

Abstract
The chimeric PAX3-FKHR transcription factor is present in a majority of alveolar rhabdomyosarcoma (ARMS), an aggressive skeletal muscle cancer of childhood. PAX3-FKHR-mediated aberrant myogenic gene expression resulting in escape from terminal differentiation program is believed to contribute in ARMS development. In skeletal muscle differentiation, activation of AKT pathway leads to myogenic gene activation and terminal differentiation. Here, we report that AKT acts, in part, by modulating PAX3-FKHR transcriptional activity via phosphorylation in the maintenance of the myogenic differentiation blockade in established mouse models of ARMS cells. We observed that low levels of AKT activity are associated with elevated levels of PAX3-FKHR transcriptional activity, and AKT hyperactivation results in PAX3-FKHR phosphorylation coupled with decreased activity once cells are under differentiation-permissible conditions. Subsequent data shows that attenuated AKT activity-associated PAX3-FKHR activity is required to suppress the function of MyoD, a key myogenic regulator of muscle differentiation. Conversely, decreased PAX3-FKHR activity results in the eradication of MyoD expression and subsequent suppression of the myogenic differentiation. Thus, AKT regulation of the PAX3-FKHR suppresses myogenic gene expression in ARMS cells, causing a failure in differentiation. Evidence is presented that provides a novel molecular link between AKT and PAX3-FKHR in maintaining myogenic differentiation blockade in ARMS.
Key words: AKT, alveolar rhabdomyosarcoma, myogenic regulator MyoD, muscle differentiation, PAX3-FKHR
Introduction
Rhabdomyosarcoma (RMS) is a malignant skeletal muscle cancer of pediatric patients, classified by two major subtypes, embryonal (ERMS) and the aggressive alveolar (ARMS). The latter responds poorly to treatment and, thus, has a poor prognosis.1 The majority of ARMS are characterized by the presence of the pathogenetic chromosomal translocation t(2;13),2,3 which generates the chimeric transcription factor PAX3-FKHR consisting of the intact DNA binding domain of PAX3 and the potent transactivation domain of FKHR (FOXO1A).3 The transcription factor PAX3 plays an essential role in regulating the skeletal muscle differentiation program.4,5 The forkhead winged-helix transcription factor FKHR also regulates the above differentiation program.6,7 The chimeric PAX3-FKHR transcription factor retains the DNA binding specificity of PAX3; however, it gains greater transcriptional activity than does PAX3 in vitro8–10 and in vivo.11 Gene expression and genome-wide binding studies have shown that PAX3-FKHR transcriptional targets largely overlap with PAX3-regulated genes and, in addition, several additional PAX3-FKHR targets.12,13 These data suggest that PAX3-FKHR acts as a dominant-acting transcriptional regulator due to its increased transactivation power and relaxed target specificity, leading to deregulated expression of downstream targets that cause ARMS phenotype.
A typical feature of ARMS also includes the myogenic phenotype with expression of MyoD,14 a bona fide target of PAX3-FKHR,13,15 which acts as a key regulator of myogenic gene expression, leading to terminal differentiation.16 However, they fail to complete the terminal myogenic differentiation program.14,17 There is a common thought that PAX3-FKHR contributes to ARMS development by sustaining the undifferentiated state through the coupling of abnormal proliferation of myogenic precursors to an escape from terminal myogenic differentiation. In support of this hypothesis, PAX3-FKHR-driven myogenic18,19 or mesenchymal stem cell (MSC)-modified myogenic cells20 expressing MyoD but no marker of terminal differentiation are proclaimed as the cells of origin of ARMS. The myogenic gene expression pattern in these cells was also similar to what is evident in ARMS.14,21 Moreover, ectopic PAX3-FKHR expression enables muscle myoblast cells to escape from low serum-induced terminal myogenic differentiation.15,22,23
The myogenic differentiation program relies on promyogenic signals, namely, the serine/threonine kinase AKT signaling pathway,24–31 which is hyperactivated as myoblasts proceed to differentiate terminally in vitro under differentiation-permissible conditions (low serum).24,28,32–34 AKT hyperactivation promotes myogenic gene expression program and, thereby, terminal differentiation. Activation of AKT signaling pathway, which is a common lesion in many cancers,35–37 is also found in ARMS.38 Moreover, studies have found that AKT targets FKHR function in normal myogenic differentiation,6,7 but its fusion derivative PAX3-FKHR abrogates such processes in ARMS.18–20,23,39 However, the molecular mechanism(s) underlying activated AKT signaling in association with PAX3-FKHR in the suppression of myogenic differentiation program in ARMS remains unknown.
We used an established murine model of ARMS cells to investigate the mechanism by which AKT/PAX3-FKHR signaling blocks terminal myogenic differentiation in ARMS cells. Here, we report that activated AKT signaling modulates PAX3-FKHR to escape terminal myogenic differentiation in ARMS cells. The data shows that the transcriptional activity of PAX3-FKHR relies on activated AKT status in ARMS cells, and that low serum-induced hyperactivated AKT disables its transactivation ability via phosphorylation in these cells. Subsequent data reveals that the AKT status adopted transcriptionally active and inactive state of PAX3-FKHR to suppress myogenic gene expression and differentiation through the inhibition of MyoD activity and expression, respectively, in ARMS cells. Together, these results suggest that AKT enforces PAX3-FKHR functional status in two modes inhibiting myogenic differentiation regulated by MyoD in ARMS cells. In addition, the ability of hyper-activated AKT to downregulate the transactivation function of PAX3-FKHR uncovers a novel clue, which could be exploited to eradicate PAX3-FKHR activity in ARMS development and, thus, may have therapeutic application.
Results
Conditional mouse models of ARMS cells are defective in terminal myogenic differentiation.
Studies by us and others have demonstrated that PAX3-FKHR-positive human ARMS cells fail to undergo terminal myogenic differentiation.17,40,41 We asked whether such a scenario is shared in mouse ARMS cell lines U20497-T (ARMS-T) and U20325 (ARMS-325), derived from PAX3-FKHR knock-in mouse models of ARMS, which contain inactivated INK4a/ARF or p53,18 respectively. Immunoblot analysis of extracts of these cells confirmed the expression of both PAX3-FKHR and myogenic regulator MyoD proteins, a typical feature of PAX3-FKHR-expressed ARMS cells,17,18,42–44 with less PAX3-FKHR and more MyoD in ARMS-325 cells than the ARMS-T cells (Fig. 1A). We then assessed the ability of ARMS-T and ARMS-325 cells in the myogenic differentiation process upon switching these cells from growth (GM) to differentiation-permissible conditions (DM). In parallel, differentiation-proficient murine C2C12 myoblasts were processed as a positive control.45 In contrast to C2C12 myoblasts, a little induction of growth arrest p21cip1 and muscle myogenin (MyoG) expression was detected in ARMS-T and AMRS-325 cells respectively; however, there was no sign of terminal muscle myosin heavy chain (MyHC) expression in these ARMS cells under DM (Fig. 1B). This data establishes that PAX3-FKHR-positive ARMS cells expressing MyoD are incapable of undergoing terminal myogenic differentiation. Impaired MyoD transcriptional activity-mediated myogenic gene expression is associated with the failure of ARMS cells to differentiate terminally.17,40,41 To verify that this is also apparent in ARMS-T and ARMS-325 cells, we evaluated MyoD transactivation ability in these ARMS cells compared with C2C12 myoblasts grown in GM or DM following transduction of lentivirus expressing with a MyoD-responsive luciferase reporter (4RE-Luc).40 The data showed a decrease in MyoD-mediated reporter gene transcription, which is more in ARMS-T-4RE-Luc than ARMS-325-4RE-Luc cells, but, as anticipated, there was an increase of it in C2-4RE-Luc cells40 grown in DM (Fig. 1C). Immunoblot analysis of these reporter cell extracts revealed that the level of MyoD protein is downregulated in these ARMS cells but induced in C2C12 cells in DM, which is strongly correlated with MyoD-dependent reporter gene transcription. Collectively, these results suggest that suppressed MyoD expression may lead to the inhibition of terminal myogenic differentiation in ARMS-T and ARMS-325 cells grown under differentiation-permissible conditions.
