

Abstract
Intracerebral hemorrhage (ICH) has been associated with inflammation and apoptosis. The CCL2-CCR2 chemotactic system is one of the major signaling pathways that induce inflammation and apoptosis. However, its role on ICH has not been investigated. We subjected wild-type, CCL2−/− and CCR2−/− mice to collagenase-induced ICH, and assessed histological and behavioral outcomes. Lack of CCL2 or CCR2 decreased the hematoma volume early after collagenase-induced ICH, but delayed its recovery. The hematoma size was accompanied by brain edema, neuronal death, and neurological scores. Although microglia activation/migration was attenuated in CCL2−/− or CCR2−/− mice 1 day after injury, more microglia were present at later time points, suggesting that alternative signaling pathways had been activated to recruit them. On the contrary, leukocyte and neutrophil infiltration was decreased in these mice, suggesting a tighter/recovered blood-brain barrier. In addition, we also found that FL- and K104Stop-CCL2 were able to restore the changes found in CCL2−/− mice, but K104A-CCL2 failed to do so. These results suggest that plasmin-mediated truncation of CCL2 may be an indispensable step to fully activate the chemokine in vivo. The data also indicate that CCL2-CCR2 signaling pathway may be a molecular target for the treatment of ICH.
Keywords: CCL2, CCR2, Intracerebral Hemorrhage
Introduction
Intracerebral hemorrhage (ICH) constitutes 10–13% of strokes and is usually accompanied with poor prognosis (Ribo and Grotta, 2006, Donnan et al., 2010). Although the ICH pathogenic events are elusive, apoptosis (Matsushita et al., 2000) and inflammation (Castillo et al., 2002) play important roles. When ICH occurs, microglia become activated and secrete inflammatory cytokines and chemokines. The blood-brain barrier (BBB) is disrupted and peripheral leukocytes infiltrate into the brain possibly via chemokine gradients (Lortat-Jacob et al., 2002, Cavcic et al., 2011). The activated microglia and infiltrated leukocytes can modulate ICH progression (Wang et al., 2003, Wang and Tsirka, 2005b, Moxon-Emre and Schlichter, 2011, Provencio et al., 2011).
Monocyte chemoattractant protein-1 (CCL2), a potent chemokine for microglia and monocytes (Yao and Tsirka, 2010, 2011b), is expressed under physiological conditions at low levels in the brain (Sheehan et al., 2007, Ma et al., 2011). Its expression is dramatically increased during excitotoxicity and stroke (Sheehan et al., 2007, Ma et al., 2011). In collagenase-induced ICH, CCL2 expression is elevated in the ipsilateral hemisphere, where it becomes proteolytically processed. Although the source of CCL2 during ICH has not yet been reported, astrocytes may be a major source based on studies on other models (Thompson and Van Eldik, 2009, Semple et al., 2010, Kooij et al., 2011, Nelson et al., 2011). By binding to its receptor CCR2, CCL2 induces the migration and accumulation of monocytes/microglia around lesion sites in brain injuries and excitotoxic lesions (Dimitrijevic et al., 2006). Previous studies in our lab showed that neuronal death and microglial accumulation were attenuated in mice lacking CCL2 or mice injected with CCL2 blocking antibody, suggesting that CCL2 contributes to death via microglial recruitment. In addition, CCL2 promotes BBB compromise (Stamatovic et al., 2003, Stamatovic et al., 2005, Dimitrijevic et al., 2006, Stamatovic et al., 2006), which may enhance leukocyte infiltration into the brain. Plasmin activity is indispensable for CCL2-induced chemotaxis and BBB compromise: (1) plasmin cleaves CCL2 at lysine (K)-104 (Sheehan et al., 2007), (2) the truncated CCL2 has a much higher chemotactic potency than the full length CCL2 (Sheehan et al., 2007, Yao and Tsirka, 2010, 2011b), and (3) the truncated CCL2, not the intact CCL2, induces BBB disruption (Yao and Tsirka, 2011b).
As CCL2 is an important inflammatory mediator and inflammation plays an important role in ICH, we asked if the CCL2/CCR2 system is critical in the ICH events. We compared the ICH outcome in wild-type (wt) mice and mice deficient for CCL2 or CCR2, or delivered back to the deficient mice the missing chemokine and evaluated ICH progression.
Materials and Methods
Animals and cultures
C57BL/6 (wild-type), CCL2−/− and CCR2−/− mice were purchased from Jackson Laboratories and maintained in the Department of Laboratory Animal Research at Stony Brook University (SBU) with free access to water and food. CCL2−/− and CCR2−/− mice have been backcrossed for 12 generations to C57BL/6. Experimental procedures were in accordance to the NIH guide for care and use of animals and the Institutional Animal Care and Use Committee at SBU.
