

Abstract
RGD peptides recognize the αvβ3 integrin, a receptor that is overexpressed on the surface of both tumour blood vessels and cancerous cells. These peptides are powerful tools that act as single antiangiogenic molecules, but recently also have been used for tumour imaging and drug targeting. We designed the molecule RAFT-(c[-RGDfK-])4, a constrained and chemically defined entity that can be produced at clinical grade quality. This scaffold was covalently coupled via a labile bridge to the proapoptotic peptide (KLAKLAK)2 (RAFT-RGD-KLA). A fluorescent, activatable probe was also introduced, allowing intracellular localization. At 2.5 μM, this molecule induced the intracellular release of an active KLA peptide, which in turn caused mitochondrial depolarization and cell death in vitro in tumour cells. In a mouse model, the RAFT-RGD-KLA peptide was found to prevent the growth of remote subcutaneous tumours. This study demonstrated that the antitumour peptide is capable of killing tumour cells in an RGD-dependent manner, thus lowering the non-specific cytotoxic effects expected to occur when using cationic cytotoxic peptides. Thus, this chemistry is suitable for the design of complex, multifunctional molecules that can be used for both imaging and therapeutics, representing the next generation of perfectly controlled, targeted drug delivery systems.
Keywords: drug vectorization, cancer, integrin αvβ3, RGD-peptide
Introduction
Selective inactivation of certain tumour-enhancing events would be expected to provide strong therapeutic benefits with the absence of detrimental side effects, as are seen with “conventional” chemotherapy. Examples of such targeting have been described for the treatment of gastrointestinal stromal tumours, renal cell carcinoma, breast cancer, colorectal cancer and non–small cell lung cancer patients harbouring epidermal growth factor receptor (EGFR) mutations (bevacizumab and erlotinib). The new highly selective molecules that are currently being tested or have recently been proven in clinical trials are either small molecules that diffuse through virtually every cell of the body, such as tyrosine kinase inhibitors (TKI), or monoclonal antibodies that bind specifically to their target and block it. Small molecules have the disadvantage of diffusing equally well in sick and normal cells, thus causing adverse effects despite their extremely high specific activity. On the other hand, the major disadvantage associated with the use of monoclonal antibodies comes from their large size and poor diffusion within solid tumours. Additionally, this diffusion problem has also prevented the successful utilization of other targeted large molecules, such as nanoparticles.
To avoid some of the therapeutic difficulties concerning the specificity, size and diffusion of the targeted molecules, we designed a molecular scaffold with a low molecular weight called RAFT(c[-RGDfK-])4. This molecule (RAFT-RGD) binds to and is endocytosed by tumour cells that overexpress the integrin αvβ3, a receptor that is easily accessible to the peptide because it is largely expressed on the surfaces of both the tumour blood vessels and tumour cells themselves. We already presented evidence that the RAFT-RGD molecule specifically targeted the αvβ3 integrin in vitro and in vivo, was rapidly endocytosed and facilitated preclinical tumour detection using nuclear or optical imaging methods (Jin et al., 2006, Jin et al., 2007, Sancey et al., 2007, Ahmadi et al., 2008, Foillard et al., 2009). These properties of RAFT-RGD make it a suitable vector for image-guided surgery of tumours (Jin et al., 2006, Sancey et al., 2007, Ahmadi et al., 2008, Sancey et al., 2009a, Keramidas et al.).
Because RAFT-RGD is rapidly internalized via the αvβ3 receptor in clathrin-coated small acidic vesicles (Sancey et al., 2009b), we decided to use it as a drug delivery system. Using an activatable disulfide bridge (Razkin et al., 2006), we linked the proapoptotic (KLAKLAK)2, (KLA) toxic peptide to the RAFT scaffold (Foillard et al., 2009). KLA is a natural antibiotic with an amphipathic structure that is known to disrupt the negatively charged mitochondrial membrane and to induce cell death if it is properly delivered by a vector into the cellular cytoplasm (Ellerby et al., 1999, Marks et al., 2005, Smolarczyk et al., 2006, Ko et al., 2009).
In the present study, we demonstrate the ability of RAFT-RGD to specifically carry and release a functional KLA peptide into three different cell lines in vitro, leading to mitochondrial depolarization. Using a quenched fluorescent probe, the distribution and subcellular release of the active KLA peptide can be followed using optical molecular imaging in vitro and in vivo. In addition, we show that repeated intraperitoneal injections of RAFT-RGD-KLA significantly reduced the growth rate of remote subcutaneous tumours, confirming the capacity of the RAFT-RGD carrier to both target these tumours after an intraperitoneal administration and effectively deliver an active cytotoxic peptide in vivo.