Figure 1.

Suppression of MyoD expression and myogenic differentiation in mouse model of ARMS cells grown under differentiation-permissible conditions. (A) Immunoblot for PAX3-FKHR, MyoD or β-actin as a loading control for extracts from ARMS-T and ARMS-325 cells grown in GM. (B) Immunoblot of extracts of ARMS-T, ARMS-325 and C2C12 myoblast cells grown in GM or DM for 2 d, probed with indicated antibodies. (C) Luciferase activity in ARMS-T-, ARMS-325- and C2C12-derivative, MyoD-responsive 4RE-Luc reporter cells grown in GM or DM for 2 d. Values are normalized to amount of protein in lysates (upper). Immunoblot probed with MyoD or β-actin antibodies to detect MyoD and loading control, respectively (lower).
Transcriptional activity of PAX3-FKHR fails to correlate its protein levels between ARMS-T and ARMS-325 cells.
We observed that the protein levels of PAX3-FKHR are greater in ARMS-T cells than ARMS-325 cells, but MyoD, a bona fide transcriptional target of PAX3-FKHR,13,15 revealed an inverse scenario (Fig. 1A). In this context, we assessed whether PAX3-FKHR differs in its transactivation ability between ARMS-T and ARMS-325 cells. In an effort to compare PAX3-FKHR transactivation ability in these ARMS cell lines, both cell lines were transduced through lentivirus expressing a PAX3-FKHR-responsive luciferase reporter (6XPRS-Luc)46 or minimal pro-moter-driven empty luciferase reporter (Em-Luc). We found a significant increase in PAX3-FKHR-mediated reporter activity only in ARMS-6XPRS-Luc cells (Fig. 2A), even though equivalent levels of PAX3-FKHR were expressed in both 6XPRS-Luc and Em-Luc ARMS cells, demonstrating the specificity of the observed effect. Interestingly, PAX3-FKHR-mediated reporter gene transcription was several fold higher in ARMS-325-6XPRS-Luc than ARMS-T-6XPRS-Luc cells, despite the fact that the PAX3-FKHR protein level was higher in the latter cells. To further validate that PAX3-FKHR is specifically mediated the reporter gene activation in ARMS-6XPRS-Luc reporter cells, PAX3-FKHR was depleted in these cells via lentivirus coexpressing its shRNA or scrambled shRNA along with GFP (green fluorescence protein). Figure 2B depicted the GFP-fluorescence image of the above viral-tranduced ARMS-6XPRS-Luc reporter cells prior to assessing PAX3-FKHR-dependnet reporter gene transcription in these cells. We found declines in reporter luciferase activity in ARMS-6XPRS-Luc cells that have received PAX3-FKHR shRNA but not scrambled shRNA (Fig. 2C). Immunoblot analysis of reporter cell extracts confirmed PAX3-FKHR knockdown by its shRNA (Fig. 2D). Together, these data indicate that PAX3-FKHR is transcriptionally more active in ARMS-325 than ARMS-T cells, and this disparity is not associated with its protein levels in the two cell lines.
Figure 2.

PAX3-FKHR transactivation ability differs to its protein status between ARMS-T and ARMS-325 cells. (A) Luciferase activity in ARMS-T- and ARMS-325-derivative Em-Luc (empty) or PAX3-FKHR-responsive 6XPRS-Luc reporter cells grown in GM medium. Values are normalized to amount of protein in lysates (upper). Immunoblot of reporter cell extracts probed with FoxO1/FKHR or β-actin antibodies to detect PAX3-FKHR and loading control, respectively (lower). (B) Level of GFP in the above ARMS-derivative, PAX3-FKHR-responsive 6XPRS-Luc reporter cells tranduced with lentivirus coexpressing GFP along with indicated shRNA grown in GM for 2 d. (C) Luciferase activity in cells as in (B) normalized to amount of protein in lysates. Error bars, ± SEM (n = 3). (D) Immunoblot of reporter cell extracts as in (C) probed with FoxO1/FKHR and β-actin antibodies.
The transactivation ability of PAX3-FKHR decreases in ARMS cells under differentiation-permissible conditions.
Although we found a correlation in the status of MyoD and PAX3-FKHR-mediated transactivation between ARMS-T and ARMS-325 cells (Figs. 1A and 2A), the level of MyoD was drastically deceased once they were transferred from GM to DM conditions (Fig. 1C). Therefore, we evaluated PAX3-FKHR-dependent reporter gene transcription in ARMS-T-6XPRS-Luc and ARMS-325-6XPRS-Luc cells grown in GM and DM. The data showed that the relative PAX3-FKHR-responsive luciferase activity was severely declined when both of these reporter cells were switched from GM to DM (Fig. 3A). This scenario was also visualized in PAX3-FKHR-responsive non-luciferase reporter GFP cells (ARMS-325-6XPRS-GFP), which were generated by transducing lentivirus expressing 6XPRS-GFP reporter gene into ARMS-325 cells, upon switching from GM to DM (Fig. 3B). We next evaluated whether the decreased PAX3-FKHR-mediated reporter gene transcription is also reflected in its endogenous target, MyoD transcription, in these ARMS cells grown in DM. Indeed, the results showed a dramatic decrease in the levels of MyoD mRNA upon switching both ARMS-T and ARMS-325 cell lines from GM to DM (Fig. 3C). In contrast, PAX3-FKHR message level remains unchanged, whether these ARMS cells were grown in GM or DM (Fig. 3D), suggesting that the observed variation in the transcriptional activity of PAX3-FKHR is not associated with its own transcription. We then considered the possibility that there may be variation in the levels of PAX3-FKHR protein in ARMS cells grown in GM vs. DM conditions. However, immunoblot analysis of extracts of these cells revealed that PAX3-FKHR protein levels were relatively equivalent in cells grown either in GM or DM (Fig. 3E). Interestingly, we observed that PAX3-FKHR is detected as a doublet, with both a faster and slower migrating form of PAX3-FKHR present in ARMS cells. Moreover, the faster migrating form of PAX3-FKHR was shifted to the slower form when these cells were switched from GM to DM. Taken together, the results demonstrate that PAX3-FKHR function, but not level, is downregulated along with a shift of its faster to slower migrating form in ARMS cells grown in DM conditions.
Figure 3.

PAX3-FKHR-dependent gene transcription is downregulated in ARMS cells grown in differentiation-permissible conditions. (A) Luciferase activity in ARMS-derivative, PAX3-FKHR-responsive 6XPRS-Luc reporter cells grown in GM or DM for 2 d. Values are normalized to amount of protein in lysates. (B) Levels of GFP in ARMS-325-6XPRS-GFP reporter cells grown as in (A). (C) Quantitative real-time PCR analysis of MyoD in ARMS-T and ARMS-325 cells cultured as in (A). (D) RT-PCR analysis of PAX3-FKHR and β-actin in the indicated ARMS cells grown as in (A). (E) Immunoblot analysis of extracts of indicated ARMS cells grown as in (A), probed with FoxO1/FKHR and β-actin antibodies. For all applicable parts, error bars are ± SEM (n = 3).
