

Background: MshB is a metal-dependent deacetylase involved in mycothiol biosynthesis.
Results: The reaction proceeds via a general acid-base pair mechanism and uses a dynamic Tyr that modulates substrate binding, chemistry, and product release.
Conclusion: The catalytic mechanism differs from a prototypical metalloprotease mechanism.
Significance: Key side chains identified in these studies can be targeted for inhibitor development.
Keywords: Enzyme Kinetics, Enzyme Mechanisms, Hydrolases, Metalloenzymes, Mutagenesis, Zinc, MshB, Metal-dependent Deacetylase, Metallohydrolase, Iron, Mycothiol, GABC Pair
Abstract
Actinomycetes are a group of Gram-positive bacteria that includes pathogenic mycobacterial species, such as Mycobacterium tuberculosis. These organisms do not have glutathione and instead utilize the small molecule mycothiol (MSH) as their primary reducing agent and for the detoxification of xenobiotics. Due to these important functions, enzymes involved in MSH biosynthesis and MSH-dependent detoxification are targets for drug development. The metal-dependent deacetylase N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) catalyzes the hydrolysis of N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside to form 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside and acetate in MSH biosynthesis. Herein we examine the chemical mechanism of MshB. We demonstrate that the side chains of Asp-15, Tyr-142, His-144, and Asp-146 are important for catalytic activity. We show that NaF is an uncompetitive inhibitor of MshB, consistent with a metal-water/hydroxide functioning as the reactive nucleophile in the catalytic mechanism. We have previously shown that MshB activity has a bell-shaped dependence on pH with pKa values of ∼7.3 and 10.5 (Huang, X., Kocabas, E. and Hernick, M. (2011) J. Biol. Chem. 286, 20275–20282). Mutagenesis experiments indicate that the observed pKa values reflect ionization of Asp-15 and Tyr-142, respectively. Together, findings from our studies suggest that MshB functions through a general acid-base pair mechanism with the side chain of Asp-15 functioning as the general base catalyst and His-144 serving as the general acid catalyst, whereas the side chain of Tyr-142 probably assists in polarizing substrate/stabilizing the oxyanion intermediate. Additionally, our results indicate that Tyr-142 is a dynamic side chain that plays key roles in catalysis, modulating substrate binding, chemistry, and product release.
Introduction
Actinomycetes, such as Mycobacterium species, are Gram-positive bacteria that contain a high GC content and a thick, hydrophobic cell wall. Pathogenic mycobacterial species are responsible for a number of infectious diseases, most notably tuberculosis and leprosy. In contrast to eukaryotes and other bacteria, these organisms do not have glutathione. Instead, they use the small molecule mycothiol (MSH)2 as their primary reducing agent and in xenobiotic metabolism for the detoxification of drugs and other toxins (1–4). MSH is probably critical for survival of mycobacteria inside the oxidative environment of activated macrophages where they reside. Consequently, enzymes in the MSH biosynthetic and MSH-dependent detoxification pathways are targets for the development of antibiotics for the treatment of diseases, such as tuberculosis (5–10).
The metalloenzyme MshB catalyzes the hydrolysis of N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside (GlcNAc-Ins) to form 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside and acetate, the fourth overall step in MSH biosynthesis (Fig. 1A). MshB is an attractive drug target because it catalyzes the rate-limiting step in MSH biosynthesis (11), it is a metalloenzyme (12–14), and the three-dimensional structure is known (15, 16). There are past successes in targeting metalloenzymes, including inhibitors of carbonic anhydrase, matrix metalloproteases, and angiotensin-converting enzyme (17–20). Because inhibitors of metalloenzymes typically contain a group that binds to the catalytic metal ion, we previously examined the cofactor preferences of MshB and found that MshB is a cambialistic metalloenzyme whose in vitro activity follows the following trend: Fe2+ > Co2+ > Zn2+ > Mn2+ > Ni2+ (14). Additionally, we found that the cofactor bound to MshB is dependent on environmental conditions (14). MshB prefers Fe2+ under anaerobic conditions regardless of the metal ion content of the medium and switches between Fe2+ and Zn2+ under aerobic conditions as the metal content of the medium is altered. MshB has a bell-shaped dependence on pH (subsaturating concentrations of substrate, V/K), indicating that there are two ionizations that are important for maximal catalytic activity (14), consistent with a reaction that proceeds through either a single bifunctional general acid-base catalyst (GABC) or GABC pair mechanism (21).
FIGURE 1.

A, reaction catalyzed by MshB. B, active site of MshB (Protein Data Bank entry 1Q74) containing a catalytic zinc ion. Only one of the four Zn2+-MshB monomers is shown. C, active site of MshB (Protein Data Bank entry 1Q7T) containing bound BOG. The structures of the two MshB-BOG monomers were overlaid, revealing two different locations for glucose binding to MshB, BOG1 and BOG2. The octyl chains of the active site BOG molecules are not observed in either monomer. GlcN-Ins, 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside.
Herein we probe the chemical mechanism of MshB. We demonstrate that residues Asp-15, His-144, Asp-146, and Tyr-142 are important for maximal catalytic activity. Our results suggest that Asp-15 functions as a general base catalyst (GBC), whereas His-144 functions as a general acid catalyst (GAC), and the catalytic metal-water/hydroxide serves as the reactive nucleophile in the reaction. Furthermore, our results indicate that Tyr-142 is important for catalytic activity. We propose that Tyr-142 is a dynamic side chain that modulates substrate binding, chemistry (via polarization of carbonyl group/stabilization of oxyanion), and product release. These insights into the chemical mechanism indicate that MshB does not follow the prototypical metalloprotease-like mechanism but instead uses a GABC pair and a dynamics to catalyze the hydrolysis of substrate.
MATERIALS AND METHODS
General Procedures
All solutions were prepared using milliQ water. Primers were purchased from Integrated DNA Technologies. Genomic DNA was purchased from ATCC. DNA sequencing was performed at the Virginia Bioinformatics Institute DNA Sequencing Facility (Virginia Tech). All chemicals were purchased from ThermoFisher Scientific, Sigma-Aldrich, and Gold Biotechnology. For kinetic experiments, solutions were prepared with reagents that did not contain extraneous metal ions and/or were treated with Chelex (Bio-Rad), and solutions were stored in “metal-free” plasticware. To maintain anaerobic conditions (Fe2+ assays), experiments were carried out in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI). Molecular graphics images were produced using the UCSF Chimera package (22).
Protein Expression and Purification
The previously reported plasmid encoding the MshB gene from Mycobacterium smegmatis containing an N-terminal His-MBP tag was used as the template for preparation of mutant plasmids (14). All mutant plasmids were prepared using the QuikChange Lightning site-directed mutagenesis kit (Stratagene). Plasmid sequences were verified by DNA sequencing. All MshB variants were expressed and purified according to published procedures (14, 23).
Briefly, cells were lysed using an Emulsiflex-C3 high pressure homogenizer (Avestin), and MshB variants were purified at 4 °C. Cell lysate was clarified by centrifugation (18,000 rpm, 4 °C) and loaded onto a pre-equilibrated (Buffer A; 30 mm HEPES, 150 mm NaCl, 1 mm triscarboxyethylphosphine or TCEP, 0.5 mm imidazole, pH 7.5) Co-IMAC column (50 ml of chelating Sepharose (GE Healthcare) charged with CoCl2). The column was washed with 150 ml of Buffer A, and His-MBP-MshB was eluted using an imidazole step gradient (200 ml each: Buffer A + 10 mm imidazole, Buffer A + 25 mm imidazole, Buffer A + 300 mm imidazole). Fractions containing His-MBP-MshB (via SDS-PAGE) were combined, concentrated (Amicon Ultra-15 centrifugal devices, Millipore), and dialyzed (Snakeskin tubing, molecular weight cut-off 10,000; Pierce) versus 2 × 4 liters of Buffer A overnight in the presence of His-TEV protease (300 μg/ml) to remove the His-MBP tag. The resulting TEV-cleaved protein was loaded onto a pre-equilibrated (Buffer A + 25 mm imidazole) Co-IMAC column. His-MBP and His-TEV remain bound to the Co-IMAC column, whereas MshB elutes in the flow-through. Fractions containing MshB (via 12% SDS-PAGE) were combined, concentrated, and dialyzed versus 2 × 4 liters of 25 mm HEPES, 1.5 mm triscarboxyethylphosphine, pH 7.5 (Slide-a-Lyzer, molecular weight cut-off 10,000; Pierce). Protein concentration was determined using the Bradford assay (Sigma). Protein aliquots were flash frozen in liquid nitrogen and stored at −80 °C.