Materials and Methods
RGD-Peptide Synthesis and Fluorescent Labelling
The different compounds were synthesized according to previously reported procedures (Boturyn et al., 2004, Foillard et al., 2009). The chemical structures are presented in Fig. 1. Four copies of the c[-RGDfK-] peptide were grafted onto the upper face of the cyclic pentapeptide RAFT backbone. On the opposite side, the peptidic sequence (KLAKLAK)2-NH2 (KLA) was grafted via a disulfide bridge and a short linker on the lysine chain (c[-KKKPGKAKPG-]) (Garanger et al., 2005). From both ends of this disulfide bridge, a quencher (QSH21®) could be added onto the first cysteine, and a Cy5 mono NHS (N-hydroxysuccinimide) ester (Amersham Biosciences, Uppsala, Sweden) could be added onto the second cysteine, as described in Fig. 1. As a negative control probe, RAFT(c[-RβADfK-])4 (RAFT-RAD) was also synthesized in a similar way.
Figure 1.

Representative picture of RAFT-(cRGD)4-QSY21®-S-S-(KLAKLAK)2-Cy5 in medium alone (A-C) or with 1 μl β-mercaptoethanol (B–D). This reducing agent induced disulfide bridge rupture and allowed the detection of the Cy5 fluorescence with the 2D-FRI imaging system before (C) and after (D) the addition of DMSO β-mercaptoethanol.
Cell Lines and Culture Conditions
The cell line HEK293(β3) is a stable human embryonic kidney cell line (HEK293) transfectant of the human β3 integrin that was cultured as described in Jin et al. (Jin et al., 2007) in DMEM supplemented with 1% glutamine, 10% fetal bovine serum (FBS), 50 units/ml penicillin, 50 μg/ml streptomycin and 700 μg/ml Geneticin (G418 sulfate, Gibco, Paisley, UK). IGROV-1, human ovarian carcinoma cells, and TS/A-pc, mouse mammary carcinoma cells, were cultured in RPMI 1640 supplemented with 1% glutamine, 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin. All cell lines were cultured at 37°C in a humidified 95% air/5% CO2 atmosphere. All of the cells described here are integrin αvβ3-positive (Jin et al., 2006, Jin et al., 2007, Sancey et al., 2007).
Colocalization Studies
The cells were cultured in 4-well Lab-Tek I chambered coverglass slides in the appropriate medium and kept at 37°C. Medium was removed and RAFT-RGD-QSY21®-S-S-KLA-Cy5 was added at 2 μM for 1 hr in DMEM without FBS and phenol red. Then red mitotracker dye (M7512, Invitrogen, Cergy Pontoise, France) was added to the cell medium at a concentration of 100 nM together with 5 μM Hoechst for 10 min. The cells were then carefully rinsed and the medium was replaced. The cells were observed in a 37°C, 5% CO2 environment on the LSM510 confocal microscope (Carl Zeiss, Jena, Germany). Confocal slices were either 0.5 or 1 μm thick. The images (8-bit colour) were processed using ImageJ software, version 1.37.
Proliferation Assay and Induction of Apoptosis
IGROV-1 cells were seeded at a density of 2.1×104 per cm2. The following day, the media were replaced with fresh media containing 0.5, 1.0, 2.5 or 5.0 μM of the appropriate compound. These media were also changed after 24 hr. The cells were harvested and analyzed at 48 hr. Three separate experiments (n = 3) were performed, and the results were expressed as the mean ± SD of the percentage of proliferation compared to the control. The cells were then fixed with 2% paraformaldehyde for 10 min, and the nuclei were stained with Hoechst 33342 at a concentration of 5 μM for 10 min to allow the quantification of apoptotic cells. The results were expressed as the percentage of apoptotic cells (at least 300 cells were counted per sample).
Mitochondrial Activity: Confocal Imaging
Mitochondrial activity was assessed using the mitochondrial potential sensor JC-1 (Invitrogen). IGROV-1 cells were cultured in 4-well Lab-Tek chamber slides and starved for 30 min before the addition of RAFT-RGD-KLA at a concentration of 1 μM for 30 to 60 min at 37°C. JC-1 was then used as described by the manufacturer at a concentration of 10 μg/ml for 10 min at 37°C. Functional mitochondria were observed after excitation at 543 nm from 565 to 615 nm (red), whereas depolarized mitochondria were observed after excitation at 488 nm from 500 to 550 nm (green). The experiment was performed on at least 20 randomly chosen cells. Three-dimensional topographies were determined using the Topo application within the LSM510 software, and the fluorescence intensities were quantified with the threshold application within the ImageJ software. In control conditions, the mean of the fluorescence intensities was arbitrary normalized to 1 for each individual imaging, and the variations observed after RAFT-RGD-KLA treatment were statistically analysed with an unpaired t-test (Statview).