Nuclear PAX3-FKHR undergoes phosphorylation in ARMS cells.
Previous studies have shown that constitutive nuclear localization of PAX3-FKHR protein is correlated with its transactivation function.9,47 However, PAX3-FKHR transcriptional activity decreased without significant alterations in the levels of mRNA or protein in both ARMS-T and ARMS-325 cells shifted from GM to DM (Fig. 3). We investigated whether altered PAX3-FKHR nuclear localization could account for the lack of PAX3-FKHR-mediated transactivation in these ARMS cells under DM. Fluorescent immunohistochemistry data revealed that the nuclear localization of PAX3-FKHR in both ARMS-T and ARMS-325 cells was similar, whether the cells were under GM or DM (Fig. 4A). This was confirmed by immunoblot analysis of the nuclear and cytoplasmic fractions of lysates from the same cells under both conditions using wild-type FKHR, which normally shuttles between the nucleus and cytoplasm,6 as a positive control (Fig. 4B). We observed a shift of PAX3-FKHR from its faster to slower migrating form when ARMS-T and ARMS-325 cells were switched from GM to DM (Fig. 3E). A phosphorylation-dependent shift in the migration of wild-type FKHR to a slower form has been demonstrated during normal myoblast differentiation.6 Therefore, we hypothesized that phosphorylation of the FKHR-derived fusion protein PAX3-FKHR might account for the shift toward a slower migrating form in these ARMS cells upon switching from GM to DM. Indeed, limited calf intestinal alkaline phosphastase (CIP) treatment of extracts of ARMS cells cultured in DM followed by immunoblotting showed a significant reduction in the amount of the slower migrating form of PAX3-FKHR and positive control wild-type FKHR (Fig. 4C). Collectively, these findings indicate that increased phosphorylation of nuclear PAX3-FKHR is triggered by DM conditions.
Figure 4.

Level of phosphorylated PAX3-FKHR is increased in the nucleus of ARMS cells under differentiation-permissible conditions. (A) Indicated ARMS cells cultured in GM or DM for 2 d are fixed and immunostained for PAX3-FKHR using PAX3 antibodies. (B) Immunoblot analysis of PAX3-FKHR and FKHR in total (TEx), nuclear (NEx) and cytoplasmic (CEx) extracts of ARMS-325 cells grown as in (A), probed with FoxO1/FKHR antibody. (C)-PAX3-FKHR, FKHR and β-actin immunoblot analysis of CIP-treated or untreated extracts from indicated ARMS cells grown in DM for 2 d.
Elevated PI3K/AKT-activation couples with increased PAX3-FKHR phosphorylation in ARMS cells under differentiation-permissible conditions.
The PI3K/AKT signaling pathway is frequently activated in many cancers,48 including ARMS.38 This pathway is also activated and acts as a promyogenic signal following the exposure of normal myoblasts to differentiation-permissible conditions.25,26 Because ARMS-T and ARMS-325 cells have an inactive terminal myogenic differentiation program (Fig. 1B), we examined the integrity of PI3/AKT activation in response to signals provoked by differentiation-permissible conditions (DM) in these cells. To this end, we checked the level of phosphorylation on Thr308 and Ser473 of AKT, a downstream effector of PI3K, both markers of activated AKT. As expected, AKT activation was detected in both ARMS-T and ARMS-325 cells under GM; however, its activation was further amplified in cells under DM (Fig. 5A), indicating a differentiation-stimuli responsive functional PI3K/AKT pathway in these tumor cells. This finding suggested that hyperactivation of AKT might be responsible for the induced PAX3-FKHR phosphorylation (Figs. 3E and 4C) in these ARMS cells under DM. PAX3-FKHR retains two of the three AKT kinase sites of wild-type FKHR located at Ser256 and Ser319.47 When the PI3K/AKT pathway was blocked by the PI3K inhibitor LY294002 (LY), decline in the level of phosphorylated form of PAX3-FKHR was visible in ARMS-T and ARMS-325 cells under DM (Fig. 5B). Immunoblot analysis confirmed the blockade of AKT activation as revealed by the reduced levels of phosphorylation on Ser473 of AKT in these LY-treated ARMS cells in DM (Fig. 5C). Therefore, these results demonstrated that differentiation-dependent AKT hyperactivation causes PAX3-FKHR phosphorylation in ARMS cells grown in DM conditions.
Figure 5.

AKT hyperactivation associates with PAX3-FKHR phosphorylation in ARMS cells under differentiation-permissible conditions. (A) Immunoblot analysis of extracts of indicated ARMS cells cultured in GM or DM for 2 d, probed with indicated antibodies. (B) Immunoblot of extracts of indicated ARMS cells cultured in GM or DM with or without LY for 2 d, probed with FoxO1/FKHR or β-actin antibodies (upper). Relative level of phosphorylated PAX3-FKHR (upper) is presented as a bar graph (lower). (C) Immunoblot analysis of cell extracts used in (B), probed with indicated antibodies.
Induced PI3K/AKT-activation disables transactivation function of PAX3-FKHR in ARMS cells under differentiation-permissible conditions.
We speculated that PI3K/AKT hyperactivation-associated PAX3-FKHR phosphorylation (Fig. 5) may be the reason for the weakened PAX3-FKHR transactivation function in ARMS cells under DM conditions (Fig. 3A). If this is true, we might expect the restoration of PAX3-FKHR transactivation function by the blockade of PI3K/AKT pathway in ARMS cells grown in DM. Note that blockade of this pathway by LY294002 (LY) inhibits PAX3-FKHR phosphorylation in these cells under DM (Fig. 5B and C). Therefore, we evaluated PAX3-FKHR-mediated transactivation in ARMS-6XPRS-Luc reporter cells grown in DM in the presence or absence of LY. The results showed the restoration of PAX3-FKHR-dependent luciferase reporter activity in LY-treated, but not untreated, ARMS-6XPRS-Luc cells under DM (Fig. 6A), suggesting that hyperactivated AKT induced the phosphorylation of PAX3-FKHR, which dampened the transactivation potency of PAX3-FKHR in ARMS cells grown in this condition. To verify that AKT hyperactivation, indeed, causes the decreased PAX3-FKHR transactivation function in ARMS cells in DM, we evaluated reporter gene transcription in ARMS-T-6XPRS-Luc cells grown in GM following overexpression of a dominant-negative form of AKT (dnAKT), a constitutive active form of it (myrAKT) or empty control through retrovirus-expressing marker enhanced GFP (eGFP). We found an increase or decrease of PAX3-FKHR-mediated luciferase activity in reporter cells that received dnAKT and myrAKT, respectively (Fig. 6B). A similar scenario was observed when the above experiment was performed in ARMS-325-6XPRS-Luc reporter cells (Fig. 6C). Fluorescence imaging depicted the expression of marker eGFP in these reporter cells tranduced with indicated retrovirus. Together, these data suggest that the levels of AKT activity are engaged in phosphor-ylation-dependent modulation of PAX3-FKHR transcriptional activation in ARMS-T and ARMS-325 cells.
Figure 6.

