

Abstract
Bone morphogenetic protein 1 (BMP1) is an astacin metalloprotease with important cellular functions and diverse substrates, including extracellular-matrix proteins and antagonists of some TGFβ superfamily members. Combining whole-exome sequencing and filtering for homozygous stretches of identified variants, we found a homozygous causative BMP1 mutation, c.34G>C, in a consanguineous family affected by increased bone mineral density and multiple recurrent fractures. The mutation is located within the BMP1 signal peptide and leads to impaired secretion and an alteration in posttranslational modification. We also characterize a zebrafish bone mutant harboring lesions in bmp1a, demonstrating conservation of BMP1 function in osteogenesis across species. Genetic, biochemical, and histological analyses of this mutant and a comparison to a second, similar locus reveal that Bmp1a is critically required for mature-collagen generation, downstream of osteoblast maturation, in bone. We thus define the molecular and cellular bases of BMP1-dependent osteogenesis and show the importance of this protein for bone formation and stability.
Introduction
Osteogenesis imperfecta (OI), also known as “brittle-bone disease” is a rare genetic collagenopathy primarily characterized by dramatically increased bone fragility causing susceptibility to numerous fractures.1,2 Individuals often show distinctive features including reduced bone mass, short stature, blue sclerae, and/or dentinogenesis imperfecta. The severity of this disorder varies from profound forms with intrauterine fractures and perinatal lethality to milder phenotypic expression such as rare fractures or even no fractures.3,4 Most OI cases are inherited in an autosomal-dominant manner and are caused by mutations in COL1A1 (MIM 120150) and COL1A2 (MIM 120160);5,6 these two genes encode for the two α chains of collagen type I, the predominant protein component of the bone matrix. For a minority of OI individuals, an autosomal-recessive inheritance is described as including underlying mutations in CRTAP7 (MIM 605497), LEPRE18 (MIM 610339), SERPINH19 (MIM 600943), PPIB10 (MIM 123841), SP711 (MIM 606633), SERPINF112 (MIM 172860), and FKBP1013 (MIM 607063). Mutations in COL1A1 or COL1A2 result in primary structural or quantitative defects of collagen I molecules, whereas genetic mutations causative for recessive forms mainly lead to defects in collagen I biosynthesis. Defects in collagen I have been associated with reduced bone mineralization and bone fragility not only in individuals suffering from congenital OI but also in elderly individuals who have developed osteoporosis.14
Type I collagen belongs to the fibril-forming collagens and is composed of a triple helix consisting of two α1(I) chains and one α2(I) chain. The α chains are synthesized at the rough endoplasmic reticulum (ER). They are highly post-translationally modified in the ER lumen and are subsequently assembled to a triple helix. This premature collagen helix, which contains globular appendages at the amino (N-) and carboxyl (C-) ends, is then transported through the trans-golgi network to the extracellular matrix (ECM), where N- and C-proteinases catalyze proteolytic cleavage of these propeptides. After this final processing step, the released mature collagen I triple-helical monomer can be assembled into highly ordered collagen fibrils.15,16
Bone morphogenetic protein 1 (BMP1) is an astacin metalloprotease,17,18 the physiological function of which has been of considerable interest for some time. This protein has been suggested to play essential roles in osteogenesis and ECM formation; it has also been described as exerting influence over dorsal-ventral patterning through the indirect activation of some TGFβ-like proteins.19–21 Of particular interest has been the role of BMP1 in proteolytic removal of the C-propeptides from procollagen precursors of the major fibrillar collagen types I–III. This processing is essential for the self-assembly of mature collagen monomers into fibrils.22 The precise functional requirement of BMP1 in vivo is unclear. To date, Bmp1 loss of function has only been analyzed in a knock-out mouse, in which it was found to be lethal around birth and for which no detailed analysis of osteogenesis was presented.23 Thus, the role of BMP1 in bone formation and organogenesis remains obscure.
Here, we describe two siblings with a high-bone-density form of OI, identify a causative mutation in BMP1, and provide detailed analysis of the effect of the mutation on BMP1 modification and secretion. We show that the hypofunctional nature of the mutation is demonstrated in vivo by using two assays that test substrate cleavage in zebrafish. We further describe a zebrafish Bmp1a mutant with skeletal defects comparable to those seen in individuals with OI, demonstrating conservation of important BMP1 function in osteogenesis across species. Our analysis of these mutants has demonstrated that loss of Bmp1a affects neither osteoblast formation nor activity but rather the ability to generate mature collagen fibrils. This finding therefore indicates that BMP1 is very much required in the process of bone formation.
Material and Methods
Whole-Exome Sequencing
Genomic DNA was enriched from exonic and adjacent splice-site sequences by the use of the Agilent SureSelect Human Exome Kit and run on the Illumina Genome Analyzer IIX Sequencer. Further data analysis was performed with an in-house bioinformatics pipeline in combination with SAMTOOLS v.0.1.7 for SNP and indel detection. In-house-developed scripts were applied for the detection of protein changes, splice-site affections, and overlaps with known variations (Ensembl build 61 and 1,000 Genomes Project release 2010_3).
Mutation Screening
The identified mutation was resequenced in an independent experiment, tested for cosegregation with the phenotype within the family, and then screened in 300 healthy control individuals from Turkey by PCR and restriction digestion (AvaI; Fermentas, St. Leon-Rot, Germany). All subjects or their legal representatives gave written informed consent for the study. The study was performed in accordance with the Declaration of Helsinki protocols and was approved by the local institutional review boards.
Generation of BMP1 Constructs
The BMP1-FLAG pcDNA3.1 construct contained full-length human cDNA of BMP1 (RefSeq accession number NM_001199.3) and was fused to a C-terminal FLAG tag; it was provided by the group of Karl E. Kadler (University of Manchester, UK) and was used for BMP1 expression studies. The identified substitution, p.Gly12Arg, was introduced by site-directed PCR mutagenesis with the use of a primer containing the specific nucleotide substitution.
Cell Culture and Transient Transfection
Human embryonic kidney (HEK) 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS, GIBCO) and antibiotics. Cells were transiently transfected with Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) and vectors containing wild-type (WT) and mutant variants of BMP1 cDNA. Transfections were performed according to the manufacturer's instructions.
Secretion Assay and Immunoblot Analysis
30 hr after transient transfection, cells and untransfected control cells were maintained in serum-free medium for 18 hr at 37°C. After starvation, supernatant-containing secreted proteins were precipitated by trichloroacetic acid, and cells were lysed with ice-cold lysis buffer. The total protein concentration of the extracts was determined by the BCA (bicinchoninic acid) Protein Assay Kit (Pierce Protein Research Products, Thermo Fischer Scientific, Rockford, IL, USA), and proteins were separated by gradient (4%–12%) SDS-PAGE (Invitrogen) under reducing conditions and transferred to a nitrocellulose membrane by immunoblotting. Immunoblots were blocked in 5% milk powder in TBS containing 0.1% Tween20, and they were probed with Flag antibody (Agilent Technologies, Waldbronn, Germany). Equal protein amounts were confirmed by β-actin detection in whole-cell lysates or by Coomassie staining of a ∼65 kDa protein in supernatant of serum-free medium. A peroxidase-conjugated secondary antibody (goat anti-mouse) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and blots were developed with an enhanced chemiluminescence system, ECL Plus (Amersham, UK); exposure on autoradiographic film (GE Healthcare, München, Germany) followed.
Zebrafish Studies
Radioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% NP-40) was used for the extraction of total protein from zebrafish larvae (6 days old) or fins (4 months old), and the protein was measured with a standard Bradford assay. Proteins (20 μg) were separated on a 6% denaturing polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with a rabbit polyclonal antibody raised against a peptide of zebrafish Collagen1a1a. Goat anti-rabbit antibody conjugated with horseradish peroxidase (HRP) was employed as a secondary antibody, and the bands were visualized with chemiluminescence detection (Millipore, Billerica, MA, USA). For the loading control, blots were stripped and then reprobed with a rabbit anti-β-actin antibody (Cell Signaling Technology, Danvers, MA, USA).