Mitochondrial Activity: FACS Analysis
IGROV-1 cells were cultured in 6-well plates and starved for 30 min before the addition of 2 μM RAFT-RGD-KLA, KLA or RAFT-RAD-KLA for 2 hr. The mitochondria were labelled with 10 μg/ml JC-1 for 30 min at 37°C. After treatment, the cells were resuspended with trypsin and carefully washed in PBS before analysis. Strongly to weakly polarized mitochondria ratios were determined compared to control conditions and then arbitrarily normalized to 100.
In Vivo Tumour Study
Ten million IGROV-1 cells were injected subcutaneously into 6-week-old female NMRI nude mice (6–8 weeks old, Janvier, Le Genest Saint Isle, France; n = 5 to 7 mice per group). Mice in the experimental groups were injected intraperitoneally five times per week with a total volume of 0.2 ml of PBS alone or a solution providing 0.12 μmol of RAFT-RGD-KLA or RAFT-RAD-KLA per mouse until day 14. Tumour length and width were measured using Vernier calipers, and the tumour volume was calculated using the standard formula: length × width squared × 0.52 (O’Reilly et al., 1997). Results are expressed as tumour volume ± S.E.M.
Immunohistochemical staining
Tumour sections, 7 μm thick, were fixed with 3.7% paraformaldehyde for 10 minutes at room temperature, then saturated for 5 minutes with 0.03% bovine serum albumin, incubated overnight at 4°C with monoclonal mouse antihuman Ki67 (1/150; Dako, Trappes, France). Immunohistochemistry was further processed using the Histostain-Plus Bulk Kit (Invitrogen). The final reaction product was visualized with diaminobenzidine. Negative controls were performed with the same sections incubated with irrelevant antibody (mouse IgG 1/30,000).
Results
RAFT-RGD-mediated delivery of KLA
The RAFT-RGD scaffold was used for the targeted delivery of the proapoptotic drug KLA into different cell lines expressing the αvβ3 integrin. KLA was covalently attached to the dye cyanine 5 (Cy5) and linked to the RAFT-RGD vector by a labile disulfide bridge. On the other side of this bridge, the RAFT-RGD moiety was coupled to QSY21®, a quencher that absorbs Cy5 fluorescence when the S-S link is intact (see Fig. 1). During internalization in the target cell, the disulfide link was reduced, leading to the appearance of a detectable Cy5 fluorescence only after the physical separation between QSY21® and KLA-Cy5 had occurred. This molecular construction was thus used to follow the delivery of the KLA-Cy5 molecule. Confocal imaging done on three different cell lines indicated that RAFT-RGD was able to efficiently release KLA-Cy5 into small vesicles (red spots). Mitochondrial labelling and colocalization experiments with KLA-Cy5 demonstrated that the released peptide reached its main target, the mitochondrion, although small endocytic vesicles containing only KLA-Cy5 could still be detected. These vesicles were mostly located in the cell periphery and represented newly formed endocytotic vesicles (Fig. 2).
Figure 2. Confocal microscopy on living cells.

The cells were starved for 30 min prior to the addition of 2 μM RAFT-(cRGD)4-QSY21®-S-S-(KLAKLAK)2-Cy5. After 1 hr, the mitochondria were labelled with Mitotracker. Confocal imaging was performed between 1 and 2 hr. Colocalization of the mitochondria (green) and (KLAKLAK)2-Cy5 (red) appears yellow. Note that the red fluorescence was not observed under excitation at 633 nm until 1 hr after addition of RAFT-(cRGD)4-QSY21®-S-S-(KLAKLAK)2-Cy5 in the medium. Pictures are representative of cells observed on three separate plates. Scale bar: 10 μm.
In vitro toxicity of RAFT-RGD-KLA
The activity of the vectorized KLA on IGROV-1 cell-proliferation and/or apoptosis was determined (Fig. 3). Cells were treated with 0 to 5 μM RAFT-RGD-KLA, RAFT-RAD-KLA or KLA, for 48 hr, and the number of viable cells was determined (Fig. 3A). In this assay KLA and RAFT-RAD-KLA had no or very moderate antiproliferative effects (p = 0.42 and p < 0.05 at 5 μM, respectively). In contrast, RAFT-RGD-KLA significantly reduced IGROV-1 proliferation (p < 0.05 at 1 μM and p < 0.01 at lower concentrations when compared with KLA).
Figure 3. RAFT-RGD-KLA-mediated cytotoxicity.