Inhibition of PI3/AKT pathway induces PAX3-FKHR transactivation in ARMS cells. (A) Luciferase activity in the indicated ARMS-6XPRS-Luc reporter cell grown in GM or DM in the presence of absence of LY for 2 d. Values are normalized to amount of protein in lysates. (B) Luciferase activity (normalized values) in ARMS-T-6XPRS-Luc cells transduced with eGFP expressing either control, myrAKT or dnAKT retroviruses grown in GM for 2 d. (C) Same as in (B), except ARMS-325-6XPRS-Luc reporter cells were used (upper). Levels of GFP in these retroviral tranduced reporter cells (bottom). For all applicable parts, error bars are ± SEM (n = 3).
Dominant-negative AKT-restored PAX3-FKHR transactivation induces MyoD expression but not its transactivation function in ARMS cells.
The results presented above suggested that AKT hyperactivation-induced deficiency of PAX3-FKHR transactivation was restored by the pharmacological blockade of AKT activity in ARMS cells grown in DM. A dominant-negative mutant of AKT (dnAKT) also enhances PAX3-FKHR transactivation in these cells under GM. However, we found that the lack of PAX3-FKHR transactivation coupled with impaired MyoD-mediated transcriptional activation was, in fact, due to its transcriptional repression in these cells under DM (Figs. 1C and 2A–C). Because MyoD is a transcriptional target of PAX3-FKHR,13,15 we investigated whether ectopic expression of dnAKT can rescue PAX3-FKHR-mediated MyoD expression and its transactivation in ARMS cells grown under DM. Therefore, we assessed PAX3-FKHR transactivation following transduction of retrovirus expressing epitope HA-tagged dnAKT or empty virus into ARMS-325-6XPRS-Luc reporter cells grown DM. The results showed that PAX3-FKHR-driven luciferase activity was increased in cells containing HA-tagged dnAKT (Fig. 7A). To ascertain that this increased PAX3-FKHR transactivation did indeed restore MyoD expression together with its transactivation function, we performed a similar experiment as above in MyoD-responsive ARMS-325-4RE-Luc reporter cells under DM conditions. Immunoblot analysis showed the induction of MyoD expression in cells expressing HA-tagged-dnAKT (Fig. 7B), which coincided with the above increased PAX3-FKHR transactivation in ARMS-325-6XPRS-luc cells grown under the same conditions. Interestingly, although MyoD was induced, its driven luciferase activity was further deceased in ARMS-325-4RE-Luc cells expressing HA-tagged-dnAKT (Fig. 7C). Together, these findings suggest that the interference of AKT signaling can rescue PAX3-FKHR mediated MyoD expression; however, it is transcriptionally inactive in ARMS cells grown in DM conditions.
Figure 7.

Dominant-negative AKT alleviates PAX3-FKHR-mediated MyoD expression exhibiting impaired transactivation in ARMS cells under differentiation-permissible conditions. (A) Luciferase activity in PAX3-FKHR-responsive ARMS-325-6XPRS-Luc reporter cells transduced with retrovirus expressing without (empty) or with HA-tagged dnAKT grown in GM or DM for 2 d. Values are normalized to amount of protein in lysates. (B) Immunoblot of extracts of MyoD-responsive ARMS-325-4RE-Luc reporter cells expressing without (empty) or with HA-tagged dnAKT via retroviral transduction, grown as in (A), probed with indicated antibodies. (C) Luciferase activity in cells used in (B). Values are normalized to amount of protein in lysates. For all applicable parts, error bars are ± SEM (n = 3).
MyoD overexpression rescues transcriptional inactive PAX3-FKHR suppressed terminal myogenic differentiation in ARMS cells.
In ARMS cells in DM, diminished PAX3-FKHR transcriptional activity failed to activate genes typically induced during myogenic differentiation, particularly muscle MyHC, a late phenotypic marker of terminal differentiation (Figs. 1B and 3A). However, the experiment was performed after 2 d in DM, which might not have been sufficient to see an effect due to the presence of residual levels of PAX3-FKHR transcriptional activity, even though this time point is sufficient to see induction in C2C12 muscle cells (Fig. 1B),49 (i.e., the effect may be delayed in ARMS). Therefore, PAX3-FKHR transcriptional activity was assessed with respect to its responsive reporter gene transcription and target MyoD-driven differentiation-associated gene expression, in ARMS-325-6XPRS-Luc reporter and parent ARMS-325 cells, respectively, grown in GM or DM for 2, 5 and 10 d. The data showed further decreases in PAX3-FKHR-driven reporter luciferase activity in cells grown in DM for 5 d, which remained low even at 10 d (Fig. 8A). However, there was no substantial change in the MyoD-induced p21cip1, MyoG and MyHC expression levels in these cells grown either 2, 5 or 10 d in DM (Fig. 8B). This finding is consistent with the fact that MyoD was absent from these cells at all three time points. These data suggest that loss of MyoD expression due to decreased PAX3-FKHR transcriptional activity blocks ARMS cells from terminal differentiation. To test this hypothesis, we generated ARMS-325-derivative ARMS-MyoD cells that ectopically overexpress MyoD (Fig. 8C). When these ARMS-MyoD cells were grown in DM for 2–10 d, remarkably, we found induced expression of p21cip1, MyoG and MyHC (Fig. 8D). Moreover, their levels of induction were enhanced with increased time of culture of these cells in DM, particularly to muscle-specific MyoG and MyHC expression. These findings indicate that MyoD overexpression overcomes the block in terminal myogenic differentiation observed in ARMS cells. Furthermore, immunoflurorescence for MyHC expression in ARMS-MyoD cells cultured in GM or DM for 10 d confirmed the differentiation phenotype as indicated by the enlargement of multinucleated cells only in DM (Fig. 8E). Taken together, these data indicate that the deficiency of PAX3-FKHR transactivation-coupled inhibition of MyoD expression is accountable for the failure of terminal differentiation in ARMS cells.
Figure 8.

Ectopic MyoD expression rescues transcriptional inactive PAX3-FKHR inability to induce terminal differentiation of ARMS cells. (A) Luciferase activity in ARMS-325-6XPRS-Luc cells grown in GM or DM for the indicated times in days. Values are expressed after protein normalization. Error bars, ± SEM (n = 3). (B) Immunoblot of extracts of ARMS-325 cells cultured as in (A), probed with indicated antibodies. (C) Immunoblot of extracts of ARMS-325 and its derivative MyoD overexpressed ARMS-MyoD cells grown in GM, probed with indicated antibodies. (D) Immunoblot of extracts of ARMS-MyoD cells cultured as in (A), probed with indicated antibodies. (E) Immunofluorescence of muscle MyHC of ARMS-MyoD cells grown in GM or DM for 10 d.