ICH Model
Mice weighting 25–35g were injected with atropine (0.6μg/g of body weight) and avertin (0.02ml/g of body weight) intraperitoneally. A burr hole was drilled (−1 mm posterior to bregma, 3 mm lateral from midline and 4 mm in depth). Collagenase (0.18U in 500nl saline)(Wang et al., 2003, Wang and Tsirka, 2005b) with or without recombinant CCL2 proteins (150ng) was injected into the brain over 5 minutes. The needle remained in place for 2min to prevent reflux. On 1, 3, 7, and 14 days post injury (dpi) mice were transcardially perfused, the brains removed, fixed, dehydrated and sectioned.
Histology
Hematoxylin/eosin (H&E) and luxol fast blue/cresyl violet staining were performed to visualize the hematoma. Briefly, for H&E sections were washed twice in PBS and kept in hematoxylin for 15min at room temperature. They were washed in tap water twice, 70% ethanol with 1% hydrochloride acid for 3 sec, followed by 3 washes in tap water and placed in eosin for 5 sec, washed in PBS, dehydrated in gradient ethanol and mounted. Luxol fast blue/Cresyl violet was also used to visualize the hematoma as described previously (Clark et al., 1998, Wang et al., 2003, Thiex et al., 2004, Wang and Tsirka, 2005b).
For Fluoro-Jade C (FJC) staining (Schmued and Hopkins, 2000) slides were incubated in 1% NaOH in 80% ethanol for 5min, washed in 70% ethanol and distilled water, and incubated in 0.06% KMnO4 for 10min. They were washed in water and transferred to 0.0001% FJC (in 0.1% acetic acid) for 10min. The slides were rinsed in water, dried at 56° C for 5min, cleared in xylene for 2min and mounted with DPX. FJC+ cells were counted in 3 sections/mouse, using 3 fields immediately adjacent to the hematoma at 400X magnification over a microscopic field of 0.01mm2. Large blood vessels were avoided. The numbers of degenerating cells were expressed as cells/mm2 (Wang et al., 2003, Wang and Tsirka, 2005b).
Hemorrhagic Injury Analysis
The injury volumes (Clark et al., 1998, Wang et al., 2003, Thiex et al., 2004, Wang and Tsirka, 2005b) in H&E-stained slides from wt, CCL2−/− and CCR2−/− mice were quantified on 30μm coronal sections using NIS-Elements D3.0. The hemorrhage areas from serial coronal slices were added and the injury volume (mm3) was calculated: Injury Volume=measured area X slice thickness (30μm).
Immunohistochemistry/immunofluorescence
Microglia/macrophages were visualized using Iba-1 antibody (1:500; Wako Inc) followed by 1:200 biotinylated anti-Rabbit or Alexa-555 anti-Rabbit antibodies for 1-hour at room temperature. The slides were stained with the ABC kit (Vector Labs) and Diaminobenzidine solution. Iba-1+ cells were counted in 5 slides using 5 random fields immediately adjacent to the hematoma at 400X magnification. Four animals/group/time point were used for quantification. Leukocytes and neutrophils were visualized by anti-CD45 (BD PharMingen) and Ly6G (R&D), respectively, both at 1:500 dilution, followed by Alexa 488-anti-rat antibody (Invitrogen).
Brain Edema Measurement
At days 1 and 7 post collagenase injection, the mice were euthanized, the brains collected and cut into two hemispheres. Cerebellum was used as internal control. The wet weights of the two hemispheres and cerebellum were obtained. The samples were dried for 24-hours and weighted (dry weight). Brain edema was calculated as: Brain Edema=(Wet Weight-Dry Weight)/Wet Weight X100. At this point we can’t differentiate between bleeding-induced water increase and secondary edema, as we measure the total water content of the tissue(Wang et al., 2003, Wang and Tsirka, 2005b).
Neurological Deficit
All mice were scored for neurological deficits 1, 3, 7, and 14 dpi using a modified 28-point scoring system (Clark et al., 1998, Wang et al., 2003, Thiex et al., 2004, Wang and Tsirka, 2005b), that includes 6 properties (body symmetry, gait, climbing, circling behavior, front limb symmetry, and compulsory circling), each graded from 0 to 4, establishing a maximum score of 24. The higher the score, the higher the neurological deficit. The person who scored the deficit was blinded to the genotypes/treatments. The individual scores of the mice tested are provided in Supplemental Figure 4.
Statistics
Results are shown as mean±SD. Student’s t-test was used to analyze differences between two groups. ANOVA was used when bigger groups were compared. Comparison with day 1 within the same genotype, *p<0.05; comparison with wild-type (for Figs. 1–3 and Table A) or CCL2−/− mice injected with FL-CCL2 (for Figs. 4–6 and Table B) at the same time points, #p<0.05.
Figure 1. The effect of CCL2 or CCR2 on injury volume.

A. Wild-type, CCL2−/−, and CCR2−/− mice were injected with collagenase and the injury volume was examined by H&E staining 0, 1, 3, 7, and 14 days later. B. Quantitative data of injury volume are mean±SD (n=4–7).
