

Abstract
Over the past three decades the pancreatic islet of Langerhans has taken center stage as an endocrine microorgan whose glucoregulatory function is highly explicable on the basis of the increasingly well understood activities of three highly interactive secretory cells. Islet dysfunction underlies both type 1 and type 2 diabetes mellitus (DM); its protection from immune attack and gluco-and lipo-toxicity may prevent the development of DM; and its replacement by non-surgical transplantation may be curative of DM. During a career marked by vision, focus and tenacity, Paul Lacy contributed substantially to the development of each of these concepts. In this review we focus on Lacy's contribution to the development of the concept of the islet as a micro-organ, how this foreshadowed our current detailed understanding of single cell function and cell-cell interactions and how this led to a reduced model of islet function encouraging islet transplantation. Next, we examine how clinical allotransplantation, first undertaken by Lacy, has contributed to a more complex view of the interaction of islet endocrine cells with its circulation and neighboring tissues, both “in situ” and after transplantation. Lastly, we consider recent developments in some alternative approaches to treatment of DM that Lacy could glimpse on the horizon but did not have the chance to participate in.
Key words: Paul Lacy, physiology of islets of Langerhans, transplantation of islets of Langerhans, insulin secretion, glucagon secretion, diabetes mellitus
Introduction
“I started my career wanting to learn about the structural basis and cell mechanics of insulin secretion and ended it trying to cure diabetes with islet transplantation. Gosh, what a surprising turn a life in science can take!” (Paul Lacy, September 1987).
Throughout his academic career at Washington University, from the early 1950s, as a newly minted Assistant Professor of Pathology, to the mid 1990s, when he retired as Kroc Professor of Pathology after a prior long term as departmental chairman, the late Paul Lacy had a focused, “islet-centric” scientific interest.1 He wished to learn as much as he could about the function, especially insulin secretion, of the pancreatic islet of Langerhans. In the first phase of that career (1955–1973), he studied the intricate in situ ultrastructure and in vitro function of the islet. He made a major contribution towards characterizing the substructure of component α, β and δ cells by techniques including selective staining and secretagogue-induced granule depletion. He identi- fied granule “emiocytosis” (exocytosis) as the key mechanism of hormone exit from islet cells. In addition, he recognized the importance of granule maturation and movement as well as Ca2+ entry and the cytoskeleton in the exocytotic process. In doing this he provided a first working model for biphasic insulin secretion. Moreover, his development of the isolated islet preparation made possible detailed enzymology, electrophysiology and living tissue microscopy.
In the second phase of his career (1973–1995), Lacy mounted an all-out scientific mission. In a heroic bench-to-bedside effort “to cure diabetes mellitus in man by human islet transplantation,” he developed and disseminated key techniques of human islet purification from cadaver donors and subsequent portal vein infusion into recipients. His specific aim was “to harvest as many pancreatic islets of Langerhans as possible, keep them healthy, make them non-antigenic, and then, by golly to transplant them into a safe space in the body,” where they'd “take up root,” “appropriately secrete insulin after a meal” and “substitute for the sick islets of the diabetic pancreas that couldn't.” With glucose-sensitive islets secreting insulin on a moment-to-moment basis “the highs and lows of blood sugar and the end-organ damage of diabetes seen after years of diabetes would be prevented.” This work culminated in the first trials of clinical trials of islet transplantation in 1990. By articulating this goal with a magical presence, a combination of a folksy Midwestern grandiloquence and a twinkle in his eye that assured even the casual listener of a self-evident truth, he raised awareness, hope and funding for a simple and elegant approach at organ replacement. However, privately, he remained keenly aware of the Achilles heel of this endeavor, namely the need for immunosuppression, the uncertainty of tissue supply and quality, and the potentially unsustainable function of islets in a foreign environment. From the early 1990s until his retirement from active science at Washington University in 1995 to pursue a love of archeology, Lacy with David Scharp, his long-term partner in the human islet transplantation adventure, concentrated on a variation on the original islet transplantation vision, xenotransplanation of much more readily available porcine islets after their encapsulation.
To celebrate the legacy of Paul Lacy's imaginative, tenacious, generous and, to be sure, “gutsy” life in science, as well as his seminal contributions to the revival of the pancreatic islet from relative investigative obscurity, this review shall focus on the influence of Lacy's seminal techniques of islet isolation, his exploration of islet function as well as his vision of curative islet transplantation. Based on a critical review of the published work of Lacy and his contemporaries and two in-depth conversations I had with him in 1987 and 2000, I shall concentrate on several features. First, how Lacy's early work on islet function motivated cellular and molecular studies of the past two decades that have culminated in complex models of stimulus-secretion coupling in individual islet cells and as well as produce a “reduced model of islet function,” which formed the underlying assumptions of the early vision of clinical islet transplantation. Second, how partial realization of that vision promoted understanding of pancreatic islets as unique micro-organs that maintain a complex internal sociobiology, intricate developmental and functional ties with their exocrine tissue neighbors, as well as important distant interactions with immune surveillance cells, which may be critical to both autoimmunity of diabetes and islet transplant rejection. The latter provides an interesting study in how a simple idea grows in complexity as it progresses from bench to bedside and then back to bench.
Part I: The Islet as a Micro-Organ
Mass isolation of islets and its bonus to physiology, the unraveling of the details of stimulus-secretion coupling in β and α cells.
Research into the isolation of islets began in the 1960s when tens of islets were liberated from rodent pancreas by enzymatic (collagenase) incubations of chopped tissue or free hand microdissection of surface islets (reviewed in reference 2), both usually from obese animals with hypertrophic islets. In a clear technical breakthrough, in 1967 Lacy and Kostianovsky3 demonstrated the ability to harvest several hundred metabolically active and structurally intact islets from the pancreas of a normal rat. Prior to digesting the fat-trimmed pancreas with collagenase, they distended and partially disrupted the enveloping acini through retrograde injection of physiological saline into the cannulated bile/pancreatic duct. The latter maneuver provided easier access of collagen to the interior of the pancreatic tissue. Islets, which are dense ovoid bodies, were separated from acini and ductal fragments by gravity sedimentation in a saline- filled cylinder or by centrifugation in a discontinuous sucrose (or later Ficoll) gradient. Using this approach, up to several hundred islets could be collected from a rodent pancreas, and several thousand islets could be collected from the pancreas of large mammal.
Success in harvesting islets and the subsequent ability to disperse these into a preparation of single cells, which could later be separated into their component types, or into preparation of cytosolic components, especially insulin granules,4 could not have come at a more propitious time. Insulin was being routinely and reliably measured by radioimmunoassay. In addition, the rudiments of a general hypothesis for stimulus-secretion coupling in β-cells were being established through several approaches. Ashcroft and Randle in Oxford and Matschinsky and group in St. Louis were providing strong evidence that glucose transport, phosphorylation and oxidation to CO2 + H2O were essential for stimulation of insulin secretion and biosynthesis.5 Grodsky and colleagues,6,7 investigating secretion of insulin by the perfused pancreas into the hepatic vein, had already shown that insulin release in response to a sustained rise in glucose was biphasic and that both phases required aerobic oxidation of glucose and adequate concentrations of extracellular Ca2+. Dean and Matthews8 using microdissected islets, demonstrated that islet cells responded to a rise in ambient glucose by depolarizing and then firing action potentials whose upstrokes appeared to be dependent on the concentration of extracellular Ca2+. As β-cell depolarization required glucose metabolism and was associated with a decrease in membrane permeability to K+, a link between β-cell metabolism and excitability was being established. Hence, an outline of a hypothesis of stimulus-secretion coupling in β-cells was emerging: glucose metabolism, by changing cell energetics was promoting electrical activity, which in turn was promoting Ca2+ entry and Ca2+-dependent exocytosis of insulin granules.
Work using all of these approaches was now made immensely easier and more disseminatable by the technique for mass isolation of islets. Lacy's lab, which had previously published a host of electron microscopic images of maturation and “emiocytosis” (surface rupture and shower-like release of contents of insulin granules in fixed tissue), demonstrated in monolayers of cultured islets that granules, in constant motion, preferentially approached the membrane after glucose stimulation; this was inhibited after application of agents that disassemble cytoskeleton.9,10 On this basis Lacy proposed that biphasic insulin secretion seen with high glucose stimulation (>10 mM) might be explained as follows. First phase insulin secretion, unaffected by cytoskeletal disrupting agents, might result from the discharge of granules already present (in contemporary terminology “docked”) at the membrane. In contrast, second phase insulin secretion, greatly decreased by cytoskeletal disrupting agents, might result from granules moving towards the membrane along cytoskeleton as their contents undergo progressive processing and aggregation. In contemporary terminology this would be while in transit from the Golgi to the plasma membrane, first by kinesin motors along microtubules, then penetrating the cortical actin cage aided by myosin V motors, and lastly attaching to the membrane via the formation of complexes between vesicle (v-, VAMP) and target (t-, SNAP23/25, syntaxin) SNARE proteins. Later experiments demonstrated small oscillatory changes in insulin secretion occurring near resting levels of glucose.10a
Within less than a decade of routine availability and dispersion of isolated islets, powerful new techniques offered the prospect of revolutionizing the entire field of stimulus-secretion coupling in endocrine cells. These included: patch clamp recording of single channel and whole cell currents underlying both the resting potential at basal levels of glucose and the complex electrical activity initiated by stimulatory glucose concentrations;11,12 optical monitoring of metabolic processes including mitochondrial membrane potential and free cytosolic ion concentration with membrane permeant fluorescent probes;13 and cloning of key transporter proteins including the ATP sensitive K channel.14 Ultimately they also included real time, single cell assays of single cell exocytosis. Most prominent among these were the following: (1) electrical measurements of increases in membrane capacitance (Cm) proportional to plasma membrane surface area in patch clamped cells, indicating increases in plasma membrane surface area due to fusion of organelles the size of insulin granules with the plasma membrane;15,16 (2) electrochemical measurements, by oxidation or reduction of discharged of both native hormone (insulin) and preloaded false transmitter (serotonin) contents17–19 of exocytosing granules, using carbon fiber electrodes whose tips are positioned at the surface of cells, over time courses very similar to the increases in Cm; and (3) optical tracking of granules with fluorescent labeled granule contents or intramembrane proteins as they approach the plasma membrane and fuse with it.
An overall consensus scheme for stimulus-secretion coupling in β-cells is presented in Figure 1A. A rise in serum glucose from a baseline level of 3 mM to stimulatory levels of 5–6 mM enhances glucose import via a moderate affinity transporter (glut-2) followed by shunting into glycolysis by glucokinase and mitochondrial oxidation (step 1), results in a small rise in cytosolic ATP and a fall in cytosolic ADP (step 2) sufficient to close down ATP-dependent K-selective ion channels (step 3). The latter consist of an inner ring of four inward rectifier K channel subunits (Kir 6.2), encompassing a pore closed by the binding of ATP, and an outer ring of sulfonylurea receptors (SUR 2), interacting with the central core in a fashion that binding of MgADP can open, while binding of sulfonylureas can, close the Kir-defined pore. The closure of K+(ATP) channels, against a background of tonically open non-selective cation channels, constitutes the basis for cell depolarization (step 4) and the activation of voltage dependent Na+, Ca2+ and K+ channels (step 5), which underlie complex electrical activity, including trains of large amplitude action potentials (APs) or long trains aborted APs riding on plateau potentials. The opening of high voltage activated (HVA) Ca2+ channels support rapid entry of Ca2+ (step 6) and Ca2+-dependent exocytosis of the contents of a small pool (at most several hundred) if insulin granules in close proximity to the plasma membrane (the readily releasble pool, RRP) (step7) and likely recruit granules from more interior regions of the cytoplasm into the RRP to refill this pool.20 Individual APsrepolarize as a result of opening of voltage-dependent K+ channels, while plateau depolarizations are likely terminated by the opening of poorly voltage-dependent but Ca2+-activated K+ channels. Incretin messengers, such glucagon-like intestinal peptide (GLP-1) released by enterochromaffin-like (EC) cells of the gut epithelium sensing newly digested nutrients, and acetylcholine released by stimulation of vagus nerve; both enhance closure of K(ATP) channels and help recruit insulin granules into a release ready pool.21,22 Current work is focusing on the contributions of distinct electrical activity patterns, novel Ca2+ currents, glucosederived second messengers other than ATP, and distinct subsets of the RRP pools, including a low Ca2+ sensitivity immediately releasable pool (IRP) likely docked near clusters of Ca2+ channels and a high Ca2+ sensitivity (HCSP) likely farther from the clusters of Ca2+ channels, to the shaping of biphasic insulin secretion.23–26
Figure 1.