For the preparation of apo-MshB (14), purified protein (≤100 μm) was incubated with 10 mm HEPES, 20 mm dipicolinic acid, 250 μm EDTA, pH 7.5, on ice. After 1 h, the protein solution was concentrated; washed (diluted with buffer and then concentrated) with 3 × 15 ml of 25 mm HEPES, 1.5 mm triscarboxyethylphosphine, pH 7.5; and run over a desalting column to remove residual dipicolinic acid/EDTA. Metal ion concentrations were determined using an ICS-3000 ion chromatography system (Dionex). Apo-MshB samples contained ≤10% metal/protein. Prior to activity measurements, apo-MshB (≤10 μm) was incubated with a stoichiometric concentration of the desired metal ion (CoCl2, FeCl2, FeCl3, MnCl2, NiCl2, ZnSO4) and incubated on ice for 30 min.
MshB Deacetylase Activity
MshB deacetylase activity was measured with the substrate N-acetyl-glucosamine (GlcNAc) (Sigma) using a fluorescamine-based assay (23). Although the GlcNAc substrate has a decreased affinity for MshB compared with the natural substrate GlcNAc-Ins (Km of 38 mm versus 340 μm), the GlcNAc moiety that undergoes the chemical transformation is conserved (12, 23). Because our primary interest is in examining the chemical step of the reaction, the commercially available GlcNAc substrate was used in these studies. In general, assay mixtures containing 50 mm HEPES, 50 mm NaCl, 1 mm triscarboxyethylphosphine or TCEP, pH 7.5, and 0–150 mm GlcNAc were pre-equilibrated at 30 °C, and reactions were initiated by the addition of enzyme (1 μm). For pH dependence experiments, the following buffers were used (all 50 mm containing 1 mm triscarboxyethylphosphine, 50 mm NaCl): MES, pH 6–6.8; Mops, pH 6.5–7.5; HEPES, pH 7.3–8.8; Bistris propane, pH 8–9; borate, pH 9–10; carbonate, pH 10–11. After incubation for various times, reactions aliquots (30 μl) were quenched by the addition of 20% TCA (10 μl), and the cleared supernatant (25 μl) was transferred into a 96-well plate, diluted with 1 m borate, pH 9 (75 μl), and reacted with fluorescamine (30 μl in CH3CN; Invitrogen). After 10 min, the fluorescence was measured (excitation 395 nm; emission 485 nm) using a SpectraMax M5 plate reader (Molecular Devices). Initial rates of product formation (<10%) were determined from these data. Equation 1 was fit to the pH rate profile, where k1 represents V/K at the pH optimum, and Ka1 and Ka2 represent dissociation constants describing the two ionizations. Equations 2 and 3 were fit to the pH rate profiles for the D15A and Y142A/F mutants, respectively, in which only a single ionization is observed. For experiments under V/K conditions, 5–50 mm GlcNAc was used as the substrate in assays. Specific concentrations of substrate used were 5 (WT, Y142F), 10 (Y142A), 20 (D15A and H144A), or 50 mm (D146A) GlcNAc.
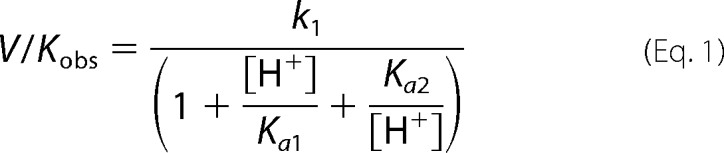 |
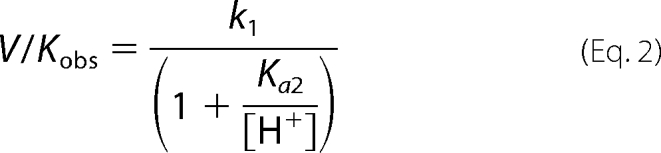 |
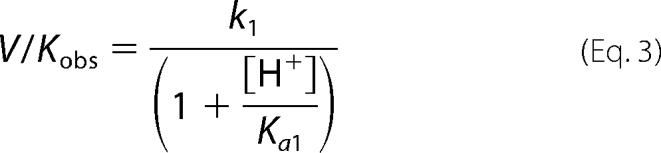 |
For determination of the steady-state parameters, deacetylase activity was measured at 6–8 different concentrations of GlcNAc (0–300 mm), and the parameters kcat, Km, and kcat/Km were obtained by fitting the Michaelis-Menten equation to the initial linear velocities using the curve-fitting program Kaleidagraph (Synergy Software), which also calculates the asymptotic S.E. values. For D146A, higher concentrations of GlcNAc (0–375 mm) were used.
Solvent viscosity assays were carried out using the microviscogens sucrose (0–35% (w/v)) and glycerol (0–35% (w/v)) and the macroviscogen Ficoll 400 (0–10% (w/v)) with a subsaturating concentration (5 mm) or saturating concentration (100 mm) of GlcNAc. The ηrel values for 0, 10, 20, 27.5, 32.5, and 35% sucrose are 1, 1.32, 1.88, 2.48, 3.06, and 3.42, respectively (24). The ηrel values for 0, 10, 20, 30, and 35% glycerol are 1, 1.3, 1.7, 2.3, and 2.9, respectively (25). The ηrel values for 0, 5, and 10% Ficoll 400 are 1, 2.2, and 4.5, respectively (26). Results for experiments examining the effect of solvent viscosity on H144A at 300 mm GlcNAc are included in supplemental Fig. S1.
Fluoride inhibition studies were carried out with 0–200 mm NaF added to the assay mixture. There was no effect on MshB activity observed in control experiments using an additional 200 mm NaCl in the assay mixture (supplemental Fig. S2). For solvent isotope effect experiments, initial rates at subsaturating substrate concentrations (5 (WT, Y142F), 10 (Y142A), 20 (D15A, H144A), or 50 mm (D146A) GlcNAc) in H2O were compared with the initial rates in ∼95% D2O. The pD values obtained for the D2O buffers using the pH meter readings were corrected by adding 0.4 to these values.
Computational Studies
Structural alignment of the six MshB monomers in available crystal structures (Protein Data Bank entries 1Q74 and 1Q7T) (15, 16) was carried out using the MatchMaker program in the UCSF Chimera package (22, 27). Potential Tyr-142 rotamers were evaluated using Chimera (22) with the Dunbrack backbone-dependent rotamer library (28). This library contains six possible Tyr rotamers that are commonly observed in proteins. A model of MshB with Tyr-142 positioned for a possible role in chemistry was prepared using Chimera by rotating Tyr-142 into the location of a Tyr rotamer in the Dunbrack library.
RESULTS
Mutations Decrease Catalytic Activity
The crystal structure of Zn2+-MshB (Fig. 1B) reveals a zinc ion in the active site bound by the side chains of three protein ligands (His-13, Asp-16, and His-147) and one to two water molecules that is surrounded by side chains typically involved in acid-base catalysis and/or stabilization of oxyanion intermediates (Asp-15, His-144, and Asp-146). Importantly, this structure reveals two potential acid-base catalysts that are in close proximity to the zinc-bound water molecules, Asp-15 and His-144; Asp-15 is located ∼2.8 Å from one zinc-bound water molecule, whereas His-144 is ∼3.4 Å away from the second zinc-bound water molecule. As a result of their close proximity to the catalytic zinc ion, these side chains are best positioned to serve as GBC/GAC in the chemical reaction. Crystal structures of a MshB·β-octyl-d-glucopyranoside (BOG) complex, which lack the catalytic metal ion (Fig. 1C), also indicate that these side chains are well positioned to interact with the substrate GlcNAc moiety (16). Additionally, the side chain of Asp-146 is located near His-13 and His-144 (2.9–3.6 Å), suggesting a potential role for Asp-146 in catalysis. On the basis of the crystal structure of the Zn2+-MshB active site (15) and the pH dependence of MshB deacetylase activity (14), the MshB-catalyzed reaction is proposed to proceed through either a single GABC or a GABC pair mechanism using the side chains of Asp-15 and/or His-144 (21).