Figure 3. CCL2 and CCR2 deficiency on iNOS expression, leukocyte infiltration, neuronal death, and neurological behavior.

A. Wild-type, CCL2−/−, and CCR2−/− mice were injected with collagenase and Iba-1 (red) and iNOS (green) levels were assessed 1, 3, 7dpi. B. Quantitative data of iNOS (mean pixel intensity) levels are mean±SD (n=3). C and D. The infiltration of leukocytes and neutrophils was examined using CD45 (C) and Ly6G (D) immunostaining, respectively, at 1, 3, 7dpi. Data are mean±SD (n=4). E. Representative images of Flour Jade C staining. F. Quantification of the number of degenerating neurons on days 1 and 7dpi. Data are mean±SD (n=4). G. Neurological scores were evaluated at 1, 3, and 7dpi. Values are mean±SD. The animal numbers for wt, CCL2−/−, and CCR2−/− at 1, 3, 7dpi were 17, 8, 13; 16, 7, 6; and 9, 12, 5, respectively.
Table 1.
Edema after ICH
| A. Wild-type, CCL2−/−, and CCR2−/− mice were subjected to ICH and water content was calculated, as described in Materials and Methods. Data are mean±SD (n=6). | ||||
|---|---|---|---|---|
| Genotype (time post ICH) | Water content (%) | |||
| Contralateral | Ipsilateral | Cerebellum | ||
| Wt | Day 1 | 76.67±0.24 | 79.67±0.82 | 74.48±0.71 |
| Day 3 | 76.92±0.57 | 78.9±0.62 | 74.94±0.75 | |
| Day 7 | 76.43±0.85 | 77.04±0.92* | 74.39±1.97 | |
| CCL2−/− | Day 1 | 76.55±0.64 | 77.87±0.50# | 74.73±0.66 |
| Day 3 | 76.53±0.29 | 78.52±0.36 | 74.89±0.30 | |
| Day 7 | 76.81±0.64 | 78.90±0.30*,# | 74.95±0.36 | |
| CCR2−/− | Day 1 | 76.80±0.32 | 78.11±0.28# | 74.58±0.30 |
| Day 3 | 76.81±0.48 | 78.63±0.60 | 74.91±0.50 | |
| Day 7 | 76.56±0.45 | 79.73±0.84*,# | 74.83±0.63 | |
| B. CCL2−/− mice were injected with collagenase and FL-, K104A-, or K104Stop-CCL2. The water content was calculated at 1, 3 and 7dpi. Data are shown as mean±SD (n=3). | ||||
|---|---|---|---|---|
| Treatment (time post ICH) | Water content (%) | |||
| Contralateral | Ipsilateral | Cerebellum | ||
| FL CCL2 | Day 1 | 76.63±0.27 | 78.52±0.30 | 74.63±0.23 |
| Day 3 | 76.56±0.33 | 78.45±0.34 | 74.67±0.04 | |
| Day 7 | 76.57±0.34 | 77.20±0.28* | 74.58±0.22 | |
| K104A CCL2 | Day 1 | 76.45±0.34 | 77.24±0.26# | 74.63±0.33 |
| Day 3 | 76.35±0.26 | 77.34±0.29# | 74.66±0.09 | |
| Day 7 | 76.51±0.30 | 78.64±0.24*,# | 74.60±0.23 | |
| K104Stop CCL2 | Day 1 | 76.73±0.21 | 80.11±0.70# | 74.60±0.10 |
| Day 3 | 76.73±0.50 | 77.25±0.06*,# | 74.63±0.25 | |
| Day 7 | 76.37±0.25 | 77.03±0.09* | 74.63±0.12 | |
Figure 4. The effect of recombinant CCL2 proteins on injury volume.