Discussion
The role of chromosomal translocation-derived chimeric transcription factor PAX3-FKHR in ARMS development is well characterized.2,3 However, studies have indicated that the mechanism of PAX3-FKHR in ARMS oncogenesis works, at least in part, by preventing myogenic cells from completing the terminal differentiation program.19,22,23 A key event in the myogenic differentiation process is the activation of the AKT signaling pathway, which acts as a promyogenic signals for myogenic gene activation.25–28 Despite activated AKT pathway in ARMS cells,38 they are defective in terminal myogenic differentiation.17,40,41 In ARMS, it is well recognized that PAX3-FKHR works by the gain of transcriptional power.2,50,51 The present study was focused on deciphering the molecular mechanism underlying activated AKT signaling in association with PAX3-FKHR transactivation in the suppression of the myogenic differentiation program in ARMS. To investigate the above mechanism, we have exploited conditional mouse models of ARMS cells.18,21
Here, we show that mouse ARMS cells reflect the defective terminal myogenic phenotype as revealed by no sign of MyHC expression similar to their human counterpart under differentiation conditions (DM).17,40,41 In this context, studies have implicated that impaired MyoD transcriptional activity-mediated myogenic gene expression, but not the absence of MyoD, is associated with the failure of ARMS cells to differentiate terminally.31,42,52 However, the data here indicated that impaired MyoD transactivation function is coupled with decreased levels of MyoD when these ARMS cells are under DM. Because MyoD is a transcriptional target of PAX3-FKHR,13,15 we evaluated the status of PAX3-FKHR transcriptional activity in these ARMS cells under DM. Interestingly, PAX3-FKHR transcriptional activity is downregulated in ARMS cells grown under DM, which coincided with decreased expression MyoD at both the mRNA and protein levels. Although, a numerous studies have delineated the transactivation function of PAX3-FKHR in attenuating myogenic differentiation,15,19,20,22,23,39,52,53 to our knowledge, downregulation of PAX3-FKHR transactivation function under DM has not been previously reported. This discovery led us to investigate the mechanism by which PAX3-FKHR transcriptional activity is downregulated in ARMS cells grown in DM. Interestingly, the data here demonstrate that decreased PAX3-FKHR transcriptional activity is not due to decreased levels of mRNA or protein nor to altered nuclear localization. However, we did perceive, during the assessment of PAX3-FKHR transactivation potency in ARMS cells, that PAX3-FKHR appears as a doublet of a faster and slower migrating form. Interestingly, the data showed the shift of PAX3-FKHR to a slower migrating form in ARMS cells grown in DM. This finding provided us a hint that the transcriptional activity of PAX3-FKHR may be modulated through its post-translational modification, which includes phosphorylation.54,55 Since induced PAX3-FKHR phosphorylation occurred when cells were grown in DM, we hypothesized that phosphorylation could be responsible for the decreased PAX3-FKHR transcriptional activity. Several studies have reported phosphorylation in the PAX3 domain of PAX3-FKHR in association with its transcriptional activity.1,54–58 However, none of the above studies reported phosphorylation-induced downregulation of transcriptional activity under differentiation conditions.
We investigated putative kinase signaling-pathway(s) induced under conditions suitable to induce terminal muscle differentiation. Our prime suspect in this context was the PI3/AKT pathway, since this pathway is activated in normal myoblasts when cultured in DM.28,29,59 In addition, PAX3-FKHR retains two consensus AKT phosphorylation sites in the FKHR domain.47 Although activated AKT is present in ARMS,38 we found hyper-activated AKT in ARMS cells grown in DM. Most significantly, the data generated using a pharmacological PI3/AKT inhibitor (LY294002) and a constitutive or dominant-negative version of AKT in ARMS cells showed that (1) the transactivation potency of PAX3-FKHR is preserved by basal activated AKT levels in cells under growth conditions, and (2) hyperactivated AKT as acts as a regulatory switch for PAX3-FKHR, changing it from a transcriptionally active to an inactive state under conditions of differentiation. In contrast, a previous study performed on NIH3T3 cells claimed that AKT activity fails to regulate the transcriptional activity of PAX3-FKHR.47 However, the previous finding does not negate our current findings, since PAX3-FHR activity and significance is cell type-specific,2,53 with a significant role almost exclusively in ARMS.2,3 Taken together, the data presented here uncover a molecular link between AKT activity and PAX3-FKHR transcriptional response, which can be modulated by manipulating the level of AKT activity.
In the context of AKT activity, one would expect that its hyperactivation would lead to terminal myogenic differentiation. This assumption was based on previous studies demonstrating that differentiation-responsive AKT activation is essential for normal myoblasts to differentiate terminally.25,29,31,33 Studies further delineated that AKT signaling promotes differentiation by activating MyoD-regulated myogenic gene transcription.29,30 Concurrently, one would also expect that loss of PAX3-FKHR transactivation function would lead to ARMS cell differentiation. This was based on a recent study that claimed PAX3-FKHR prevents myogenic differentiation of ARMS cells through repression of myogenin expression.39 Their results showed that siRNA-mediated PAX3-FKHR silencing induces myogenin, a downstream MyoD target, to cooperate with it in late gene expression during terminal differentiation16 and late muscle MyHC expression in ARMS cells. However, the expression of MyoD, a direct target of PAX3-FKHR,13,15 was not significantly downregulated by PAX3-FKHR siRNA in their study in ARMS cells. In contrast, a previous study showed that PAX3-FKHR can transactivate myogenin independent of MyoD.60 Moreover, a recent study demonstrated that PAX3-FKHR activates MyoD expression, but at the same time attenuates the MyoD-mediated terminal myogenic program.15 Our data demonstrates that the restoration of PAX3-FKHR activity mediated induction of MyoD by dnAKT is incompetent in gene activation function in ARMS grown in DM, supporting that PAX3-FKHR activity mediated simultaneous induction and inhibition of MyoD regulated the myogenic program. Hence, PAX3-FKHR silencing by its siRNA might induce myogenin expression that cooperates with residual MyoD, leading to expression of late gene in MyoD-mediated myogenic differentiation in ARMS. Our data, however, indicate that hyperactivation of AKT or its activity led to loss of PAX3-FKHR transactivation potency and was not associated with induced expression of either myogenin or MyHC in ARMS cells grown in DM, but expression of MyoD is lost. In this scenario, the absence of MyoD along with myogenin, the latter mediated directly by the loss of PAX3-FKHR transcriptional activity and/or coupled with MyoD eradication, eventually led to the inhibition of MyHC expression. Importantly, restoration of MyoD expression in ARMS cells that possess transactivation-incompetent PAX3-FKHR repairs the defective terminal myogenic program under differentiation conditions. The data further underlines the role of MyoD in the activation of genes, such as growth arrest p21cip1 and muscle early myogenin followed by late MyHC.
In summary, our findings lead to the hypothesis that AKT tailors the PAX3-FKHR mediated block in muscle differentiation through both inhibitions of MyoD expression and function in ARMS cells (Fig. 9). Central to this perspective is our finding that AKT-dependent phosphorylation manipulates the transcriptional activity of PAX3-FKHR in ARMS cells. While activated AKT sustains PAX3-FKHR transcriptional activity-mediated block of MyoD activity and arrests differentiation of ARMS cells, hyperactivated AKT-mediated switching to transactivation-incompetent PAX3-FKHR fails to rescue the differentiation arrest through loss of MyoD expression in these cells. Thus, the activated AKT status could be critical to the control of PAX3-FKHR-positive ARMS cells to maintain undifferentiated state by preventing terminal differentiation.
Figure 9.
