

Abstract
Expression of Fibroblast Growth Factor 2 (FGF2) is believed to be a contributing factor to the growth of a number of tumor types, including hepatocellular carcinoma (HCC). However, the potential of monoclonal antibodies that neutralize FGF2 for treatment of cancer patients has not yet been explored in clinical trials. We therefore generated a novel monoclonal antibody (mAb), GAL-F2, specific for FGF2 and characterized its properties in vitro and in vivo. GAL-F2 binds to a different epitope than several previous anti-FGF2 mAbs tested: this novel epitope was defined using chimeric FGF1/FGF2 proteins and alanine scanning mutagenesis and shown to comprise amino acids in both the amino and carboxy regions of FGF2. GAL-F2 blocked binding of FGF2 to each of its four cellular receptors, strongly inhibited FGF2-induced proliferation and downstream signaling in HUVEC, and inhibited proliferation and downstream signaling in two HCC cell lines. Moreover, GAL-F2, administered at 5 mg/kg i.p. twice weekly, potently inhibited growth of xenografts of the SMMC-7721, HEP-G2 and SK-HEP-1 human HCC cell lines in nude mice, and in some models had a strong additive effect with an anti-VEGF mAb or sorafenib. Treatment with GAL-F2 also blocked angiogenesis and inhibited downstream cellular signaling in xenografts, indicating its anti-tumor mechanism of action. Our report supports clinical testing of a humanized form of the GAL-F2 mAb for treatment of HCC and potentially other cancers.
Keywords: FGF2, FGF receptor, VEGF, liver cancer, xenograft
Introduction
The Fibroblast Growth Factor (FGF) family plays important roles in embryonic development, tissue repair, angiogenesis and the growth of certain tumors (1, 2). The FGF family has 22 known members in humans, including FGF2 (also called basic FGF). Human FGF2 is an 18 kDa non-glycosylated polypeptide consisting of 146 amino acids in the mature form derived from a 155 aa precursor (3). The precursor does not encode a signal sequence, but FGF2 is secreted by an unconventional pathway independent of the ER-Golgi complex (4).
There are only four FGF receptors, designated FGFR1 – FGFR4, with the various FGFs binding the different FGFRs to varying extents (5). The FGF receptors are structurally related transmembrane tyrosine kinases: each consisting of an extracellular domain (ECD) comprising three immunoglobulin-like domains (D1, D2 and D3), a single transmembrane helix, and an intracellular kinase domain (6). Two alternative exons can be utilized for the second half of the D3 domain, leading to forms denoted IIIb and IIIc (5). In addition to binding all the receptors FGFR1–4 with high affinity, FGF2 binds to heparin sulfate proteoglycans with lower affinity.
FGF2 stimulates proliferation of fibroblasts and is involved in tissue remodeling and regeneration (3). FGF2 also induces migration, proliferation and differentiation of endothelial cells (7) so is a potent angiogenic factor (2). FGF2 is believed to play a role in cancer, both by stimulating angiogenesis and tumor cells directly (2). FGF2 is strongly expressed in most gliomas (8), contributes to progression of prostate tumors (7), and is a key factor for the growth of melanomas (9). Overexpression of FGF2 and/or correlation with clinical features or outcome has also been reported for pancreatic cancer (10), and other types of cancer (11, 12).
The role of FGF2 in hepatocellular carcinoma (HCC; hepatoma) has been extensively studied and recently reviewed (13). Hepatomas are characterized by neovascularization, and angiogenesis plays a pivotal role in their growth, with FGF2 being an important pro-angiogenic factor (14). Higher serum level of FGF2 is an independent predictor of poor clinical outcome in HCC patients (15). FGF2 is overexpressed in HCC (16), and correspondingly FGF2 and FGFRs are widely expressed by HCC cell lines (17, 18). FGF2 antisense RNA induced the loss of tumorigenicity of SK-HEP-1 HCC xenografts in nude mice (19). An anti-FGF2 mAb inhibited proliferation of many HCC cell lines, and administering the anti-FGF2 mAb locally at the site of the tumor inhibited growth of KIM-1 HCC xenografts (18).
Monoclonal antibodies (mAbs) against various growth factors or their receptors including VEGF, EGF receptor, and HER2 are now being used to treat various types of cancer with considerable success. The association of FGF2 expression with many types of cancer and especially HCC suggests that FGF2 may also be an excellent target for a therapeutic mAb. A number of anti-FGF2 mAbs have previously been developed and shown to neutralize various activities of FGF2 in vitro and in some cases in vivo, including the mAbs DG2 (20), bFM-1 (21), 1E6 (22), 254F1 (23), FB-8 (24) and 3H3 (25). Of these, 3H3 is especially interesting, as it was reported to suppress growth of U87MG and T98G glioma xenografts and HeLa cell xenografts (26). However, to our knowledge, no anti-FGF2 mAb has been entered into clinical trials. With a view toward enhancing interest in FGF2 as an important therapeutic target, in this study we have developed and characterized a new mAb that has a unique epitope on FGF2.
Materials and Methods
Cell lines and mAb reagents
HEP-G2 (ATCC HB-8065) and SK-HEP-1 (ATCC HTB-52) were obtained from ATCC, and SMMC-7721 from Cell Bank of Chinese Academy of Sciences, but were not independently authenticated. Anti-FGF2 mAbs were purchased: 3H3 (Calbiochem), bFM-1 (Millipore), FB-8 (Abcam). The 3H3 mAb was also purified from its hybridoma, a kind gift of Dr. Akira Hori (Takeda Pharmaceutical Company), and anti-VEGF mAb A.4.6.1 was purified from its hybridoma (ATCC HB-10709).