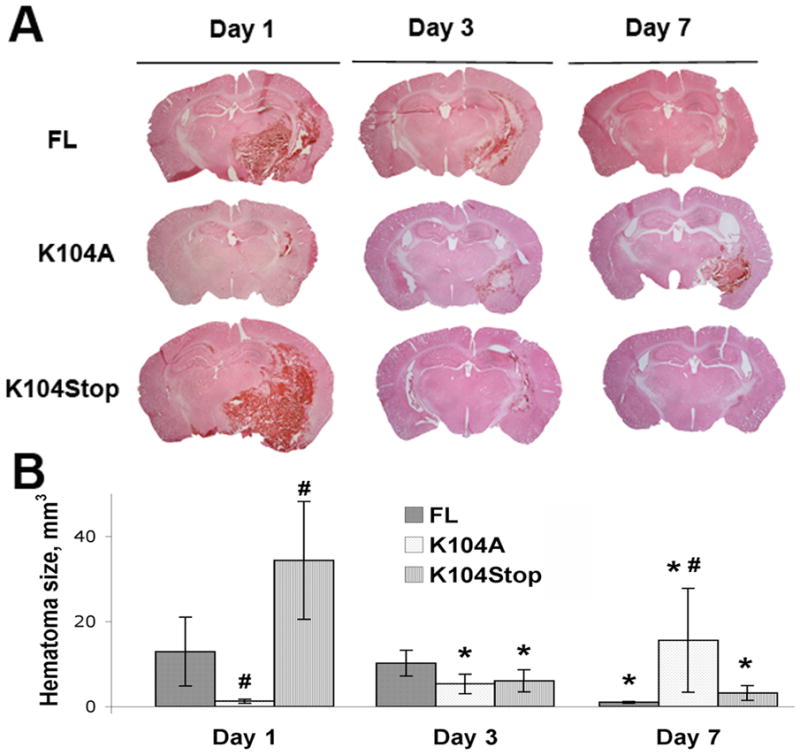
A. CCL2−/− mice were injected with collagenase and FL-, K104A-, or K104Stop-CCL2 and the injury volume was examined by H&E staining 1, 3, 7dpi. B. Quantitative data of injury volume are mean±SD (n=4).
Figure 6. Recombinant CCL2 proteins rescue ICH properties in CCL2−/− mice.

A. CCL2−/− mice were injected with collagenase and FL-, K104A-, or K104Stop-CCL2. 1, 3, 7dpi the brains were collected and stained for Iba-1 and iNOS. B. Quantitative data of iNOS (mean pixel intensity) are mean±SD (n=3). C and D. The infiltration of leukocytes and neutrophils was examined using CD45 (C) and Ly6G (D) immunostaining, respectively, at 1, 3, 7dpi. Data are mean±SD (n=4). E. Degenerating neurons were visualized using FluoroJade C on days 1 and 7dpi and quantified. Data are mean±SD (n=3). F. Neurological scores were evaluated at 1, 3, and 7dpi. Values are mean±SD. The animal numbers for CCL2−/− mice infused with FL, K104A, and K104Stop-CCL2 at 1, 3, 7dpi were 8, 8, 3; 9, 6, 4; and 11, 6, 3, respectively.
Results
CCL2 or CCR2 deficiency and hematoma volume
Because the CCL2-CCR2 chemokine system is critical for microglial recruitment (Yao and Tsirka, 2010), which play both detrimental and beneficial roles during ICH (Wang et al., 2003, Wang and Tsirka, 2005b, Wang and Dore, 2007), we examined whether lack of CCL2 or CCR2 can affect the hematoma size. We induced hemorrhage in wt mice and CCL2−/− or CCR2−/− mice and examined the outcome at 1, 3, 7, and 14 dpi. The hematoma was visualized by H&E (Figure 1) and luxol fast blue/cresyl violet staining (Supplement Figure 1). In wt mice the hematoma was very large (28.42mm3) at 1 dpi, and decreased over time (22.2 mm3 at 3 dpi and 4.26 mm3 at 7 dpi, and 0.61 mm3 at 14 dpi). In CCL2−/− and CCR2−/− mice the hematoma size followed a different timecourse. It was smaller at 1dpi (5.01 and 7.27 mm3, respectively), peaked at 3dpi (12.48 and 20.2 mm3) and decreased slowly (11.68 and 19.17 mm3 at 7 dpi and 4.25 and 4.72 at 14 dpi, respectively). These data suggest that CCL2-CCR2 signaling may affect the progression and resolution of the hematoma.
Microglia/Macrophage recruitment and leukocyte infiltration during ICH in CCL2−/− or CCR2−/− mice
We visualized microglia/macrophages recruited to the ICH using Iba-1, a microglia/macrophage marker. There was no difference in baseline microglia numbers among the genotypes (Figure 2). As reported (Wang et al., 2003, Wang and Tsirka, 2005b, Wang and Dore, 2007), Iba-1+ cells showed large cell bodies and short processes in wt mice at 1dpi and their numbers increased slightly (Figure 2), indicating the transformation towards an activated state. In CCL2−/− and CCR2−/− mice, neither the numbers nor the morphology of Iba-1+ cells changed. The Iba-1+ cell density increased at 3dpi in wt mice and CCL2−/− or CCR2−/− mice (Figure 2), suggesting that alternative (CCL2- and CCR2-independent) signaling events may be activated. Although more microglia/macrophages were observed in the penumbra by 7dpi, the intensity of the Iba-1 signal decreased, suggesting potentially that the microglia had started reverting to a resting state. CCL2−/− and CCR2−/− mice by 7dpi, however, showed high density and strong staining of microglia/macrophages, suggesting that the signaling pathway(s) that had been activated in 3dpi was still functional at 7dpi.
Figure 2. CCL2 and CCR2 deficiency affects microglial/macrophage activation after ICH.