To distinguish between these two possible mechanisms, we used a combination of mutagenesis and kinetic experiments. We prepared a series of MshB constructs wherein active site side chains (Asp-15, His-144, and Asp-146) were mutated to Ala using site-directed mutagenesis, and the steady-state parameters for these constructs were determined using the substrate GlcNAc (Table 1 and supplemental Fig. S3). Results from these experiments indicate that removal of the Asp-15, His-144, and Asp-146 side chains lead to an overall decrease in catalytic activity, suggesting that these side chains are important for maximal catalytic activity. Specifically, the D15A mutation leads to a modest (<2-fold) increase in Km, an ∼80-fold decrease in kcat, and a 222-fold decrease in kcat/Km. The H144A mutation leads to a modest increase in the value of Km to ∼50 mm but decreases the values of kcat and kcat/Km 60- and 100-fold, respectively. The D146A mutant could not be saturated with [GlcNAc] of 375 mm; therefore, the steady-state parameters for this mutant are estimated values. The D146A mutation results in a >10-fold increase in Km and >100-fold decrease in kcat/Km.
TABLE 1.
Steady-state kinetic parameters of MshB mutants
| MshBa,b | Km | kcat | kcat/Km | WT activity |
|---|---|---|---|---|
| mm | min−1 | m−1s−1 | % | |
| WTc | 38 ± 4 | 46 ± 2 | 20 ± 1 | |
| D15A | 52 ± 11 | 0.29 ± 0.02 | 0.09 ± 0.01 | 0.5 |
| Y142A | 22 ± 2 | 1.8 ± 0.05 | 1.36 ± 0.10 | 6.8 |
| Y142F | 9 ± 0.5 | 0.57 ± 0.01 | 1.05 ± 0.05 | 5.3 |
| Y142Q | 17 ± 2 | 1.2 ± 0.04 | 1.17 ± 0.11 | 5.9 |
| H144A | 57 ± 8 | 0.69 ± 0.04 | 0.20 ± 0.02 | 1.0 |
| D146A | >400 | >2 | ∼0.08d | <0.4 |
a Apo-MshB was incubated with stoichiometric Zn2+ for 30 min prior to activity measurement.
b Substrate used GlcNAc.
c Data adapted from Ref. 14.
d Estimated from slope of initial linear region on V versus [S] plot.
Solvent Viscosity Effects
To aid in deciphering the rate-limiting step for MshB deacetylation, we examined the effect of solvent viscosity on the rate of MshB-catalyzed deacetylation. Solvent microviscosity slows the rates of steps that involve movement of small molecules, such as substrate binding and product release, as well as conformational changes in proteins (24–26, 29–31). In contrast, rates for internal processes, such as chemistry, proceed independent of solvent microviscosity. To control for changes in activity that arise from nonspecific interactions, we used multiple microviscogens (sucrose, glycerol) as well as the macroviscogen Ficoll 400. Macroviscogen alter solvent viscosity but do not slow the rates of diffusion of small molecules and are used to control for changes in activity that arise from nonspecific interactions (24, 25, 29–31).
We measured the effect of sucrose and glycerol (microviscsogens), as well as Ficoll 400 (macroviscogen), on MshB activity. Results from experiments with WT MshB (Fig. 2 and supplemental Table S1) indicate that both kcat and kcat/Km are significantly slowed in the presence of the microviscogens sucrose and glycerol. For effects on kcat, the slopes observed in the presence of glycerol and sucrose are 0.74 ± 0.13 and 0.62 ± 0.11, respectively. The finding that the plots for assays in the presence of glycerol and sucrose have comparable slopes suggests that the microviscogens are affecting the diffusion of small molecules and/or a conformational change in the protein and not simply having nonspecific effects on MshB (e.g. dielectric constant). For effects on kcat/Km, the slopes for assays in the presence of glycerol and sucrose are 1.72 ± 0.12 and 1.70 ± 0.28, respectively. Again, the finding that the plots for assays in the presence of glycerol and sucrose have comparable slopes suggests that the microviscogens are affecting the diffusion of small molecules and/or a conformational change in the protein and not having nonspecific effects on MshB. These initial plots of kcat or kcat/Km versus ηrel show some deviation from linearity, and therefore, additional analyses were carried out on these data (below). As expected, the slopes of the plots examining the effect of Ficoll 400 on kcat (0.03 ± 0.006) and kcat/Km (−0.086 ± 0.02) indicate that these parameters are unaffected by the macroviscogen Ficoll 400.
FIGURE 2.

A and B, effect of solvent viscosity on kcat (A) and kcat/Km (B). The effects of the microviscogens sucrose (0–35% (w/v)) and glycerol (0–35% (w/v)) on WT MshB are depicted as open and filled circles, respectively, whereas the macroviscogen Ficoll 400 (0–10% (w/v)) is denoted as open squares. Data for the Y142F mutant in the presence of sucrose is depicted by open triangles. Plots C and D are used to determine if there is a conformational change associated with product release and/or substrate binding. Apo-MshB was incubated with stoichiometric Zn2+. After 30 min, the enzyme was diluted into assay buffer (50 mm HEPES, 1 mm TECP, 50 mm NaCl, pH 7.5) containing substrate, and the initial rates for the deacetylation of a subsaturating (5 mm) or saturating (100 mm) concentration of GlcNAc were measured as described under “Materials and Methods.” The slopes are provided in supplemental Table S1.
For rate-limiting steps where product release is associated with conformational changes in the protein, a plot of log kcat versus log ηrel is linear, whereas for rate-limiting steps where product release occurs in the absence of a structural rearrangement, a plot of log kcat versus ηrel2 is linear (29, 31). To gain insights into whether product release in MshB is dependent on a conformational change in the protein, we examined which of these plots better describe our data (Fig. 2, C and D, and supplemental Table S1). Although both plots comparably describe data obtained using glycerol as the viscogen, the sucrose data are better described by the log kcat versus log ηrel plot, suggesting that product release in MshB may be coupled to a conformational change in the protein. The finding that kcat/Km is also better described by log kcat/Km versus log ηrel (supplemental Fig. S4) may suggest that there is also a conformational change coupled with substrate binding to MshB.
We examined the effect of solvent viscosity on the activity of the D15A and H144A mutants (Fig. 3 and supplemental Table S2) to determine if product release remains rate-limiting for these catalytically impaired mutants. Once again we observe that kcat is significantly slowed in the presence of glycerol and sucrose. These data are clearly better described by a plot of log kcat versus log ηrel (Fig. 3A) compared with a plot of log kcat versus ηrel2 (Fig. 3B), suggesting that the rate-limiting step in these mutants is a conformational change that is coupled to product release. (Note that because we are unable to reach substrate saturation with the D146A mutant, the effect of solvent viscosity could only be determined under kcat/Km conditions; supplemental Fig. S4 and Table S3).
FIGURE 3.