FGF and FGFR reagents
Purified human FGF2 was purchased from R&D Systems and also prepared as described below. Mouse FGF2 was purchased from ProSpec-Tany TechnoGene. Flag-FGF2 was prepared by fusion of the Flag peptide MDYKDDDDK to the amino terminus of the 155 amino acid form of FGF2 and expression in the pET22b(+) vector (EMD Biosciences), which provides a pelB leader sequence for secretion into the medium. Flag-FGF1 and Flag-FGF1-FGF2 chimeric proteins were prepared similarly to Flag-FGF2, after fusion of the appropriate regions from human FGF1 and FGF2 genes. Flag-FGF2 alanine substitution mutants were prepared by in vitro mutagenesis. FGF2, Flag-FGF2 and the variant forms were purified using heparin-Sepharose C L-6B beads (Amersham Biosciences); expression levels and final concentrations were determined using an ELISA kit (R&D Systems). GST-FGF2 was prepared by inserting the FGF2 gene (amino acids 1–155) into the pGEX4T-1 vector (GE Healthcare Life Sciences), which contains the GST gene for linkage upstream of an inserted gene. After induction and cell lysis, GST-FGF2 was purified using a column of glutathione agarose (Sigma-Aldrich).
Peptides consisting of FGF2 amino acids 30–44 (KRLYCKNGGFFLRIH), 102–118 (FFFERLESNNYNTYRSR) and 137–155 (SKTGPGQKAILFLPMSAKS) were synthesized (SynBioSci) with a cysteine at the N or C-terminus for conjugation to keyhole limpet hemocyanin (KLH). FGF2-Fc and the FGFR-Fc fusion proteins were prepared by linkage of DNA encoding FGF2 (with a signal peptide) or the extracellular domains of FGFR1IIIc (amino acids 1–375), FGFR2IIIc (amino acids 1–377) and FGFR4 (amino acids 1–368) to the human Ig Fc region (amino acids 216 to 446) in the pDisplay vector (Invitrogen), expression in mammalian 293F cells (Invitrogen) and purification using a protein A/G column; FGFR3IIIc (amino acids 23–375)-Fc protein was from R&D Systems.
Generation of GAL-F2 mAb to human FGF2
Balb/c mice (5 to 6-week-old female) were immunized by injections in their rear footpads at 1-week intervals: 10 times with GST-FGF2 in monophosphoryl lipid A/ trehalose dicorynomycolate (MPL/TDM, Sigma-Aldrich) followed by 4 times with mixtures of FGF2-Fc and the three KLH-conjugated FGF2 synthetic peptides listed above in MPL/TDM. Three days after the final injection, popliteal lymphoid cells were fused with P3/X63-Ag8U1 mouse myeloma cells as described (27). Ten days after the fusion, hybridoma culture supernatants were screened for ability to capture FGF2 using the Flag-FGF2 binding ELISA described below. Selected mAbs were then screened for blocking activity in the FGFR1-Fc/FGF-Flag binding ELISA described below. The GAL-F2 hybridoma was chosen for further work and cloned twice by limiting dilution. The isotype of GAL-F2 was determined using isotype specific antibodies (BioRad).
ELISAs
Each step of each assay was performed by room temperature incubation with the appropriate reagent for 1 hour, except the initial plate coating step was done overnight at 4°C. Between each step, plates were washed 3 times in PBS containing 0.05% Tween 20. Data points were generally in triplicate. To measure binding of mAbs to FGF2 or to chimeric FGF1–FGF2 or alanine-substituted FGF2, ELISA wells were coated with goat anti-mouse IgG, blocked with 2% BSA, incubated with hybridoma supernatant during screening or with various concentrations of purified GAL-F2 or other anti-FGF2 mAb to be tested, and then incubated with the relevant form of FGF2 fused to Flag peptide. The bound Flag-FGF2 was detected by addition of HRP-anti-Flag M2 antibody (Sigma-Aldrich) and then TMB substrate.
To measure inhibition of FGF2 binding to FGFRs, ELISA wells were coated with goat anti-human IgG-Fc, blocked with BSA, and incubated with 0.5 μg/ml of FGFR1IIIc-Fc, FGFR2IIIc-Fc, FGFR3IIIc-Fc or FGFR4-Fc. The wells were then incubated with increasing concentrations of GAL-F2 or another anti-FGF2 mAb, together with Flag-FGF2 (0.1 μg/ml), which was detected by addition of HRP-anti-Flag M2 antibody (Sigma-Aldrich) and TMB.
To compare binding of GAL-F2 to human and mouse FGF2, ELISA wells were coated with 50 μg/ml of heparin (Sigma-Aldrich) and then incubated with 0.3 μg/ml of either human or mouse FGF2 so the heparin could capture the FGF2, followed by blocking with BSA. The wells were then incubated with increasing concentrations of GAL-F2 mAb; bound mAb was detected by addition of HRP-goat anti-mouse IgG-Fc and TMB.
For the competitive binding assay, heparin-coated ELISA wells were incubated with 0.5 μg/ml of human FGF2 followed by blocking with BSA. The wells were then incubated with increasing concentrations of various anti-FGF2 mAbs plus 0.5 μg/ml of biotinylated GAL-F2 (Bio-GAL-F2). The bound Bio-GAL-F2 mAb was detected by addition of HRP-streptavidin/TMB.
Proliferation and signaling assays
For the assays using human umbilical vascular endothelial cells (HUVEC; Cambrex), the cells were grown in EMB-2 Endothelial Growth Medium with FCS and endothelial cell growth supplements (Cambrex). HUVEC (5 × 103 cells/100 μl/well) were incubated in EMB-2 /1% FCS/ 0.1% BSA overnight, followed by incubation in EMB-2 / 0.1% FCS / 0.1% BSA (EMB assay media) for 24 hr. The cells were then incubated again in EMB assay media with 10 ng/ml FGF2 plus various concentrations of GAL-F2 or other anti-FGF2 mAbs for 2 days, and the level of cell proliferation was determined by addition of WST-1 (Roche Bioscience) for 16 hr. For the signaling assay, HUVEC were incubated in EMB assay media; then 10 ng/ml FGF2 and/or 10 μg/ml mAb was added for 10 min and the cells lysed for western blotting. In this assay only, we tested a genetically engineered form of GAL-F2, which has similar binding and functional properties to GAL-F2 and which will be described in detail elsewhere.