A. Wild-type, CCL2−/−, and CCR2−/− mice before (day 0), or after ICH (1, 3, 7 dpi) were perfused, and microglial/macrophage activation was examined by Iba-1 DAB-staining. B. Quantitative data of Iba-1+ cells. Data are shown as mean±SD (n=4).
Microglial activation was also evaluated using inducible nitric oxide synthase (iNOS) as a marker: iNOS expression was observed at 1dpi, peaked at 3dpi and decreased by 7dpi in wt mice (Figures 3A and B). The expression of iNOS at 1 and 3dpi in CCL2−/− or CCR2−/− mice was similar to wt mice, but by 7dpi the mice still displayed high level of iNOS, which co-localized with Iba-1 at the border of hematoma (Figure 3A).
CCL2 can compromise BBB integrity (Stamatovic et al., 2003; 2005; 2006; Dimitrijevic et al., 2006; Yao and Tsirka, 2011a, b) thus allowing the infiltration of peripheral leukocytes into the brain. We assessed inflammatory cells infiltration using CD45, a pan-leukocyte marker, and Ly6G, a neutrophil marker. Infiltrated CD45+ cells were observed around the hematoma early after ICH (1 and 3dpi, Figure 3C), probably due to collagenase-induced disruption of blood vessels. By 7dpi, the CD45+ signal increased in wt mice, although the hematoma size was smaller, suggesting infiltration of peripheral blood cells at this time point. In CCL2−/− and CCR2−/− mice the CD45+ signal was still low by 7dpi (Figure 3C), suggesting little-to-no infiltration of leukocytes. Neutrophils showed accumulation in wt brains early after injury (1 and 3dpi) and the number decreased significantly by 7dpi (Figure 3D). This neutrophil infiltration time-course was consistent with previous reports (Gong et al., 2000, Xue and Del Bigio, 2000a, b, Peeling et al., 2001, Wang and Tsirka, 2005a). CCL2 and CCR2 deficient mice showed only low neutrophil cell infiltration in the brain, which did not increase drastically during the ICH time course (Figure 3D). Previously we have shown that CCL2 compromises BBB integrity in a CCR2-dependent manner and there is no difference in baseline BBB leakage among wt, CCL2−/− and CCR2−/− mice under physiological conditions (Yao and Tsirka, 2011b). Together our data suggest that in the absence of CCL2 and/or CCR2 the compromise of BBB may be delayed in collagenase-induced ICH.
Effects of CCL2-CCR2 deficiency on edema, neuronal death, and neurological behavior
Brain edema is an important clinical complication of ICH. We measured the water content in the animals after ICH. Significant increase of brain water was found in the ipsilateral hemisphere in wt mice 1dpi (Table 1A). Mirrored by the hematoma volume, the water content in the ipsilateral side decreased at later time points (3 and 7dpi), suggesting a recovery from ICH. CCL2−/− and CCR2−/− mice showed a trend that followed the progress of ICH. The water contents in the contralateral hemisphere and the cerebellum (the internal control), however, were not affected by the ICH.
ICH-induced neuronal death was visualized using FJC, a dye that specifically labels degenerating neurons (Schmued et al., 1997, Wang and Tsirka, 2005a, b, Bian et al., 2007). Wt mice showed many FJC+ cells (387/mm2) at 1dpi and the number decreased at 7dpi (142/mm2, Figures 3E and F), as the animals were recovering. CCL2−/− mice showed a lower number (229/mm2) of degenerating neurons at 1dpi, but the numbers increased at 7dpi (295/mm2). CCR2−/− mice followed a similar trend as CCL2−/− mice (200 and 303 FJC+ cells/mm2 at 1 and 7dpi, respectively), suggesting that the number of degenerating neurons correlated with hematoma size.
Furthermore, we assessed neurobehavioral changes after ICH. For wt mice the neuronal deficit score was high early after injury but decreased at 7dpi (from 10.9 and 10.1 at 1 and 3dpi, respectively, representing the severity of the injury to 4.7 at 7dpi). CCL2−/− mice showed low neuronal deficit score at 1dpi (7.3) and higher scores at 3 (9) and 7dpi (10.2) (Figure 3G). CCR2−/− mice followed the same trend as CCL2−/− (Figure 3G). The neurological scores decreased for all animals at 14dpi (albeit remaining still higher in CCL2−/− and CCR2−/− mice) indicative of an active recovery process.