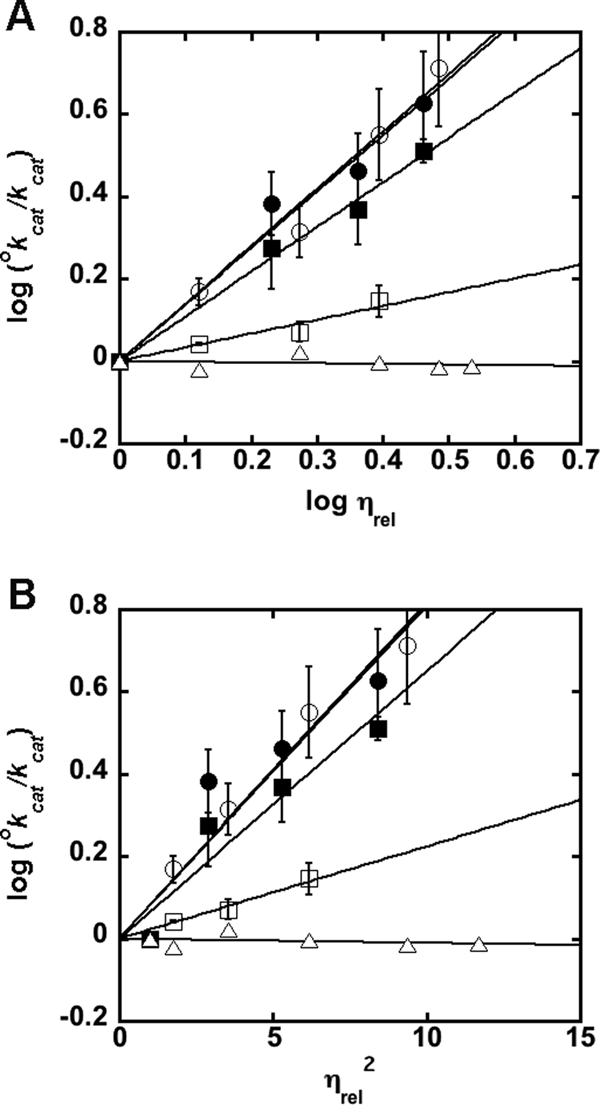
Effect of solvent viscosity on kcat of MshB mutants. The effects of the microviscogens sucrose (0–35% (w/v)) and glycerol (0–35% (w/v)) on WT MshB are depicted as open and closed symbols, respectively. MshB mutants examined are D15A (circles), H144A (squares), and Y142F (triangles). Plots A and B are used to determine if a conformational change is associated with product release/substrate binding. Apo-MshB was incubated with stoichiometric Zn2+. After 30 min, the enzyme was diluted into assay buffer (50 mm HEPES, 1 mm TECP, 50 mm NaCl, pH 7.5) containing substrate and the initial rates for the deacetylation at saturating concentrations (100 mm) of GlcNAc were measured as described under “Materials and Methods.” The slopes of the fits are provided in supplemental Table S2. The effect of glycerol on H144A activity using 300 mm GlcNAc is included in supplemental Fig. S1.
Role of Metal-Water/Hydroxide
We previously examined the ability of divalent metal ions to serve as cofactors for MshB and found that the overall activity follows the following trend: Fe2+ > Co2+ > Zn2+ > Mn2+ > Ni2+ (14). Although these results confirm the importance of the metal cofactor for activity, the specific role of the metal ion in the MshB-catalyzed reaction has not been elucidated. Therefore, we set out to probe the role of the metal ion in catalysis by MshB.
Fluoride is often used to probe the identity of the reactive nucleophile for metallohydrolases (32–35). Specifically, fluoride inhibits (uncompetitive) enzymes that utilize a metal-bound water or hydroxide as the reactive nucleophile. Therefore, we measured the MshB-catalyzed deacetylation of GlcNAc in the presence of various concentrations of NaF (0–200 mm). Results from these experiments (supplemental Fig. S5) show that NaF acts as an uncompetitive inhibitor of the deacetylation reaction consistent with the metal-water/hydroxide serving as the reactive nucleophile in the MshB-catalyzed reaction.
MshB activity (V/K) exhibits a bell-shaped dependence on pH with two ionizations having pKa values of ∼7.3 (pKa1) and 10.5 (pKa2) (14). There is an increase in activity with increasing pH for the ionization described by pKa1, whereas there is a decrease in activity with increasing pH for the ionization described by pKa2. Because the metal-bound water could be reflected in one of these pKa values, we examined the pH dependence of deacetylase activity for MshB reconstituted with various divalent metal ions. Results from these experiments are shown in Table 2. These results confirm that MshB activity follows the trend: Fe2+ > Co2+ > Zn2+ > Mn2+ > Ni2+. Furthermore, these findings indicate that the identity of the metal ion does not significantly alter pKa1 and has only a modest effect on pKa2 (0.6 pH units). These results rule out the metal-water as the source of pKa1, whereas the small effect on pKa2 (0.6 pH units) suggests that it is unlikely that this pKa reflects ionization of the metal-water either.
TABLE 2.
Effect of metal ions on pH dependence of wild-type MshB
| Me2+-MshBa,b | pKa1 | pKa2 | V/K at pH optimum |
|---|---|---|---|
| m−1s−1 | |||
| Zincc | 7.4 ± 0.07 | 10.5 ± 0.06 | 48 ± 1.5 |
| Cobalt | 7.2 ± 0.09 | 11.0 ± 0.09 | 56 ± 2.3 |
| Iron | 7.2 ± 0.09 | 10.5 ± 0.08 | 63 ± 2.3 |
| Nickel | 7.1 ± 0.03 | 11.1 ± 0.03 | 7.3 ± 0.1 |
| Manganese | 7.2 ± 0.12 | 11.0 ± 0.12 | 8.0 ± 0.4 |
a Apo-MshB was incubated with stoichiometric metal for 30 min prior to activity measurement.
b Substrate 5 mm GlcNAc.
c Data adapted from Ref. 14.
Mutations Alter pH Dependence
To gain further insights into the chemical mechanism of MshB, we probed the identities of the ionizations observed in the wild-type (WT) MshB pH profile under subsaturating concentrations of substrate (V/K). We chose to focus on the parameter V/K rather than V because V/K examines the reaction of free enzyme with free substrate through the first irreversible step, chemistry, and therefore will provide information about the chemical mechanism of the enzyme. In contrast, results from solvent viscosity experiments (Figs. 2 and 3) indicate that that the parameter kcat (V) reflects a conformational change coupled to product release and therefore does not provide information about the chemical step of the reaction (the pH profile under V conditions is included in supplemental Fig. S6). We examined the pH dependence of the MshB Ala mutants (Asp-15, His-144, and Asp-146), and the results from these experiments are summarized in Table 3 and Fig. 4. These results confirm that the side chains of Asp-15, His-144, and Asp-146 are important for catalytic activity because the rate at the pH optimum is decreased ∼180 to 530-fold. Importantly, we observe that pKa1 is lost in the D15A mutant (Fig. 4), suggesting that pKa1 reflects ionization of Asp-15 in the WT enzyme. Although the H144A and D146A mutations decrease MshB activity, the pKa values observed for these mutants are the same as those observed for the WT protein, suggesting that neither His-144 nor Asp-146 is responsible for the ionizations observed in the WT protein.
TABLE 3.
pH dependence of Zn2+-MshB variants
| MshBa,b | pKa1 | pKa2 | V/K at pH optimum | WT activity |
|---|---|---|---|---|
| m−1s−1 | ||||
| WTc | 7.4 ± 0.07 | 10.5 ± 0.06 | 48.2 ± 1.5 | % |
| D15A | 10.5 ± 0.13 | 0.091 ± 0.003 | 0.2 | |
| Y142A | 7.1 ± 0.10 | 0.96 ± 0.03 | 2.0 | |
| Y142F | 7.0 ± 0.05 | 0.65 ± 0.01 | 1.3 | |
| H144A | 7.1 ± 0.10 | 10.5 ± 0.09 | 0.27 ± 0.01 | 0.6 |
| D146A | 7.2 ± 0.10 | 10.4 ± 0.10 | 0.18 ± 0.01 | 0.4 |
a Apo-MshB was incubated with stoichiometric metal for 30 min prior to activity measurement.
b Substrate 5 (WT, Y142F), 10 (Y142A), 20 (D15A, H144A), or 50 mm (D146A) GlcNAc.
c Data adapted from Ref. 14.