For proliferation assays of HEP-G2 and SMMC-7721, cells were seeded in 96-well plates in DMEM / 10% FCS. After allowing the cells to adhere overnight, the cells were incubated for 4 days in DMEM / 0.1% BSA with or without mAb, and WST-1 was added for 30 min. For the signaling assay, the cells were incubated in DMEM/ 10% FCS overnight, followed by incubation in DMEM/0.1% BSA for 24 hr, and then 10 μg/ml mAb was added for various time periods.
For all western blots, whole cell lysate was separated on a 4–20% Tris-Glycine SDS gel, transferred onto a PVDF membrane (GE Healthcare) and immunostained with specific antibodies: mouse anti-p-Erk1/2 and rabbit anti-p-Akt(Ser473), anti-Akt and anti-Erk1/2 (Cell Signaling Technology); and mouse anti-Hsp70 (Santa Cruz Biotechnology) as a sample loading control. Immunostained protein bands were detected with Amersham ECL Plus Western Blotting Detection Reagents (GE Healthcare).
Xenograft models
Animal experiments were conducted in accordance with U.S. Public Health Service policy. Human HCC cell lines were grown in complete DMEM medium and harvested in PBS. Female 5 to 6-week-old athymic nude mice were injected s.c. with 107 SMMC-7721 cells or 2 × 106 HEP-G2 cells in 0.1 ml PBS in the dorsal area. For SK-HEP-1, female Scid mice were similarly injected with 5 × 106 cells. When the tumor sizes reached ~100 mm3, mice were grouped randomly (n = 5–7 / group) and GAL-F2 mAb (100 μg in 0.1 ml, equivalent to 5 mg/kg body weight) was administered i.p. twice per week. In some experiments, A4.6.1 mAb was administered in the same dosage regimen instead of or in addition to GAL-F2. For experiments using sorafenib, the sorafenib (LC Laboratories) was dissolved in 1:1 ethanol and cremophor EL (Sigma-Aldrich) and then diluted in water to 2 mg/ml on the day of treatment. A dose of 0.2 ml of diluted sorafenib (equivalent to 20 mg/kg) was administered orally on five consecutive days of each week. When used, cisplatin (Sigma-Aldrich) in PBS was injected i.p. at 6 mg/kg once per week. Tumor volumes were determined twice weekly by measuring in two dimensions, length (a) and width (b), and calculating volume as V = ab2/2. Statistical analysis was performed by Student's t test applied to the final timepoint.
For the signaling assay, whole cell lysates of SMMC-7721 xenografts were prepared by homogenization of tumor pieces in lysis buffer, and the western blots run and developed as described above for the cell lines. For evaluation of angiogenesis, cryostat 5-μm-thick sections of HEP-G2 xenografts were stained with antibody to mouse CD31 (Dianova, Germany) to detect blood vessels. The bound primary antibodies were detected by using the Vectastain Elite ABC kit (Vector Labs). Developed sections were counterstained with Mayer's hematoxylin (Fisher Scientific), dehydrated and mounted.
Results
Generation and binding properties of anti-FGF2 mAbs
To obtain an optimal anti-FGF2 mAb, mice were immunized with GST-FGF2, FGF2-Fc or KLH-conjugated synthetic FGF2 peptides alone or in various combinations, and the resulting mAbs were screened for efficacy at blocking FGF2 binding to FGFR1. Through screening some thousands of mAbs from about 20 hybridoma fusions, the GAL-F2 mAb was selected after an immunization regimen of 10 weekly injections with GST-FGF2 followed by 4 injections with mixtures of FGF2-Fc and the peptides. GAL-F2 is of the IgG2b isotype.
In an ELISA assay, GAL-F2 bound to FGF2 as well or better than three previously developed anti-FGF2 mAbs that were available for comparison (Fig. 1A): bFM-1 (21), FB-8 (24) and 3H3 (25). Moreover, in a competition assay, only GAL-F2 blocked its own binding to FGF2 (Fig. 1B), so that among these mAbs, GAL-F2 has a unique epitope. GAL-F2 did not bind to denatured FGF2 in a western blot (not shown), suggesting that GAL-F2 recognizes a conformational epitope. GAL-F2 also did not display detectable binding to the other member of the FGF subfamily to which FGF2 belongs, FGF1 (see below).
Figure 1.
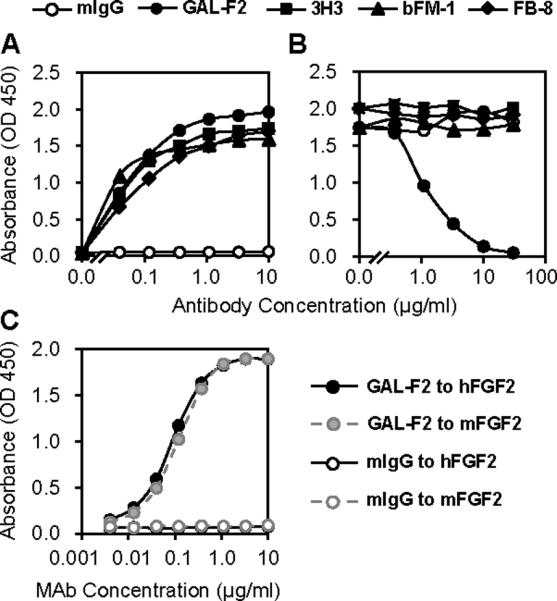
A, Binding of GAL-F2, other anti-FGF2 mAbs and negative control mAb mIgG to human FGF2 measured by ELISA; B, Competitive binding ELISA of anti-FGF2 mAbs against biotinylated GAL-F2; C, Binding ELISA of GAL-F2 to human FGF2 (hFGF2) and mouse FGF2 (mFGF2). In C, the curves for control mIgG binding superimpose and cannot be distinguished.
To help interpret the results of the xenograft experiments described below, we determined the ability of GAL-F2 to bind to mouse FGF2. In an ELISA assay, GAL-F2 bound to mouse FGF2 indistinguishably from human FGF2 (Fig. 1C). The cross-reactivity of GAL-F2 with mouse FGF2 may seem surprising because the mAb was generated by immunization with human FGF2 in mice. However, human and mouse FGF2 have high sequence homology, and the low level expression of FGF2 in tissues of the adult animal may make it relatively easy to break tolerance. In any case, the good reactivity of GAL-F2 with FGF2 from mouse and probably other species will facilitate animal studies of this mAb.