Infusion of FL- and plasmin-truncated CCL2 restores wt-phenotype in CCL2−/− mice
Plasmin cleaves CCL2 at K104 enhancing in vitro its chemotactic potency and BBB-compromising activity (Sheehan et al., 2007, Yao and Tsirka, 2010, 2011b). To investigate whether CCL2 activation is involved in ICH, we examined whether CCL2 becomes truncated during ICH. Two bands corresponding to full-length (FL) and truncated (Cleaved) CCL2 were detected 6 hours after ICH in the ipsilateral, but not the contralateral, hemisphere, suggesting CCL2 activation after ICH (Supplemental Figure 2). We then infused different recombinant, previously characterized CCL2 proteins (Yao and Tsirka, 2010, 2011a, b) into the brains of CCL2−/− mice and examined the outcome. Since collagenase does not cleave CCL2 (Supplemental Figure 3), we injected CCL2 together with collagenase on day 0 and assessed the pathological changes at 1, 3, and 7dpi. In CCL2−/− mice infused with cleaved K104Stop-CCL2 (constitutively active CCL2), we observed a larger hematoma at 1dpi (34.35mm3) that decreased at 3 and 7dpi (6.08 and 3.21mm3, respectively) (Figure 4). Infusion of K104A-CCL2 (plasmin uncleavable CCL2) (Yao and Tsirka, 2010) did not affect the CCL2−/− hematoma characteristics. Like K104Stop-CCL2, FL-CCL2 infusion induced a large hematoma (12.92mm3) at 1dpi and the volume decreased to 1.01mm3 at 7dpi (Figure 4). FL-CCL2-infused mice had a smaller hematoma at 1dpi, compared to K104Stop-CCL2-injected mice, suggesting that K104Stop-CCL2 is more potent than FL-CCL2.
Infusion of FL- or K104Stop-CCL2 to CCL2−/− mice restores wt ICH characteristics
Infusion of K104Stop-CCL2 significantly increased microglia/macrophage density in the peri-injury site at 1dpi (Figure 5). Infusion of FL- and K104A-CCL2 did not enhance the density of microglia/macrophages adjacent to the hematoma at 1dpi and the cells showed long processes and lightly-stained cell body, indicating a resting state. By 3dpi, microglia/macrophages exhibited a fully activated morphology (were amoeboid with short processes and intensely-stained cell body) and the numbers of microglia/macrophages increased significantly in all groups (Figure 5). This is consistent with the observation that microglia/macrophages were fully activated in CCL2−/− at 3dpi (Figure 2). By 7dpi in mice injected with FL- and K104Stop-CCL2 a dramatic decrease of Iba-1+ numbers was evident, and the cells returned to the resting or semi-activated state (Figure 5). Mice infused with K104A-CCL2 showed consistent activation and high density of microglia/macrophages around the injury site.
Figure 5. Recombinant CCL2 proteins change microglial/macrophage activation pattern after ICH.

A. CCL2−/− mice were injected with collagenase and FL-, K104A-, or K104Stop-CCL2. The brains were collected at 1, 3, 7 dpi and DAB-stained with Iba-1 antibody. B. Quantitative data of Iba-1+ cells. Data are shown as mean±SD (n=4).
The levels of iNOS were similar between wt and CCL2−/− mice (Figure 3). When the recombinant CCL2 proteins were infused, iNOS levels changed: uncleavable K104A-CCL2 infusion induced an increase of iNOS expression over time (Figure 6A, B), whereas infusion of K104Stop-CCL2 resulted in iNOS decrease over time. Similar to K104Stop-CCL2, FL-CCL2 decreased iNOS expression at 7dpi (Figure 6A, B). Together, our data suggest that FL- or K104Stop-CCL2 infusion can revert the microglial activation state observed in CCL2−/− mice.
We examined the CCL2 protein infusion effect into CCL2−/− mice on leukocyte and neutrophil infiltration after ICH. As shown in Figure 6C, infusion of FL-CCL2 or K104Stop-CCL2 yielded a slow increase in CD45+ cell infiltration at later time points in the brain parenchyma. The infusion of K104A-CCL2 did not affect the CD45+ cell infiltration at any time point examined. Similarly, infusion of FL- and K104Stop-CCL2 significantly enhanced neutrophil infiltration at early time points after ICH and decreased by 7dpi (Figure 6D). K104A-CCL2 infusion did not yield dramatic changes in Ly6G levels (Figure 6D), again suggesting that K104A-CCL2 was not functional active.
We also quantified the effects of these recombinant proteins on edema, neuronal death and behavior of the CCL2−/− mice. Infusion of FL- or K104Stop-CCL2 into the animals resulted in increase of the water content measurements in the ipsilateral hemisphere at the early timepoints after ICH, but was decreased at 7dpi, whereas the infusion of K104A-CCL2 led to increase of water content and accompanying edema over time (Table 1B).
Similarly, the numbers of degenerating neurons were high early after the infusion of FL- or K104Stop-CCL2, but decreased significantly at 7dpi (Figure 6E). These data suggest that it is the truncated fully active CCL2 (K104Stop-CCL2) that more efficiently reverted the phenotype of CCL2−/− mice.