FIGURE 4.
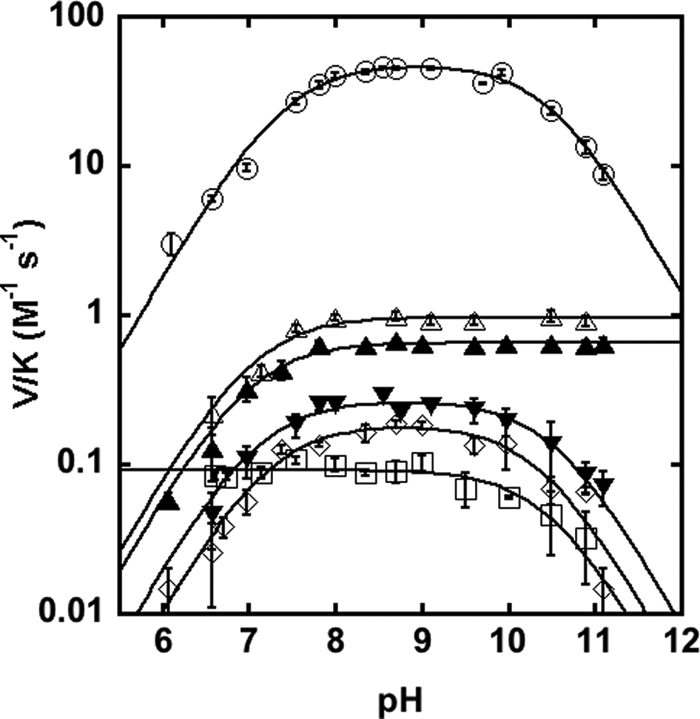
pH rate profiles. Shown is the effect of pH on the deacetylation of GlcNAc by MshB under subsaturating substrate concentrations (V/K). The pH dependence of the MshB-catalyzed reaction is shown for WT (○), D15A (□), Y142A (△), Y142F (▴), H144A (▾), and D146A (♢). Assays were measured at 30 °C with subsaturating (5–50 mm) concentrations of GlcNAc as described under “Materials and Methods.” The pKa values were determined by fitting the equation including two ionizations (Equation 1) or one ionization (Equation 2 or 3) and are shown in Table 3.
Examination of Tyr-142
Because we could not identify the source of pKa2 using metal substitution or our initial mutagenesis experiments (D15A, H144A, and D146A), we examined the MshB crystal structures more closely for additional possible sources of this ionization. Importantly, the side chain of Tyr-142 appears to be a dynamic side chain, moving several Å in the Zn2+-MshB and MshB-BOG complex structures. An overlay of the six MshB monomers (four Zn2+-MshB and two MshB-BOG) is shown in Fig. 5A. The Tyr-142 side chain is found in four different locations in the six monomers. The locations in red, orange, yellow, and purple are observed in the four Zn2+-MshB monomers, whereas the blue and green locations are observed in MshB-BOG monomers. The locations of these side chains and rotation of Tyr-142 in chimera both indicate that the Tyr-142 side chain is unobstructed and can move freely (360°) from being on the MshB surface/solvent exposed (purple) to various locations in the active site.
FIGURE 5.

MshB active site. A, overlay of the six MshB monomers. The locations of the active site side chains (His-13, Asp-15, Asp-16, His-144, Asp-146, and His-147) do not vary significantly, whereas the location of the Tyr-142 side chain (rainbow-colored) varies in the structures. A model of the Zn2+-MshB active site with Tyr-142 rotated into a catalytic location is represented by the gray wire. B, potential Tyr rotamers. The six Tyr rotamers (1–6) from the Dunbrack library are displayed as wires. The six rotamers fall into three general locations with two orientations of the phenyl ring at each location. The location of the purple side chain in A is represented by the gray stick. C, Zn2+-MshB active site with Tyr-142 rotated into a catalytic location (position of rotamer 5).
Additionally, we examined the Tyr rotamers using the Dunbrack database in Chimera (22, 28). Tyr rotamers are most commonly observed in three general locations with two orientations of the aromatic ring at each location, yielding a total of six possible rotamers (Fig. 5B). Examination of these locations reveals that two of the locations (rotamers 1–4) are observed in the Zn2+-MshB structures, whereas the third location (rotamers 5 and 6) lies between the Tyr-142 positions observed in the MshB-BOG structures. A model of MshB with Tyr-142 in this third location is shown in Fig. 5C (gray wire in Fig. 5A). In this rotamer, the hydroxyl group of Tyr-142 is within hydrogen bonding distance of the metal-water, which would be capable of participating in catalysis. Therefore, we examined the effect of the Y142A mutant on catalytic activity.
The Y142A mutation significantly alters the steady-state parameters (Table 1). This mutation leads to a ∼2-fold decrease in Km, 26-fold decrease in kcat, and a 15-fold decrease in kcat/Km. To further examine the role of Tyr-142 in catalysis, we examined the Y142F (sterics, hydrophobicity) and Y142Q (hydrogen bonding) mutants. The Y142F mutation leads to a 4-fold decrease in Km, an ∼80-fold decrease in kcat, and a 20-fold decrease in kcat/Km, whereas the Y142Q mutation leads to a 2-fold decrease in Km, a ∼40-fold decrease in kcat, and a 15-fold decrease in kcat/Km. Additionally, we observe that the ionization described by pKa2 is no longer observed in the pH profiles for the Y142A or Y142F mutants (Fig. 4). These results suggest that pKa2 reflects ionization of Tyr-142 in WT MshB.
Furthermore, we find that the solvent viscosity effect under kcat conditions described for MshB in WT and mutant (D15A and H144A) enzymes is lost in the Y142F mutant (Figs. 2 and 3), whereas the solvent viscosity effect under kcat/Km conditions described for WT is also significantly diminished in the Y142F mutant (Fig. 2B and supplemental Table S3 and Fig. S4).
Tyr-142 Dynamics Modulate Access to Active Site
Mutation of Tyr-142 to Ala, Phe, or Gln leads to a ≤4-fold decrease in Km, suggesting that Tyr-142 plays a role in substrate binding. Because the location of Tyr-142 varies from being on the surface of the protein (purple) to inside the active site, the Tyr-142 side chain may modulate access to the active site. To probe if the side chain Tyr-142 affects access to the enzyme active site, we examined the surfaces of each overlaid monomer (Fig. 5A) with the two BOG molecules (Fig. 1C) as a model for GlcNAc-Ins binding. The results are shown in Fig. 6 and suggest that the structural changes that occur to MshB upon movement of Tyr-142 appear to alter access to the active site. A and C of Fig. 6 represent the same structures from two different viewpoints, with the side chain of Tyr-142 shown in dark gray. In the four Zn2+-MshB monomers (red, orange, yellow, and purple), both sugar molecules are visible, indicating that these Tyr conformations would allow for substrate binding and product release. In the two MshB-BOG monomers (blue and green), the sugar molecules become obscured, indicating that the movement of substrate and product in/out of the active site would be hindered in these Tyr conformations. Fig. 6B is a representation of the viewpoint depicted in Fig. 6A, wherein the side chain of Tyr-142 has been truncated to Ala (dark gray) in attempts to visualize the changes that occur upon the Y142A mutation. Similarly, Fig. 6D is a representation of the viewpoint depicted in Fig. 6C, wherein the side chain of Tyr-142 has been truncated to Ala (dark gray). The information in Fig. 6, B and D, predicts that there should be better access to the active site in the Y142A mutant because the sugar molecules become more visible.
FIGURE 6.

MshB active site accessibility. The panels show the surfaces of the six MshB monomers from the overlaid structures in A for two different viewpoints. The surface colors correspond to locations of Tyr-142 shown in A, and the dark gray is the Tyr-142 side chain. The substrate GlcNAc-Ins is presumed to bind in the location of the two BOG molecules (colored in red and light gray) that are observed in the MshB-BOG structures (Fig. 1C). A, viewpoint 1 with the Tyr-142 side chain shown in dark gray. B, viewpoint 1, where the Tyr-142 side chain was truncated to Ala (dark gray). C, viewpoint 2 with the Tyr-142 side chain shown in dark gray. D, viewpoint 2, where the Tyr-142 side chain was truncated to Ala (dark gray).