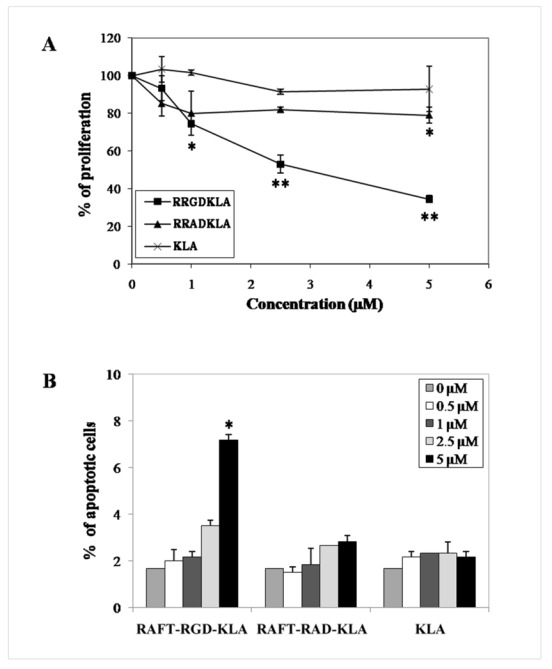
(A) IGROV-1 cells were treated for 48 hr with 0.5, 1.0, 2.5 or 5.0 μM of each compound, and the number of viable cells was counted. The results are expressed as the percentage of cells compared to untreated control. (B) The cells were then fixed, and their nuclei were labelled using Hoechst 33342. The number of apoptotic nuclei was counted. The results are expressed as the mean ± SD (n = 3). * P < 0.05, ** P < 0.01.
To further characterize the proapoptotic effect of RAFT-RGD-KLA, apoptosis induced by the different compounds was quantified by counting the number of fragmented nuclei after Hoechst staining (Fig. 3B). In agreement with the data presented in Fig. 3A, at 5 μM, RAFT-RGD-KLA significantly enhanced apoptosis compared to all other groups (p < 0.05). These experiments on proliferation and apoptosis were also performed on the other two cell lines (TS/A-pc and HEK293(β3)) and produced similar results.
The mitochondrial activity in the IGROV-1 cells was then studied in the presence of RAFT-RGD-KLA at a concentration of 1 μM using the green monomer compound JC-1. This compound fluoresces red when it is in its “J-aggregated” form in polarized healthy mitochondria (Reers et al., 1991, Smiley et al., 1991), while it fluoresces green when in it is in its monomeric dispersed form, a hallmark of the collapsing of mitochondria during apoptosis or after metabolic stress. Two hours after the addition of 1 μM RAFT-RGD-KLA to the culture medium of the IGROV-1 cells, mitochondrial depolarization was observed (Fig. 4A), as indicated by an increase in the green fluorescence intensity from 1 (control condition) to 3.62 ± 2.99 (mean of fluorescence intensity/pixel) after treatment (p < 0.05). The red fluorescence intensity was similar in both conditions (1 to 1.05 ± 0.30, p = 0.6).
Figure 4. Mitochondrial activity in IGROV-1 cells.

(A) Example of confocal imaging of polarized (red, ex. 543 nm) and depolarized (green, ex. 488 nm) mitochondria in IGROV-1 living cells before and after treatment with RAFT-RGD-KLA at a concentration of 1 μM. Corresponding 3D surface topographies from confocal imaging are represented below. Treatment with RAFT-RGD-KLA induced an increase in green fluorescence, i.e., mitochondria depolarization (p < 0.05). Topography scale: low (in blue) to high fluorescence intensity (in red). Angle of view: lower right corner. (B) Polarized to depolarized mitochondria ratio from JC-1 staining of IGROV-1 cells after 2 hr of treatment with PBS (control) or 2 μM RAFT-RGD-KLA, KLA or RAFT-RAD-KLA. RAFT-RGD-KLA was found to significantly decrease the mitochondrial ratio compared to the control condition (p = 0.002 ** and p = 0.095, respectively).
Using the same model, mitochondrial activity was studied by flow cytometry in the presence of 2 μM RAFT-RGD-KLA, RAFT-RAD-KLA or KLA for 2 hr (Fig. 4B). Mitochondrial ratios were defined as polarized (i.e., functional mitochondria)/depolarized mitochondria and normalized to 100 in the control condition. The presence of RAFT-RGD-KLA significantly decreased the mitochondrial ratio compared to the control (44.43 ± 8.24 vs. 100 ± 11.33) or RAFT-RAD-KLA-treated groups (61.01 ± 11.29) (p < 0.01 and p < 0.05, respectively).
Reduction of tumour growth in vivo after intraperitoneal injection of RAFT-RGD-KLA
The toxicity of RAFT-RGD-KLA was confirmed in vivo on mice bearing IGROV-1 flank tumours (Fig. 5). The animals were treated repeatedly with intraperitoneal (IP) injections of PBS or 0.12 μmol of RAFT-RGD-KLA or RAFT-RAD-KLA daily beginning on the day following cell implantation. Fifteen days later, tumour growth was significantly reduced by RAFT-RGD-KLA compared to all of the other groups (p ≤ 0.0001). Moreover, no significant difference was observed after RAFT-RAD-KLA treatment compared to the control group (p = 0.6536).
Figure 5. Inhibition of tumour growth in vivo by RAFT-RGD-KLA.