Furthermore, the infusion of FL-CCL2 or K104Stop-CCL2 resulted in a better behavioral over time (10.25 at 1dpi, 8.25 at 3dpi and 5.75 at 7dpi for FL-CCL2, and 11.73 at 1dpi, 8.67 at 3dpi, and 4.25 at 7dpi for K014Stop-CCL2). K104A-CCL2 infusion yielded a low score at 1dpi (5.78) and it increased to 7.67 and 9.75 at 3 and 7dpi, respectively (Figure 6F). Collectively, our data showed that infusion of FL- or K104Stop-CCL2 changed the ICH outcome in CCL2−/− mice and made it comparable to that of wt mice after ICH, underscoring the crucial role of plasmin-mediated truncation of CCL2 on its biological activities in vivo.
Discussion
Hemorrhagic stroke, which represents a small percentage of stroke incidents (Qureshi et al., 1997), is an acute and devastating clinical event. Inflammation plays an important role in ICH pathophysiology (Gong et al., 2001, Wang et al., 2003, Wang and Tsirka, 2005b, Xi et al., 2006, Wang, 2010). Activated microglia and infiltrated macrophages accumulate in the peri-hematoma region (Wang et al., 2003, Wang and Tsirka, 2005b, Wang and Dore, 2007). Here we report that CCL2 and CCR2 affect the progression of ICH, as deficiency in the chemokine and chemokine receptor significantly decreased hematoma size at early time points but delayed the recovery. The size of hematoma was paralleled by the water content in the ipsilateral hemisphere and neuronal death at the peri-hematoma region. Consistently, neurobehavioral deficit scores were also mirrored by hematoma size. These data suggest that mice deficient for CCL2 or CCR2 have an initially milder but longer-lasting injury after ICH.
Microglia, as the brain resident immune cells, play a dual role during brain injury: they promote neuronal death by secreting a variety of cytotoxic factors, such as TNF-α and IL-1β (Kim and de Vellis, 2005), and they phagocytose cell debris and injured neurons to enhance recovery (Elkabes et al., 1996). Which role they undertake after ICH is dependent on the time after injury and their local environment. Our data suggest that Iba-1+ cells may play a detrimental role at early time points (1dpi) in the collagenase-induced ICH model, although their role later is unclear due to the activation of alternative signaling cascades. It should be noted that Iba-1 staining identifies both brain-resident microglia and infiltrating macrophages. We do make the assumption that since collagenase acts locally, the infiltration of macrophages will most likely take place around the site of collagenase injection. Presence of activated Iba1+ cells further away from the lesion area may signify the activation and recruitment of microglia. Immunohistochemistry for iNOS, a functional marker of proinflammatory microglia/macrophages, reveals that the activation of microglia may be directly related to the tissue damage.
Unexpectedly, at later time points after ICH the knockout mice showed higher density of Iba-1+ cells in the peri-hematoma region, suggesting that signaling pathways distinct from the CCL2-CCR2 system may function to attract microglia/macrophages. We are currently studying the mechanisms underlying such alternative signaling pathways in the chemokine/chemokine receptor KO mice after ICH.
It has been shown that leukocyte infiltration correlates with disruption of BBB (Gurney et al., 2006, Candelario-Jalil et al., 2007, Reijerkerk et al., 2008, Kuang et al., 2009). Consistent with our previous report that CCL2−/− or CCR2−/− mice have a ‘tighter’ BBB, infiltration of peripheral cells was low in these mice at all the time points examined. Since CD45 is also expressed by brain resident microglia, although at low levels, resident microglia may contribute to the CD45+ signal we detected. The number of neutrophils, however, was decreased in both knockout mice early after ICH, suggesting that CCL2-CCR2 may regulate neutrophil infiltration directly or indirectly. Consistent with previous reports that neutrophils are fast-responding cells (Gong et al., 2000, Xue and Del Bigio, 2000a, b), longer (7dpi) timepoints revealed no differences among the three genotypes.
The infusion of CCL2 proteins showed that FL- and K104Stop-CCL2 restored wt phenotype in CCL2−/− mice, whereas plasmin-resistant K104A-CCL2 failed, suggesting that plasmin-mediated truncation is an indispensable step to fully activate CCL2 in vivo. This is consistent with our in vitro data that plasmin-truncated CCL2 has higher biological activities (Yao and Tsirka, 2010, 2011b). Although here we did not infuse recombinant CCL2 proteins in wt mice, we have previously shown (Yao and Tsirka, 2011b) that when we infuse FL- or active CCL2 (K104Stop-CCL2) in wt animals the integrity of BBB is more readily disrupted. Therefore we would expect that upon overexpression/administration of CCL2 active recombinant proteins into wt mice we would observe a bigger hematoma early after ICH and a delayed recovery.