Proposed schemes of stimulus-secretion coupling in β- and α-cells of the islet. (A) Stimulus-secretion coupling in β-cells. Digestion of food in the gut both increases blood glucose and releases incretins (acetylcholine and glucagon-like intestinal peptide), which trigger insulin secretion. GLP-1 via a G-protein coupled receptor stimulates the production of cAMP which serves to activate protein kinase A or epac, while acetylcholine activates via a G-protein coupled muscarinic receptor of CaM kinase ii. (B) Stimulus-secretion coupling in α-cells. A rise in serum glucose and release of contents of insulin granule inhibits glucagon secretion. In contrast, hypoglycemia rapidly stimulates glucagon release by relieving glucose inhibition of exocytosis, and by activating central nervous system stress pathways that turn release catecholamines (CA) from adrenergic nerve fibers and adrenal medullary chromaffin cells. It is likely that CAs priming the docking of glucagon-containing granules into a readily-releasable pool.
Recently, Lacy's early experiments on movement of insulin granules have attracted intense interest. These approaches have been improved and made more quantitative using enhanced optical techniques (including evanescent wave or total internal reflectance microscopy, confocal microscopy and two photon excitation microscopy) for tracking the movement of near membrane granules up to the membrane and their subsequent fusion with the plasma membrane and release of their into the extracellular space (ECS). Specific tags for exocytosis include red or green fluorescent protein labeled peptides, including C-peptide of derived from pro-insulin, intrinsic granule membrane proteins (syncollin, or phogrin), granule membrane attached proteins (tissue plasminogen activator), and even insulin bound Zn2+ released into the ECS with decondensation of crystalline insulin granule core and then detected by a bath applied fluorescent indicator.27–31 Endocytosis can be tracked by the filling of granule exocytotic figures with otherwise excluded fluorescent dextran, applied as a fluid phase tracer or with quenching of pH-sensitive fluorescence on granule re-acidification after their recapture. With simultaneous capacitance and amperometry recordings from the same cell it is becoming possible to ask what fraction of attempted exocytoses lead to exit of the crystalline insulin (with its associated Zn2+) into the extracellular space, as opposed to more transient and restricted fusion pore formation with diffusion of more soluble contents, including peptides such as C-peptide or small molecular weight molecules such ATP and GABA.32 The latter two likely play autocrine or paracrine regulatory roles in the islet rather than true endocrine roles systemically. As anticipated by Lacy, some evidence suggests that “newcomer” granules, perhaps refilling the HCSP, are the major contributors to second phase insulin secretion. Careful application of these approaches may yield evidence as to whether granules from distinct granule pools differentially release soluble vs. condensed contents, and how rapidly granule membrane is retrieved and resorted.30,31
In addition, stimulus-secretion coupling by α-cells has also received intensive study using single cell techniques presented above. Outlines have emerged as to how tonic glucagon secretion results at very low levels of glucose and insulin, how glucagon secretion is curtailed in response to a post-prandial rise in glucose and how glucagon secretion reappears at high levels of glucose or in the presence of high concentrations of adrenalin.32–34 As shown in Figure 1B, α-cells exhibit similar K+(ATP) channels as found in β-cells, though most are closed at low levels of glucose. In addition, they have an abundance of non-selective cation channels, as well as standard voltage dependent Na+ channels and low voltage activated (LVA or T-type) Ca2+ channels. At 2–3 mM glucose α-cells are depolarized and fire spontaneous action potentials that trigger Ca2+ entry through LVA Ca2+ channels; as a result, they exocytose glucagon-containing granules. Raising extracellular glucose towards or above 5 mM closes down more K+(ATP) channels (step 1–3) and further depolarizes the α-cells into a voltage range (near −35 mV) (step 4) where Na+ and T-type Ca2+ channels are inactivated (step 5) and AP amplitude is decreased or its activity even ceases, thereby reducing Ca2+ entry (step 6) and glucagon granule exocytosis. In addition, secretory products of the β-cell, including GABA, Zn2+ as well as insulin, likely suppress glucagon secretion. The best studied of these is GABA: it binds to and opens ionotropic GABA-A receptor channels, which carry Cl- current and clamp the membrane potential of the α-cell near the Cl- equilibrium potential thereby reducing α-cells electrical activity and glucagon release. Lastly, at supraphysiological levels of glucose, where all remaining K+(ATP) channels are closed, or where large concentrations of adrenalin is released by the chromaffin cells of the adrenal medulla, HVA Ca2+ channels are augmented, α-cells may develop plateau depolarizations to −20 mV (step 5a) that are sufficient to tonically activate HVA Ca2+ channels and induce Ca2+ entry (step 6a) as well as recruit more granules into a readily releasable pool, thereby promoting a phase of glucagon release (step 7a), such as that seen during diabetic ketoacidosis or other systemic stresses.
Much less is known about stimulus-secretion coupling in somatostatin secreting d cells. Early reports (e.g., a recent one using human cells) suggest that the resting membrane potential is dependent on sulfonylurea- but not glucose-dependent K+(ATP) channels, that closure of the latter channels sets off Na+ and Ca2+ dependent action potentials, and that exocytosis is Ca2+ entry-dependent but slow to start and continuing for up to a second after current through HVA Ca2+ channnels has ceased.35
The extensive effort at understanding the cell biology and biophysics of stimulus-secretion coupling underlying normal insulin secretion has yielded a better understanding of two rare conditions congenital conditions.36–38 Congenital hyperinsulinemia of infancy is now thought most often to be due to the loss of function mutation of K+(ATP) channels (hyposensitivity to MgADP). In contrast, neonatal diabetes, is now thought most often to be due to gain of function K+(ATP) channels (mutations of Kir6.2). However, the rather prevalent state of disordered insulin secretion in man, type 2 diabetes mellitus (DM 2) where β-cells of islets, though reduced in number, remain largely intact, has been much less tractable. Some evidence suggests that in DM 2 β-cells have underlying variations in key coupling factors in cell metabolism (e.g., glucokinase) or key channel subunits e.g., SUR of K+(ATP). Other evidence suggests that increases in free fatty acids (ffas), resulting from developing obesity, may downregulate key regulatory features or even dissociate Ca2+ channels from secretory granules.39 However, to date, there has been no systematic study of glucose-induced depolarization, electrical activity or exocytosis in single cells from DM 2 islets. It is likely that progress will continue to be slow. DM 2 pancreases are not easily procured and often do not readily yield their islets. Also, in a disease that progresses slowly, major heterogeneity in basic defects underlying pathophysiology, may be present and superimposed on ageing-dependent “loss of vigor” of key regulatory features. Lastly, as we have learned from type 1 DM, even the most promising of small animal models may not really replicate the human disease.40 However, investigation may be facilitated by cryopreservation/thaw of islets, which preserves whole islet and single cell function, to promote storage, sharing and comparative testing of available tissue.41
In addition to enhancing our understanding of individual endocrine cell types in the islet, studies begun by Lacy, and now ongoing world-wide, have demonstrated just how interrelated cells in the islet micro-organ are. Lacy's early ultra-structural work42 vigorously continued by Orci and collaborators, demonstrated complex endocrine-endocrine cell and endocrine cellendothelial cell contacts implying complex “social interactions” of component cells. These include contiguity of β-cells with α- and δ-cells, inter-β cell gap junctions, structural polarization of β-cells between capillaries, and alignment of complex endothelial cell pores with regions of preferred exocytosis in β-cells. Electrophysiological studies have shown that electrical coupling of β-cells contribute to their co-ordinated electrical activity43 while secretion studies on single β-cells vs. those in small clusters have demonstrated the supra-linear augmentation of secretion resulting from cell-cell coupling. Furthermore, studies of the functional islet microvasculature have provided evidence for differential perfusion of islets. In many species the afferent circulation burrows into the core of the islet, where the majority of β- and δ-cells reside, and then branches as to return to the mantle where the majority of α-cells reside. This anatomical organization should permit β-cell secretions to inhibit α-cell secretion of glucagon and δ-cell secretion of somatostatin to inhibit both α- and β-cells.
Using an idea proposed by Snell in 1957 to explain the delayed growth or destruction of weakly antigenic transplanted tumors, in the early 1980s Lacy championed the concept of long-distance interactions between cells of the islet micro-organ and the immune system.44,45 The major immunological players in this interaction are (1) immune surveillance cells traversing the islet and recognizing unusual or foreign antigens and (2) immune affector cells returning from lymph nodes with instructions to damage or kill cells bearing the unusual or foreign antigens. Simply put, Lacy's idea was that presentation of allo- or auto-antigens by the wandering “passenger leukocytes,” now called dendritic cells/macrophages (or DC/MΦs) was critical for both autoimmunity of type 1 diabetes mellitus and islet transplant rejection, respectively. In the case of autoimmunity of diabetes, the autoantigens bound by the major histocompatibility complex (MHC) at the surface of the DC/MΦs were thought to be present at the surface of the β-cell. Contemporary candidates range from insulin, to processed viral antigens that mimic other β-cells proteins e.g., Coxsackie enterovirus antigens mimicking peptides derived from glutamic acid decarboxylase (GAD 64), or rotovirus antigens mimicking peptides derived from tyrosine phosphatase Ia (reviewed in ref. 46)In the case of islet transplant rejection, the alloantigens were thought to be present on the surface of capillaries of the allografted islet. Ultimately this leads to local activation of specifically pre-selected cytotoxic lymphocytes (CTLs) in the draining lymph nodes causing them to home in on the islets, destroy target cells and reduce islet function.
Passenger macrophages have come of age.47 Our current understanding is that when new specific antigens appear at the surface of foreign capillaries or β-cells undergoing destruction, they are taken up and processed by DC/MΦs (see Fig. 2). Antigen processed in the lysosomal pathway is ultimately presented on MHC Class II sites on these cells, while antigen processed in the proteosome pathway is presented on MHC class I. Surface antigen presentation converts immature, tolerogenic DC/MΦs to mature immunogenic DC/MΦs. After migration to local lymph nodes, mature DC/MΦs, via their liganded MHC class II sites and co-stimulatory sites bind to clonally selected sentinel (CD4+) T-helper (TH) cells, which express on their surface T-cell receptors (TCR) for MHC class I-peptide complex and co-stimulating molecules. The binding of surface T-cell receptors (TCRs) converts members of the TH cell population from a largely regulatory to a largely autoreactive phenotype (TH1), which begin to divide and secrete inflammatory cytokines, such as IFNγ. In addition, mature DC/MΦs via their MHC Class I/peptide complex also to CD8+ cells. The latter, plus the cytokines secreted by the activated TH1 cells, promote the maturation of CD8+ cells into cytotoxic T lymphocytes (CTLs).