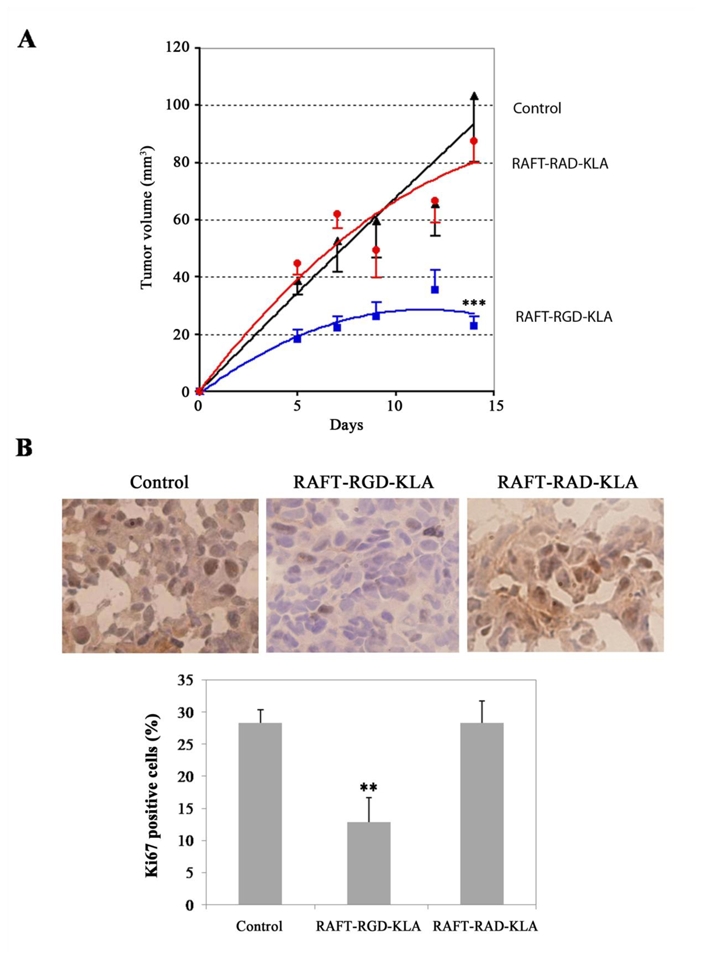
(A) At day 0, IGROV-1 cells were implanted subcutaneously in the right legs of nude mice. Mice were treated daily for 14 days with an IP injection of 200 μl PBS (control) or with solutions containing 0.12 μmol RAFT-RGD-KLA or RAFT-RAD-KLA. Tumour volumes are expressed as the mean tumour volume ± S.E.M. (n = 5–7 per group; *** P < 0.0001). (B) Ki67 nuclear protein detected by immunostaining on frozen tumour sections from control mice, mice treated by RAFT-RGD-KLA or RAFT-RAD-KLA, as indicated. Histogram: the percentage of positive cells for Ki67 was determined after counting the number of positively stained cells per 1,000 cells on the tissue section. The results are expressed as the mean ± SD; ** P < 0.01 for comparisons between RAFT-RGD-KLA and control.
This antitumour activity was associated with a marked decrease in tumour cell proliferation. This was demonstrated on tumour sections extracted at day 14, after immunostaining with an antibody to the Ki67 nuclear antigen. Indeed, the percentage of cycling, Ki67-positive cells was 28.30 ± 2.07% and 28.33 ± 3.37% for the control or the RAFT-RAD-KLA group, respectively, whereas only 12.87 ± 3.83% of the cells were cycling in the RAFT-RGD-KLA group (p < 0.01) (Fig. 5B). RAFT-RGD-KLA treatment was also associated with a slightly increased level of cleaved caspase-3 staining in the tumours (data not shown).
Discussion
Our aim was to demonstrate that RAFT-RGD, a multimeric targeting vector capable of detecting tumours and their metastasis (Keramidas et al., 2010), can also be used for the delivery of a toxic peptide in vivo.