N-glycosidase Assay
For the N-glycosylation studies, 20 μg of whole-cell lysates of transiently transfected and untransfected HEK 293T cells were either treated with N-glycosidase F (PNGaseF) (New England BioLabs, Frankfurt, Germany) or left untreated. The enzyme reaction was performed according to the manufacturer's instructions. Proteins were subjected to SDS-PAGE and analyzed by anti-Flag immunoblotting. Equal protein amounts were confirmed by β-actin detection.
Fish Lines and Mapping
Embryos were obtained by natural crosses and were staged as per Parichy et al.24 The microwaved (med tt281), dino (chd tt250), and frilly fins (frf tm317a, frf tf5, frf tp34, and frf ty68) alleles that we used have been described previously25 and were isolated in an N-ethyl-N-nitrosourea (ENU) screen in Freiburg (frf fr24). frf tm317 was used for all analyses. The Tg(sp7:mCherry) transgenic line has been reported previously.26 We performed genetic mapping by crossing frf+/− and med+/− fish to the WIK strain and by subjecting the F2 progeny to simple-sequence-length polymorphism (SSLP) meiotic mapping as outlined by Geisler.27 Heat shocks were performed at 37°C for 1 hr at 30 and 56 hr postfertilization (hpf).
Microscopy
Fluorescent images were taken on an Olympus Fluoview confocal microscope, whereas brightfield and Nomarski micrographs were taken on a Zeiss Axioimager or a Leica MZ16FA. For live imaging, larvae were anesthetized in Tricaine and mounted in 3% methyl cellulose or 1% low-melting-point agarose. All whole-mount stainings using alizarin red, in situ hybridization, or immunodetection were cleared in glycerol prior to mounting. Stained ultrathin sections of the fins were employed for transmission electron microscopy (TEM) on a Jeol JEM-1010 electron microscope.
RNA and DNA Isolation, cDNA Synthesis, and Sequencing
Trizol (Invitrogen, CA, USA) was used for the isolation of RNA from WT or mutant larvae, and SuperscriptIII Reverse Transcriptase (Invitrogen, CA, USA) was used for the generation of cDNA by reverse transcription. RT-PCR was used for the amplification of bmp1a cDNA from all frilly fins mutated alleles and corresponding siblings. Similarly, col1a1a cDNA was amplified from the microwaved mutant allele and siblings. Resulting PCR fragments were purified and sequenced directly. To identify mutations at the genomic level, we extracted larval genomic DNA and directly sequenced the region of interest from amplified PCR products.
DNA Construct Generation
Gateway cloning technology and entry constructs from the Tol2Kit28 were used for the generation of zebrafish and Myc-tagged human Bmp1 heat-shock constructs. Bmp1 coding regions were cloned into a middle entry vector via standard cloning methods or through a BP Gateway reaction.
DNA and RNA Injections
DNA and RNAs were diluted in Danieau buffer and Phenol red before being injected into 1-cell embryos by a Pico-Injector (Harvard Apparatus, MA, USA). Sense RNAs for overexpression or rescue were transcribed from cDNAs and cloned into pCS2+. Plasmids were linearized with NotI, and capped mRNA was synthesized with mMessage mMachine SP6 kit (Ambion, Applied Biosystems, Austin, TX, USA). Full-length cDNA for BMP1 was amplified from a HeLa-cell cDNA library and cloned into pCS2+. A primer harboring the p.Gly12Arg signal-peptide substitution was used for the generation of the mutant BMP1 cDNA version by PCR. chordin RNA was transcribed as previously reported.29 chordin and BMP1 RNA were injected at a concentration of 60 and 450 pg, respectively. To generate adult dino mutants, we rescued early patterning defects by injecting 30 pg of chordin RNA. Tol2 RNA was generated as published.30
In Situ Hybridization
Single- and double-stranded-RNA in situ hybridizations were performed and developed with either chromogenic substrate31 or fluorescent tyramide signal amplification.32 Probes for sp7, osteopontin, and collagen10a1 were synthesized as described.33 The bmp1a probe was generated with EcoRI by linearizing clone IMAGp998H0417161 from ImaGenes (Berlin, Germany) and was transcribed with T7 Polymerase. In situ hybridization of 8 dpf (days postfertilization) larvae required extended 40 min proteinase K (15 μg/ml) digestions at room temperature, 48 hr hybridization in the antisense probe, and 2 days of signal development at room temperature.
Skeletal and Matrix Staining
Bones of larvae and adult fish were stained with alizarin red alone or in combination with the cartilage stain, alcian blue, as described by Walker et al.34 and Spoorendonk et al.26 For microscopic analysis of fibrillar collagen organization, fins and larvae were fixed in 4% paraformaldehyde and embedded in 1% agarose for cryosectioning. Sections were stained with picrosirius red as previously described35 and were visualized by birefringence under polarization filters.
Antibody Staining
Myc-tagged BMP1 proteins were detected by whole-mount immunofluorescent staining with the 9E10 monoclonal antibody (Santa Cruz, CA, USA). 3 dpf embryos were fixed in 4% paraformaldehyde overnight at 4°C and were then washed with 0.1% PBS Triton X-100. Embryos were permeabilized by incubation in 100% acetone for 7 min at −20°C, were rewashed, and were blocked overnight in PBS/Triton with 0.5% goat serum and 0.1% dimethyl sulfoxide. After extensive washing, the embryos were incubated with Alexa488-conjugated secondary antibodies (Invitrogen) diluted in blocking solution. Finally, embryos were rewashed before they were cleared in glycerol. Osteoblasts of the adult fins were stained with the zns5 monoclonal antibody (obtained from the Zebrafish International Resource Center [Eugene, OR, USA]) either in the whole mount or after cryosectioning. They were then counterstained with either alizarin red (for the whole mounts) or DAPI (for the cryosections) as per previous methods.36
Retinoic-Acid Treatment
We purchased all-trans retinoic acid (RA) from Sigma (MI, USA), and we made a 1 mM stock solution by dissolving it in ethanol. Larvae were treated with 1 μM RA diluted in egg water at 4 dpf. The controls were exposed to an equivalent amount of just the carrier (ethanol). Fresh solution was added every other day until the fish were fixed at 11 dpf for alizarin-red processing.
MicroCT Scanning of Adult Fish
Adult fish were euthanized in Tricaine and scanned immediately at 40 kV, 130 μA with a Siemens Inveon PET-CT (positron emission tomography-computed tomography) scanner. The images were reconstructed and analyzed with Inveon Acquisition Workplace 1.4 and Inveon Research Workplace 3.0, respectively. Bone mineral density was calculated with the density-phantoms standards supplied by the company.
Results
A Homozygous BMP1 Mutation Causes High Bone Mineral Density and Multiple Fractures
We used whole-exome sequencing to identify the causative mutation underlying an autosomal-recessive form of bone fragility in a consanguineous family from Turkey. Both affected individuals (Figures 1A and 1B and Table 1) presented with multiple fractures after minimal trauma occurred in their second year of life. Interestingly, despite recurrent fractures, bone-density measurements showed values high above the normal range in both individuals (Figures 1A and 1B). The male index individual had >15 fractures before bisphosphonate treatment was initiated; this treatment was administered on the basis of the hypothesis that the individuals had an OI-like disease with a high rate of fractures, including vertebral fractures, as a result of impaired bone material with high production rates and high bone turnover. Osteoclastic activities were elevated and measured by deoxypyridinoline excretion. These clinical and biochemical findings were not typical of a classical form of osteopetrosis. After treatment began, we observed increased bone mass, reduced fractures, and improved vertebral structures. When the bisphosphonate treatment was completed, fracture rates again began to increase. Increased bone mass and a reduced fracture rate were also observed in his affected sister while she was undergoing bisphosphonate therapy. High Z scores were seen before and during therapy.
Figure 1.