Solvent Isotope Effects
Because the proposed reactions involve GABC, we examined if a solvent isotope effect is observed for the MshB-catalyzed reaction. Solvent isotope effects of 2–4 are typically observed for reactions that proceed through GBC, whereas inverse solvent isotope effects are observed for reactions that proceed through GAC (36). We measured the pH dependence of the solvent isotope effect for the WT protein under V/K conditions (Fig. 7A and Table 4). Results from these experiments reveal a small normal solvent isotope effect on V/K in WT MshB. This finding suggests that either 1) the chemistry step reflected in V/K has contributions from both GAC and GBC or 2) proton transfer is not a significant rate-determining step under these conditions.
FIGURE 7.

Solvent isotope effect of MshB variants. The solvent isotope for MshB was measured in H2O (open symbols) and 95% D2O (closed symbols) at 30 °C. Solvent isotope effects were measured under subsaturating substrate concentrations of GlcNAc. The values for V/K were measured using 5 or 20 mm GlcNAc as described under “Materials and Methods.” A, WT MshB in H2O (○) and 95% D2O (●). B, D15A in H2O (□) and 95% D2O (■). C, H144A in H2O (▿) and 95% D2O (▾). D, Y142F in H2O (△) and 95% D2O (▴). The pKa values were determined by fitting an equation including one or two ionizations (Equations 1–3) to these data and are shown in Table 4. Fits for H2O data are represented with dashed lines, and D2O data are represented with solid lines.
TABLE 4.
Solvent isotope effects of Zn2+-MshB variants
| MshBa,b | pKa1 | pKa2 | V/KH2O/V/KD2O |
|---|---|---|---|
| WTc (H2O) | 7.4 ± 0.1 | 10.5 ± 0.1 | 1.2 |
| WT (D2O) | 7.6 ± 0.1 | 10.6 ± 0.1 | |
| D15A (H2O) | 10.4 ± 0.3 | 0.8 | |
| D15A (D2O) | 10.3 ± 0.3 | ||
| Y142F (H2O) | 7.1 ± 0.05 | 1.0 | |
| Y142F (D2O) | 7.3 ± 0.05 | ||
| H144A (H2O) | 7.1 ± 0.1 | 10.5 ± 0.1 | 5.4 |
| H144A (D2O) | 7.4 ± 0.4 | 10.9 ± 0.4 |
a Apo-MshB was incubated with stoichiometric metal for 30 min prior to activity measurement.
b Substrate 5 (WT, Y142F), 10 (Y142A), or 20 mm (D15A, H144A) GlcNAc.
c Data adapted from Ref. 14.
To aid in deciphering between these two possibilities and to gain additional insights into the chemical mechanism, we also measured the pH dependence of the solvent isotope effect for the MshB mutants D15A, Y142F, and H144A under V/K conditions. The results from these experiments are shown in Fig. 7 and Table 4. We observe a slight inverse solvent isotope effect of 0.8 for the D15A mutant (Fig. 7B), whereas there is no solvent isotope effect for the Y142F mutant (Fig. 7D). The most striking observation is the large solvent isotope effect for the H144A mutant (Fig. 7C) of ∼5. There is no significant difference in the pKa values observed between H2O and D2O for any of the mutants examined.
DISCUSSION
MshB Uses Key Protein Side Chains and Metal-Water/Hydroxide as Reactive Nucleophile
Results from mutagenesis studies (Tables 1 and 3) indicate that active site side chains Asp-15, Tyr-142, His-144, and Asp-146 are all important for catalytic activity. The locations of Asp-15 and His-144 (Fig. 1, B and C) suggest that these side chains may function as GABC in the chemical mechanism, whereas the location of Tyr-142 in the model (Fig. 5C) suggests that this side chain may play a role in polarization of substrate and/or stabilization of the oxyanion intermediate. The location of Asp-146 suggests that this side chain does not directly participate in the chemical mechanism but plays an indirect role via interaction(s) with the side chain(s) of His-13 and/or His-144 (Fig. 1, B and C). Titration of apo-D146A with Zn2+ (supplemental Fig. S7) suggests that the loss of activity observed for this mutant cannot be attributed to decreased binding of the catalytic Zn2+ ion via interaction with the zinc ligand H13 under the reaction conditions.
Previous results have shown that the activity of MshB is dependent on the identity of the catalytic metal ion (14). Herein we demonstrate that NaF is an uncompetitive inhibitor of MshB (supplemental Fig. S5). These results are consistent with MshB using a metal-bound water/hydroxide as the reactive nucleophile in the reaction.
Parameter kcat Reflects Conformational Change Coupled to Product Release
Results from solvent viscosity experiments indicate that the parameter kcat/Km is inhibited by microviscogens (sucrose, glycerol), not macroviscogens (Ficoll 400). This is expected, given that the parameter kcat/Km reflects substrate binding through the first irreversible step (i.e. chemistry) and suggests that kcat/Km is partially limited by substrate association in WT MshB. The finding that the effect of solvent viscosity on kcat/Km is diminished in the Y142F mutant (Fig. 2B and supplemental Table S3) may suggest that the Tyr-142 side chain is involved in the conformational change coupled to substrate binding or that chemistry becomes more rate-limiting under kcat/Km conditions in catalytically slow mutants.
Results from solvent viscosity experiments also indicate that the parameter kcat is inhibited by the microviscogens glycerol and sucrose, suggesting that kcat reflects a step that involves the diffusion of small molecules (i.e. substrate binding, product release) and/or a conformational change in MshB. Because both the WT and mutant data are better described by plots of log kcat versus log ηrel compared with plots of log kcat versus ηrel2, the rate-limiting step for MshB is probably a conformational change that is associated with product release or substrate binding. The finding that the value of kcat is similar with the substrates GlcNAc-Ins (0.49 s−1) and GlcNAc (0.77 s−1), whereas Km values for these substrates vary significantly (340 μm and 38 mm, respectively) is consistent with the hypothesis that kcat reflects a conformational change that is associated with product release in WT MshB rather than simple product dissociation (12, 23).
The findings that 1) mutation of Tyr-142 (Ala, Gln, Phe) results in a 15–20-fold decrease in kcat, 2) the Y142F mutant is the only MshB variant examined where the value of the parameter kcat is unaffected by changes in solvent viscosity, 3) the location of the Tyr-142 side chain appears to be dynamic in available crystal structures, and 4) the value of kcat is identical for the D15A and Y142F mutants suggest that the Tyr-142 side chain, probably the hydroxyl group, is involved in the conformational change that is associated with product release. The slopes for log kcat versus log ηrel plots range from 0 to 1 depending on degree of coupling of active site to solvent (25). The finding that the slopes for WT MshB obtained in the presence of glycerol and sucrose are 0.72 ± 0.09 and 0.81 ± 0.06, respectively, suggests that there is significant coupling between the dynamics of MshB and solvent molecules. This is expected, given the role of Tyr-142 in modulating the dynamics of MshB and its observed location(s) in the enzyme (Figs. 5 and 6). Furthermore, results from kinetics data support a role for Tyr-142 in product release because the parameter kcat is significantly decreased upon mutation of Tyr-142 (Table 1). Specifically, we observe that the value of kcat for the Tyr-142 mutants follows the trend Ala > Gln > Phe for substitution at position 142, suggesting that the product release becomes slower with increasing hydrophobicity of the side chain (supplemental Fig. S8; correlation plots for Km also provided). It is likely that the rate acceleration with Tyr at this position arises from a combination of factors (i.e. hydrophobicity, hydrogen bonding interactions, and length of side chain).