Receptor blocking activities of GAL-F2
We next tested the ability of GAL-F2 to inhibit binding of FGF2 to its four receptors FGFR1–4. FGFR-Fc fusion proteins were bound via anti-IgG-Fc antibody to ELISA plates, which were incubated with FGF2-Flag together with the anti-FGF2 mAb; bound FGF2-Flag was detected with a labeled anti-Flag mAb. The IIIc isoforms of the receptors FGFR1–3 were used, because FGF2 does not bind well to all of the IIIb forms (5). GAL-F2 strongly inhibited binding of FGF2 to the FGFRs, with IC50 < 0.5 μg/ml in each case (Fig. 2). Relative to other anti-FGF2 mAbs tested, GAL-F2 was somewhat better than 3H3 at inhibition of binding to each of the receptors, while comparable to bFM-1 for FGFR1–3 and substantially better for FGFR4 (Fig. 2).
Figure 2.
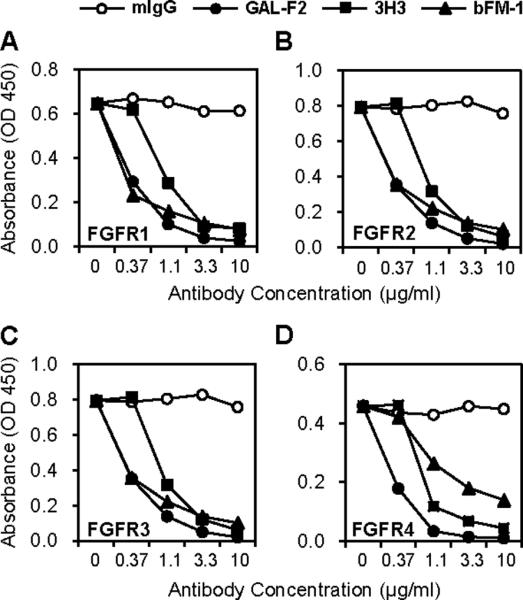
Inhibition of binding of FGF2 to each FGFR by GAL-F2 and other anti-FGF2 mAbs, measured by ELISA. A, FGFR1IIIc; B, FGFR2IIIc; C, FGFR3IIIc; D, FGFR4.
Inhibition of cellular proliferation and downstream signaling by GAL-F2
In principle, the ability of GAL-F2 to block binding of FGF2 to its receptors means that the mAb should neutralize all biological activities of FGF2. However, because small amounts of residual FGF2 binding might still be sufficient to transduce a signal, we tested the inhibitory ability of GAL-F2 in bioassays. GAL-F2 inhibited the FGF2-induced proliferation of HUVEC with an IC50 of approximately 0.1 μg/ml and complete inhibition at 1 μg/ml (Fig. 3A), indicating anti-angiogenic activity. In this assay, GAL-F2 was comparable to bFM-1 but somewhat more potent than 3H3. FGF2 treatment of HUVEC increased phosphorylation of the downstream effector molecules Akt and Erk1/2 (Fig. 3B, lines 1 and 3 respectively), without significantly affecting total Akt and Erk1/2 levels. This induced phosphorylation was strongly inhibited by GAL-F2 but not by a negative control mAb (Fig. 3B).
Figure 3.
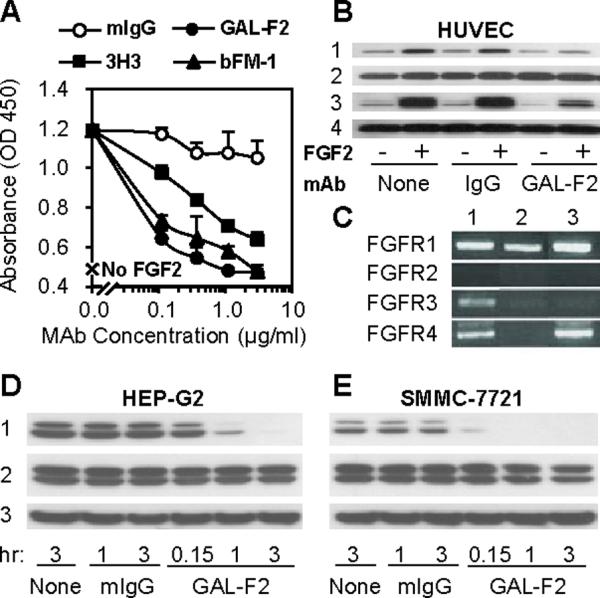
A, Inhibition of FGF2-induced proliferation of HUVEC by GAL-F2 and other anti-FGF2 mAbs. The error bars are S.D. B, Western blot of HUVEC incubated with or without FGF2 and GAL-F2. The blot was stained with mAbs detecting p-Akt (1), Akt (2), p-Erk1/2 (3), Erk1/2 (4). By densitometry, the relative intensities of the p-Akt bands, normalized to FGF2+ with no mAb, were from left to right: 32, 100, 35, 87, 14, 23. C, RT-PCR analysis of FGFR mRNA expression in HCC cell lines HEP-G2 (1), SMMC-7721 (2) and SK-HEP-1 (3), using primers specific for each of the FGFRs. D, E, Western blot of HEP-G2 and SMMC-7721 cells incubated with or without GAL-F2. The blot was stained with mAbs detecting p-Erk1/2 (1), Erk1/2 (2) and Hsp70 (3).
We also tested the ability of GAL-F2 to inhibit proliferation and downstream signaling in two hepatocellular carcinoma cell lines, HEP-G2 and SMMC-7721. These cell lines each express one or more FGFRs (Fig. 3C) and produce FGF2 themselves (see below), so no exogenous FGF2 was added for these experiments. GAL-F2 moderately inhibited proliferation of these cell lines in a concentration-dependent manner, with 20 – 25% inhibition obtained at 20 μg/ml mAb (data not shown). Moreover, GAL-F2 (10 μg/ml) strongly inhibited phosphorylation of Erk1/2, with essentially complete inhibition achieved after 1 hr in SMMC-7721 cells and 3 hr in HEP-G2 cells (Fig. 3D, E; line 1); phospho-Akt could not be detected in these cell lines even in the absence of mAb (not shown). Hence, activation of the MAP kinase pathway in these cell lines is completely dependent on autocrine stimulation by FGF2 under the conditions tested.