Nanomedicine is on the cusp of being able to provide us with several types of new therapeutic systems to fight cancer. Nanoparticles are currently imparting a “second life” to the chemotherapeutic arsenal. Indeed, nanoparticles not only help with drug solubilisation and bioavailability but also assist in the transport of drugs across barriers and with the release of drugs to tumour sites while sparing healthy tissues from the damage that causes the major side effects of chemotherapy. Several drugs are currently being encapsulated such as doxorubicin, paclitaxel, adryamycin and taxol (for a recent review see Kedar et al., 2010). However, the tumour specificity of these nanoparticles is due to the enhanced permeability and retention phenomenon (EPR effect), which allows for their passive accumulation into tumours. Specific, ligand-mediated, active targeting of nanoparticles is still a major issue, and it is possible that this cannot be expected by simply adding a tumour-specific ligand onto the surface of the particles (Ferrari, 2010). In addition, despite some promising results, the delivery of large “bio”-drugs such as DNA, siRNA or peptides by nanoparticles is still very limited, which may be related to our poor ability to achieve an active targeting that would allow the binding, internalization, release and adapted intracellular trafficking of these unstable molecules. We thus choose a parallel approach, based on the use of our multifunctional tumour targeting RAFT-RGD molecule. Because it is actively internalized once attached to the integrin receptor (Sancey et al., 2009b) and then releases the drug or molecular probe into the cytoplasm of the target cells (Garanger et al., 2005, Jin et al., 2006, Razkin et al., 2006) due to the presence of a labile disulfide bridge between the RAFT-RGD vector and the cargo, we hypothesized that RAFT-RGD could efficiently deliver a toxic KLA peptide to tumour cells.
RAFT-RGD-KLA allows the internalization and release of KLA into intracellular vesicles, but a fraction of the peptide can escape these vesicles and reach its main target, the mitochondria. Indeed, we observed that 90 to 95% of the mitochondria were decorated with the Cy5-labelled KLA peptide in our conditions. In addition, the released KLA peptide was still active and toxic because it was disturbing the mitochondrial potential, as demonstrated by the membrane potential sensitive dye JC-1. At low doses, the released drug initiated mitochondrial depolarization, which could result in the inhibition of cellular proliferation and the induction of apoptosis when as little as 2.5 μM of peptides was provided to the IGROV-1 human cancer cells in vitro.
The amphipathic KLA peptide not very toxic (Borgne-Sanchez et al., 2007). It is supposed to induce apoptosis by disrupting the negatively charged mitochondrial membrane, but not the zwitterionic plasma membrane of eukaryotic cells. However, a very similar peptide was shown to induce also necrosis after disruption of the plasma membrane (Papo et al., 2006). The vectorization of this proapoptotic peptide (or of its D-diastereomer) has previously been reported using antibody-targeted delivery (Marks et al., 2005), fusion to the cell-penetrating peptide TAT (Kwon et al., 2008) or poly-arginine (Law et al., 2006), infection with a recombinant adenovirus vector (Oshikiri et al., 2006) and by fusion to a protein transduction domain (Mai et al., 2001) or to an integrin αvβ3 targeting RGD-peptide (Ellerby et al., 1999, Smolarczyk et al., 2006). In our experiment, the growth of human IGROV-1 xenografts was significantly reduced after intraperitoneal treatment with KLA (0.12 μmol, daily) conjugated to RAFT-RGD. In vivo, the reduction of tumour growth involved a diminution in the number of cycling cells and a poor induction of apoptosis. However, apoptosis was detected in vitro with low doses of molecules (2.5 μM). This dose was significantly less than the previously reported 80 to 100 μM concentrations of protected peptides of the D-form (Borgne-Sanchez et al., 2007). This suggests that the use of a multimeric RGD-presenting scaffold, plus the -NH2 modification on the C-terminal extremity of the KLA peptide (Foillard et al., 2008) and the introduction of a labile disulfide bridge, are sufficient to provide active peptide delivery to the mitochondria of target cells after an intraperitoneal injection in vivo. The natural L-diastereomers are known to lose their activity in serum and to be cleaved by trypsin and proteinase K (Papo et al., 2002). Diastereomers containing L or D amino acids have several functional differences with regard to their capacity to permeate zwitterionic membranes (Papo and Shai, 2003). In addition, it has been shown that RGD-mediated systemic delivery of a toxic cationic peptide called tachylepsin can induce tumour killing (Chen et al., 2001); however, its mechanism of action could be indirect and may involve the activation of the classic complement pathway (Chen et al., 2005). For the compound used in this study, the use of an NH2-protected, L-isomer of KLA was found to be specific and required the binding of the molecule to its receptor because RAFT-RAD-KLA had no effect in vitro or in vivo. Thus, it is possible to rule out a non-specific mechanism of action such as complement activation or direct permeation of the plasma membrane by KLA.
RAFT-RGD was necessary to concentrate enough KLA motifs on the cell surface and to induce their internalization within clathrin-coated vesicles (Sancey et al., 2009b) into which the disulfide link would be reduced and the KLA moiety released. The KLA peptide could then help the molecule to destabilize the vesicles and reach the mitochondria. In vivo, the amount of intact KLA in contact with the mitochondria was not sufficient; rather, the peptide reduced cellular proliferation by decreasing the energy supply. To augment the concentration of active peptide reaching the target cells, it would be interesting to encapsulate and protect RAFT-RGD-KLA in multi-stage delivery systems such as stealth liposomes or nanovesicles. As discussed earlier, this would allow the accumulation of a large quantity of intact RAFT-RGD-KLA by the EPR effect. When the capsule collapsed within the tumour, it would then release the targeted delivery system to the immediate vicinity of the tumour cell surface, where the RAFT-RGD would then efficiently mediate the intracellular uptake and delivery of KLA.