Figure 2.
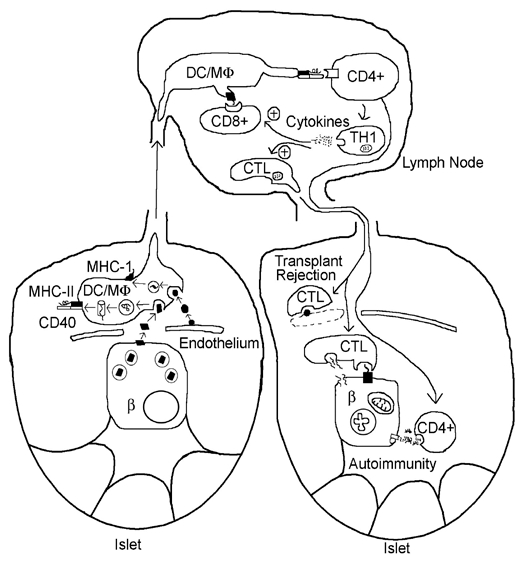
immune responses evoked by β-cells: concept of passenger leukocyte (dendritic cell/macrophage, DC/MΦ) as key intermediary in the development of islet autoimmunity and islet transplant rejection. See text for discussion. More recent evidence suggests that the homing of CTLs specific for insulin results from the presentation of antigen (insulin fragments) by endothelial cells.
During their circulation, the CD8+-derived CTLs are attracted into islets by chemokines released by the local inflammatory response to β-cells and as well as by MHC Class I/peptide complexes expressed on the surfaces of β-cells themselves. CTL form “immunological synapses” with the β-cells, thereby promoting β-cell apoptosis, secondary to release of granzymes and channel-forming perforins.
Lacy's “reduced model of islet function and dysfunction”: A bit of oral history concerning some assumptions underlying the vision of islet transplantation.
“I'm a simple pathologist; I believe what I can see and touch.” (Paul Lacy, September 1987). That's how Lacy began describing the concept of islet function that guided his work in our first extended conversation in 1987. Admitting it didn't encompass all that was known about islets, he thought it was fairly simple and utilitarian: “good for a practicing pathologist and would-be transplanter.”
Lacy conceived of islets of Langerhans as microperfused and autonomically innervated micro-organs containing at least three different types of endocrine cells each intercommunicating via intimate contacts and internal vasculature; this idea would later be called by Pipeleers “the bio-sociology of the islet”.48 However, Lacy saw the β-cell as the key cell in the islet because its hypoglycemia-inducing secretion, insulin, unlike the α-cell's hyperglycemia-inducing secretion, glucagon, could not be duplicated by any other hormone (such as adrenaline). Lacy was convinced that the β-cell's unusual biphasic response to a sustained rise in glucose was critical to its function. An early spike of insulin secretion, lasting 5–10 minutes, from a small pool of “emiocytosis-ready” insulin granules, was critical in saturating insulin receptors in liver and causing a decrease in glucose release from, and increase in glucose entry into that depot store. An ever increasing second or dome phase of insulin release, lasting up to hours, from a more slowly mobilized pool of maturing insulin granules, traveling along microtubules to the vicinity of plasma membrane surface49,50 was critical for steady state glucose uptake into muscle and fat. He recognized that insulin secretion was primed by the digestive tract via both a vagal reflex and endocrine secretions from gut lining. However, since biphasic secretion was preserved after islets were isolated into a denervated, non-perfused state and even after they had spread out into a monolayer in culture, Lacy conjectured that from a practical standpoint, if an islet were in a “safe, well perifused place” innervation, incretin priming and even intact internal capillary beds might not be essential for the maintenance of an intrinsic secretory pattern.
Lacy saw islets and islet cells as “plastic” rather than static tissue. Over their extended function, islets would need to expand during developmental demand for increased insulin output (e.g., pregnancy) probably through hypertrophy. Also being a tissue with high metabolic turnover, some cells would get worn-out and need to be replaced with new ones, probably through the infrequent mitoses of the healthiest cells.51 In fact, in experimental diabetes produced by partial pancreatectomy of the growing rodents, symptomatic disease only developed after the need for insulin outstripped the ability of the remnant hypertrophic islets to secrete it in a hyperglycemic environment.52,53 In contemporary parlance this is now called “functional islet reserve”.54 Hence, protecting the function of islets on a longterm basis required a sufficient complement of functioning islets to maintain near physiological circulating glucose and prevent the exposure of islets to prolonged hyperglycemia. However, he saw no reason why these processes would not occur even in environments foreign to islets provided the islets were in contact with the bloodstream and remained metabolically intact.
Lacy viewed isolated islets, lacking surrounding acinar tissue, as potentially poorly antigenic, provided passenger immune cells could be removed from islet vasculature and intrinsic endothelial cells were lost or replaced by “host” endothelium as functional capillary supply was re-hooked. (This assumption has finally been tested experimentally and supported.55) Hence, given a choice of transplanting a whole pancreas, which required vascular anastomoses that could immediately clot, as well as complex exocrine drainage exocrine draining, vs. transplanting isolated islets into accessible site, in his opinion the latter was clearly more preferable.
Lastly, Lacy viewed the generalized dysfunction in diabetic mellitus from a “β-cell-centric” perspective, one that was not nearly as popular before the mid-1990s as it is now. That is, if dysglycemia were controlled sufficiently early on most of the long-range consequences of diabetes, including retinopathy, neuropathy and nephropathy, might be prevented, ameliorated or even partially reversed. However, he thought that fine regulation of serum glucose could only achieved with a rapid feedback-controlled system requiring glucose-recognition by the insulin-dispenser emptying directly into the central blood stream, where it is needed, rather than into peripheral sites such as skin, where it would only be slowly absorbed. Hence for him islet transplantation into vasculature, or sites capable of intense vascularization, would be the most preferable source of insulin administration and the most physiological approach for insulin replacement therapy. Since long-term poor glucose control bred islet dysfunction, it would be necessary to transplant a full complement (literally a “pancreas'-worth”) of well-functioning islets and to have as many of them as possible continue to function for as long as possible.
Hence, for Lacy there were three essential prerequisites for islet transplantation. By the late 1980s good progress had been made towards satisfying at least two of these.
“Islet greediness” or the efficient harvest, in minimally ischemic fashion, as many islets as possible from a donor pancreas and then “ keeping as many as possible, as happy as possible, for as long as possible.” In the case of “islet harvest” from large animal pancreases, the solution was an ingenious bipartite isolation perfusion chamber designed by post-doctoral fellow Camillo Ricordi56 in to which the enzyme-infused pancreas was placed. By 1988, Ricordi, Scharp and Lacy were able to isolate as many as 800,000 islet equivalent from pancreatic of brain-dead human donors and nearly 400,000 islet equivalents from canine and pig pancreases. Islet preparations from these species were often >90% pure, and >90% viable, as assessed by light refraction and vital dye exclusion. Furthermore, in vitro islets of all species functioned well during perifusion as well post-transplantation even after several weeks in low temperature culture or after cryopreservation/thaw.
-
Delivery of isolated islets to an easily accessible quasi-pancreatic site. Soon after the first successful high yield isolations from rodent pancreases, Ballinger, Scharp and Lacy57 began their heroic efforts of transplanting small quantities of islets into a variety of sites: under the capsule of kidney; under the capsule of spleen; intra-peritoneally, on the omentum; and later even intratesticularly.58 The first experiments, in which islets were placed under the capsule of the kidney, demonstrated “proof of principle” that islets could secrete insulin and restore glycemia to syngeneic rats treated with streptozotocin, a β-cell specific toxin.
While technically a 2–3 ml volume of packed islets, obtained from a human donor, would easily be accommodated under the capsule of a single human kidney, practically subcapsular implantation required major surgery. Hence a less invasive approach to clinical islet transplantation was desirable. In addition, Lacy realized that insulin secretion into a site where first phase release would affect early uptake of glucose by liver was highly desirable from a physiological standpoint.
In 1973 Ballinger, Scharp and Lacy first reported the reestablishment of near euglycemia and normal fasting insulinemia in streptozotocin-treated rats by injection/embolization of 400–600 hand picked, syngeneic islets in the portal vein, where glucose-avid hepatocytes would have primary access to glucose for rapid deposition as glycogen. Furthermore, within 2–12 weeks, polyuria, polydypsia and hyperglycemia were abolished and thereafter islets sustained their function for many months. Histological, long term transplanted islets (>5 months) contained well granulated α-, β- and δ-cells that projected across the vascular space formed and formed direct contacts and even functional complexes with hepatocytes.59 In fact, neighboring hepatocytes showed increased glycogen and lipid deposits. (Quite recently this finding has been shown to have clinical implications in that in human patients who have received infusion of islets into the portal vein, the delayed development of regions of hepatic steatosis has been used to track the presence of functional islets.60) By the mid-1980s clinical interventional radiology was nearly ready to tackle tissue infusions into human portal veins. However, the key piece of data missing from all of the islet transplantation trials in animals, and only provided years later,61 was a detailed study of the metabolic state of the transplanted islets, including their oxygen consumption, and their abilities to undergo biphasic insulin secretion and to revascularize. As discussed below, this turned out to be truly critical issue in the preparation and survival of islet transplants.
-
Making islets non-immunogenic. In early rodent experiments, transplantation was performed syngeneically (between genetically identical individuals) or into a host with little capacity for transplant rejection. To further reduce the probability of rejection a single does of anti-lymphocyte serum or monoclonal antibody was infused into the host prior to transplant. However, for transplantation into a non-immunocompromised host of nonidentical genetic background, either islets had to be rendered non-immunogenic or clinical immunosuppression would be needed. Lacy's vision did not include chronic immunosuppression because he was fully cognizant of the vast array of systemic side-effects of contemporary immunosuppressants as well as their potential for very specific effects on islet metabolism. Lacy was convinced that “islets, as metabolic factories, won't thrive with high doses of catabolic drugs like corticosteroids nearby”.
Lacy spent much of the 1980s developing approaches to reduce islet immunogenicity by attempting to deplete isolated islets of dendritic macrophages (DC/MΦs) and remnant capillary endothelial cells. His approaches, including extended culture of islets at subphysiological temperatures and/or high oxygen tension, treatment with anti-dendritic cell antibody as well as islet cryopreservation and thaw were not only seminal in islet transplantation but also in raising general “immunological consciousness” about dendritic cells. Currently, making donor islets less immunogenic and recipients less reactive remains a field of active bench-to-bedside inquiry.62
Part II: The Practice of Islet Transplantation to Cure Diabetes
The experience of the first 20 years.
The proof of practicality of all great visions resides in the details of the success of wideranging daily application. Concerning islet transplantation, Lacy, ever the ebullient public enthusiast, remained the private cool-headed realist. He knew that rodent “diabetes” was not human diabetes mellitus and believed that the transition of islet transplant from short lived, inbreed caged rodent to long-lived, free ranging and genetically diverse man would be difficult. However, he also freely admitted that the only way to investigate and circumvent the shortcomings of human islet transplantation was to perform the “in vivo” human experiments and take what was learned at the bedside back to bench for improvement. In addition he noted that “what's worth doing never comes easy.” Nevertheless, by the late 1980s, a little over a decade after he authored a master plan for clinical islet transplantation,63 Lacy supervised the first partially successful transplantation.