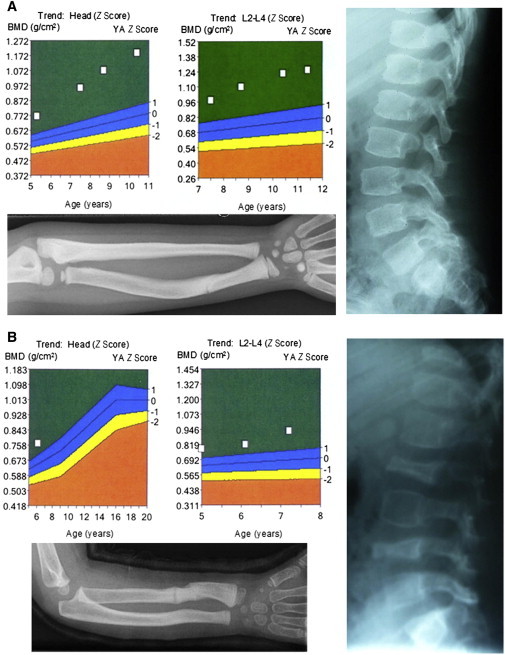
Two Siblings with Autosomal-Recessive Bone Fragility and High-Bone-Mass Phenotype
(A and B) Clinical data of both individuals are shown. Above, diagrams illustrate Z scores of bone-mineral-density measurements of the head and vertebrae L2–L4, respectively, indicating highly increased levels of bone mineral density. X-rays show fractured and bent forearms (below) and spinal columns (right) of individuals. High-radiation X-rays of the forearm and spinal column of individual 1 indicate an intense bone density. Vertebrae of individual 2 are flattened and irregularly formed (B).
Table 1.
Clinical Features of Both Individuals
| Findings | Individual IV:1 | Individual IV:2 |
|---|---|---|
| Age at first visit (years) | 5.0 | 1.9 |
| Age at last visit (years) | 11.4 | 7.5 |
| Age at start of bisphosphonate treatment (years) | 5.4 | 2.8 |
| Age at end of bisphosphonate treatment (years) | 10.4 | 7.5 |
| Birth length and birth weight | normal | normal |
| Confirmed prenatal fractures | none | none |
| Age at first fracture (months) | 23 | 14 |
| Color of sclera | white | white |
| Dentinogenesis imperfecta | no | no |
| Hypermobility of joints | no | no |
| Cardial impairments | none | none |
| Hearing impairment | no | no |
| Old fractures of extremitiesa | yes | yes |
| Vertebral fracturesa | yes | yes |
| Bowing of upper extremitiesa | no | no |
| Bowing of lower extremitiesa | antecurvation of both tibiae | no |
| Shortening of upper extremitiesa | no | no |
| Shortening of lower extremitiesa | no | no |
| Weight at first visit in kg/BMI (SD) | 23.6 (+1.8) | 10.5 (−0.4) |
| Weight at end of bisphosphonate treatment in kg/BMI (SD) | 44.0 (+1.7) | 22.0 (+0.8) |
| Height at first visit in cm (SD) | 112.0 (+0.1) | 82.3 (−0.7) |
| Height at end of bisphosphonate treatment in cm (SD) | 139.0 (−0.6) | 112.2 (−2.6) |
| Retarded gross motor functions | no | no |
| Mobility at first visit (BAMF score) | 8 | 7 |
| Mobility at last visit (BAMF score) | 9 | 9 |
| Intelligence | normal | normal |
| Calcium levela (mmol/l) [range] | 2.43 [2.20–2.65] | 2.28 [2.20–2.65] |
| Alkaline phosphatase at first visit (U/l) [range] | 133 [<269] | 107 [<281] |
| Alkaline phosphatase at last visit (U/l) [range] | 116 [<300] | 114 [<300] |
| Procollagen-1-C-peptidea (marker for osteoblastic activity) (μg/l) [range] | 170 [193–716] | 141 [225–676] |
| Deoxypyridinoline/creatinine (marker for osteoclastic activity) at first visit (nM/mM) [mean ± SD] | 58.66 [16.5 ± 5.0] | 63.9 [19.5 ± 7.2] |
| Deoxypyridinoline/creatinine (marker for osteoclastic activity) at end of bisphosphonate treatment (nM/mM) [mean ± SD] | 17.6 [14.2 ± 5.3] | 29.9 [19.5 ± 7.2] |
The following abbreviations are used: SD, standard deviation; BMI, body mass index; and BAMF, brief assessment of motor function.
At first presentation.
The exome of the proband was enriched by the Agilent SureSelect Human Exome kit and was run on an Illumina Genome Analyzer IIX. Over 90% of the exonic sequences had coverage of at least 20× (Figure 2A), and the mean coverage was 72×. We took advantage of the parental consanguinity (Figure 2B), and we used linkage analysis to determine larger stretches of homozygosity in the exome by using identified variants throughout the exome as haplotype blocks. In addition to filtering variants for their location within the identified stretches of homozygosity, we considered those variants that were not annotated in dbSNP132 or the 1,000 Genomes Database to be possibly causative; this reduced the number of putative variants to three (Table S1, available online). The relevant alteration was the homozygous c.34G>C substitution located in the excellent functional candidate gene, BMP1. Sanger sequencing confirmed that both affected individuals were homozygous for the c.34G>C mutation, whereas both parents were heterozygous. In addition, it was detected neither in 300 healthy Turkish control individuals nor in over 2,400 exomes covering the c.34G>C position (Exome Variant Server, National Heart, Lung, and Blood Institute Exome Sequencing Project [ESP], Seattle, WA; Figure 2C).
Figure 2.
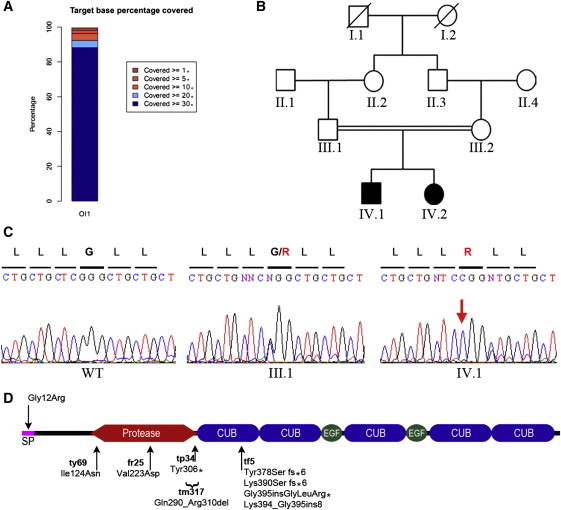
Whole-Exome Sequencing and Filtering Identify Mutation in BMP1
(A) Statistical overview of target-base coverage during sequencing process. Over 90% of identified variations were covered more than 20×.
(B) Pedigree structure of the consanguineous Turkish family.
(C) Sequence chromatograms of the identified c.34G>C BMP1 mutation predicted to substitute the glycine at position 12 with arginine. The c.34G>C mutation was found to be heterozygous (middle panel) in both parents and homozygous in both individuals (right panel).
(D) Schematic view of BMP1 domain structure. The locations of identified mutations in humans (above) and zebrafish (below) are shown. Note that the frf tf5 mutation generates multiple splice isoforms. The following abbreviation is used: SP, signal peptide.
