1038/nprot.2006.239. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Pilger R, Sitte N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med. 2000;28:64–74. doi: 10.1016/s0891-5849(99)00207-5. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein CJ. Cell size, nuclear content, and the development of polyploidy in the Mammalian liver. Proc Natl Acad Sci USA. 1967;57:327–334. doi: 10.1073/pnas.57.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa D, Hashimoto N, Hoshima M, Yamaguchi T, Sawai K, Nakamura Y, et al. Analysis of numerical aberrations of specific chromosomes by fluorescent in situ hybridization as a diagnostic tool in breast cancer. Cancer. 1996;77:2064–2069. doi: 10.1002/(SICI)1097-0142(19960515)77:10<2064::AID-CNCR15>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Persons DL, Robinson RA, Hsu PH, Seelig SA, Borell TJ, Hartmann LC, et al. Chromosome-specific aneusomy in carcinoma of the breast. Clin Cancer Res. 1996;2:883–888. [PubMed] [Google Scholar]
- Mukherjee AB, Costello C. Aneuploidy analysis in fibroblasts of human premature aging syndromes by FISH during in vitro cellular aging. Mech Ageing Dev. 1998;103:209–222. doi: 10.1016/s0047-6374(98)00041-4. [DOI] [PubMed] [Google Scholar]
- Mukherjee AB, Alejandro J, Payne S, Thomas S. Age-related aneuploidy analysis of human blood cells in vivo by fluorescence in situ hybridization (FISH) Mech Ageing Dev. 1996;90:145–156. doi: 10.1016/0047-6374(96)01762-9. [DOI] [PubMed] [Google Scholar]
- Geigl JB, Langer S, Barwisch S, Pfleghaar K, Lederer G, Speicher MR. Analysis of gene expression patterns and chromosomal changes associated with aging. Cancer Res. 2004;64:8550–8557. doi: 10.1158/0008-5472.CAN-04-2151. [DOI] [PubMed] [Google Scholar]
- Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nat Rev Drug Discov. 2003;2:803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Saretzki G, Docke W, Lotze C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence. Exp Cell Res. 1995;220:186–193. doi: 10.1006/excr.1995.1305. [DOI] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- Samper E, Flores JM, Blasco MA. Restoration of telomerase activity rescues chromosomal instability and premature aging in Terc−/− mice with short telomeres. EMBO Rep. 2001;2:800–807. doi: 10.1093/embo-reports/kve174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MA, Kim SH, Campisi J. Reversible manipulation of telomerase expression and telomere length. Implications for the ionizing radiation response and replicative senescence of human cells. J Biol Chem. 2002;277:28609–28617. doi: 10.1074/jbc.M203747200. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- Wong SH, Knight JA, Hopfer SM, Zaharia O, Leach CN, Jr, Sunderman FW., Jr Lipoperoxides in plasma as measured by liquid-chromatographic separation of malondialdehyde-thiobarbituric acid adduct. Clin Chem. 1987;33 (2 Part 1:214–220. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.