By infusing, via an umbilical vein catheter, 800,000 nearly pure islets, harvested from 1.4 pancreata of brain dead patients, into the portal vein of an insulin-dependent diabetic renal transplant patient, already immunosuppressed with low doses of cyclosporine and prednisone, his team succeeded in maintaining the patient off exogenous insulin for 12 days.64 Within months, protégés, such as Ricordi, achieved more long term success with larger infusions of islets into other renal transplant patients treated with a steroid-free immunosuppression regime based on tacrolimus. In spite of the continuing dearth of tissue supply and the need for immunosuppression, the arduous race for the cure was on. Spurred by bursts of euphoria, especially one engendered by the “Edmonton protocol” utilizing sirolimus + tacrolimusbased, steroid-free maintenance immunosuppression occurred. Repeated infusions of a pancreas' worth of freshly isolated islets to brittle (hypoglycemia-prone) insulin dependent type 1 diabetics (DM 1) without renal allografts,65 were also attempted. To date, nearly a thousand patients have been transplanted with varying success using variants of the techniques presented the seminal paper by the Lacy team. We shall review this limited success, the clues these surgical “experiments” have offered us about engrafting islets, and how the return from bedside back to bench has allowed valuable clues to be tested individually and rigorously.
The most active islet transplantation centers (Edmonton, Miami, Minnesota and an international consortium) have reviewed their results after adoption of the Edmonton protocol now including daclizumab induction.66–68 Transplanted patients as a group had DM 1 for an average of 25 years, were of near ideal weight, had at least 50% normal creatinine clearance and experienced recurrent, severe hypoglycemia. Pancreases used as tissue sources were generally selected for cold ischemia time of less than 12 hours. The initial transplants consisted of 400,000 islet equivalents (IE) (∼5–10 cc of tissue of 30–90% islet purity), either freshly prepared or cultured for 24–72 hours, and determined to be both bacteria- and endotoxin-free. Transplantation was achieved by percutaneous transhepatic intraportal infusion of a small volume of heparinized solution. Up to two booster infusions were later given bringing the average cumulative total to 800,000 IE.
While 44–82% of patients were insulin independent at the end of the first year, only 7.5% remained so at the end of the fifth year, despite apparent 80% graft survival as determined by C-peptide secretion. Never-the-less, patients returning to insulin dependence had better glycemic control (HbA1c 7 vs. 9) with fewer episodes of hypoglycemia, the latter due to either reduced insulin requirement, improved glucose counter-regulation or some correction of symptomatic awareness of hypoglycemia. Chronic complications in transplanted patients included mouth ulcers, anemia, diarrhea, ovarian cysts, acne and increased need for treatment with antihypertensives and cholesterol lowering statin drugs. In addition, there were tendencies towards worsening of renal function and development of proteinuria, perhaps as a result of the immunosuppressive drugs used in the face of underlying diabetic glomerulosclerosis. Also, development of de novo donor specific antibodies in patients on continued immunosuppression, raised concerns about possible predisposition to worsened outcomes in subsequent major organ transplants. In spite of case reports (reviewed in reference 69) there are no controlled studies to indicate whether islet transplantation slows progression of long-term systemic complications of DM such as cardiomyopathy, macro- or micro-angiopathy, proliferative retinopathy, or peripheral/autonomic neuropathy. Hence results to date have caused some to question whether islet transplantation is ready for “prime-time” or just remains experimental and properly reserved as adjunctive therapy to renal transplantation in brittle diabetics with end-stage renal disease.
More fundamentally, the rapid declines in islet function post-transplant have caused even the key enthusiasts to reexamine the original vision more critically. However, as with all important new therapies, severe challenges tend to provoke useful responses. In the case of islet transplantation, perhaps the most intriguing challenge is the issue of whether, as Lacy had originally speculated, immunosuppression, might be the true limiting step or Achilles' heal of the entire process. Specifically, recent data suggest that in test organisms (and perhaps patients) with a history of autoimmune DM, T cell depletion produced by immunosuppressives such as FK and rapamycin might indirectly result in the homeostatic expansion of the remaining T-cells, such as those autoreactive to the β-cell, and thereby cause a recrudescence of autoimmune DM, now however masquerading as transplant failure. If so, should patients be specifically chosen for lack of recurrent or recent autoimmunity? Alternatively, would pre- or concurrent drug treatment of recipients to reduce memory T cells prevent expansion of anti-islet autoreactivity?
What islet transplantation has further taught us about the islet as a micro-organ.
Islet transplantation is still “a work in progress.”70,71 In retrospect, the overwhelming concern with obtaining sufficient islet mass for transplantation had caused “myopia” in the early transplant vision. On the one hand, there was a general lack of intimate knowledge and standardization of the metabolic state of islets being produced from the source pancreases and then transplanted after variable time in culture. On the other hand, there was little understanding of the stability of the islets as micro-organs outside of their physiological milieu and especially in the portal vein. For example, traditionally there was nearly universal reliance on optical measures for quantitation immediate “viability,” loose standards for quantitation of yield (the islet equivalent), and, at best, modest standards for effective “in vitro” secretion after acute culture (2- to 3-fold increases vs. >10-fold increase in the best quality islets). All of this led to the acceptance of mediocre quality tissue for transplantation. With the US Food and Drug Administration demanding quality assurance and standardization of the transplant “product or device” prior to its infusion, these features are now under active scrutiny from both the retrospective and prospective point of view. The condition of the pre-transplant islet and its dependence on the quality of the source pancreas as well as the treatment of islets during and after harvest are now tracked by every transplantation center.
In the case of the pre-transplant islet, first it was necessary to understand pre-harvest, which source of pancreases are most likely to prove the best tissue sources. Variables include donor age, adiposity, insulin reserve, and potential injury in the immediate pre-harvest state (e.g., during cytokine storm with brainstem death). There is general recognition of a central conundrum in islet harvestment: islets from the pancreases of young (<35) and lean donors, likely to be the most robust islets, are technically difficult to isolate, while those from pancreases of older (>50) and obese donors are somewhat larger and easier to liberate. Second, it was critical to minimize cold ischemia time of the pancreas both pre- and post-isolation. While islets are highly metabolic and do poorly under hypoxia, pancreases used for islet preparations are very often “left over” organs, offered for harvesting after of heart, kidneys and lungs have been obtained or after the pancreas has been rejected for whole organ transplantation. Cold ischemia time is often well beyond an optimum of 6–8 hours. Recent improvements include the use of oxygenated fluorocarbon solution transport. Moreover, the potency of, and exposure time to, enzymes in the islet isolation solutions, needed to be standardized and insured. Lastly, beyond the general consideration of whether the best islet products are shaggy vs. round and nearly fresh vs. cultured for several days, there was a critical need for individual aliquots of islets to be carefully evaluated and standardized prior to transplantation. Advances, which may prove critical to the standardizing harvested islets, are the rapid use of laser scanning cytometry and measurement of O2 consumption on exposure to glucose to characterize islets, fluorescent-activated sorting of islet cells and optical assay of mitochondrial membrane potential in product β-cells, and rapid assessment of insulin release by single islets using microfluidic perfusion chambers and secretion assays other than the very time consuming radio-immunoassay.
In the case of the transplanted islets, especially ones introduced into the portal vein, their post-transplant courses appear far from smooth. Following Paul Robertson's apt description, there is increasing evidence that the islet in the portal vein is a “stranger in a strange land” and suffers on a number of accounts (see Fig. 3). However, increasing recognition of key issues has stimulated a variety of approaches to ameliorating some of them.
Figure 3.
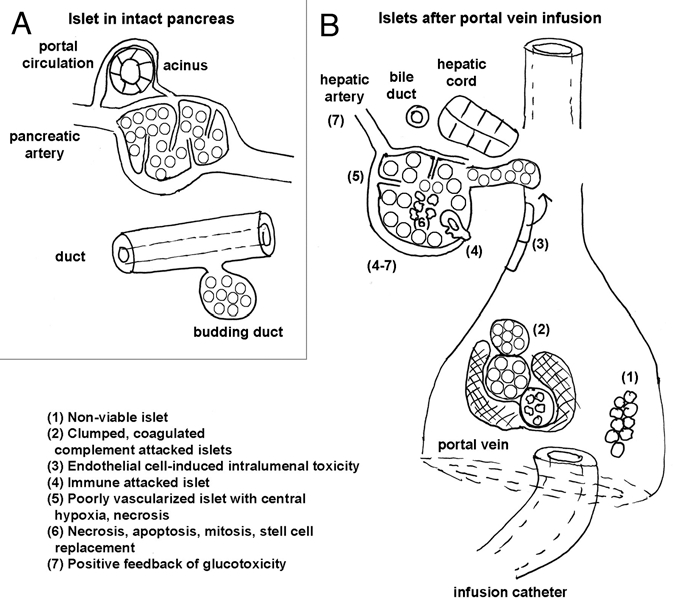
Complexities and hidden pitfalls inherent in islet transplantation, especially into the portal circulation. (A) The islet's sheltered, nurtured existence in its native environment, where its intrinsic microcirculation is intact, its portal circulation allows it to increase its blood flow by borrowing from acinar supply post-prandially, and its intrinsic replenished with cells or replacement as a micro-organ, when worn out. (B) The islet's precarious situation as a “stranger” post-transplantation (see text box legend).
Ischemic damage during procurement or culture results in an initial dearth of functional islets; a conservative estimate is that 30% of islets have necrotic cores at the time of infusion.
Immediate islet loss due to coagulation induced by tissue factors resulting from islet prep impurities (acinar and duct fragments) or portal vein thrombosis resulting from vessel injury during infusion.72,73
Ongoing loss of 30–40% of viable islets after introduction into the portal vein, even in the absence of portal thrombosis. An instant blood-mediated inflammatory/thrombotic reaction (IBMIR) has been identified,74 where stress + hypoxia-induced surface and soluble tissue factor, a cytokine receptor subfamily, results in expression of FVIIa, and macrophage chemoattractant factor. This results in platelet coagulation, complement activation and leukocyte infiltration of islets. In addition, hepatocyte and produces Kupffer cell activation and damages islet cells via INOS and cyclooxygenase 2 generation, even in absence of portal thrombosis. There is encouraging evidence that this reaction may be inhibited, among other ways, by preincubation or co-administration with nicotinamide and inactivated TFVIIa and or by coating islets with autologous endothelial cells (obtained from peripheral venous scrapings and expanded in vitro to coat islets).
Rapid onset of apoptosis may be induced by several factors. The combination of IBMIR and apoptosis may produce loss of 70% of infused islet tissue in 3 days. Here again, genetic and pharmacological inhibition has been possible in the forms of ex vivo transduction of islets with X-linked inhibitor of apoptosis (repressor of terminal caspase pathway).