Functional Effects of the p.Gly12Arg Substitution in BMP1
The mutation is predicted to substitute arginine for a conserved glycine residue (p.Gly12Arg) within the signal peptide of BMP1 (Figure 2D); this signal peptide is essential for the protein's localization to the ER, correct posttranslational glycosylation, and secretion.37 Indeed, we found that in contrast to the Flag-tagged WT BMP1 that is transiently expressed in HEK 293T cells, the p.Gly12Arg signal-peptide variant BMP1 showed a drastically reduced secretion capacity (Figure 3A). Moreover, we detected a predominant additional lower-molecular-weight band for mutant BMP1 in immunoblots from cell lysates. Results from N-glycosidase treatment of WT and mutant-BMP1-transfected HEK 293T cells indicated that the lower band in untreated lysates represented a nonglycosylated form of BMP1 (Figure 3B). Interestingly, deficits in BMP1 glycosylation can also negatively impact secretion.37 These data thus indicate that p.Gly12Arg BMP1 is inefficiently secreted and has diminished posttranslational glycosylation, which might contribute to its impaired secretion. To assess whether the amino acid substitution in the signal peptide causes a reduction in extracellular proteolytic activity, we analyzed dorsal-ventral patterning of the zebrafish embryo—a process which is extremely sensitive to levels of the Bmp1 target, Chordin—as an in vivo assay for BMP1 function. Injection of WT BMP1 RNA evoked a mild ventralization of embryos, whereas the mutant RNA had quantitatively reduced activity (Figures 3C–3E and 3I). Exogenous chordin RNA dorsalizes embryos (Figures 3F and 3I), an effect efficiently reversed by WT BMP1 RNA but not by the mutant RNA (Figures 3G–3I). These data demonstrate that the p.Gly12Arg substitution compromises BMP1 activity in vivo and imply that the human enzyme can cleave the targets of its zebrafish counterpart.
Figure 3.
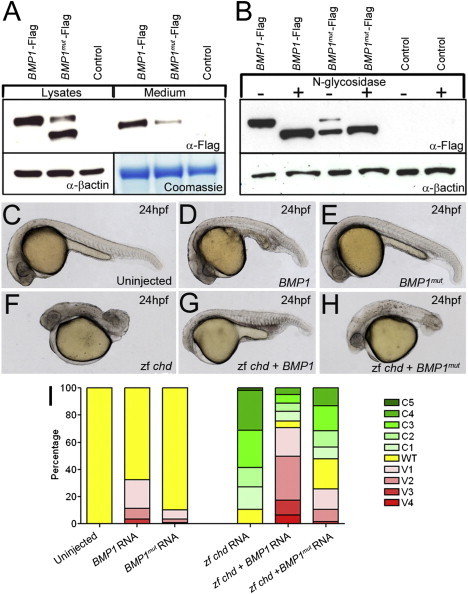
A Signal-Peptide Substitution in BMP1 Causes Secretion and Glycosylation Defects In Vitro and Loss of Protease Activity In Vivo
(A) Immunoblot of HEK 293T cells transfected with either Flag-tagged WT BMP1 (BMP1-Flag; lanes 1 and 4) or p.Gly12Arg-substituted BMP1 (BMP1mut-Flag; lanes 2 and 5) and untransfected control cells (control; lanes 3 and 6). Immunoblotting shows that the p.Gly12Arg protein (lanes 1–3) isolated from cell lysates had increased mobility; reduced amounts of this BMP1 protein were secreted into the medium (lanes 4–6).
(B) Immunoblot of lysates of HEK 293T cells transfected with either Flag-tagged WT BMP1 (BMP1-Flag; lanes 1 and 2) or p.Gly12Arg-substituted BMP1 (BMP1mut-Flag; lanes 3 and 4) and untransfected controls (control; lanes 5 and 6). After being harvested, lysates were either treated with N-glycosidase (lanes 2, 4, and 6) or left untreated (lanes 1, 3, and 5). The predominant mutant-BMP1 band with increased mobility migrates at the same rate as deglycosylated WT BMP1.
(C–I) The p.Gly12Arg-substituted BMP1 exhibits reduced Chordinase activity in vivo. Lateral views of uninjected 24 hpf zebrafish embryos (C) and embryos injected with RNA encoding either WT BMP1 (BMP1; D and G) or p.Gly12Arg signal-peptide variant BMP1 (BMP1mut; E and H). Chordinase activity was assessed by its ability to ventralize WT embryos (C–E) or rescue dorsalized (chordin RNA injected) embryos (F–H). In both assays, the mutant BMP1 showed reduced ability to counteract either the endogenous or exogenous Chordin (quantified in I).
Phenotypic Characterization of the Zebrafish frilly fins Mutant
We analyzed the zebrafish frilly fins (frf−/−) mutant, which was initially described as causing a phenotype25 characterized by a ruffled larval fin (Figures 4A and 4B) as well as a shortened body axis and malformed craniofacial structures and fin shape (Figures 4C and 4D). We observed a striking reduction in ossification of vertebrae from 6 dpf to 11 dpf (Figures 4E and 4F; data not shown). The osteopenia persisted in such a manner that at 15 dpf, the anterior vertebrae had partially ossified but were misshapen (Figures 4I and 4J). The fact that overall length and growth of mutant larvae were not reduced at this stage argues against a general developmental delay (data not shown). By 25 dpf, all frf mutant vertebrae appeared to have ossified (Figures S1E and S1F), but fusions could be seen between some vertebrae (Figures S1G and S1H). Fins appeared hypomorphic at all stages, and delayed development of bony rays (lepidotrichia) appeared at 25 dpf (Figures S1E and S1F). Adult fins had reduced numbers of lepidotrichia, which appeared wavy, underwent limited bifurcation, and were often fused to adjacent rays (Figures 4K and 4L). The presence of calluses is suggestive of spontaneous fracturing during fin outgrowth (Figure 4L). We generated maternal-zygotic mutant embryos from five alleles; the fact that none were dorsalized indicates that Bmp1a is dispensable for dorsal-ventral patterning (data not shown).
Figure 4.
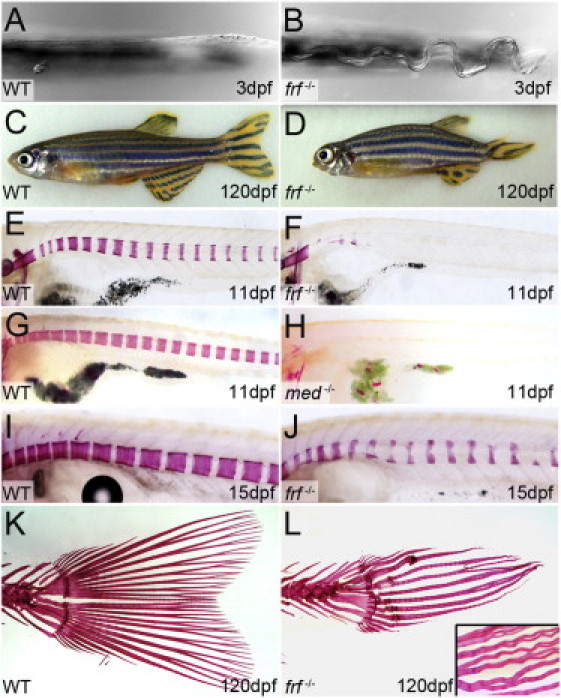
The Zebrafish frilly fins Mutant Displays Larval Fin-Fold Ruffling and Osteogenesis Defects
(A and B) Ventral view of the posterior medial fin fold at 3 dpf in a frilly fins (frf) (B) larva showing undulations in the fin fold, which normally has a linear morphology (A).
(C and D) Compared to the siblings, 4-month-old frf−/− adults (D) are short and display axis defects, body curvature, and fin and craniofacial dysmorphogenesis.
(E–L) Alizarin-red staining of frf−/− (F, J, and L), microwaved (med−/−) (H), and WT siblings (E, G, I, and K) at 11 dpf (E–H), 15 dpf (I and J), and 4 months (K and L). Both frf and med display reduced ossification of the vertebrae (F and H), whereas nascent vertebrae are osteopenic and dysmorphic (J). Tail fins in frf−/− have lost the WT fan shape (K) and display fracture calluses, reduced bifurcations (L), and crinkled lepidotrichia, which often fuse to each other (L; inset).