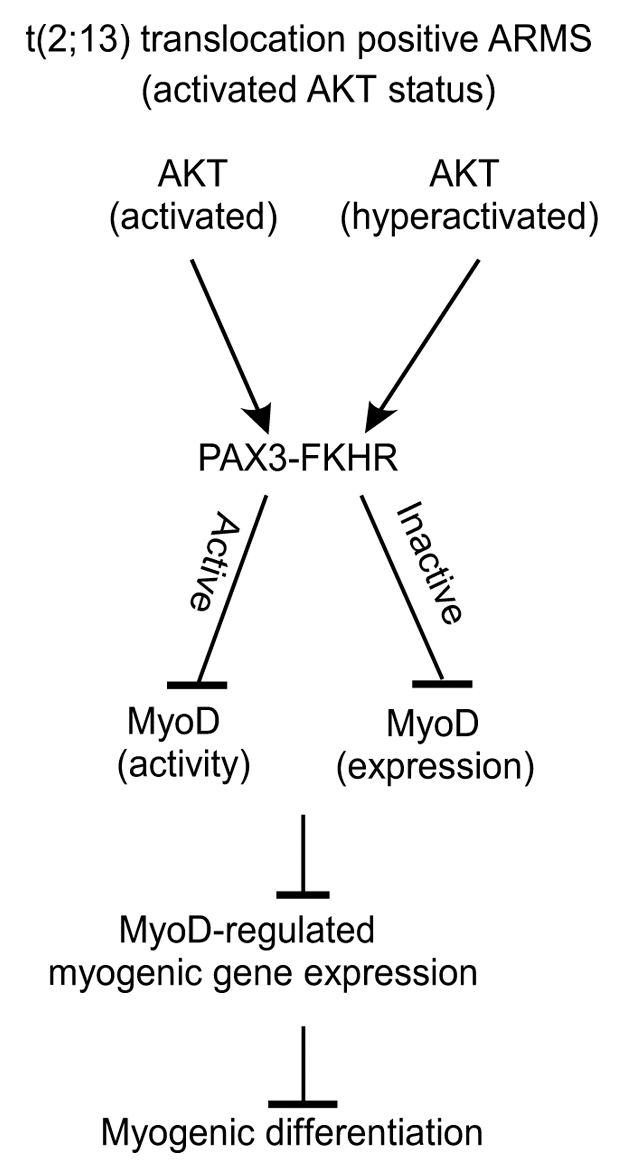
Model depicting AKT-directed dual strategy in the suppression of MyoD-driven myogenic differentiation through PAX3-FKHR in ARMS cells.
Materials and Methods
Cell culture.
The following cell lines were used in this study: mouse ARMS U20497T (ARMS-T) and U20325 (ARMS-325); mouse C2C12 myoblasts, 293FT, Phoenix-ampho and HeLa. Mouse ARMS cell lines U204097T (PAX3-FKHR+/+, INK4a/ARF−/−) and U20325 (PAX3-FKHR+/+, p53−/−) were established from tumors of the indicated conditional mouse ARMS strains as described previously in references 18 and 61. C2C12 myoblasts, HeLa, 293FT and Phoenix-ampho cell lines were described previously in reference 49. Except C2C12 cells, all cells were grown in DMEM supplemented with 10% FBS (growth medium, GM). C2C12 myoblasts were grown in growth medium (GM, 20% FBS), and cells were induced to differentiate by incubating in differentiation-permissible (DM) medium as described in reference 45. Cells were treated with 25 µM of LY294002 (purchased from Calbiochem) or control DMSO vehicle. Stable cell populations were generated through lentiviral/retroviral transduction followed by antibiotic selection.
Plasmids.
PAX3-FKHR responsive lentiviral pLA-6XPRS-Luc reporter luciferase vector was generated by PCR amplification of 6XPRS-9 binding sites from vector 6XPRS9-LucS (kindly provided by Frederic G. Barr62) followed by cloning into the pLA-4RE-Luc vector49 that lack the MyoD-responsive 4RE element. Reporter pLA-6XPRS-GFP vector was generated by replacing the Luc reporter from pLA-6XPRS-Luc with PCR amplified entire coding region of GFP from lentiviral pLVR-GFP vector. Lentiviral pLV-shRNAPAX3-FKHR-GFP and pLV-shRNAscramble-GFP vectors were generated in the following way: a 64 oligonucleotide small hairpin loop template for shRNA representing 19 bp oligos against PAX3-FKHR mRNA or control scrambled was initially cloned in pLV-shRNA (bleo)63 vector to obtain pLV-shRNA PAX3-FKHR (bleo) and pLV-shRNA scrambled (bleo) vectors, respectively. Finally, the bleomycin (bleo)-resistant gene was replaced with the entire coding region of GFP by sub-cloning. Retroviral pBabe-eGFP (eGFP, enhanced green fluorescence protein), pBabe-myrAKT-eGFP and pBabe-dnAKT-eGFP vectors were generously received from Eugene Kandel. Retroviral pBabe-HA-myrAKT was generated by sub-cloning the SnaB1/EcoR1 fragment from pCMV5-HA-myrAKT (Addgene) into pBabe (puro) vector. Retroviral pBabe-MyoD was described in a previous study in reference 63. All PCR-derived sequences were confirmed by sequencing.
Retroviruses and lentiviruses production and transduction.
Retroviruses and lentivirus were generated following transient transfection of retroviral vectors or lentiviral vectors in Phoenixampho and 293FT packaging cell lines, respectively, as previously described in references 49 and 63. Viral supernatants were collected 48 h post-transfection by passing through 0.45 µm syringe. Viruses were diluted with growth medium containing 8 µg/ml of freshly prepared polybrene (Sigma) and tranduced into target cells for 8 h three consecutive times. Where appropriate, antibiotic selection was performed 48 h post-transduction until untransduced cells died. Viral titers were routinely assessed by GFP florescence in HeLa cells transduced with serial dilution of GFP expressing retroviral and lentiviral stock.
Generation of stable population of cells.
We generated ARMS-T-4RE-Luc and ARMS-325-4RE-Luc reporter cell populations containing MyoD-responsive 4RE-luc luciferase reporter vector via lentivirus transduction followed by selection with puromycin. Similarly, puromycin-resistant ARMS-T-6XPRS-Luc and ARMS-325-6XPRS-Luc cell populations containing the PAX3-FKHR-responsive 6XPRS-Luc reporter vector or empty Em-luc vector were generated. ARMS-325-6XPRS-GFP cell populations were generated by transducing lentivirus expressing 6XPRS-GFP reporter gene. Puromycinresistant populations of MyoD overexpressed ARMS-325 (ARMS-MyoD) cells were generated by retroviral transduction. Lentivirus co-expressing PAX3-FKHR shRNA and GFP or control scrambled shRNA and GFP were transduced into ARMS-T-6XPRS-Luc and ARMS-325-6XPRS-Luc reporter cells. All cells were maintained at 37°C, 5% CO2 in a humidified atmosphere.
Antibodies.
The following antibodies were purchased and used in this study: FoxO1/FKHR (C29H4), Phosphor-Akt (Ser473) (D9E), Phosphor-Akt (Thr308) (C31E5E), and Akt (pan) (C67E7) (Cell Signaling); MyoD (M318, C20) and p21cip1 (C19) (Santa Cruz Biotechnology); Myogenin (BD Bioscience); MyHC (MF20) (Developmental Hybridoma Bank), HA-Peroxidase (H6533) and β-actin-Peroxidase (clone RG-96) (Sigma-Aldrich).
Cell extracts, western blotting and CIP treatment.
Preparation of total, nuclear and cytoplasmic cell extracts and immunoblotting were described previously in reference 45. For immunoblot analysis, the signal was detected using an ECL-Plus reagent (GE Healthcare), and the image was retrieved and analyzed by Alpha Innotech FluorChem® HD2 Imager (R&D Systems). For CIP treatment, 50 µg of total cell extracts was incubated with 50 U of Calf Intestine alkaline Phosphatase (CIP) (Roche) at 37°C for 1 h followed by immunoblot analysis.
Luciferase reporter assay.
The ARMS derivative PAX3-FKHR-responsive 6XPRS-Luc and MyoD-responsive 4RE-Luc luciferase reporter cells were seeded in a 12-well plate (5 × 104 cells per well) and grown in GM or DM for different time periods. Luciferase assay was performed as described previously in reference 45, and luciferase activity was determined in triplicate using a luciferase assay system according to the manufacture's instruction (Promega) and normalized by total protein.