Epitope mapping
To locate the epitope of GAL-F2, we first took advantage of the fact that GAL-F2, as well as the anti-FGF2 mAbs 3H3 and bFM-1, does not bind to FGF1 (Fig. 4A). We therefore made two chimeric proteins: FGF2(1–79)/FGF1(68–155) in which the amino terminal half of FGF2 is fused to the carboxy terminal half of FGF1, and FGF1(1–41)/ FGF2(45–155) in which the amino terminal part of FGF1 is linked to the carboxy part of FGF2, where the numbers in parentheses indicate the amino acids that come from each protein. Both constructs had the Flag peptide fused to the amino terminus as a tag. As seen in Fig. 4A, GAL-F2 did not bind well to either of these constructs, suggesting that its epitope contains amino acids in both the amino region (before residue 45) and carboxy region (after residue 79) of FGF2. In contrast, the 3H3 mAb bound to FGF2(1–79)/FGF1(68–155), indicating its epitope is in the amino region, whereas the bFM-1 mAb bound to FGF1(1–41)/FGF2(45–155), so its epitope is more in the carboxy region. The three different epitopes of GAL-F2, 3H3 and bFM-1 are consistent with the inability of these mAbs to compete for binding (Fig. 1B).
Figure 4.

Relative binding of GAL-F2, bFM-1 and 3H3 to FGF2 or FGF1 or chimeric FGF2-FGF1 proteins (A), and of GAL-F2 to alanine mutants of FGF2 (B), measured by ELISA. Binding of each mAb to wildtype (WT) FGF2 is set as 100%. The means of triplicate values are shown; there was little variation between triplicates. C, Ribbon diagram of the crystallographic structure of FGF2 complexed with the extracellular D2 and D3 domains of FGFR2 (PDB ID 1IIL, ref. 28), with the indicated amino acids shown in space-filling form (R31, Y33, K55 and S152 in yellow; F40L and M151, which eliminate GAL-F2 binding, in red). The leucine mutant was used at F40 because the alanine mutant could not be expressed.
To more precisely delineate the epitope of GAL-F2, we measured binding to FGF2 variants in which individual amino acids were replaced by alanine. From a larger number of variants tested, substitution at FGF2 amino acid positions 31, 33, 40 and 55 in the amino region and 151 and 152 near the carboxy terminus substantially reduced or eliminated GAL-F2 binding (Fig. 4B). At least one of the mAbs bFM-1 and 3H3 bound well to each of the variants, verifying their conformational integrity (not shown). On the known crystallographic structure of FGF2 in complex with the D2 and D3 domains of FGFR2IIIc (28), these 6 amino acids lie close together in 3D space (Fig. 4C). Moreover, the epitope they form is located at the interface of FGF2 and FGFR2 (6), providing an explanation for why GAL-F2 blocks binding of FGF2 to its receptors.
GAL-F2 demonstrated potent anti-tumor activity in vivo
We tested the ability of GAL-F2 to inhibit xenografts from HCC cell lines, when administered alone or in combination with one of three other relevant agents: sorafenib, approved for treatment of HCC; cisplatin, widely used to treat HCC; and the anti-VEGF A4.6.1 mouse precursor mAb of bevacizumab (29), currently being tested in clinical trials for HCC. The three HCC cell lines used, which were selected because of their ability to grow well as xenografts, all produced similar amounts of VEGF, approximately 200 pg/ml in 7-day culture media, but varying amounts of FGF2: SMMC-7721 (~70 pg/ml), HEP-G2 (~400 pg/ml), SK-HEP-1 (~500 pg/ml). For the xenograft studies, GAL-F2 mAb was administered i.p. twice weekly at 5 mg/kg after tumor size had reached ~100 mm3, as was A4.6.1 when used.
GAL-F2 strongly inhibited growth of SMMC-7721 xenografts (Fig. 5A, p = 0.003), although not as well as A6.4.1 (Fig. 5B); however, GAL-F2 added to the inhibitory effect of A4.6.1 (Fig. 5B, p = 0.03 for GAL-F2 + A4.6.1 vs A4.6.1 alone) and in fact the combination caused tumor regression. Cisplatin had only a modest effect on growth of xenografts from this cell line when used alone or added to GAL-F2 (Fig. 5C; p = 0.08 for cisplatin vs mIgG, and p = NS for GAL-F2 + cisplatin vs GAL-F2). Sorafenib at 20 mg/kg five times per week, somewhat higher than the typical dose in human patients (400 mg fixed dose once or twice daily), also had only a modest effect used alone or with GAL-F2 (Fig. 5D), and indeed in another experiment the effect was even less pronounced, so its efficacy in this model is doubtful.
Figure 5.

Inhibition of growth of SMMC-7721 (A – D), HEP-G2 (E – G) and SK-HEP-1 (H) HCC xenografts by the indicated agents compared to negative control mAb mIgG. In the legends, “Both” indicates that GAL-F2 and the other listed agent were both administered. The means of groups of 5–7 mice are shown; the error bars are S.E.M. GAL-F2 and A.4.6.1 were administered i.p. at 5 mg/kg twice per week, cisplatin at 6 mg/kg i.p. once per week, and sorafenib orally at 20 mg/kg five times per week. Panels A and D are from the same experiment but shown separately for greater visual clarity.