Thus, this study present evidence that RAFT-RGD can deliver a peptide intracellularly. This is an important proof of concept, although it is necessary to improve the overall cytotoxicity of these molecules for further possible therapeutic applications.
Acknowledgments
We thank Corine Tenaud and Mélanie Guidetti for their technical assistance. This study was supported by the ARC (Association pour la Recherche sur le Cancer), the INCA (Institut National du Cancer), the CLARA (Canceropole Lyon Auvergne Rhône-Alpes), the University Joseph Fourier, INSERM (Institut National de la Santé et de la Recherche Médicale) and the ANR (Agence Nationale pour la Recherche).
Footnotes
Declaration of Interest
All the authors declare that there is no conflict of interest
References
- Ahmadi M, Sancey L, Briat A, Riou L, Boturyn D, Dumy P, Fagret D, Ghezzi C, Vuillez JP. Chemical and Biological Evaluations of an (111) In-Labeled RGD-Peptide Targeting Integrin Alpha(V) Beta(3) in a Preclinical Tumor Model. Cancer Biother Radiopharm. 2008;23:691–700. doi: 10.1089/cbr.2008.0528. [DOI] [PubMed] [Google Scholar]
- Borgne-Sanchez A, Dupont S, Langonne A, Baux L, Lecoeur H, Chauvier D, Lassalle M, Deas O, Briere JJ, Brabant M, Roux P, Pechoux C, Briand JP, Hoebeke J, Deniaud A, Brenner C, Rustin P, Edelman L, Rebouillat D, Jacotot E. Targeted Vpr-derived peptides reach mitochondria to induce apoptosis of alphaVbeta3-expressing endothelial cells. Cell Death Differ. 2007;14:422–435. doi: 10.1038/sj.cdd.4402018. [DOI] [PubMed] [Google Scholar]
- Boturyn D, Coll JL, Garanger E, Favrot MC, Dumy P. Template assembled cyclopeptides as multimeric system for integrin targeting and endocytosis. J Am Chem Soc. 2004;126:5730–5739. doi: 10.1021/ja049926n. [DOI] [PubMed] [Google Scholar]
- Chen J, Xu XM, Underhill CB, Yang S, Wang L, Chen Y, Hong S, Creswell K, Zhang L. Tachyplesin activates the classic complement pathway to kill tumor cells. Cancer Res. 2001;65:4614–4622. doi: 10.1158/0008-5472.CAN-04-2253. [DOI] [PubMed] [Google Scholar]
- Chen Y, Xu X, Hong S, Chen J, Liu N, Underhill CB, Creswell K, Zhang L. RGD-Tachyplesin inhibits tumor growth. Cancer Res. 2001;61:2434–2438. [PubMed] [Google Scholar]
- Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28:181–188. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foillard S, Jin ZH, Garanger E, Boturyn D, Favrot MC, Coll JL, Dumy P. Synthesis and biological characterisation of targeted pro-apoptotic peptide. Chembiochem. 2008;9:2326–2332. doi: 10.1002/cbic.200800327. [DOI] [PubMed] [Google Scholar]
- Foillard S, Sancey L, Coll JL, Boturyn D, Dumy P. Targeted delivery of activatable fluorescent pro-apoptotic peptide into live cells. Org Biomol Chem. 2009;7:221–224. doi: 10.1039/b817251j. [DOI] [PubMed] [Google Scholar]
- Garanger E, Boturyn D, Jin Z, Dumy P, Favrot MC, Coll JL. New multifunctional molecular conjugate vector for targeting, imaging, and therapy of tumors. Mol Ther. 2005;12:1168–1175. doi: 10.1016/j.ymthe.2005.06.095. [DOI] [PubMed] [Google Scholar]
- Jin ZH, Josserand V, Foillard S, Boturyn D, Dumy P, Favrot MC, Coll JL. In vivo optical imaging of integrin alphaV-beta3 in mice using multivalent or monovalent cRGD targeting vectors. Mol Cancer. 2007;6:41. doi: 10.1186/1476-4598-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin ZH, Josserand V, Razkin J, Garanger E, Boturyn D, Favrot MC, Dumy P, Coll JL. Noninvasive optical imaging of ovarian metastases using Cy5-labeled RAFT-c(-RGDfK-)4. Mol Imaging. 2006;5:188–197. [PubMed] [Google Scholar]
- Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine. 2010 doi: 10.1016/j.nano.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Keramidas M, Josserand V, Righini CA, Wenk C, Faure C, Coll JL. Intraoperative near-infrared image-guided surgery for peritoneal carcinomatosis in a preclinical experimental model. Br J Surg. 2010;97:737–743. doi: 10.1002/bjs.6986. [DOI] [PubMed] [Google Scholar]