A second zebrafish fin mutant, microwaved (med), displays a phenotype similar to that of frf mutants—delayed ossification at 11 dpf (Figures 4G and 4H) and undulation of the larval fin (Figures S1I and S1J).25 As with frf mutants, all vertebrae of med mutants eventually ossify (Figures S1A–S1D). To compare the two mutants in more detail, we quantified bone density in the adults by microCT analysis (Figures 5A–5D) and assessed both vertebral and lepidotrichial bone. Both frf and med distal fin rays had reduced bone density when they were compared to their respective siblings, but the bone of the vertebrae had divergent phenotypes. Like the lepidotrichial bone, the vertebrae of med mutants also had reduced bone density; surprisingly, however, frf-mutant adult vertebrae had increased bone density (Figure 5E) similar to that seen in the human individuals with the BMP1 mutation, whereas med mutants displayed traits similar to those seen in individuals with classical OI. Meiotic mapping revealed close linkage between the med locus and zebrafish col1a1a on linkage group 3 (data not shown). Sequencing of col1a1a cDNA (GenBank accession number BC063249.1) from med−/− mutants identified a G>A transition predicted to substitute a highly conserved glutamic acid with a lysine at position 888 (p.Glu888Lys; Figures S1K–S1N). Thus, the microwaved mutant constitutes a zebrafish model of classical OI.
Figure 5.
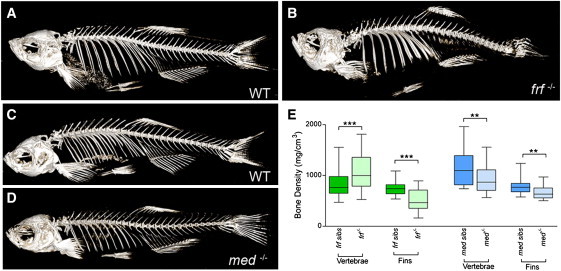
CT Analysis of frilly fins and microwaved Bone Reveals Altered Adult Bone Densities
(A–D) microCT analysis of bone density in a 7-month-old frf−/− mutant (B) and its WT sibling (A) as well as a med−/− mutant (D) and its WT sibling (C). Note that although the med mutant looks overtly normal (D), the frf mutant skeleton displays axial curvature and defects in the head skeleton (B).
(E) Box plots of density measurements derived from microCT analysis of mutants and sibling vertebrae and fin lepidotrichia. med mutants have osteopenia of both lepidotrichia in the fins and vertebrae (blue boxes). Although frf mutants also display osteopenia of the fins, they show an increased bone density in the vertebrae (green boxes). The Mann-Whitney U test was performed for comparing densities between mutants and siblings for each bone type (∗∗∗ denotes p < 0.001; ∗∗ denotes p < 0.01; and n = 8 for all data sets). Boxes indicate the median and the 25th and 75th percentiles, whereas the whiskers display the largest and smallest values.
The Zebrafish frilly fins Mutant is Caused by a bmp1 Mutation
We next mapped the frf mutant to an interval that contains bmp1a on linkage group 8 (Figure S2A). Sequencing of the bmp1a cDNA (GenBank accession number BC163535.1) from five frf alleles identified two missense mutations causing the substitutions p.Ile124Asn and p.Val223Asp (Figure 2D and Figures S2F and S2H), which are within the protease domain and which affect conserved amino acids (Figures S2K and S2L); a nonsense mutation that truncates the protein at the end of the proteolytic domain (p.Tyr306∗; Figure 2D and Figure S2G); and two splice-site mutations, one (associated with frf tm317) leading to the deletion of 21 amino acids from the proteolytic domain (p.Gln290_Arg310del; Figure 2D and Figures S2B, S2C, and S2I) and another (associated with frf tf5) generating four main erroneous splicing products (p.Tyr378Serfs∗6, p.Lys390Serfs∗6, p.Gly395insGlyLeuArg∗, and p.Lys394_Gly395ins8; Figure 2D and Figures S2D, S2E, and S2J). We were able to rescue the frf larval fin phenotype by injecting a DNA construct driving zebrafish Bmp1a expression from a heat-shock promoter, further supporting the conclusion that frilly fins represents a bmp1a mutant (Figures S3A–S3C).
Bmp1 Function in Bone Formation and Development
To understand the function of Bmp1 in bone formation, we characterized zebrafish bmp1a gene expression and the frf mutant phenotype in more detail. Zebrafish bmp1a is expressed in osteoblasts. During larval stages, we observed strong expression in fin mesenchyme cells within the fin fold at a stage contemporaneous with the appearance of the fin-fold defect in frf mutants (Figures 6A and 6B). In addition, we noted expression in the floor plate and hypochord, branchial arches and operculum (Figures 6A and 6C), and sites of bone formation in the head. We confirmed expression of bmp1a in osteoblasts by double-fluorescent in situ hybridization with the zebrafish osteoblast maker col10a1. We found consistent overlap of both mRNAs in osteoblasts on the operculum and cleithrum (Figures 6E and 6E″; data not shown). We also noted expression in clusters of cells arranged metamerically adjacent to the notochord, a location and arrangement consistent with osteoblasts of the vertebral column (Figure 6D).
Figure 6.
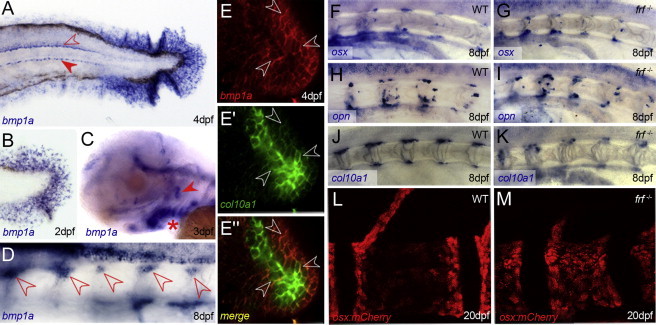
bmp1a Is Expressed in Osteoblasts, which Appear Normally Differentiated in frilly fins
(A–D) In situ hybridization of bmp1a at 4 dpf (A), 2 dpf (B), 3 dpf (C) and 8 dpf (D). Expression is seen in fin mesenchyme cells of the fin fold (A and B), floor plate and hypochord (A, open and filled arrowheads, respectively), branchial arches (C, asterisk), and operculum (C, arrowhead) and perichordal cells of the anterior notochord (D, arrowheads).
(E–E″) Confocal images of double-fluorescent in situ hybridizations showing coexpression of bmp1a (E and E″; red) and the osteoblast marker collagen10a1 (E′ and E″; green) in osteoblasts on the operculum at 4 dpf. Most cells express both markers (three cases highlighted by arrowheads), and central, more mature osteoblasts express slightly higher levels of col10a1.
(F–K) Perichordal expression of osteoblast markers is not disrupted in frf mutants. Lateral images of anterior notochord of 8 dpf WT (F, H, and J) and frf−/− (G, I, and K) larvae hybridized with probes for sp7 (F and G), osteopontin (H and I), and collagen10a1 (J and K).
(L and M) Confocal images of sp7:mCherry-expressing osteoblasts on vertebrae of WT (L) and frf mutant (M) larvae at 20 dpf. There is no reduction in osteoblast numbers in the mutant.
To determine whether the compromised ossification in frilly fins mutant larvae is due to insufficient generation of osteoblasts, we analyzed expression of sp7, osteopontin, and collagen10a1 by in situ hybridization in frf mutants. Osteoblasts were found to be normal in number and location, consistent with the interpretation that loss of bmp1a does not disrupt osteoblast generation, number, localization, or differentiation (Figures 6F–6K). We confirmed the presence of normal osteoblast numbers by first crossing frf into the sp7:mcherry transgenic line,26 which labels osteoblasts on skeletal structures. Compared to WT cells, mCherry-positive cells did not decrease in number in the frf mutant vertebrae or fins, even at the earliest times that such cells are visible (Figures 6L and 6M and Figure S4A and S4B). We obtained a similar result by immunostaining with the osteoblast-specific zns5 antibody, although we did observe altered cellular morphology—osteoblasts appeared more cuboidal in the mutant than did the flattened cells in the WT bone (Figures 7A–7D). The latter effect was also seen in the lepidotrichia when imaged by transmission electron microscopy (Figures 7E and 7F). However, no significant change in osteoblast number was observed in fin rays (Figure S4C). To test whether this altered-morphology phenotype is concomitant with a loss of osteoblast activity, we exposed frf mutants to RA, which was previously described as stimulating osteoblast activity and causing precocious hyperossification of the entire vertebral column.26,33 Unlike in WT (Figures 7G and 7H), ossification in frf mutants was almost completely refractory to RA treatment (Figures 7I and 7J), consistent with a defect in the ability of osteoblasts to effectively generate osteoid, downstream of osteoblast differentiation and activity.