GAL-F2 also strongly inhibited growth of HEP-G2 xenografts (Fig. 5E, p < 0.001), and was as effective as A4.6.1 for this cell line (Fig. 5F). Importantly, GAL-F2 and A4.6.1 had a very strong combined effect (Fig. 5F, p = 0.006 for GAL-F2 + A4.6.1 vs GAL-F2 and p = 0.002 for GAL-F2 + A4.6.1 vs A4.6.1). Cisplatin had little activity in this model (not shown), but sorafenib and GAL-F2 were about equally effective (Fig. 5G), and again there was a very strong combined effect (Fig. 5G, p = 0.003 for GAL-F2 + sorafenib vs GAL-F2 and p = 0.006 for GAL-F2 + sorafenib vs sorafenib). Finally, we also tested GAL-F2 against the SK-HEP-1 cell line, which is widely considered to be an HCC line, although it seems to be from a liver adenocarcinoma of endothelial origin (30). GAL-F2 had a substantial inhibitory effect on SK-HEP-1 xenografts (Fig. 5H, p < 0.001), but A4.6.1 had no effect alone or when added to GAL-F2 (data not shown). The inability of GAL-F2 to completely suppress growth of any of the xenografts when used as a single agent may be due to redundancy in the FGF family or to growth signals provided by other growth factors.
The toxicology of GAL-F2 has not been formally studied because a humanized form of this mAb rather than GAL-F2 itself will become a clinical candidate. However, it is encouraging that during the extensive xenograft studies described above, no obvious signs of toxicity due to GAL-F2 were observed, such as lethargy or weight loss. These results are meaningful because GAL-F2 binds to mouse FGF2 as well as human FGF2.
Mechanism of Action
To gain insight into the mechanism by which GAL-F2 inhibits growth of xenografts, we compared xenografts from mice treated with GAL-F2 to xenografts from mice treated with negative control mAb mIgG. In SMMC-7721 xenografts, treatment with GAL-F2 (5 mg/kg twice/week for 2 weeks) inhibited phosphorylation of the downstream effector molecule Erk1/2 in the FGF signaling pathway by 2 to 3-fold, but had no effect on total Erk1/2 or Hsp70, as seen in a western blot (Fig. 6A). In a separate experiment, we showed that GAL-F2 treatment (5 mg/kg twice/week for 3.5 weeks) almost completely blocks angiogenesis in HEP-G2 xenografts, by staining tumor tissue slices with an anti-CD31 antibody recognizing endothelial cells (Fig. 6B). These and previous results (Fig. 3D, E) support both inhibition of angiogenesis and direct inhibition of cell proliferation as mechanisms of the anti-tumor action of GAL-F2.
Figure 6.
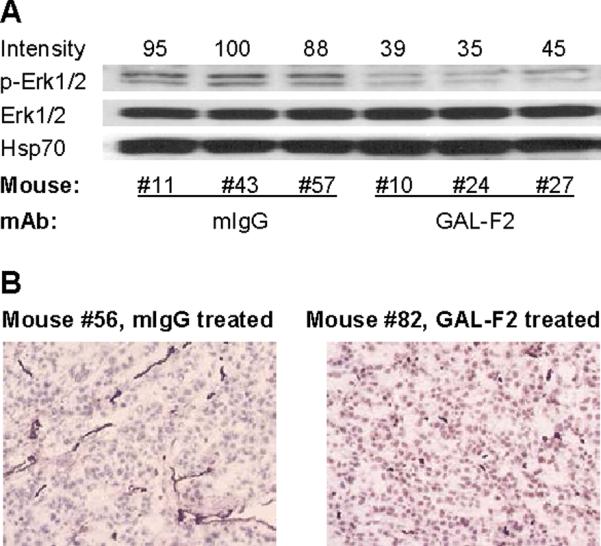
A, Western blot of whole cell lysates from SMMC-7221 xenografts in individual mice treated with GAL-F2 mAb or control mouse mAb mIgG, stained with antibodies to the indicated proteins. The relative intensities of the p-Erk1/2 bands determined by densitometry, normalized to the highest value (mouse #43), are shown above the blot. B, Representative fields from cryostat sections of HEP-G2 xenografts from mice treated with GAL-F2 or mIgG, stained with anti-CD31 mAb to detect blood vessels, counterstained with Mayer's hematoxylin, and photographed at 100×.
Discussion
Primary liver cancer, of which hepatocellular carcinoma (HCC) is the predominant form, is a fairly common type of malignancy in Western countries, with about 20,000 new cases in the U.S. in 2009, very comparable to the incidence of ovarian cancer (31). Largely due to the high prevalence of hepatitis B and C, liver cancer is much more common in Asian countries, making it is the third leading cause of cancer death worldwide (32). The prognosis for HCC is very poor, with a 5-year survival rate in the U.S. of less than 10%. Surgery is a common treatment for liver cancer but only provides cures when the cancer is in its earliest stages. Systemic or intra-arterial chemotherapy, often based on 5-fluorouracil and cisplatin, has very limited efficacy in prolonging survival (33). Sorafenib (Nexavar®), a small-molecule inhibitor of multiple tyrosine kinase receptors including Kit, Flt-3, VEGFR1-3 and PDGFR-β, was recently approved for treatment of unresectable hepatocellular carcinoma, making it the first and currently only targeted therapy approved for HCC. However, sorafenib is not curative and only extends overall patient survival by about 3 months. Hence, new drugs directed against other molecular targets in HCC are certainly needed.
Targeted therapies under development in HCC have recently been thoroughly reviewed (13, 33). A number of drugs that inhibit multiple tyrosine kinases associated with angiogenesis including VEGF receptors are in various phases of clinical trials for HCC. Of these, brivanib (34) is especially relevant because it inhibits the FGF receptors FGFR1-3 in addition to VEGFR1-3 and is in a Phase III trial for HCC. However, despite sometimes promising early results, the success of such drugs is by no means assured: the multikinase inhibitor sunitinib (Sutent®), already approved for treatment of renal cell carcinoma, failed in a Phase III trial for HCC (35). One explanation for such mixed results is the toxicity associated with the relatively broad specificity of multikinase inhibitors, which may make it impossible to achieve drug levels sufficient to fully shut down the target pathways.
Because of their greater specificity, monoclonal antibodies generally have fewer side effects than small molecule antineoplastic drugs. However, only a limited number of mAbs are currently being developed in HCC, according to the ClinicalTrials.gov website. Of these the most interesting are cetuximab (Erbitux®) to EGFR and bevacizumab (Avastin®) to VEGF, because they have already been approved and are widely used for other cancers. Nonetheless, despite elevated levels of EGFR expression found in HCC, the results of clinical trials of Erbitux in HCC have not been striking (33).