Functional revascularization and its maintenance. Data from rodents suggests that long term, even under the best conditions in syngeneic rodents, less than 75% of transplanted islets remain intact. Islets retrieved from any engrafted site displayed decrease oxidative utilization of glucose, decreased insulin contents, and decreased regulated insulin release. However, intrapancreatically- transplanted islets seem to suffer the least while intraportally-infused islets seem to suffer the most.75 Islets recovered after intra-portal infusion often have poorly perfused cores, with intra-islet pO2 being 5–10 torr after implantation into the portal vein vs. 40 torr in the intact pancreas.76 Tissue hypoxia may acutely raise cytosolic [Ca2+] and contribute to constituitive insulin release, while decreasing glucose-induced insulin release, which requires vigorous aerobic, including mitochondrial, metabolism. Moreover, long-term this hypoxia may be detrimental to survival of β-cells predisposing them to stressinduced apoptosis, especially as β-cells lack a hypoglycemia/hypoxia inducible mitochondrial inner membrane protein that protects other cells that rapidly oxidize glucose, such as neighboring α-cells and cardiac myocytes.77 In humans and nonhuman primates, islets introduced into the portal circulation become enveloped in thrombus and then are slowly incorporated subendothelially in the vessel wall to be revascularized by the vaso-vasorum.75 This long term revascularization, achieved by an arduous, islet-threatening process, is likely to be suboptimal, in spite of evidence that islets secrete angiogenic factors, such as vascular endothelial cell growth factor-A (VEGF-A) and angiopoietin-1. Two novel approaches focusing on improving the islet vascularization are being developed. The first is based on tissue engineering and consists of co-culture of islets with bone marrow derived mesangial stem cells and endothelial cells to provide adequate source of angiogenic factors as well as promote invasion of endothelial cells and the sprouting of chimeric vessels from the islet. The second is pre-transplant treatment of islets with prolactin, a known angiogenic stimulant to islets.78
Loss of neural innervation prevents the neurally-mediated increase in islet perfusion during increased oxygen demand of glucose utilization occurring with stimulus-secretion coupling. This is likely not reversible given the geography of transplanted islets.
-
Poor islet cell proliferation combined with unlikely islet neogenesis should militate replacement of apoptotic β-cells and result in progressive decline in the number of functional islets. Clinically, attempts at slowing apoptosis and promoting islet neogenesis in patients with chronic allograft dysfunction have included administering exenatide, a long-acting analog of GLP-1.
In the future magnetic resonance imging, now being developed with targeted iron oxide “contrast agents” for use in rodents, may permit monitoring of islet engraftment and vascularization, immune rejection, glucotoxicity and the role of islet purity in ultimate longevity, thereby helping to evaluate current, as well as predict future, allograft function.79
What auto-transplantation of partially purified islets and whole pancreas transplant have taught us about the islet as a micro-organ.
In addition, valuable lessons have been learned about the viability of the islet as a micro-organ when the results of transplantation of purified islets are compared with two other replacement techniques. They are (1) auto transplantation of pancreatic tissue, in patients with chronic pancreatitis and (2) simultaneous renal pancreatic transplantation, in diabetics with renal failure two competitor islet replacement techniques. First, from the experience of the Robertson's group it is clear that rapidly prepared, partially purified islet harvests autotransplanted into the portal vein can provide of reduction in fasting glucose for as long as 13 years. Here reduction in glycosylated hemoglobin is highly correlated with the mass of initial islet infused. Glucagon secretion in response to arginine, but not to hypoglycemia, is also well preserved.80 From this experience, as predicted by Lacy, the functional quantity of transplant mass is critical to its subsequent success and, as suggested by the islet transplantation studies, the portal circulation might be less than ideal for stable counter-regulatory function in glucose control. Surprisingly, in light of Lacy's reduced model of islet function, the mass of co-transplanted acinar and ductal tissues did not seem to be an impediment to subsequent islet function. Perhaps the trade-off point, between the trauma of islet purification and the avoidance of potential for tissue inflammatory response by less extensive purification of islets, needs to be shifted. As suggested by tissue culture studies, including those of pancreatic discards after islet isolation,81,82 cells within fragments of ductal and acinar tissue liberated from their restraining matrix, might actually transdifferentiate and bud off islets, or else secrete factors that protect islets or enhance their revascularization during hyperglycemia. Interestingly, when the Edmonton group carefully compiled the characteristics of infused islet tissue and compared this with the survival of the subsequent transplant, after two years transplantation they found a significant positive correlation between the function of the intra-portally administered allografts and the number of ductal (cytokeratin-19 positive) cells in the infusate. In the same study, optical estimates of islet purity did not correlate well with insulin staining of tissue and long-term outcome of graft did not correlate well with post-isolation in vitro glucoseinduced insulin secretion.
Second, despite the feared arduousness of the procedure, beginning in the 1990s pancreas transplantation became generally successful. New techniques for organ harvesting reduced exocrine damage; bladder drainage of ductal secretions permitted easy of assessment of amylase secretion as an index of exocrine function; subsequent enteric drainage avoided complications of intermittent metabolic acidosis and volume depletion; and improved HLA matching and immunosuppressive regimes (based on tacrolimus, rather cyclosporine), improved graft survival.83 Simultaneous renal/pancreatic transplantation had threeyear survival rate of 70–80%, or similar to most other types of organ transplantation, and provided a means of effective control of glycemia and autonomic insufficiency. Solitary pancreatic transplantation could reverse lesions of diabetic nephropathy and as well as macro-vascular changes. In this case Lacy may have been too pessimistic: in the face of well-preserved islet mass and adequate attachment to central vascular supply, a limited degree of chronic rejection and cytotoxicity from immunosuppressant therapy may not be as swift and potent islet killer as he suspected.
Part III: Looking Over the Horizon at Emerging Alternative Approaches and Perspectives on the Roads not Taken
My second extended conversation with Lacy occurred in 2000 in one of his visits to Washington University after his retirement from active scientific inquiry. Lacy was now the thoughtful contributor to advisory boards in academia and industry and an elder statesman in the ever expanding diabetes community. His views were much more philosophical. As far as curing diabetes by islet transplantation, he realized that several limiting problems had emerged. These included the likely insurmountable problem of scarcity of human tissue; the vagaries of immunology and immunosuppression; and the nagging complexity of transplanting this “simple” micro-organ, including whether the portal vein was an ideal target site. While he had considered alternative sites for transplantation, including intraperitoneal (omental) and even intramuscular, paralleling its increasingly use for parathyroid transplantation, he was more concerned that realistically transplantation would probably require a new approach: use of encapsulated surrogate islet tissue. However, he voiced a more fundamental concern: over the length of his career the scope of diabetes had changed from an autoimmune disease of a few immunologically unfortunate individuals into an environmentally induced epidemic of over-calorization in a sedentary population,84 an impending public health catastrophe that no program of islet replacement would effectively serve. Put another way, in 50 years, the bad genetic background underlying the bulk of diabetes had shifted from the tendency toward autoimmunity, which always made life miserable, to the tendency towards metabolic thriftiness, which in the past undoubtedly had a clear selection advantage. Though in no way discounting DM 1 as a major disease, Lacy wondered how drastically different his career in diabetes research would be if he were starting out in 2000. Would he largely be investigating how to prevent islet toxicity due to chronic, excessive nutrient fuel rather than trying to replace islet function? To conclude this tribute we shall refer to some of Lacy's thoughts, as well as recent developments, concerning these areas.
Identifying a surrogate transplant tissue for an alternative transplantation approach.
If not human islets for transplantation, what then? As we have emphasized, Lacy subscribed to the following working definition of β-cells: a cell population that (1) senses glucose over a physiological range of 3–7 mM and responds by secreting insulin, (2) interacts appropriately with glucagon-secreting α-cells and somatostatin-secreting δ-cells and (3) regulates its own mass by precisely balancing of mitosis and apoptosis over years while still being capable of undergoing an occasionally growth spurt (such as in pregnancy). Hence β-cells could only be replaced by β-cells. If scarcity of human tissue was the critical issue, this could be circumvented with xenotransplantation using stable islets from pigs, which are long-lived organisms with lower sensitivity to destruction by autoimmunity85,86 and whose insulin, differing from human by only 1 amino acid, had been successfully used to treat diabetics for decades. At this time he preferred xenotransplantation to de novo tissue engineering using transformed clonal cell lines or elaborated stem cells and perhaps even to tissue extracted from life-support brain dead humans. Specifically he thought that pigs could be raised in a sterile environment (now in development in Minnesota and New Zealand), screened and genetically modified to reduce risks of xeno-zoonosis.87 The ability to harvest pancreases from health animals rather than life-supported cadavers would substantially reduce the problems of using ischemic, cytokine-activated tissue likely exposed pre-harvest to a variety of pathogens difficult to test for, wide fluctuations in pre-harvest organ perfusion and the possible effects intravascular coagulation causing micro-angiopathy. The issue of islet xenoimmunology and transplantation might be addressed (1) by genetic engineering to produce organs that minimize host immune or (2) by “immunoisolation” of transplanted tissues. “Knock out the enzymes that trigger reactions or surround the islets with a semi-permeable membrane giving them access to nutrients but not antibodies or immune cells. Then hopefully they will secrete small molecules like insulin and not be a source of retrovirus or oncogenes.” Currently, it is possible knock out of α 1,3 galactosyltransferase,85 an antigen responsible for antibody response and to engineer inhibitors of the complement system cascade. Lacy was enthusiastic about encapsulation of islets in a selectively permeable matrix.88,89 “With introduction into the peritoneal cavity, they could even be retrieved and exchanged for new ones if they simply wore out.” The major question in his mind was whether to use embryonic, foetal, neonatal or young or adult pigs as islet sources. Lacy's new vision of islet transplantation was actually addressing two “brave new world” areas, xenotransplantation and encapsulation, which have gained increasing interest of the past decade.
In the early 1990s, Lacy and Scharp,90 along with similarly-minded investigators, had begun work on islet encapsulation. Since the mid-1990s, individually encapsulated islets (in microspheres) or small clusters of encapsulated islets (in cylindrical diffusion capsules) had been introduced free into the bloodstream or into the peritoneal cavity. Since then, islets have also been seeded onto hollow fiber semi-permeable membranes to form a bio-hybrid device resembling a modern dialyzer and perfused as an arteriovenous shunt.89 Generally, the encapsulating medium has consisted of a biocompatible alginate whose surface is cross-linked with calcium to form capsules that are then coated with poly-L-lysine (PLL). Key problems using this approach have been loss of islet function and continuing need for immunosuppression, two likely related phenomena. Over time, the beads often collapse, and the PLL permits growth of fibroblasts attracting CD4+ T cells, B cells and macrophages and causing the accumulation of cytokines, chemokines, complement and immunoglobulins, all of which impair function of the encapsulated islets.91 While implantation of these capsules has improved glucose control in animal studies, it often proved dif- ficult to demonstrate significant insulin secretion by retrieved beads. However, there has been recent good news in the form of better understanding of why encapsulated islets are poor secretors as well as the design of better beads.
Transplantation of porcine islets in the absence of encapsulation has also been examined, with resultant reports of prolonged reversal of diabetes reported in non-human primates after intraportal infusion of either adult or neonatal porcine islets.92,93 While heavy immunosuppressive targeting of T cells was used to delay eventual T-cell mediated rejection, genetic engineering to eliminate hyper-acute rejection of solid organs, was not attempted. The ability to make pig transgenics, however, suggests that IBMIR might be “engineered away”. The issue of the relative advantage of neonatal vs. adult islets remains: while neonatal islets do not function immediately, they are less immunogenic and have potential for growth. Encouragingly, thus far no transmission of porcine endogenous retroviruses has been reported.
Finally I took the risk and asked about his views of the then developing research with cell based therapies for β-cell replacement: engineered clonal β-cells or harvesting/differentiating stem cells to be β-cell-like. His responses were bits of wry skepticism delivered in a mildly irritated tone. First, he asked how trustable one might expect a tumor cell to be regarding slow reproduction and stably secretion vs. reversion to uncontrolled proliferation and unpredictable or episodic secretion. Second, he asked how one would actually identify the stem cell for an adult β-cell and if one started far back in development would one simply be growing foregut teratomas for “a heck of a long time before figuring anything out.”