Figure 7.
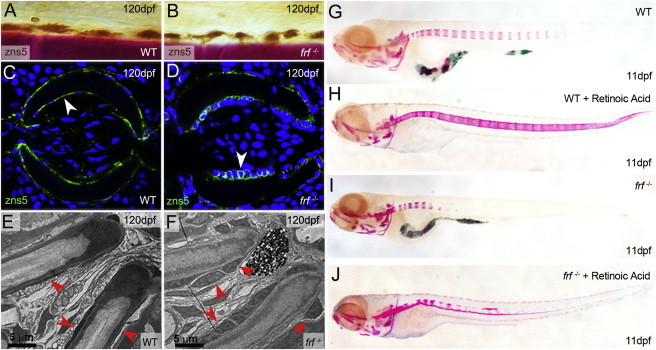
Osteoblasts in frilly fins Mutants Have Altered Morphology but Cannot Hyperossify upon RA Treatment
(A–D) Immunohistochemical (A and B) and immunofluorescent (C and D) staining of osteoblasts with the zns5 antibody (brown stain in A and B; green stain in C and D) in WT (A and C) and frf−/− (B and D) fins at 120 dpf. (A) and (B) display lateral views of fin rays counterstained with alizarin red (bone is in red), whereas (C) and (D) are transverse sections of fin rays counterstained with DAPI (blue). There is no loss in number of zns5+ osteoblasts in frf−/− mutants. However, the osteoblasts display an altered morphology when viewed in cross section; they appear more cuboidal where they are normally flat cells that maintain intimate contact with the bone surface (arrowheads in C and D).
(E and F) Electron micrographs of transverse sections of adult fin rays. Osteoblasts are indicated with red arrowheads and appear flat in WT fins (E) yet more cuboidal in frf−/− fins (F).
(G–J) Alizarin-red staining of 11 dpf WT (G and H) and frf−/− larvae (I and J) treated with (H and J) or without (G and I) RA for enhancing osteoblast activity. Despite normal numbers of differentiated osteoblasts in frf mutants, these cells are unable to mineralize the notochord efficiently upon RA stimulation; this suggests a defect downstream of osteoblast differentiation.
Bmp1 could potentially affect the ossification process through proteolytic cleavage processes involving a number of targets. Among these, its ability to cleave and inactivate the BMP2/4 inhibitor Chordin is well documented and has the potential to affect the ossification process.38,39 To test whether the frf phenotypes could be due to an excess of uncleaved Chordin, we generated frf−/− chordin−/− double-mutant adults (in which the early ventralized phenotype was rescued by chordin mRNA injection). As previously described,40 Chordin function is dispensable after gastrulation for axial skeleton generation and patterning (Figures S5A and S5C), yet it remains possible that elevated levels might perturb osteogenesis. However, Chordin loss at late larval stages failed to rescue the frf defect (Figures S5B and S5D). In addition to Chordin cleavage, Bmp1 also plays a role in generating mature Collagen I through the cleavage of the C-terminal propeptide domain (Figure 8A). Indeed, the inability of RA to rescue the ossification process in frf mutants and the similarity between the frf and med vertebrae phenotypes suggest that the major requirement for Bmp1 in bone formation is the generation of mature Collagen I. Using the picrosirius-red staining method to enhance birefringency of highly ordered fibrillar collagen,35 we analyzed collagen fibrillogenesis. At all stages analyzed and in both fins and vertebrae, we found a significant reduction in birefringence in frf−/− mutants; this indicates a loss of fibrillar-collagen structure (Figures 8B–8G). Ultrastructural imaging of fibrillar collagen in the fins at 6 dpf by transmission electron microscopy revealed disruption to the normal periodic collagen fibril (Figure 8H) in the mutant (Figure 8I). Immunoblot analysis, employing an antibody raised against zebrafish Col1a1a, demonstrated compromised C-propeptide removal in frf−/− mutants at both larval and adult stages (Figures 8J and 8K).
Figure 8.
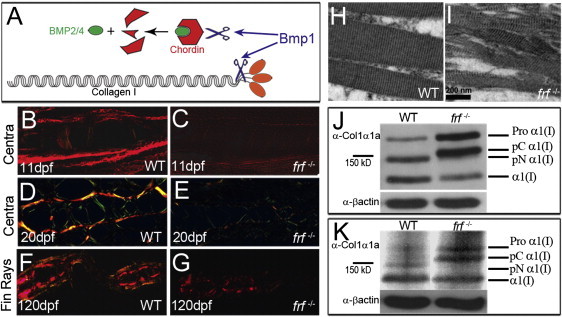
frf Displays Defects in Fibrillar Collagen Order and Col1a1a Processing
(A) Major proteolytic roles of Bmp1 include removing the C-propeptide (orange ovals) of pro-Collagen I and cleaving the BMP2/4 inhibitor, Chordin (red hexagon), to release free BMP2/4 (green oval).
(B–G) Picrosirius-red stained sagittal (B–E) and transverse (F and G) sections of WT (B, D, and F) and frf−/− (C, E, and G) larvae at 11 dpf (B and C), 20 dpf (D and E), and 4 months (F and G). Sections viewed under polarized light reveal the reduced collagen-fiber-associated birefringency in the Centra region (B–E) and fin rays (F and G) in frf−/− mutant (C, E, and G) and WT (B, D, and F) larvae.
(H and I) Transmission electron micrographs of longitudinal sections of WT (H) and frf−/− (I) larval medial fins at 6 dpf show loss of structured collagen fibers in the mutant.
(J and K) Immunoblots of protein extracted from WT (lane 1) and frf−/− mutant (lane 2) larvae probed with an antibody directed against zebrafish Collagen1α1a (upper panels in both J and K) or an antibody against β-actin as a loading control (lower panels). The four possible Collagen1α1 forms are indicated on the right; these forms include Procollagen1α1 retaining both C- and N- terminal propetides (Pro α1[I]), mature collagen α1(I) retaining neither propeptide (α1[I]), a form retaining only the N-propeptide (pN α1[I]), and a form retaining only the C-propeptide (pC α1[I]). In 6 dpf (J) and 4-month-old (K) frf−/− mutants, the two forms retaining the C-propeptide predominate.
Because the frf larval fin phenotype is most likely due to loss of Bmp1-mediated collagen processing, we used a test for such processing as an in vivo assay to assess the activity of BMP1 with the p.Gly12Arg substitution. Toward this end, we injected into frf embryos DNA constructs containing a heat-shock promoter upstream of either human WT BMP1 coding sequences or a mutant version bearing the p.Gly12Arg substitution. This assay demonstrated that BMP1 with the p.Gly12Arg substitution had a significantly reduced ability to rescue the fin defect (Figures S3A–S3E); this finding suggests that p.Gly12Arg causes a reduced ability to augment in vivo the deficit in frf C-propeptidase activity.
Discussion
In this study, we identify a homozygous missense mutation substituting an amino acid in the signal peptide of BMP1 in a Turkish consanguineous family with autosomal-recessive high-bone-density OI. BMP1 is known to have several functions in different pathways, including the proteolytic processing of the procollagen I C-propeptide for the generation of mature collagen type I. On the basis of this information and given the fact that the majority of OI cases are associated with defects in COL1A genes or in genes involved in collagen I biosynthesis,3,5 we propose BMP1 as a highly relevant gene in this OI-disease context.