Perhaps more promising have been recent clinical trials of Avastin in HCC (33), including a combination study of Avastin and the EGFR inhibitor erlotinib (Tarceva®) (36). Additional clinical studies in HCC of Avastin alone or in combination with erlotinib or other drugs are ongoing. The rationale for a trial of Avastin in combination with a humanized anti-FGF2 mAb is especially compelling. There is abundant evidence for “cross-talk” between FGF2 and VEGF, and these growth factors can synergize to induce angiogenesis (reviewed in refs. 2 and 13). More specifically, FGF2 and VEGF synergize in tumor growth and angiogenesis of HCC (37). Importantly, upregulation of FGF2 expression is an important mechanism of resistance to Avastin and other anti-VEGF drugs (38 – 40). It is therefore plausible that co-treatment with an anti-FGF2 mAb would increase the efficacy of Avastin and delay or overcome the development of resistance to it. To provide support for this concept, we compared treatment with a combination of GAL-F2 and the anti-VEGF mouse precursor mAb of Avastin against treatment with each agent alone, in three HCC xenograft models. For the two models in which the anti-VEGF mAb was effective, addition of GAL-F2 significantly increased that efficacy.
Because FGF2 was discovered more than three decades ago (41) and its involvement in angiogenesis has been known much longer than that of VEGF, it is legitimate to question why no anti-FGF2 mAb has been developed clinically, and whether this indicates that something is “wrong” with FGF2 as a cancer target. However, the reasons for this lack of development activity appear to be largely historical. The first neutralizing mAbs to FGF2 were described in 1989 – 1991 at a time when the early failures of mouse mAbs in treating cancer had led to discouragement with the mAb approach. In addition, the lack of a conventional signal sequence in the FGF2 precursor, the low level of FGF2 in serum, and the existence of intracellular forms of FGF2, all raised questions for a time as to whether secreted FGF2 is physiologically relevant. By the time that humanization and other technologies had solved the immunogenicity problem and made mAbs desirable for treating cancer and other diseases, while the biological importance of secreted FGF2 had been demonstrated, FGF2 had lost the “novelty factor” that would make it appealing to pharmaceutical companies. Indeed, patents on the early anti-FGF2 mAbs were either not obtained or will soon expire, making commercial development of those mAbs almost impossible in practice. Finally, the involvement of angiogenesis in the growth of so many tumors initially left uncertain the types of cancer for which an anti-FGF2 mAb should be tested, increasing the risk and expense of any contemplated development plan. Only recently have a number of studies, including this one, provided data that supports HCC as an excellent first indication for clinical trials of an anti-FGF2 mAb. Hence, a humanized form of the GAL-F2 mAb will be worth testing for the treatment of HCC and potentially other types of cancer.
Acknowledgments
Financial support: NIH grants 5R44CA101283-03 and 1R43CA144086-01 (K. Jin Kim)
Abbreviations
- mAb
monoclonal antibody
- HCC
hepatocellular carcinoma
Footnotes
Conflict of interest: The authors own stock in Galaxy Biotech, LLC
References
- 1.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–78. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Okada-Ban M, Thiery JP, Jouanneau J. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:263–7. doi: 10.1016/s1357-2725(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 4.Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J Cell Physiol. 1992;151:81–93. doi: 10.1002/jcp.1041510113. [DOI] [PubMed] [Google Scholar]
- 5.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–97. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 6.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–37. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Dow JK, deVere White RW. Fibroblast growth factor 2: its structure and property, paracrine function, tumor angiogenesis, and prostate-related mitogenic and oncogenic functions. Urology. 2000;55:800–06. doi: 10.1016/s0090-4295(00)00457-x. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi JA, Mori H, Fukumoto M, Igarashi K, Jaye M, Oda Y, et al. Gene expression of fibroblast growth factors in human gliomas and meningiomas: demonstration of cellular source of basic fibroblast growth factor mRNA and peptide in tumor tissues. Proc Natl Acad Sci USA. 1990;87:5710–14. doi: 10.1073/pnas.87.15.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Becker D. Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat Med. 1997;3:887–93. doi: 10.1038/nm0897-887. [DOI] [PubMed] [Google Scholar]
- 10.Yamanaka Y, Friess H, Buchler M, Beger HG, Uchida E, Onda M, et al. Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res. 1993;53:5289–96. [PubMed] [Google Scholar]
- 11.Barclay C, Li AW, Geldenhuys L, Baguma-Nibasheka M, Porter GA, Veugelers PJ, et al. Basic fibroblast growth factor (FGF2) overexpression is a risk factor for esophageal cancer recurrence and reduced survival, which is ameliorated by coexpression of the FGF2 antisense gene. Clin Cancer Res. 2005;11:7683–91. doi: 10.1158/1078-0432.CCR-05-0771. [DOI] [PubMed] [Google Scholar]
- 12.Boelaert K, McCabe CJ, Tannahill LA, Gittoes NJ, Holder RL, Watkinson JC, et al. Pituitary tumor transforming gene and fibroblast growth factor-2 expression: potential prognostic indicators in differentiated thyroid cancer. J Clin Endocrinol Metab. 2003;88:2341–47. doi: 10.1210/jc.2002-021113. [DOI] [PubMed] [Google Scholar]
- 13.Finn RS. Development of molecularly targeted therapies in hepatocellular carcinoma: where do we go now? Clin Cancer Res. 2010;16:390–7. doi: 10.1158/1078-0432.CCR-09-2084. [DOI] [PubMed] [Google Scholar]