Gene expression analysis.
Total RNA was isolated from ARMS-T and ARMS-325 cells grown in GM and DM using TRIzol reagent according to manufacturer's instruction (Invitrogen). RNA was then transcribed into cDNA using Superscript III reverse transcriptase (Invitrogen) as instructed by the manufacturer. Semi-quantitative PCR and quantitative real-time PCR (qRT-PCR) analysis were performed as described in our recent study in reference 63. For quantitation, target mRNA level was normalized to the constitutively expressed β-actin mRNA. The cycle threshold value (CT) for target and β-actin mRNAs for each sample was calculated. A normalized target value was then derived by subtracting the amount of target mRNA by that of β-actin (ΔCT) and the changes of target mRNA expression were quantified using delta-delta CT method. All reactions were performed in triplicate.
Immunofluorescence.
Cells cultured in GM and DM were washed with PBS, fixed with 4% paraformadehyde and permeabilized with 0.1% TX-100 in PBS for 30 min. After blocking with donkey normal serum, cells were incubated with primary antibody. Alexa-Fluor 488 (green) or 594 (red) conjugated secondary antibodies (Invitrogen) were used to detect the bound primary antibody in ARMS-325 and ARMS-T cells respectively. Nuclei were stained with DAPI. Images were documented using fluorescence microscope Leica DMI 4000 B (Leica microsystems).
Primers and oligonucleotide.
Primers for cloning
6XPRS-9 binding sites: Forward: CCG CTC GAG CGG ATC GAT AAT TCG AGC TCG AC; Reverse: TCC CCC GGG GGA GCC CAA GGT CGA AGC TTA
GFP: Forward: AAC TGC AGA ACC AAT GCA TTG GAT GGT GAG CAA GGG C; Reverse: CGG GAT CCC GTT ACT TGT ACA GCT CGT CCA TG
Primers for RT-PCR
PAX3-FKHR: Forward: AGA CAT TTA CAC CAG GGA GGA; Reverse: CTG TCA TGA TGG GAG AAA CTC
MyoD: Forward: CCG CCG CCT GAG CAA AGT GA; Reverse: CCG GAG GCG ACT CTG GTG GT
β-actin: Forward: CAC ACT GTG CCC ATC TAC G; Reverse: TGC TTG CTG ATC CAC ATC
Oligonucleotides for RNA interference
PAX3-FKHR: CCT CTC ACC TCA GAA TTC A
Scramble: ATC TAT CCG ACC ACC TAT C
Acknowledgements
This work was supported by Public Health Service grant AR051502 to A.K.M. from National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). We would like to thank Frederic G. Barr (Laboratory of Pathology, National Cancer Institute) and Eugene Kandel (Roswell Park Cancer Institute) for providing reagents. We would also like to thank Munmun Mal for experimental assistance and Catherine Burkhart (Cleveland BioLabs) for discussion and comments on the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Naini S, Etheridge KT, Adam SJ, Qualman SJ, Bentley RC, Counter CM, et al. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. 2008;68:9583–9588. doi: 10.1158/0008-5472.CAN-07-6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linardic CM. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 2008;270:10–18. doi: 10.1016/j.canlet.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mercado GE, Barr FG. Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: recent advances. Curr Mol Med. 2007;7:47–61. doi: 10.2174/156652407779940440. [DOI] [PubMed] [Google Scholar]
- 4.Ridgeway AG, Skerjanc IS. Pax3 is essential for skeletal myogenesis and the expression of Six1 and Eya2. J Biol Chem. 2001;276:19033–19039. doi: 10.1074/jbc.M011491200. [DOI] [PubMed] [Google Scholar]
- 5.Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell. 1997;89:127–138. doi: 10.1016/S0092-8674(00)80189-0. [DOI] [PubMed] [Google Scholar]
- 6.Bois PR, Grosveld GC. FKHR (FOXO1a) is required for myotube fusion of primary mouse myoblasts. EMBO J. 2003;22:1147–1157. doi: 10.1093/emboj/cdg116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hribal ML, Nakae J, Kitamura T, Shutter JR, Accili D. Regulation of insulin-like growth factor-dependent myoblast differentiation by Foxo forkhead transcription factors. J Cell Biol. 2003;162:535–541. doi: 10.1083/jcb.200212107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennicelli JL, Edwards RH, Barr FG. Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc Natl Acad Sci USA. 1996;93:5455–5459. doi: 10.1073/pnas.93.11.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, et al. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15:1522–1535. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sublett JE, Jeon IS, Shapiro DN. The alveolar rhabdomyosarcoma PAX3/FKHR fusion protein is a transcriptional activator. Oncogene. 1995;11:545–552. [PubMed] [Google Scholar]
- 11.Relaix F, Polimeni M, Rocancourt D, Ponzetto C, Schäfer BW, Buckingham M. The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain-of-function phenotype with ligand-independent activation of Met signaling in vivo. Genes Dev. 2003;17:2950–2965. doi: 10.1101/gad.281203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- 13.Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010;70:6497–6508. doi: 10.1158/0008-5472.CAN-10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonin PN, Scrable H, Shimada H, Cavenee WK. Muscle-specific gene expression in rhabdomyosarcomas and stages of human fetal skeletal muscle development. Cancer Res. 1991;51:5100–5106. [PubMed] [Google Scholar]
- 15.Graf Finckenstein F, Shahbazian V, Davicioni E, Ren YX, Anderson MJ. PAX-FKHR function as pangenes by simultaneously inducing and inhibiting myogenesis. Oncogene. 2008;27:2004–2014. doi: 10.1038/sj.onc.1210835. [DOI] [PubMed] [Google Scholar]
- 16.Cao Y, Kumar RM, Penn BH, Berkes CA, Kooperberg C, Boyer LA, et al. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006;25:502–511. doi: 10.1038/sj.emboj.7600958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tapscott SJ, Thayer MJ, Weintraub H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 1993;259:1450–1453. doi: 10.1126/science.8383879. [DOI] [PubMed] [Google Scholar]
- 18.Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18:2614–2626. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Schwartz J, Wang C. Comparative analysis of paired- and homeodomain-specific roles in PAX3-FKHR oncogenesis. Int J Clin Exp Pathol. 2009;2:370–383. [PMC free article] [PubMed] [Google Scholar]
- 20.Ren YX, Finckenstein FG, Abdueva DA, Shahbazian V, Chung B, Weinberg KI, et al. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. 2008;68:6587–6597. doi: 10.1158/0008-5472.CAN-08-0859. [DOI] [PubMed] [Google Scholar]
- 21.Nishijo K, Chen QR, Zhang L, McCleish AT, Rodriguez A, Cho MJ, et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. 2009;69:2902–2911. doi: 10.1158/0008-5472.CAN-08-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epstein JA, Lam P, Jepeal L, Maas RL, Shapiro DN. Pax3 inhibits myogenic differentiation of cultured myoblast cells. J Biol Chem. 1995;270:11719–11722. doi: 10.1074/jbc.270.20.11719. [DOI] [PubMed] [Google Scholar]