The identified substitution, p.Gly12Arg, is located in exon 1, which encodes the signal peptide of BMP1. Although it remains to be determined whether the signal-peptide variant results in a reduction of BMP1 enzymatic activity per se, bioinformatic analysis predicted a disruption of the signal peptide and therefore suggested a failure of intracellular protein sorting of premature BMP1 and an exclusion from the secretory pathway. Subsequently, we confirmed in in vitro assays that the amino acid substitution leads to severely reduced post-translational N-glycosylation of the mutant protein and impaired protein secretion (Figures 3A and 3B). Our results agree with those published in a previous study (Garrigue-Antar et al.37), which showed the importance of N-glycosylation for secretion and stability of BMP1. Compromised BMP1 secretion thus reduces the availability of BMP1 in the extracellular matrix, potentially leading to the insufficient processing of substrates, including the procollagen I C-propeptide. Indeed, we were able to demonstrate measurably reduced processing of two substrates by p.Gly12Arg-substituted BMP1 in two in vivo assays in zebrafish. Exogenous Bmp1 has been described as cleaving the dorsal determinant, Chordin, and leading to ventralization of zebrafish embryos.41 We exploited this finding to show that the p.Gly12Arg-substituted version of BMP1 was less efficient at ventralizing the embryo than the WT BMP1 (Figure 3). In addition, unlike the WT BMP1, the p.Gly12Arg-substituted BMP1 was unable to measurably rescue the larval fin ruffling of the bmp1a zebrafish mutant (Figure S3); larval fin ruffling is associated with defective collagen-rod formation. Thus, we have shown that p.Gly12Arg leads to both reduced secretion and subsequent reduced processing of the substrates Chordin and Collagen I.
Such reduced C-propeptide cleavage predicts the assembly of procollagen instead of mature collagen into collagen I fibrils. Immunoblot analysis of both larval and adult zebrafish protein samples demonstrated that forms retaining the C-propeptide predominate upon reduction of Bmp1 function (Figure 8). Unfortunately, no material was available to show this effect in the affected individuals. We hypothesize that this results in an impairment of the collagen matrix within the bone structure and could be the major cause in the underlying pathomechanism. Supporting this hypothesis, use of the birefringent collagen stain, picrosirius red, demonstrated less ordered collagen-fiber structure in zebrafish bmp1a mutants (Figure 8). Interestingly, recent studies have assumed that collagen C-propeptides assembled within collagen fibrils would either increase the intrafibrillar spacing or directly serve as nucleators of mineralization.15 This might explain the high bone mineral density we noted in our individuals (Figure 1 and Table 1). Our findings provide evidence that insufficient collagen processing caused by p.Gly12Arg most likely leads to ectopic accumulation of minerals in the bone. However, this bone is nonetheless structurally compromised, leading to fragility. The precise pathophysiology of increased bone mineralization upon defective collagen processing remains unclear and needs to be investigated further.
We note similar defects in bone formation in the zebrafish frilly fins mutant, and we show that these defects correspond to mutations in bmp1a. Alizarin-red staining demonstrated delayed ossification at larval stages and malformation of adult skeletal structures with evidence of fractures (Figure 4). Quantification of mineral content by microCT analysis demonstrated a higher mineral content of mature bone, as seen in the individuals. The reason for the divergent mineral-content phenotypes between the larval-stage frilly fins mutant and the adult-stage mutant is currently unclear. We hypothesize that there is a difference in mineralization rate between the two stages and that mature collagen is rate limiting during the initial deposition in larval stages, whereas in adult stages, the retained telopeptide gradually induces increased mineralization through a mechanism that has not been determined. Accordingly, the only location found to have reduced mineralization in adult frilly fins is the distal fin, a site of bone deposition.
As the first described animal model with reduced Bmp1 function in an adult, frilly fins was used for the investigation of the role of this protease in bone formation. We showed that although generation and patterning of osteoblasts is unaffected (Figure 6), there was an intriguing alteration in morphology of the osteoblasts—they adopted a cuboidal shape (Figure 7). Although this might indicate a defect in the osteoblasts themselves, we favor the interpretation that this is a result of reduced adhesion to the compromised bone matrix. Supporting this is the in vitro observation that osteoblasts cultured on bone matrix lacking collagen appeared rounded compared to flattened cells cultured on a purified mineralized collagen matrix.42
The role of Bmp1 (and other Tolloid-related proteins) in dorsal-ventral patterning through Chordin cleavage is well documented. We could, however, conclusively show that this was not the relevant substrate underlying the frilly fins bone phenotype (Figure S5). In fact, multiple lines of evidence from our analysis support the interpretation that the major role of Bmp1 in ossification is removal of the C-propeptide from Collagen I. First, we note similarity of frf to the microwaved mutant, which we identified as a collagen1a1a mutant (Figure 4). Second, frf larvae are not able to hyperossify the vertebral column upon retinoic-acid stimulation of the osteoblasts (Figure 7), most consistent with a defect that is considerably downstream in the process of osteoid formation. Finally, we show compromised Collagen-I processing and higher-order structure in frf biochemically and histologically (Figure 8).
While we were preparing this manuscript, Martinez-Glez et al.43 described a homozygous missense mutation causing an alteration in the protease domain of BMP1 in two affected individuals from an Egyptian family affected by severe autosomal-recessive OI. In line with our findings, concomitant abnormal procollagen I C-propeptide processing was described. Comparison of the individuals' phenotypes in both studies showed the following differences: affected individuals in the Egyptian family presented with classical autosomal-recessive OI, whereas our individuals, as well as the zebrafish model, presented with bone fragility associated with an increase in bone mineral density. Interestingly, Lindahl K. et al.15 very recently described COL1A1 mutations affecting the BMP1 C-propeptide cleavage site; also, the individuals in this study presented with an increased-mineralization OI phenotype very similar to our individuals, suggesting that impaired BMP1-related collagen C-propeptide cleavage (either by mutations in BMP1 or mutations affecting the BMP1 cleavage site in COL1A1) causes a distinct form of OI. In contrast, the missense mutation described by Martinez-Glez et al.43 might have different functional consequences and thereby cause phenotypic variability and differences in the severity of the disease.
All together, our combined data in humans and zebrafish define the molecular and cellular bases of BMP1-dependent osteogenesis and show the importance of this protein for bone formation and stability. These data, in both humans and zebrafish, support the finding that deficits in removal of the C-propeptide from Collagen I result in autosomal-recessive OI with high bone mineral density.15
Acknowledgments
We are grateful to all family members that participated in this study, Esther Milz for excellent technical assistance, Karin Boss for critically reading the manuscript, and Kaicheng Liang from the Singapore Bioimaging Consortium for microCT imaging. This work was supported by the German Federal Ministry of Education and Research by grant 01GM0880 (SKELNET) to B.W. The authors would like to thank the National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project and the following ongoing studies that produced and provided exome-variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women's Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010).
Contributor Information
Bernd Wollnik, Email: bwollnik@uni-koeln.de.