- 14.Ribatti D, Vacca A, Nico B, Sansonno D, Dammacco F. Angiogenesis and anti-angiogenesis in hepatocellular carcinoma. Cancer Treatment Rev. 2006;32:437–44. doi: 10.1016/j.ctrv.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Poon RT, Ng IO, Lau C, Yu WC, Fan ST, Wong J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am J Surg. 2001;182:298–304. doi: 10.1016/s0002-9610(01)00708-5. [DOI] [PubMed] [Google Scholar]
- 16.Kin M, Sata M, Ueno T, Torimura T, Inuzuka S, Tsuji R, et al. Basic fibroblast growth factor regulates proliferation and motility of human hepatoma cells by an autocrine mechanism. J Hepatol. 1997;27:677–87. doi: 10.1016/s0168-8278(97)80085-2. [DOI] [PubMed] [Google Scholar]
- 17.Asada N, Tanaka Y, Hayashido Y, Toratani S, Kan M, Kitamoto M, et al. Expression of fibroblast growth factor receptor genes in human hepatoma-derived cell lines. In Vitro Cell Dev Biol Anim. 2003;39:321–328. doi: 10.1290/1543-706X(2003)039<0321:EOFGFR>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 18.Ogasawara S, Yano H, Iemura A, Hisaka T, Kojiro M. Expressions of basic fibroblast growth factor and its receptors and their relationship to proliferation of human hepatocellular carcinoma cell lines. Hepatology. 1996;24:198–205. doi: 10.1053/jhep.1996.v24.pm0008707262. [DOI] [PubMed] [Google Scholar]
- 19.Maret A, Galy B, Arnaud E, Bayard F, Prats H. Inhibition of fibroblast growth factor 2 expression by antisense RNA induced a loss of the transformed phenotype in a human hepatoma cell line. Cancer Res. 1995;55:5075–79. [PubMed] [Google Scholar]
- 20.Reilly TM, Taylor DS, Herblin WF, Thoolen MJ, Chiu AT, Watson DW, et al. Monoclonal antibodies directed against basic fibroblast growth factor which inhibit its biological activity in vitro and in vivo. Biochem Biophys Res Commun. 1989;164:736–43. doi: 10.1016/0006-291x(89)91521-0. [DOI] [PubMed] [Google Scholar]
- 21.Matsuzaki K, Yoshitake Y, Matuo Y, Sasaki H, Nishikawa K. Monoclonal antibodies against heparin-binding growth factor II/basic fibroblast growth factor that block its biological activity: invalidity of the antibodies for tumor angiogenesis. Proc Natl Acad Sci USA. 1989;86:9911–15. doi: 10.1073/pnas.86.24.9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aonuma M, Yoshitake Y, Nishikawa K, Tanaka NG. Different antitumor activities of anti-bFGF neutralizing antibodies: heparin-binding domain provides an inefficient epitope for neutralization in vivo. Anticancer Res. 1999;19:4039–44. [PubMed] [Google Scholar]
- 23.Rege AA, Bjercke RJ, Erichsen D, Owens R, Stephan CC, Brock TA. Development of novel monoclonal antibodies for the analysis of functional sites in FGF-2. Growth Factors. 1999;16:161–9. doi: 10.3109/08977199909002126. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn H, Köpff C, Konrad J, Riedel A, Gessner C, Wirtz H. Influence of basic fibroblast growth factor on the proliferation of non-small cell lung cancer cell lines. Lung Cancer. 2004;44:167–74. doi: 10.1016/j.lungcan.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Hori A, Sasada R, Matsutani E, Naito K, Sakura Y, Fujita T, et al. Suppression of solid tumor growth by immunoneutralizing monoclonal antibody against human basic fibroblast growth factor. Cancer Res. 1991;51:6180–84. [PubMed] [Google Scholar]
- 26.Takahashi JA, Fukumoto M, Kozai Y, Ito N, Oda Y, Kikuchi H, et al. Inhibition of cell growth and tumorigenesis of human glioblastoma cells by a neutralizing antibody against human basic fibroblast growth factor. FEBS Lett. 1991;288:65–71. doi: 10.1016/0014-5793(91)81004-r. [DOI] [PubMed] [Google Scholar]
- 27.Chuntharapai A, Kim KJ. Generation of monoclonal antibodies to chemokine receptors. Methods Enzymol. 1997;288:15–27. doi: 10.1016/s0076-6879(97)88004-4. [DOI] [PubMed] [Google Scholar]
- 28.Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci USA. 2001;98:7182–7. doi: 10.1073/pnas.121183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–4. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 30.Heffelfinger SC, Hawkins HH, Barrish J, Taylor L, Darlington GJ. SK HEP-1: a human cell line of endothelial origin. In Vitro Cell Dev Biol. 1992;28A:136–42. doi: 10.1007/BF02631017. [DOI] [PubMed] [Google Scholar]
- 31.American Cancer Society . Cancer Facts and Figures 2009. American Cancer Society; Atlanta, GA: 2009. [Google Scholar]
- 32.Garcia M, Jemal A, Ward EM, Center MM, Hao Y, Siegel RL, et al. Global Cancer Facts & Figures 2007. American Cancer Society; Atlanta, GA: 2007. [Google Scholar]
- 33.Huynh H. Molecularly targeted therapy in hepatocellular carcinoma. Biochem Pharmacol. 2010;80:550–60. doi: 10.1016/j.bcp.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 34.Huynh H, Ngo VC, Fargnoli J, Ayers M, Soo KC, Koong HN, et al. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin Cancer Res. 2008;14:6146–53. doi: 10.1158/1078-0432.CCR-08-0509. [DOI] [PubMed] [Google Scholar]
- 35.Pfizer Discontinues Phase 3 Trial of Sutent® in Advanced Hepatocellular Carcinoma. Pfizer; New York, NY: Apr 22, 2010. [Google Scholar]
- 36.Thomas MB, Morris JS, Chadha R, Iwasaki M, Kaur H, Lin E, et al. Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J Clin Oncol. 2009;27:843–50. doi: 10.1200/JCO.2008.18.3301. [DOI] [PubMed] [Google Scholar]
- 37.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology. 2002;35:834–42. doi: 10.1053/jhep.2002.32541. [DOI] [PubMed] [Google Scholar]
- 38.Dempke WC, Heinemann V. Resistance to EGF-R (erbB-1) and VEGF-R modulating agents. Eur J Cancer. 2009;45:1117–28. doi: 10.1016/j.ejca.2008.11.038. [DOI] [PubMed] [Google Scholar]
- 39.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gospodarowicz D. Purification of a fibroblast growth factor from bovine pituitary. J Biol Chem. 1975;250:2515–20. [PubMed] [Google Scholar]