- 23.Roeb W, Boyer A, Cavenee WK, Arden KC. PAX3-FOXO1 controls expression of the p57Kip2 cell cycle regulator through degradation of EGR1. Proc Natl Acad Sci USA. 2007;104:18085–18090. doi: 10.1073/pnas.0708910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez I, Tripathi G, Carter EJ, Cobb LJ, Salih DA, Lovett FA, et al. Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol-3-kinase pathways in myogenesis. Mol Cell Biol. 2004;24:3607–3622. doi: 10.1128/MCB.24.9.3607-22.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang BH, Aoki M, Zheng JZ, Li J, Vogt PK. Myogenic signaling of phosphatidylinositol-3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc Natl Acad Sci USA. 1999;96:2077–2081. doi: 10.1073/pnas.96.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang BH, Zheng JZ, Vogt PK. An essential role of phosphatidylinositol-3-kinase in myogenic differentiation. Proc Natl Acad Sci USA. 1998;95:14179–14183. doi: 10.1073/pnas.95.24.14179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaliman P, Viñals F, Testar X, Palacín M, Zorzano A. Phosphatidylinositol-3-kinase inhibitors block differentiation of skeletal muscle cells. J Biol Chem. 1996;271:19146–19151. doi: 10.1074/jbc.271.32.19146. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Jiang B, Ensign WY, Vogt PK, Han J. Myogenic differentiation requires signalling through both phosphatidylinositol-3-kinase and p38 MAP kinase. Cell Signal. 2000;12:751–757. doi: 10.1016/S0898-6568(00)00120-0. [DOI] [PubMed] [Google Scholar]
- 29.Serra C, Palacios D, Mozzetta C, Forcales SV, Morantte I, Ripani M, et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell. 2007;28:200–213. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson EM, Rotwein P. Selective control of skeletal muscle differentiation by Akt1. J Biol Chem. 2007;282:5106–5110. doi: 10.1074/jbc.C600315200. [DOI] [PubMed] [Google Scholar]
- 31.Wilson EM, Tureckova J, Rotwein P. Permissive roles of phosphatidylinositol-3-kinase and Akt in skeletal myocyte maturation. Mol Biol Cell. 2004;15:497–505. doi: 10.1091/mbc.E03-05-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cabane C, Coldefy AS, Yeow K, Dérijard B. The p38 pathway regulates Akt both at the protein and transcriptional activation levels during myogenesis. Cell Signal. 2004;16:1405–1415. doi: 10.1016/j.cellsig.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Rotwein P, Wilson EM. Distinct actions of Akt1 and Akt2 in skeletal muscle differentiation. J Cell Physiol. 2009;219:503–511. doi: 10.1002/jcp.21692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/MCB.20.11.3951-64.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 36.Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des. 2010;16:34–44. doi: 10.2174/138161210789941865. [DOI] [PubMed] [Google Scholar]
- 37.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol-3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 38.Cen L, Hsieh FC, Lin HJ, Chen CS, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer. 2007;97:785–791. doi: 10.1038/sj.bjc.6603952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kikuchi K, Tsuchiya K, Otabe O, Gotoh T, Tamura S, Katsumi Y, et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun. 2008;365:568–574. doi: 10.1016/j.bbrc.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 40.Lee MH, Jothi M, Gudkov AV, Mal AK. Histone methyltransferase KMT1A restrains entry of alveolar rhabdomyosarcoma cells into a myogenic differentiated state. Cancer Res. 2011;71:3921–3931. doi: 10.1158/0008-5472.CAN-10-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, et al. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson J, Gordon A, Pritchard-Jones K, Shipley J. Genes, chromosomes and rhabdomyosarcoma. Genes Chromosomes Cancer. 1999;26:275–285. doi: 10.1002/(SICI)1098-2264(199912)26:4<275::AIDGCC1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. 1999;4:34–44. [PubMed] [Google Scholar]
- 44.Merlino G, Helman LJ. Rhabdomyosarcoma—working out the pathways. Oncogene. 1999;18:5340–5348. doi: 10.1038/sj.onc.1203038. [DOI] [PubMed] [Google Scholar]
- 45.Mal A, Sturniolo M, Schiltz RL, Ghosh MK, Harter ML. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 2001;20:1739–1753. doi: 10.1093/emboj/20.7.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chalepakis G, Fritsch R, Fickenscher H, Deutsch U, Goulding M, Gruss P. The molecular basis of the undulated/Pax-1 mutation. Cell. 1991;66:873–884. doi: 10.1016/0092-8674(91)90434-Z. [DOI] [PubMed] [Google Scholar]
- 47.del Peso L, González VM, Hernández R, Barr FG, Núñez G. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene. 1999;18:7328–7333. doi: 10.1038/sj.onc.1203159. [DOI] [PubMed] [Google Scholar]
- 48.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mal AK. Histone methyltransferase Suv39 h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 2006;25:3323–3334. doi: 10.1038/sj.emboj.7601229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charytonowicz E, Cordon-Cardo C, Matushansky I, Ziman M. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009;279:126–136. doi: 10.1016/j.canlet.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 51.Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- 52.Scuoppo C, Riess I, Schmitt-Ney M, Allegra P, Forni PE, Bersani F, et al. The oncogenic transcription factor PAX3-FKHR can convert fibroblasts into contractile myotubes. Exp Cell Res. 2007;313:2308–2317. doi: 10.1016/j.yexcr.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 53.Linardic CM, Naini S, Herndon JE, 2nd, Kesserwan C, Qualman SJ, Counter CM. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. 2007;67:6691–6699. doi: 10.1158/0008-5472.CAN-06-3210. [DOI] [PubMed] [Google Scholar]
- 54.Amstutz R, Wachtel M, Troxler H, Kleinert P, Ebauer M, Haneke T, et al. Phosphorylation regulates transcriptional activity of PAX3/FKHR and reveals novel therapeutic possibilities. Cancer Res. 2008;68:3767–3776. doi: 10.1158/0008-5472.CAN-07-2447. [DOI] [PubMed] [Google Scholar]
- 55.Zeng FY, Dong H, Cui J, Liu L, Chen T. Glycogen synthase kinase 3 regulates PAX3-FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem Biophys Res Commun. 2010;391:1049–1055. doi: 10.1016/j.bbrc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation and transformation. Cell. 2004;117:421–426. doi: 10.1016/S0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 57.Dietz KN, Miller PJ, Hollenbach AD. Phosphorylation of serine 205 by the protein kinase CK2 persists on Pax3-FOXO1, but not Pax3, throughout early myogenic differentiation. Biochemistry. 2009;48:11786–11795. doi: 10.1021/bi9012947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dietz KN, Miller PJ, Iyengar AS, Loupe JM, Hollenbach AD. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int J Biochem Cell Biol. 2011;43:936–945. doi: 10.1016/j.biocel.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tureckova J, Wilson EM, Cappalonga JL, Rotwein P. Insulin-like growth factor-mediated muscle differentiation: collaboration between phosphatidylinositol-3-kinase-Akt-signaling pathways and myogenin. J Biol Chem. 2001;276:39264–39270. doi: 10.1074/jbc.M104991200. [DOI] [PubMed] [Google Scholar]
- 60.Zhang L, Wang C. Identification of a new class of PAX3-FKHR target promoters: a role of the Pax3 paired box DNA binding domain. Oncogene. 2007;26:1595–1605. doi: 10.1038/sj.onc.1209958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taniguchi E, Nishijo K, McCleish AT, Michalek JE, Grayson MH, Infante AJ, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene. 2008;27:6550–6560. doi: 10.1038/onc.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xia SJ, Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene. 2004;23:6864–6871. doi: 10.1038/sj.onc.1207850. [DOI] [PubMed] [Google Scholar]
- 63.Lee MH, Jothi M, Gudkov AV, Mal AK. Histone methyltransferase KMT1A restrains entry of alveolar rhabdomyosarcoma cells into a myogenic differentiated state. Cancer Res. 2011;71:3921–3931. doi: 10.1158/0008-5472.CAN-10-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]