Ionization of Asp-15 and Tyr-142 Is Important for Maximal Activity
Interestingly, pKa1 is not observed in the D15A mutant (Fig. 4), suggesting that this pKa reflects ionization of the Asp-15 side chain in WT MshB. Because the ionization described by pKa1 leads to an increase in activity with increasing pH, Asp-15 most likely functions as a GBC in the reaction to deprotonate the metal-bound water, activating it for attack of the carbonyl group on substrate. Structural data available support this mechanism because the side chain of Asp-15 shares a hydrogen bond to one of the metal-waters in the MshB crystal structure (Fig. 1B). The magnitude of the decrease in activity observed for the D15A mutant (∼530-fold) is smaller than that observed for side chains that are bifunctional GABC (103–105-fold) (37–39), and consequently, this side chain is probably part of a GABC pair that catalyzes the deacetylation reaction. The pKa of Asp-15 is probably elevated due to the environment of the active site, which includes an adjacent Asp-16 side chain.
Results from pH studies also reveal that the ionization described by pKa2 is lost in the Y142F and Y142A mutants (Fig. 4), suggesting that the side chain of Tyr-142 is responsible for pKa2 in WT MshB. The ionization described by pKa2 leads to a decrease in activity with increasing pH, indicating that the Tyr must remain protonated for maximal activity. This suggests two possible roles for Tyr-142 in the chemical reaction: 1) GAC to protonate the amine leaving group or 2) to polarize the carbonyl substrate/stabilize the oxyanion intermediate. Due to the magnitude of the decrease in activity at the pH optima (V/K) compared with the H144A mutant (74- versus 179-fold), the distance between the Tyr-142 hydroxyl and metal-water (2.15 Å) in the catalytic model depicted in Fig. 5C, and the orientation of Tyr-142 relative to the presumed substrate binding site (Figs. 1C and 6), it seems most likely that Tyr-142 functions in the latter role to polarize the carbonyl group on substrate/stabilize the oxyanion intermediate. The modest change that is observed for pKa2 upon substitution of the active site metal ion (Table 2) may also be explained by this ionization reflecting Tyr-142 because this side chain is proposed to hydrogen-bond to the metal-water and therefore would be sensitive to the identity of the bound metal ion. The assignment of pKa2, reflecting the ionization of Tyr-142, is preferred over assignment of pKa2 as the metal-water because 1) the pKa is lost in the Y142A/F mutants (compared with a modest shift in pKa2 observed in the metal substitution experiments), and 2) it seems unlikely that substitution of Tyr-142 with Phe and Ala would both markedly increase the pKa of the metal-water by several pH units given the differences in hydrophobicity between the active sites containing Phe versus Ala at this position.
The decrease in activity of H144A mutant (∼180-fold) and location of the side chain (Fig. 1, B and C) are consistent with His-144 serving as the GAC in the reaction working in concert with Asp-15 as part of a GABC pair. The magnitudes of the activity decreases for the D15A (530-fold) and H144A (∼180-fold) mutants are more consistent with the loss of a single GBC or GAC that functions as part of a GABC pair rather than the loss of residues that function as bifunctional GABC (103–105-fold) (37–39). Additionally, results from solvent isotope effect experiments are consistent with a mechanism that uses a GABC pair. Specifically, these results suggest that there is no significant solvent isotope effect observed in the WT protein because it includes contributions from both GAC and GBC. In the D15A mutant where the proposed GBC has been removed, there is a small inverse solvent isotope effect (∼0.8) observed that is consistent with a reaction that proceeds via GAC. In the H144A mutant, where the proposed GAC has been removed, there is a large solvent isotope effect (∼5) observed that is consistent with a reaction that proceeds via GBC. Taken together, these results support a model in which Asp-15 and His-144 function as a GABC pair in the WT MshB-catalyzed reaction.
Chemical Mechanism
Together these data suggest that MshB proceeds via the chemical mechanism shown in Fig. 8. In this mechanism, the carbonyl group on substrate replaces one of the metal-waters. Upon binding, the catalytic metal ion and the side chain of Tyr-142 polarize the substrate carbonyl group. The side chain of Asp-15 functions as a GBC to activate the metal-water for attack on the carbonyl substrate. The catalytic metal ion and Tyr-142 stabilize the resulting oxyanion tetrahedral intermediate. Finally, the side chain of His-144 functions as a GAC to facilitate breakdown of the tetrahedral intermediate. This proposed mechanism using a GABC pair is similar to the mechanism used by the metal-dependent deacetylase LpxC (UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase) and unlike prototypical metalloprotease carboxypeptidase A and the histone deacetylase enzymes, which use a single bifunctional GABC for catalysis (21).
FIGURE 8.

Proposed mechanism for MshB.
Dynamics of Tyr-142 Play Critical Role in Catalysis
The changes in Km that are observed in the Tyr-142 mutants, solvent viscosity effects, and the overlay of structures with BOG molecules (Fig. 6) are consistent with Tyr-142 playing a role in substrate binding and product release, whereas the activity data indicate that Tyr-142 plays a role in the chemical mechanism. These different roles for MshB in catalysis can be described using the structures in Fig. 6. We hypothesize that when Tyr-142 is located in the red and orange positions, the active site is open and allows for substrate binding. Movement of Tyr-142 (counterclockwise) toward the catalytic metal ion blocks access to the active site and allows for participation of Tyr-142 in catalysis (model lies between blue and green positions). Following participation in chemistry, Tyr-142 continues moving counterclockwise to the purple and then red locations, allowing for product release. Results from solvent viscosity experiments are consistent with a role for Tyr-142 assisting in substrate binding and product release, whereas effects on kcat/Km and V/K support a role for Tyr-142 in chemistry. Together, our findings suggest that the dynamic Tyr-142 plays a critical role in catalysis by modulating substrate binding, chemistry, and product release.
Supplementary Material
Acknowledgments
Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 RR001081).
This work was supported by Jeffress Memorial Trust Grant J960 (to M. H.).

This article contains supplemental Tables S1–S3 and Figs. S1–S8.