Thomas J. Carney, Email: tcarney@imcb.a-star.edu.sg.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ENSEMBL, http://www.ensembl.org
Exome Variant Server, http://snp.gs.washington.edu/EVS/
PolyPhen, http://coot.embl.de/PolyPhen
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Byers P.H., Cole W.G. Osteogenesis Imperfecta. In: Royce P., Steinmann B., editors. Connective Tissue and its Heritable Disorders: Molecular, Genetic, and Medical Aspects. Second Edition. John Wiley & Sons; Hoboken, NJ: 2002. pp. 385–430. [Google Scholar]
- 2.Sillence D.O., Rimoin D.L. Classification of osteogenesis imperfect. Lancet. 1978;1:1041–1042. doi: 10.1016/s0140-6736(78)90763-8. [DOI] [PubMed] [Google Scholar]
- 3.Basel D., Steiner R.D. Osteogenesis imperfecta: Recent findings shed new light on this once well-understood condition. Genet. Med. 2009;11:375–385. doi: 10.1097/GIM.0b013e3181a1ff7b. [DOI] [PubMed] [Google Scholar]
- 4.Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 5.Marini J.C., Forlino A., Cabral W.A., Barnes A.M., San Antonio J.D., Milgrom S., Hyland J.C., Körkkö J., Prockop D.J., De Paepe A. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pollitt R., McMahon R., Nunn J., Bamford R., Afifi A., Bishop N., Dalton A. Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I-IV. Hum. Mutat. 2006;27:716. doi: 10.1002/humu.9430. [DOI] [PubMed] [Google Scholar]
- 7.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 8.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Dijk F.S., Nesbitt I.M., Zwikstra E.H., Nikkels P.G., Piersma S.R., Fratantoni S.A., Jimenez C.R., Huizer M., Morsman A.C., Cobben J.M. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lapunzina P., Aglan M., Temtamy S., Caparrós-Martín J.A., Valencia M., Letón R., Martínez-Glez V., Elhossini R., Amr K., Vilaboa N., Ruiz-Perez V.L. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker J., Semler O., Gilissen C., Li Y., Bolz H.J., Giunta C., Bergmann C., Rohrbach M., Koerber F., Zimmermann K. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mann V., Ralston S.H. Meta-analysis of COL1A1 Sp1 polymorphism in relation to bone mineral density and osteoporotic fracture. Bone. 2003;32:711–717. doi: 10.1016/s8756-3282(03)00087-5. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl K., Barnes A.M., Fratzl-Zelman N., Whyte M.P., Hefferan T.E., Makareeva E., Brusel M., Yaszemski M.J., Rubin C.J., Kindmark A. COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum. Mutat. 2011;32:598–609. doi: 10.1002/humu.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myllyharju J., Kivirikko K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Bond J.S., Beynon R.J. The astacin family of metalloendopeptidases. Protein Sci. 1995;4:1247–1261. doi: 10.1002/pro.5560040701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sterchi E.E., Stöcker W., Bond J.S. Meprins, membrane-bound and secreted astacin metalloproteinases. Mol. Aspects Med. 2008;29:309–328. doi: 10.1016/j.mam.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge G., Greenspan D.S. Developmental roles of the BMP1/TLD metalloproteinases. Birth Defects Res. C Embryo Today. 2006;78:47–68. doi: 10.1002/bdrc.20060. [DOI] [PubMed] [Google Scholar]
- 20.Ge G., Greenspan D.S. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J. Cell Biol. 2006;175:111–120. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kessler E., Takahara K., Biniaminov L., Brusel M., Greenspan D.S. Bone morphogenetic protein-1: The type I procollagen C-proteinase. Science. 1996;271:360–362. doi: 10.1126/science.271.5247.360. [DOI] [PubMed] [Google Scholar]
- 22.Canty E.G., Kadler K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki N., Labosky P.A., Furuta Y., Hargett L., Dunn R., Fogo A.B., Takahara K., Peters D.M., Greenspan D.S., Hogan B.L. Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development. 1996;122:3587–3595. doi: 10.1242/dev.122.11.3587. [DOI] [PubMed] [Google Scholar]
- 24.Parichy D.M., Elizondo M.R., Mills M.G., Gordon T.N., Engeszer R.E. Normal table of postembryonic zebrafish development: Staging by externally visible anatomy of the living fish. Dev. Dyn. 2009;238:2975–3015. doi: 10.1002/dvdy.22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Eeden F.J., Granato M., Schach U., Brand M., Furutani-Seiki M., Haffter P., Hammerschmidt M., Heisenberg C.P., Jiang Y.J., Kane D.A. Genetic analysis of fin formation in the zebrafish, Danio rerio. Development. 1996;123:255–262. doi: 10.1242/dev.123.1.255. [DOI] [PubMed] [Google Scholar]
- 26.Spoorendonk K.M., Peterson-Maduro J., Renn J., Trowe T., Kranenbarg S., Winkler C., Schulte-Merker S. Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development. 2008;135:3765–3774. doi: 10.1242/dev.024034. [DOI] [PubMed] [Google Scholar]
- 27.Geisler R. Mapping and cloning. In: Nusslein-Volhard C., Dahm R., editors. Zebrafish: A practical approach. Oxford University Press; Oxford: 2002. pp. 175–212. [Google Scholar]
- 28.Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P., Chien C.B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 29.Rentzsch F., Zhang J., Kramer C., Sebald W., Hammerschmidt M. Crossveinless 2 is an essential positive feedback regulator of Bmp signaling during zebrafish gastrulation. Development. 2006;133:801–811. doi: 10.1242/dev.02250. [DOI] [PubMed] [Google Scholar]
- 30.Balciunas D., Wangensteen K.J., Wilber A., Bell J., Geurts A., Sivasubbu S., Wang X., Hackett P.B., Largaespada D.A., McIvor R.S., Ekker S.C. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006;2:e169. doi: 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 32.Brend T., Holley S.A. Zebrafish whole mount high-resolution double fluorescent in situ hybridization. J. Vis. Exp. 2009;25:1229. doi: 10.3791/1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laue K., Jänicke M., Plaster N., Sonntag C., Hammerschmidt M. Restriction of retinoic acid activity by Cyp26b1 is required for proper timing and patterning of osteogenesis during zebrafish development. Development. 2008;135:3775–3787. doi: 10.1242/dev.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker M.B., Kimmel C.B. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem. 2007;82:23–28. doi: 10.1080/10520290701333558. [DOI] [PubMed] [Google Scholar]
- 35.Borges L.F., Gutierrez P.S., Marana H.R., Taboga S.R. Picrosirius-polarization staining method as an efficient histopathological tool for collagenolysis detection in vesical prolapse lesions. Micron. 2007;38:580–583. doi: 10.1016/j.micron.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Brown A.M., Fisher S., Iovine M.K. Osteoblast maturation occurs in overlapping proximal-distal compartments during fin regeneration in zebrafish. Dev. Dyn. 2009;238:2922–2928. doi: 10.1002/dvdy.22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrigue-Antar L., Hartigan N., Kadler K.E. Post-translational modification of bone morphogenetic protein-1 is required for secretion and stability of the protein. J. Biol. Chem. 2002;277:43327–43334. doi: 10.1074/jbc.M207342200. [DOI] [PubMed] [Google Scholar]
- 38.Scott I.C., Blitz I.L., Pappano W.N., Imamura Y., Clark T.G., Steiglitz B.M., Thomas C.L., Maas S.A., Takahara K., Cho K.W., Greenspan D.S. Mammalian BMP-1/Tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev. Biol. 1999;213:283–300. doi: 10.1006/dbio.1999.9383. [DOI] [PubMed] [Google Scholar]
- 39.Lee K.S., Kim H.J., Li Q.L., Chi X.Z., Ueta C., Komori T., Wozney J.M., Kim E.G., Choi J.Y., Ryoo H.M., Bae S.C. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell. Biol. 2000;20:8783–8792. doi: 10.1128/mcb.20.23.8783-8792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher S., Halpern M.E. Patterning the zebrafish axial skeleton requires early chordin function. Nat. Genet. 1999;23:442–446. doi: 10.1038/70557. [DOI] [PubMed] [Google Scholar]
- 41.Muraoka O., Shimizu T., Yabe T., Nojima H., Bae Y.K., Hashimoto H., Hibi M. Sizzled controls dorso-ventral polarity by repressing cleavage of the Chordin protein. Nat. Cell Biol. 2006;8:329–338. doi: 10.1038/ncb1379. [DOI] [PubMed] [Google Scholar]
- 42.Baslé M.F., Grizon F., Pascaretti C., Lesourd M., Chappard D. Shape and orientation of osteoblast-like cells (Saos-2) are influenced by collagen fibers in xenogenic bone biomaterial. J. Biomed. Mater. Res. 1998;40:350–357. doi: 10.1002/(sici)1097-4636(19980605)40:3<350::aid-jbm2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 43.Martínez-Glez V., Valencia M., Caparrós-Martín J.A., Aglan M., Temtamy S., Tenorio J., Pulido V., Lindert U., Rohrbach M., Eyre D. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum. Mutat. 2012;33:343–350. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.