However, over the past ten years substantial progress has been made on harvesting/differentiating/engineering β-cell-like entities. While I'd anticipate Lacy would still have a healthy skepticism regarding these endeavors, he'd probably maintain a real interest in them. Certainly, he'd be very pleased to learn of new evidence supporting his contention that the best way to replace a β-cell is with another real β-cell. Based on genetic linkage tracing analysis, the major source for steady state β-cell replacement as well as proliferation after partial pancreatectomy, or targeted ablation is from extant ones by simple cell division. Furthermore, it is now proposed that cell division is the source of islet mass expansion in pregnancy, after gastric bypass surgery, and perhaps in partial reversal/recovery from autoimmune diabetes.94,95 Unfortunately, it has also become clearer that currently used immunosuppressive drugs inhibit regenerative restoration of reduced β-cell mass by cell division. However, being an astute biologist, he'd likely anticipate that there is still some room for a facultative endocrine progenitor cell in the adult pancreas. With partial pancreatic duct ligation causing acinar degeneration and duct proliferation, cells expressing the transcription factor neurogenin-3 (NGn3) can be harvested and then injected into explanted embryonic pancreas to differentiate into mature islet endocrine cells, recapitulating their role in the embryonic mode of endocrine pancreatic development.96–98 This would tend to downplay potential roles of bone marrow, spleen and liver as primary sources of stem cell for adult islet regeneration but suggest that going several steps back to attempting might not be entirely farfetched.99
Assuredly, he'd likely be interested in some tantalizing developments suggesting the possibilities of (1) differentiating an embryonic stem (i.e., pluripotential cells derived from the inner mass of the blastocyst) through a pancreatic endodermal stage, which then could be matured into pro-endocrine cells, and (2) reprogramming of adult pancreatic exocrine cells into β-cells by inducing the re-expression of several transcription factors.100 In the first case, one such recent attempt has made use of an initial induction factor, activin A and culminated in the maturation of foregut-like tissue after several month of implantation/maturation in mice made diabetic with streptozotocin. Improved glycemic control was achieved, though C-peptide secretion from these cells are not well regulated. In the second case, the transcription factors are NGn3, Pdx1 and Mafa.
Lastly, he'd likely give a critical eye to a recently reported conditionally transformed (reversibly immortalized) nontumorigenic human β-cell clone (NAKT-15). These reverted cells display a large complement of insulin granules, hormone processing enzymes and β-cell transcription factors (e.g., Pdx-1) as well as impressive increases in insulin and C-peptide secretion in response to 25 mM glucose, and control blood glucose for up to 30 weeks in combined immunodeficient mice made diabetic with streptozotocin.101 One might almost here Lacy say that we're not there yet, but a little closer.102
Interrupting islet gluco-and lipo-toxicity and prevention of type 2 DM.
In the 1990s the previously vague issue of glucotoxicity, which Lacy and his endocrinology colleague David Kipnis recognized in the 1960s, started to be more clearly objectified. While short periods (few hours) of hyperglycemia may be stimulus for β-cell mitosis and neogenesis, prolonged hyperglycemia has an opposite effect.101 Several lines of experiments revealed that islets incubated with even twice normal glucose concentration (11 mM) for as little as one week showed increased rates of insulin release at low (1.7–3 mM) glucose yet failed to increase their secretion further at elevated (16.7 mM) glucose, despite having adequate insulin stores and pro-insulin synthesis and hyperglycemia-induced increases in glucose oxidation.103 β-cells had “tuned out” to glucose, but why? The situation was clearly worsened by incubation with 28 mM glucose, where insulin synthesis was massively downregulated. More recent data suggest that in partially pancreatectomized rats with elevated serum glucose concentrations, β-cells in remnant islets actually dedifferentiate and lose mRNA coding for most of the proteins in the cascade for glucose signaling. Surprisingly, they also upregulate mRNA coding for anti-oxidant and anti-apoptotic processes (e.g., glutathione peroxidase, superoxide dismutase and catalase), which are normally present at quite low levels, and they are partially protected against corticosteroid-induced apoptosis. 104 In a sense, this may represent a balance state of “don't overuse it and don't rapidly loose it.” Islets need this protection as hyperglycemia, per se, may cause β-cell damage. Prolonged exposure to elevated glucose may result, among other features, in (1) secretion of cytokines, such as IL-1γ that induce nitric oxide synthetase and the production of nitric oxide, which is toxic to β-cells, probably by inducing both apoptosis and an inflammatory response; and (2) over-synthesis of pro-insulin to be trafficked through the endoplasmic reticulum (ER) resulting in “ER stress.”105
With this as a basis, attempts have been made in experimental models to protect β-cells from toxicity and potentially reverse diabetes.106,107 These include rendering patients truly euglycemic by insulin therapy (as opposed to glucose control by an oral hypoglycemic agent or a peripheral insulin sensitizer); administration of long-acting analogs of glucagon-like peptide 1 (GLP-1), which in addition to enhancing glucose-induced insulin release also appears to stimulate β-cell proliferation and inhibit apoptosis; and/or administration of a natural soluble IL-1 receptor antagonist, IL-1Ra. Other approaches include administration of anti-CD3 antibodies to halt autoimmune destruction and bone marrow engraftment at first detection of autoimmune DM.108 It is worth recalling that as the metabolic syndrome (or what we might call “hypertensive diabesity”) develops in glucotoxic individuals, dyslipidemia and β-cell lipotoxicity also occurs. The latter combination is what appears to be most injurious to isletsin that saturated fatty acids exert pro-apoptotic effects through production of ceramide that injures mitochondria while low density lipoproteins appear to be apoptosis-inducing through c-Jun N-terminal kinase. However, both glucotoxicity and lipotoxicity, if caught early, might be pharmacologically reversible as well as epidemiologically preventable.
Epilogue
Paul Lacy was a scientist with enormous drive and focused vision. When others asked “wherefore and how” he responded with an emphatic “why not,” and galvanized others to enthusiastically pursue practical answers with him. He seemed to be guided by the dictim of Albert Einstein that “nature, though subtle, is never treacherous.” This spirit has been transmitted to his long term scientific partner David Scharp and former postdoc Camillo Ricordi who vigorously pursue Lacy's vision of curative islet transplantation.
Lastly, Paul Lacy conducted his career with intellectual honesty, openness and generosity of spirit to colleagues with diverse interests in islet biology and immunology as well as to generation of medical student, whom he taught and inspired at Washington University. While studying the sometimes illusive islet, he personified an ethos of “no man is an island entire of himself.” That personal legacy may be as memorable as his scientific one.
Acknowledgements
This work was supported by NIH DK020579 (DRTC, Washington University in St. Louis. I thank Phillip Blevins (Teaching Support for Medical Education Division, Washington University in St. Louis) and Alon Friedman (Advanced Teacher Education Institute, Shlomi, Israel) for help with preparation of figures.
Footnotes
Previously published online: www.landesbioscience.com/journals/islets/article/12156
References
- 1.Latta P, Ricordi C, Scharp D. Paul E. Lacy MD, PhD, February 7, 1924–February 15, 2005. Am J Transplant. 2005;5:976–977. doi: 10.1111/j.1600-6143.2005.00929.x. [DOI] [PubMed] [Google Scholar]
- 2.Hellerstrom C. The life story of the pancreatic B cell. Diabetologia. 1984;26:393–400. doi: 10.1007/BF00262208. [DOI] [PubMed] [Google Scholar]
- 3.Lacy PE, Kostianovsky M. A method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–39. doi: 10.2337/diab.16.1.35. [DOI] [PubMed] [Google Scholar]
- 4.Howell SL, Fink CJ, Lacy PE. Isolation and properties of secretory granules from rat islets of Langerhans I. Isolation of a secretory granule fraction. J Cell Biol. 1969;41:154–161. doi: 10.1083/jcb.41.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashcroft SJ, Weerasinghe LC, Randle PJ. Interrelationship of islet metabolism, adenosine triphosphate content and insulin release. Biochem J. 1973;132:223–231. doi: 10.1042/bj1320223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grodsky GM, Bennett LL. Cation requirements for insulin secretion in the isolated perfused pancreas. Diabetes. 1966;15:910–913. doi: 10.2337/diab.15.12.910. [DOI] [PubMed] [Google Scholar]
- 7.Curry DL, Bennett LL, Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83:572–584. doi: 10.1210/endo-83-3-572. [DOI] [PubMed] [Google Scholar]
- 8.Dean PM, Mathews EK, Sakamoto Y. Pancreatic islet cells: effects of monosaccharides, glycolytic intermediates and metabolic inhibitors on membrane potential and electrical activity. J Physiol. 1975;246:459–478. doi: 10.1113/jphysiol.1975.sp010899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kostianovsky M, McDaniel ML, Still MF, Codilla RC, Lacy PE. Monolayer cell culture of adult rat islets of Langerhans. Diabetologia. 1974;10:337–344. doi: 10.1007/BF02627736. [DOI] [PubMed] [Google Scholar]
- 10.Lacy PE, Howell SL, Young DA, Fink CJ. A new hypothesis of insulin secretion. Nature. 1968;219:1177–1180. doi: 10.1038/2191177a0. [DOI] [PubMed] [Google Scholar]
- 10a.Marchetti P, Scharp DW, Mc Lear M, Gingerich R, Finke E, Olack B, et al. Pulsatile insulin secretion from isolated human pancreatic islets. Diabetes. 1994;43:827–830. doi: 10.2337/diab.43.6.827. [DOI] [PubMed] [Google Scholar]
- 11.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta cell. Prog Biophy Mol Biol. 1991;54:87–141. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 12.Misler S, Barnett DW, Gillis KG, Pressel DM. Electrophysiology of stimulus-secretions coupling in human beta-cells. Diabetes. 1992;41:1221–1228. doi: 10.2337/diab.41.10.1221. [DOI] [PubMed] [Google Scholar]