- Ko YT, Falcao C, Torchilin VP. Cationic liposomes loaded with proapoptotic peptide D-(KLAKLAK)(2) and Bcl-2 antisense oligodeoxynucleotide G3139 for enhanced anticancer therapy. Mol Pharm. 2009;6:971–977. doi: 10.1021/mp900006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MK, Nam JO, Park RW, Lee BH, Park JY, Byun YR, Kim SY, Kwon IC, Kim IS. Antitumor effect of a transducible fusogenic peptide releasing multiple proapoptotic peptides by caspase-3. Mol Cancer Ther. 2008;7:1514–1522. doi: 10.1158/1535-7163.MCT-07-2009. [DOI] [PubMed] [Google Scholar]
- Law B, Quinti L, Choi Y, Weissleder R, Tung CH. A mitochondrial targeted fusion peptide exhibits remarkable cytotoxicity. Mol Cancer Ther. 2006;5:1944–1949. doi: 10.1158/1535-7163.MCT-05-0509. [DOI] [PubMed] [Google Scholar]
- Mai JC, Mi Z, Kim SH, Ng B, Robbins PD. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001;61:7709–7712. [PubMed] [Google Scholar]
- Marks AJ, Cooper MS, Anderson RJ, Orchard KH, Hale G, North JM, Ganeshaguru K, Steele AJ, Mehta AB, Lowdell MW, Wickremasinghe RG. Selective apoptotic killing of malignant hemopoietic cells by antibody-targeted delivery of an amphipathic peptide. Cancer Res. 2005;65:2373–2377. doi: 10.1158/0008-5472.CAN-04-2594. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- Oshikiri T, Miyamoto M, Hiraoka K, Shichinohe T, Kawarada Y, Kato K, Suzuoki M, Nakakubo Y, Kondo S, Dosaka-Akita H, Kasahara N, Katoh H. Transcriptional targeting of adenovirus vectors with the squamous cell carcinoma-specific antigen-2 promoter for selective apoptosis induction in lung cancer. Cancer Gene Ther. 2006;13:856–863. doi: 10.1038/sj.cgt.7700953. [DOI] [PubMed] [Google Scholar]
- Papo N, Oren Z, Pag U, Sahl HG, Shai Y. The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. J Biol Chem. 2002;277:33913–33921. doi: 10.1074/jbc.M204928200. [DOI] [PubMed] [Google Scholar]
- Papo N, Seger D, Makovitzki A, Kalchenko V, Eshhar Z, Degani H, Shai Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006;66:5371–5378. doi: 10.1158/0008-5472.CAN-05-4569. [DOI] [PubMed] [Google Scholar]
- Papo N, Shai Y. New lytic peptides based on the D,L-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry. 2003;42:9346–9354. doi: 10.1021/bi027212o. [DOI] [PubMed] [Google Scholar]
- Razkin J, Josserand V, Boturyn D, Jin ZH, Dumy P, Favrot M, Coll JL, Texier I. Activatable fluorescent probes for tumour-targeting imaging in live mice. ChemMedChem. 2006;1:1069–1072. doi: 10.1002/cmdc.200600118. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- Sancey L, Ardisson V, Riou LM, Ahmadi M, Marti-Batlle D, Boturyn D, Dumy P, Fagret D, Ghezzi C, Vuillez JP. In vivo imaging of tumour angiogenesis in mice with the alpha(v)beta (3) integrin-targeted tracer (99m)Tc-RAFT-RGD. Eur J Nucl Med Mol Imaging. 2007;34:2037–2047. doi: 10.1007/s00259-007-0497-z. [DOI] [PubMed] [Google Scholar]
- Sancey L, Dufort S, Josserand V, Keramidas M, Righini C, Rome C, Faure AC, Foillard S, Roux S, Boturyn D, Tillement O, Koenig A, Boutet J, Rizo P, Dumy P, Coll JL. Drug development in oncology assisted by noninvasive optical imaging. Int J Pharm. 2009a;379:309–316. doi: 10.1016/j.ijpharm.2009.05.034. [DOI] [PubMed] [Google Scholar]
- Sancey L, Garanger E, Foillard S, Schoehn G, Hurbin A, Albigès-Rizo C, Boturyn D, Souchier C, Grichine A, Dumy P, Coll JL. Clustering and internalization of integrin avb3 with a tetrameric RGD-synthetic peptide. Mol Ther. 2009b;5:837–843. doi: 10.1038/mt.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolarczyk R, Cichon T, Graja K, Hucz J, Sochanik A, Szala S. Antitumor effect of RGD-4C-GG-D(KLAKLAK)2 peptide in mouse B16(F10) melanoma model. Acta Biochim Pol. 2006;53:801–805. [PubMed] [Google Scholar]