- MSH
- mycothiol
- MshB
- N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase
- GlcNAc-Ins
- N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside
- GABC
- general acid-base catalysis
- GBC
- general base catalyst
- GAC
- general acid catalyst
- IMAC
- immobilized metal ion affinity chromatography
- TEV
- tobacco etch virus
- MBP
- maltose-binding protein
- BOG
- β-octyl-d-glucopyranoside
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
REFERENCES
- 1. Newton G. L., Buchmeier N., Fahey R. C. (2008) Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 72, 471–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jothivasan V. K., Hamilton C. J. (2008) Mycothiol. Synthesis, biosynthesis, and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 25, 1091–1117 [DOI] [PubMed] [Google Scholar]
- 3. Rawat M., Av-Gay Y. (2007) Mycothiol-dependent proteins in actinomycetes. FEMS Microbiol. Rev. 31, 278–292 [DOI] [PubMed] [Google Scholar]
- 4. Fan F., Vetting M. W., Frantom P. A., Blanchard J. S. (2009) Structures and mechanisms of the mycothiol biosynthetic enzymes. Curr. Opin. Chem. Biol. 13, 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gammon D. W., Steenkamp D. J., Mavumengwana V., Marakalala M. J., Mudzunga T. T., Hunter R., Munyololo M. (2010) Conjugates of plumbagin and phenyl-2-amino-1-thioglucoside inhibit MshB, a deacetylase involved in the biosynthesis of mycothiol. Bioorg. Med. Chem. 18, 2501–2514 [DOI] [PubMed] [Google Scholar]
- 6. Gutierrez-Lugo M. T., Baker H., Shiloach J., Boshoff H., Bewley C. A. (2009) Dequalinium, a new inhibitor of Mycobacterium tuberculosis mycothiol ligase identified by high-throughput screening. J. Biomol. Screen. 14, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gutierrez-Lugo M. T., Bewley C. A. (2008) Natural products, small molecules, and genetics in tuberculosis drug development. J. Med. Chem. 51, 2606–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Metaferia B. B., Fetterolf B. J., Shazad-Ul-Hussan S., Moravec M., Smith J. A., Ray S., Gutierrez-Lugo M. T., Bewley C. A. (2007) Synthesis of natural product-inspired inhibitors of Mycobacterium tuberculosis mycothiol-associated enzymes. The first inhibitors of GlcNAc-Ins deacetylase. J. Med. Chem. 50, 6326–6336 [DOI] [PubMed] [Google Scholar]
- 9. Nicholas G. M., Eckman L. L., Newton G. L., Fahey R. C., Ray S., Bewley C. A. (2003) Inhibition and kinetics of mycobacterium tuberculosis and mycobacterium smegmatis mycothiol-S-conjugate amidase by natural product inhibitors. Bioorg. Med. Chem. 11, 601–608 [DOI] [PubMed] [Google Scholar]
- 10. Bhave D. P., Muse W. B., 3rd, Carroll K. S. (2007) Drug targets in mycobacterial sulfur metabolism. Infect. Disord. Drug Targets 7, 140–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Newton G. L., Av-Gay Y., Fahey R. C. (2000) N-Acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) is a key enzyme in mycothiol biosynthesis. J. Bacteriol. 182, 6958–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Newton G. L., Ko M., Ta P., Av-Gay Y., Fahey R. C. (2006) Purification and characterization of Mycobacterium tuberculosis 1-d-myo-Inosityl-2-acetamido-2-deoxy-α-d-glucopyranoside deacetylase, MshB, a mycothiol biosynthetic enzyme. Protein Expr. Purif. 47, 542–550 [DOI] [PubMed] [Google Scholar]
- 13. Nicholas G. M., Eckman L. L., Kovác P., Otero-Quintero S., Bewley C. A. (2003) Synthesis of 1-d- and 1-l-myo-inosityl 2-N-acetamido-2-deoxy-α-d-glucopyranoside establishes substrate specificity of the Mycobacterium tuberculosis enzyme AcGI deacetylase. Bioorg. Med. Chem. 11, 2641–2647 [DOI] [PubMed] [Google Scholar]
- 14. Huang X., Kocabas E., Hernick M. (2011) The activity and cofactor preferences of N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) change depending on environmental conditions. J. Biol. Chem. 286, 20275–20282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maynes J. T., Garen C., Cherney M. M., Newton G., Arad D., Av-Gay Y., Fahey R. C., James M. N. (2003) The crystal structure of 1-d-myo-inosityl 2-acetamido-2-deoxy-α-d-glucopyranoside deacetylase (MshB) from Mycobacterium tuberculosis reveals a zinc hydrolase with a lactate dehydrogenase fold. J. Biol. Chem. 278, 47166–47170 [DOI] [PubMed] [Google Scholar]
- 16. McCarthy A. A., Peterson N. A., Knijff R., Baker E. N. (2004) Crystal structure of MshB from Mycobacterium tuberculosis, a deacetylase involved in mycothiol biosynthesis. J. Mol. Biol. 335, 1131–1141 [DOI] [PubMed] [Google Scholar]
- 17. Supuran C. T. (2010) Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 20, 3467–3474 [DOI] [PubMed] [Google Scholar]
- 18. Lia N. G., Shib Z. H., Tang Y. P., Duan J. A. (2009) Selective matrix metalloproteinase inhibitors for cancer. Current Medicinal Chemistry 16, 3805–3827 [DOI] [PubMed] [Google Scholar]
- 19. Drag M., Salvesen G. S. (2010) Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 9, 690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White R. J., Margolis P. S., Trias J., Yuan Z. (2003) Targeting metalloenzymes. A strategy that works. Curr. Opin. Pharmacol. 3, 502–507 [DOI] [PubMed] [Google Scholar]
- 21. Hernick M., Fierke C. A. (2010) Mechanisms of metal-dependent hydrolases in metabolism. in Comprehensive Natural Products II (Mander L. N., Lui H.-W. B., eds) pp. 547–581, Elsevier Science Publishing Co., Inc., New York [Google Scholar]
- 22. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 23. Huang X., Hernick M. (2011) A fluorescence-based assay for measuring N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase activity. Anal. Biochem. 414, 278–281 [DOI] [PubMed] [Google Scholar]
- 24. Hall R. S., Xiang D. F., Xu C., Raushel F. M. (2007) N-Acetyl-d-glucosamine-6-phosphate deacetylase. Substrate activation via a single divalent metal ion. Biochemistry 46, 7942–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raber M. L., Freeman M. F., Townsend C. A. (2009) Dissection of the stepwise mechanism to β-lactam formation and elucidation of a rate-determining conformational change in β-lactam synthetase. J. Biol. Chem. 284, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneck J. L., Briand J., Chen S., Lehr R., McDevitt P., Zhao B., Smallwood A., Concha N., Oza K., Kirkpatrick R., Yan K., Villa J. P., Meek T. D., Thrall S. H. (2010) Kinetic mechanism and rate-limiting steps of focal adhesion kinase-1. Biochemistry 49, 7151–7163 [DOI] [PubMed] [Google Scholar]
- 27. Meng E. C., Pettersen E. F., Couch G. S., Huang C. C., Ferrin T. E. (2006) Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 7, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dunbrack R. L. (2002) Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 12, 431–440 [DOI] [PubMed] [Google Scholar]
- 29. Arnett S. O., Gerratana B., Townsend C. A. (2007) Rate-limiting steps and role of active site Lys-443 in the mechanism of carbapenam synthetase. Biochemistry 46, 9337–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ricci G., Caccuri A. M., Lo Bello M., Rosato N., Mei G., Nicotra M., Chiessi E., Mazzetti A. P., Federici G. (1996) Structural flexibility modulates the activity of human glutathione transferase P1–1. Role of helix 2 flexibility in the catalytic mechanism. J. Biol. Chem. 271, 16187–16192 [DOI] [PubMed] [Google Scholar]
- 31. Raber M. L., Arnett S. O., Townsend C. A. (2009) A conserved tyrosyl-glutamyl catalytic dyad in evolutionarily linked enzymes. Carbapenam synthetase and β-lactam synthetase. Biochemistry 48, 4959–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen G., Edwards T., D'souza V. M., Holz R. C. (1997) Mechanistic studies on the aminopeptidase from Aeromonas proteolytica. A two-metal ion mechanism for peptide hydrolysis. Biochemistry 36, 4278–4286 [DOI] [PubMed] [Google Scholar]
- 33. Javid-Majd F., Blanchard J. S. (2000) Mechanistic analysis of the argE-encoded N-acetylornithine deacetylase. Biochemistry 39, 1285–1293 [DOI] [PubMed] [Google Scholar]
- 34. Todd M. J., Hausinger R. P. (2000) Fluoride inhibition of Klebsiella aerogenes urease. mechanistic implications of a pseudo-uncompetitive, slow-binding inhibitor. Biochemistry 39, 5389–5396 [DOI] [PubMed] [Google Scholar]
- 35. Harris M. N., Ming L. J. (1999) Different phosphate binding modes of Streptomyces griseus aminopeptidase between crystal and solution states and the status of zinc-bound water. FEBS Lett. 455, 321–324 [DOI] [PubMed] [Google Scholar]
- 36. Schowen K. B., Schowen R. L. (1982) Solvent isotope effects of enzyme systems. Methods Enzymol. 87, 551–606 [PubMed] [Google Scholar]
- 37. Hernick M., Gennadios H. A., Whittington D. A., Rusche K. M., Christianson D. W., Fierke C. A. (2005) UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J. Biol. Chem. 280, 16969–16978 [DOI] [PubMed] [Google Scholar]
- 38. Cha J., Auld D. S. (1997) Site-directed mutagenesis of the active site glutamate in human matrilysin. Investigation of its role in catalysis. Biochemistry 36, 16019–16024 [DOI] [PubMed] [Google Scholar]
- 39. Wetterholm A., Medina J. F., Rådmark O., Shapiro R., Haeggström J. Z., Vallee B. L., Samuelsson B. (1992) Leukotriene A4 hydrolase. Abrogation of the peptidase activity by mutation of glutamic acid-296. Proc. Natl. Acad. Sci. U.S.A. 89, 9141–9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.