- 13.Grynkiewicz G, Poenie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescent properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 14.Seino S. ATP-sensitive potassium channels: model of heteromultimeric potassium channel receptor assemblies. Annual Rev Physiol. 1999;61:337–361. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- 15.Gillis KD, Misler S. Single cell assay of exocytosis from pancreatic islet B cells. Pfluegers Arch-EJP. 1992;420:121–123. doi: 10.1007/BF00378654. [DOI] [PubMed] [Google Scholar]
- 16.Ammala C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicted by action potentials and voltage clamp calcium currents in individual mouse pancreatic beta cells. J Physiol. 1993;472:655–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith PA, Duchen MR, Ashcroft FM. A fluorometric and amperometric study of calcium and secretion in islolated mouse pancreatic beta-cells. Pfluegers Arch-EJP. 1995;430:808–818. doi: 10.1007/BF00386180. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Misler S. Amperometric detection of quantal secretion from patch-clamped rat pancreatic beta cells. J Biol Chem. 1996;271:270–277. doi: 10.1074/jbc.271.1.270. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Shen H, Atkinson MA, Kennedy RT. Detection of exocytosis at individual pancreatic beta cells by amperometry at a chemically modified microelectrode. Proc Natl Acad Sci. 1995;92:9608–9612. doi: 10.1073/pnas.92.21.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barg S, Ma X, Eliasson L, Galvanovskis J, Goepel SO, Obermuller S, et al. Fast exocytosis with few calcium channels in insulin secreting mouse pancreatic beta cells. Biophys J. 2001;81:3308–3323. doi: 10.1016/S0006-3495(01)75964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gromada J, Bokvist K, Ding WG, Holst JJ, Nielsen JH, Rorsman P. Glucagon-like peptide 7–37 amide stimulated exocytosis in human pancreatic beta-cells by both proximal and distal steps in stimulus-secretion coupling. Diabetes. 1998;47:57–165. doi: 10.2337/diab.47.1.57. [DOI] [PubMed] [Google Scholar]
- 22.Silva AM, Dickey AS, Barnett DW, Misler S. Ion channels underlying stimulus-secretion coupling and its cell-cell variety in beta cell of transplantable porcine islets of Langerhans. Channels. 2009;3:91–100. doi: 10.4161/chan.3.2.7865. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Gillis KD. A highly Ca-sensitive pool of granules is regulated by glucose and protein kinase in insulin-secreting INS-1 cells. J Gen Physiol. 2004;124:641–651. doi: 10.1085/jgp.200409081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misler S, Zhou Z, Dickey AS, Silva AM, Pressel DM, Barnett DW. Electrical activity and exocytotic correlates of biphasic insulin secretion from beta-cell of canine islets of Langerhans. Channels. 2009;3:1811–1893. doi: 10.4161/chan.3.3.8972. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen MG, Sherman A. Newcomer insulin secretory granules as a highly calcium sensitive pool. Proc Natl Acad Sci USA. 2009;106:7432–7436. doi: 10.1073/pnas.0901202106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thurmond DC, Gonelle-Gispert C, Furukawa M, Halban PA, Pessin JE. Glucose stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimidesensitive factor attachment protein receptor protein) complex. Mol Endocrinol. 2003;17:732–742. doi: 10.1210/me.2002-0333. [DOI] [PubMed] [Google Scholar]
- 27.Barg S, Olofsson CS, Schreiver-Abein J, Wendt A, Gebre-Medhin S, Renstroem E, et al. Delay between fusion pore opening and peptide release from large dense core vesicles in neuroendocrine cells. Neuron. 2002;33:287–299. doi: 10.1016/s0896-6273(02)00563-9. [DOI] [PubMed] [Google Scholar]
- 28.Ma L, Bindokas VP, Kuznetsov A, Rhodes C, Hays L, Edwardson JM, et al. Direct imaging shows that insulin granule exocytosis occurs by complete vesicle fusion. Proc Natl Acad Sci. 2004;101:9266–9271. doi: 10.1073/pnas.0403201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohara-Imaizumi M, Nakamichi Y, Tanaka T, Ishida H, Nagamatsu S. Imaging exocytosis of single insulin secretory granules with evanescent wave microscopy. J Biol Chem. 2002;277:3805–3808. doi: 10.1074/jbc.C100712200. [DOI] [PubMed] [Google Scholar]
- 30.Michael DJ, Geng X, Cawley NX, Loh YP, Rhodes CJ, Drain P, et al. Fluorescent cargo proteins in pancreatic beta cells: design determines secretion kinetics as exocytosis. Biophys J. 2004;87:3–5. doi: 10.1529/biophysj.104.052175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michael DJ, Xiong W, Geng X, Drain P, Chow RH. Human insulin vesicle dynamics during pulsatile secretion. Diabetes. 2007;56:1277–1288. doi: 10.2337/db06-0367. [DOI] [PubMed] [Google Scholar]
- 32.Braun M, Wendt A, Birnir B, Broman J, Elliasson L, Galvanovskis J, et al. Regulated exocytosis of GABA-containing synaptic microvesicles in pancreatic betacells. J Gen Physiol. 2004;123:191–204. doi: 10.1085/jgp.200308966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goepel SO, Kanno T, Barg S, Weng X-G, Gromada J, Rorsman P. Regulation of glucagon release in mouse alpha-cells by KATP channels and inactivation of TTX-sensitive Na channels. J Physiol. 2000;528:509–520. doi: 10.1111/j.1469-7793.2000.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wendt A, Birnir B, Busschard K, Gromada J, Salehi A, Sewing S, et al. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes. 2004;53:1038–1045. doi: 10.2337/diabetes.53.4.1038. [DOI] [PubMed] [Google Scholar]
- 35.Braun M, Ramracheya R, Amisten S, Bengtsson M, Moritoh Y, Zhang Q, et al. Somatostatin release, electrical activity, membrane currents and exocytosis in human pancreatic delta cells. Diabetologia. 2009;52:1566–1578. doi: 10.1007/s00125-009-1382-z. [DOI] [PubMed] [Google Scholar]
- 36.Ashcroft FM. ATP-sensitive channnelopathties: focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nichols CG. K-ATP channnels as molecular sensors of cell metabolism. Nature. 2006;4449:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 38.Hattersley AT, Ashcroft FM. Activating mutations of Kir6.2 and neonatal diabetes: clinical syndromes, new clinical insights and new therapy. Diabetes. 2005;54:2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 39.Atkinson MA. Thirty years of investigating the autoimmune basis for type 1 diabetes: why can't we prevent or reverse this disease? Diabetes. 2005;54:1253–1263. doi: 10.2337/diabetes.54.5.1253. [DOI] [PubMed] [Google Scholar]
- 40.Hoppa MB, Collins S, Ramracheya R, Hodson L, Amisten S, Zhang Q, et al. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca channels from secretory granules. Cell Metabolism. 2009;10:455–465. doi: 10.1016/j.cmet.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misler S, Dickey A, Barnett DW. Maintenance of stimulus-secretion coupling and single beta-cell function in cryopreserved-thawed human islets of Langerhans. Pfluegers Arch-EJP. 2005;450:395–405. doi: 10.1007/s00424-005-1401-y. [DOI] [PubMed] [Google Scholar]
- 42.Williamson JR, Lacy PE, Grieder JW. Ultrastructural changes in islets of the rat produced by tolbutamide. Diabetes. 1961;10:460–469. doi: 10.2337/diab.10.6.460. [DOI] [PubMed] [Google Scholar]
- 43.Benninger RKP, Zhang M, Head S, Satin Ls, Piston DW. Gap junction coupling and calcium waves in pancreatic islets. Biophys J. 2008;95:5048–5061. doi: 10.1529/biophysj.108.140863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janney CG, Davie JM, Lacy PE, Finke EH. Characterization of lymphocytes from rejected and non-rejected islet xenografts. Transplantation. 1982;33:585–587. doi: 10.1097/00007890-198206000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Lacy PE, Finke EH. Activation of intra-islet cells causes destruction of islet cells. Am J Path. 1991;138:1183–1190. [PMC free article] [PubMed] [Google Scholar]
- 46.Wong FS, Janeway CA. Insulin-dependent diabetes mellitus and its animal models. Curr Opinion Immunol. 1999;11:643–647. doi: 10.1016/s0952-7915(99)00031-x. [DOI] [PubMed] [Google Scholar]
- 47.Janeway CA. How the immune system protects the host from infection. Microbes Infect. 2001;3:1167–1171. doi: 10.1016/s1286-4579(01)01477-0. [DOI] [PubMed] [Google Scholar]
- 48.Pipeleers D. The bio-sociobiology of pancreatic beta cells. Diabetologia. 1987;30:277–291. doi: 10.1007/BF00299019. [DOI] [PubMed] [Google Scholar]
- 49.Lacy PE, Howell SL, Young DA, Fink CJ. A new hypothesis of insulin secretion. Nature. 1968;219:1177–1180. doi: 10.1038/2191177a0. [DOI] [PubMed] [Google Scholar]
- 50.Lacy PE, Young DA, Fink CJ. Studies on insulin secretion in vitro from isolated islets of the rat pancreas. Endocrinology. 1968;83:1155–1161. doi: 10.1210/endo-83-6-1155. [DOI] [PubMed] [Google Scholar]
- 51.Bonner-Weir S. Beta-cell turnover: its assessment and implications. Diabetes. 2001;50:20–24. doi: 10.2337/diabetes.50.2007.s20. [DOI] [PubMed] [Google Scholar]
- 52.Martin JM, Gregor WH, Lacy PE, Evens RG. The effect of hyperglycemia upon islet regeneration in rats. Diabetes. 1963;12:538–544. doi: 10.2337/diab.12.6.538. [DOI] [PubMed] [Google Scholar]
- 53.Martin JM, Lacy PE. The prediabetic period in partially pancreatectomized rats. Diabetes. 1963;12:238–242. [Google Scholar]
- 54.Teuscher AU, Kendall DM, Smets YPC, Leone JP, Sutherland DER, Robertson RP. Successful islet autotransplantation in humans: functional insulin secretory reserve as an estimate of surviving islet cell mass. Diabetes. 1998;47:324–330. doi: 10.2337/diabetes.47.3.324. [DOI] [PubMed] [Google Scholar]
- 55.Pericin M, Althage A, Freigang S, Hengartner H, Rolland E, Dupraz P, et al. Allogeneic b-cells correct diabetes and resist immune rejection. Proc Natl Acad Sci USA. 2002;99:8203–8206. doi: 10.1073/pnas.122241299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW. Automated method for the isolation of human pancreatic islets. Diabetes. 1988;3:413–420. doi: 10.2337/diab.37.4.413. [DOI] [PubMed] [Google Scholar]
- 57.Ballinger WF, Lacy PE. Transplantation of intact pancreatic islets in rats. Surgery. 1972;72:175–186. [PubMed] [Google Scholar]
- 58.Kemp CB, Knight MJ, Scharp DW, Ballinger WF, Lacy PE. Effect of transplantation site on the results of pancreatic islet isografts in diabetic rats. Diabetologia. 1973;9:486–491. doi: 10.1007/BF00461694. [DOI] [PubMed] [Google Scholar]
- 59.Kemp CB, Knight MJ, Scharp DW, Ballinger WF, Lacy PE. Transplantation of isolated pancreatic islets into the portal vein of diabetic rats. Nature. 1973;244:447–453. doi: 10.1038/244447a0. [DOI] [PubMed] [Google Scholar]
- 60.Pipeleers-Marichal MA, Pipeleers DG, Cutler J, Lacy PE, Kipnis DM. Metabolic and morphologic studies on intraportal islet transplanted rats. Diabetes. 1976;25:1041–1051. doi: 10.2337/diab.25.11.1041. [DOI] [PubMed] [Google Scholar]
- 61.Markmann JF, Rosen M, Siegelman ES, Soulen MC, Deng S, Barker CF, et al. Magnetic resonance—defined periaortal stenosis following intraportal islet transplantation. Diabetes. 2003;52:1501–1594. doi: 10.2337/diabetes.52.7.1591. [DOI] [PubMed] [Google Scholar]
- 62.Inveradi L, Kenyon NS, Ricordi C. Islet transplantation: immunological perspectives. Current Opin. Immunol. 2003;15:507–511. doi: 10.1016/s0952-7915(03)00115-8. [DOI] [PubMed] [Google Scholar]
- 63.Lacy PE. Workshop on pancreatic islet cell transplantation in diabetes. Diabetes. 1978;27:427–429. doi: 10.2337/diab.27.4.427. [DOI] [PubMed] [Google Scholar]
- 64.Scharp DW, Lacy PE, Santiago JV, McCullough CS, Weide LG, Falqui L, et al. Insulin independence after islet transplantation into Type I diabetic patient. Diabetes. 1990;39:515–518. doi: 10.2337/diab.39.4.515. [DOI] [PubMed] [Google Scholar]
- 65.Shapiro AMJ, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type I diabetes mellitus using a glucocorticoid free immunosuppressive regimen. N Engl J Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 66.Froud T, Ricordi C, Baidal DA, Hafiz MM, Ponte G, Cure P, et al. Islet transplantation in diabetes mellitus using cultured islets and steroid free immunosuppression: Miami experience. Am J Transplant. 2005;5:2037–2046. doi: 10.1111/j.1600-6143.2005.00957.x. [DOI] [PubMed] [Google Scholar]
- 67.Ryan FA, Paty BW, Senior PA, Brgan D, Alfadhli E, Kneteman NM, et al. Five year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–2069. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- 68.Shapiro AMJ, Ricordi C, Hering BJ, Aucincloss H, Lindblad R, Robertson RP, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2005;355:1318–1330. doi: 10.1056/NEJMoa061267. [DOI] [PubMed] [Google Scholar]
- 69.Fiorina P, Shapiro AMJ, Ricordi C, Secchi A. The clinical impact of islet transplantation. Am J Transplant. 2008;8:1990–1997. doi: 10.1111/j.1600-6143.2008.02353.x. [DOI] [PubMed] [Google Scholar]
- 70.Ricordi C, Strom TB. Clinical islet transplantation: advances and immunological challenges. Nature. 2004;4:259–268. doi: 10.1038/nri1332. [DOI] [PubMed] [Google Scholar]
- 71.Robertson RP. Islet transplantation as a treatment for diabetes—a work in progress. N Engl J Med. 2004;350:694–705. doi: 10.1056/NEJMra032425. [DOI] [PubMed] [Google Scholar]
- 72.Carlsson PO, Andersson A, Carlsson C, Hellerstrom C, Hoglund E, King A, et al. Engraftment and growth of transplanted pancreatic islets. Ups J Med Sci. 2000;105:107–123. doi: 10.1517/03009734000000058. [DOI] [PubMed] [Google Scholar]
- 73.Jansson L, Carlsson PO. Graft vascular function after transplantation of pancreatic islets. Diabetologia. 2002;45:749–763. doi: 10.1007/s00125-002-0827-4. [DOI] [PubMed] [Google Scholar]
- 74.Bennet W, Groth CG, Larsson R, Nilsson B, Korsgren O. Isolated human islets trigger an instant blood mediated inflammatory reaction: implications for intraportal islet transplantation as a treatment for patients with type 1 diabetes Ups. J Med Sci. 2000;105:125–140. doi: 10.1517/03009734000000059. [DOI] [PubMed] [Google Scholar]
- 75.Johansson U, Rasmusson I, Niclou SP, Forslund N, Gustavsson L, Nilsson B, et al. Formation of composite endothelial cell-mesenchymal stem cell-islets: a novel approach to islet vascularization. Diabetes. 2004;57:2393–2401. doi: 10.2337/db07-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mattsson G, Jannson L, Nordin A, Andersson A, Carisson PO. Evidence of functional impairment of syngeneically transplanted mouse pancreatic islets retrieved from the liver. Diabetes. 2004;53:948–954. doi: 10.2337/diabetes.53.4.948. [DOI] [PubMed] [Google Scholar]
- 77.Wang J, Cao Y, Chen Y, Chen Y, Gardner P, Steiner DF. Pancreatic beta cells lack a low glucose and O2-inducible mitochondrial protein that augments cell survival. Proc Natl Acad Sci USA. 2006;103:10636–10641. doi: 10.1073/pnas.0604194103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Korsgren O, Lundgren T, Felldin M, Foss A, Isakson B, Permert J, et al. Optimizing islet engraftment is critical for successful islet transplantation. Diabetologia. 2008;51:227–232. doi: 10.1007/s00125-007-0868-9. [DOI] [PubMed] [Google Scholar]
- 79.Medarova Z, Moore M. A MRI as a tool to monitor islet transplantation. Nat Rev Endocrinol. 2009;5:444–452. doi: 10.1038/nrendo.2009.130. [DOI] [PubMed] [Google Scholar]
- 80.Paty BW, Ryan EA, Shapiro AMJ, Lakey JRT, Robertson RP. Intra-hepatic islet transplantation in type 1 diabetic patient does not restore hypoglycemic hormonal counter-regulation or symptom recognition after insulin independence. Diabetes. 2002;51:3428–3434. doi: 10.2337/diabetes.51.12.3428. [DOI] [PubMed] [Google Scholar]
- 81.Street CN, Lakey JRT, Shapiro AJM, Imes S, Rjotte RV, Ryan EA, et al. Islet graft assessment in the Edmonton protocol: Implications for predicting longterm clinical outcome. Diabetes. 2004;53:3107–3114. doi: 10.2337/diabetes.53.12.3107. [DOI] [PubMed] [Google Scholar]
- 82.Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Diabetes. 2000;97:7999–8004. doi: 10.1073/pnas.97.14.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burke GW, Ciancio G, Sollinger HW. Advances in pancreas transplantation. Transplantation. 2004;77:62–67. doi: 10.1097/01.tp.0000126929.71923.77. [DOI] [PubMed] [Google Scholar]
- 84.Ferrannini E, Camastra S, Gastaldelli A, Sironi AM, Natali A, Muscelli E, et al. Beta-cell function in obesity: Effects of weight loss. Diabetes. 2004;53:26–33. doi: 10.2337/diabetes.53.suppl_3.s26. [DOI] [PubMed] [Google Scholar]
- 85.Dufane D, Gianello P. Pig islets for xenotransplantation. Curr Opin Nephr Hypertens. 2009;18:4950–5000. doi: 10.1097/MNH.0b013e328331a8e3. [DOI] [PubMed] [Google Scholar]
- 86.Hunter P. Xeno's paradox. EMBO Reports. 2009;10:554–557. doi: 10.1038/embor.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garavenko O, Croxson MC, Irgang M, Karlas A, Denner J, Elliot RB. Monitoring for the presence of potentially xenotic viruses in recipients of pig islet xenotransplantation. Clin Microbiology. 2004;42:5336–5353. doi: 10.1128/JCM.42.11.5353-5356.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lacy PE. Treating diabetes with transplanted cells. Scientific American. 1995 doi: 10.1038/scientificamerican0795-50. [DOI] [PubMed] [Google Scholar]
- 89.Lanza RP, Chick WL. Encapsulated cell therapy. Scientific American: Science and Medicine. 1995 [Google Scholar]
- 90.Scharp DW, Swanson CJ, Olack BJ, Latta PP, Hegre OD, Doherty EJ, et al. Protection of encapsulated human islets implanted without immunosuppression in patients with type I or type II diabetes and in nondiabetic control subjects. Diabetes. 1994;43:1167–1170. doi: 10.2337/diab.43.9.1167. [DOI] [PubMed] [Google Scholar]
- 91.Schneider S, Feilen PJ, Brunnenmeier F, Minnemann T, Zimmermann H, Zimmermann U, et al. Long-term graft function of adult rat and human islets encapsulated in novel alginate-based microcapsules after transplantation in immunocompetent diabetic mice. Diabetes. 2005;54:687–693. doi: 10.2337/diabetes.54.3.687. [DOI] [PubMed] [Google Scholar]
- 92.Hering BJ, Wijkstrom M, Graham ML, Hærdstedt M, Aasheim TC, Jie T, et al. Prolonged diabetes reversal after intraportal xenotransplantation of wild-type porcine islets in immunosuppressed nonhuman primates. Nature Medicine. 2006;12:301–303. doi: 10.1038/nm1369. [DOI] [PubMed] [Google Scholar]
- 93.Hering BJ, Walawalkar N. Pig-to-nonhuman primate islet xenotransplantation. Transplant Immunol. 2009;38:204–212. doi: 10.1016/j.trim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 94.Dor Y, Brown J, Martines OI, Melton DA. Adult pancreatic beta cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–47. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 95.Zhou Q, Melton DA. Pathways to new beta cells Cold Spring Harbor Symposium on Quantitative Biology. 2008;73:175–181. doi: 10.1101/sqb.2008.73.002. [DOI] [PubMed] [Google Scholar]
- 96.Seaberg RM, Smukler SR, Kieffer TJ, Enikolopov G, Asghar Z, Wheeler MB, et al. Clone identification of multipotent precursors from adult mouse pancreas that generated neural and pancreatic lineages. Nature Biotechnol. 2004;22:1115–1124. doi: 10.1038/nbt1004. [DOI] [PubMed] [Google Scholar]
- 97.Todorov I, Omori K, Pascual M, Rawson J, Nair I, Valiente L, et al. Generation of human islets through expansion and differentiation of non-islet pancreatic cells discarded after islet isolation. Pancreas. 2006;32:130–138. doi: 10.1097/01.mpa.0000202945.78331.93. [DOI] [PubMed] [Google Scholar]
- 98.Hao E, Bjorn T, Itkin-Ansari P, Lakey Jonathan RT, Ifat G, Monosov E, et al. Beta-cell differentiation from non-endocrine epithelial cells of the adult human pancreas. Nature Medicine. 2006;12:310–316. doi: 10.1038/nm1367. [DOI] [PubMed] [Google Scholar]
- 99.Lechner A, Yang YG, Blacken RA, Wang L, Nolan AL, Habener JF. No evidence for significant transdifferentiation of bone marrow into pancreatic beta cells in vivo. Diabetes. 2004;56:616–623. doi: 10.2337/diabetes.53.3.616. [DOI] [PubMed] [Google Scholar]
- 100.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton A. In vivo reprogramming of adult pancreatic exocrine cells to beta cells. Nature. 2008;455:627–633. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Narushima M, Kobayashi N, Okitsu T, Tanaka Y, Li S, Chen Y, et al. A human beta-cell line for transplantation therapy to control type 1 diabetes. Nature Biotechnology. 2005;23:1274–1282. doi: 10.1038/nbt1145. [DOI] [PubMed] [Google Scholar]
- 102.Weir GC, Bonner-Weir S. Beta cell precursors—a work in progress. Nature Biotechnology. 2004;22:1095–1097. doi: 10.1038/nbt0904-1095. [DOI] [PubMed] [Google Scholar]
- 103.Weir GC, Bonner-Weir S. Five stages of evolving beta cell dysfunction during its progression to diabetes. Diabetes. 2004;53:16–21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- 104.Elzirik DL, Korbutt GS, Hellerstrom C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the beta cell function. Am Soc Clin Invest. 1992;90:1263–1268. doi: 10.1172/JCI115989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Laybutt DR, Glandt M, Xu G, Ahn YB, Trivedi N, Bonner-Weir S, et al. Critical reduction in Beta cell mass results in two distinct outcomes over time. J Biological Chem. 2003;278:2997–3005. doi: 10.1074/jbc.M210581200. [DOI] [PubMed] [Google Scholar]
- 106.Donath MY, Halban PA. Decreased beta-cell mass I diabetes: significance, mechanisms and therapeutic implications. Diabetologia. 2004;47:581–589. doi: 10.1007/s00125-004-1336-4. [DOI] [PubMed] [Google Scholar]
- 107.Pileggi A, Fenjves ES, Klei D, Ricordi C, Pastori RL. Protecting pancreatic Betα-cells. Life. 2004;56:387–394. doi: 10.1080/15216540400006469. [DOI] [PubMed] [Google Scholar]
- 108.Nichols J, Cooke A. Overcoming self-destruction in the pancreas. Current Opinions in Biotechnology. 2009;20:511–515. doi: 10.1016/j.copbio.2009.09.009. [DOI] [PubMed] [Google Scholar]