Due to its early identification and high potency for chemoattraction of mononuclear cells, the CCL2-CCR2 axis has been extensively studied. Some reports suggest that in ischemic stroke, inhibition of the CCL2-CCR2 system ameliorates injury by reducing brain edema, leukocyte infiltration and expression of inflammatory mediators (Dimitrijevic et al., 2007, Tsukuda et al., 2011), indicating a detrimental role of CCL2-CCR2 axis. It should be noted that inflammation can be both detrimental and beneficial, depending on the timing and environment. Other reports showed that the CCL2-CCR2 interaction is important for the recruitment of neural stem cells to the ischemic brain (Andres et al., 2011). In hemorrhagic stroke, we found that deletion of CCL2 or CCR2 significantly decreased the injury volume and brain edema and enhanced neurological deficit score at early time points, but delayed the recovery and clearance of the hematoma. Consistent with the ischemic studies, our data also support such a complex role of the CCL2-CCR2 system in ICH. The early beneficial role of lack of CCL2 is probably due to decreased cytokine & chemokine secretion, leading to attenuated neuronal death. The delayed recovery, however, may be caused by the dysregulation of inflammatory response. The activation of alternative chemokine signaling pathways and over-activation/accumulation of microglia/macrophages may prolong the detrimental phase of inflammation or prevent the transition from detrimental phase to beneficial phase. Thus, further investigations should focus on the time course of CCL2-CCR2 activation, the compensation of inhibiting CCL2-CCR2 activation, and the potential molecular mechanisms regulating its expression.
Supplementary Material
The effect of CCL2 and CCR2 on hematoma size. Luxol Fast Blue/Cresyl Violet staining was used to visualize the hematoma.
ICH induces truncation of CCL2. Six hours after ICH, the contralateral and ipsilateral hemispheres were collected and subjected to western blot using anti-CCL2 antibody (Abcam). Two bands corresponding to intact and truncated CCL2 were detected in the ipsilateral but not the contralateral hemisphere. Actin was used as a loading control.
Collagenase does not cleave mouse CCL2. Recombinant FL-CCL2 was incubated with collagenase for 24hrs at 37° C, resolved on 15% SDS-PAGE and visualized using anti-6His and anti-CCL2 antibodies.
Individual neurological scores for the animals subjected to ICH. The neurological scores for each category and each animal are presented.
Acknowledgments
We thank Dr M. Frohman and Tsirka lab members for advice and discussions. This work was partially supported by a SigmaXi grant-in-aid (to YY) and NIH R0142168 (to SET).
Footnotes
The authors declare no conflicts of interest.
References
- Andres RH, Choi R, Pendharkar AV, Gaeta X, Wang N, Nathan JK, Chua JY, Lee SW, Palmer TD, Steinberg GK, Guzman R. The CCR2/CCL2 Interaction Mediates the Transendothelial Recruitment of Intravascularly Delivered Neural Stem Cells to the Ischemic Brain. Stroke. 2011 doi: 10.1161/STROKEAHA.110.606368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian GL, Wei LC, Shi M, Wang YQ, Cao R, Chen LW. Fluoro-Jade C can specifically stain the degenerative neurons in the substantia nigra of the 1-methyl-4-phenyl-1,2,3,6-tetrahydro pyridine-treated C57BL/6 mice. Brain Res. 2007;1150:55–61. doi: 10.1016/j.brainres.2007.02.078. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J Neurochem. 2007;100:1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- Castillo J, Davalos A, Alvarez-Sabin J, Pumar JM, Leira R, Silva Y, Montaner J, Kase CS. Molecular signatures of brain injury after intracerebral hemorrhage. Neurology. 2002;58:624–629. doi: 10.1212/wnl.58.4.624. [DOI] [PubMed] [Google Scholar]
- Cavcic A, Tesovic G, Gorenec L, Grgic I, Benic B, Lepej SZ. Concentration gradient of CXCL10 and CXCL11 between the cerebrospinal fluid and plasma in children with enteroviral aseptic meningitis. Eur J Paediatr Neurol. 2011 doi: 10.1016/j.ejpn.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Clark W, Gunion-Rinker L, Lessov N, Hazel K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke. 1998;29:2136–2140. doi: 10.1161/01.str.29.10.2136. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006;26:797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Hankey GJ, Davis SM. Intracerebral haemorrhage: a need for more data and new research directions. Lancet Neurol. 2010;9:133–134. doi: 10.1016/S1474-4422(10)70001-6. [DOI] [PubMed] [Google Scholar]
- Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Boulis N, Qian J, Turner DE, Hoff JT, Keep RF. Intracerebral hemorrhage-induced neuronal death. Neurosurgery. 2001;48:875–882. doi: 10.1097/00006123-200104000-00037. discussion 882–873. [DOI] [PubMed] [Google Scholar]
- Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- Gurney KJ, Estrada EY, Rosenberg GA. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Kooij G, Mizee MR, van Horssen J, Reijerkerk A, Witte ME, Drexhage JA, van der Pol SM, van Het Hof B, Scheffer G, Scheper R, Dijkstra CD, van der Valk P, de Vries HE. Adenosine triphosphate-binding cassette transporters mediate chemokine (C-C motif) ligand 2 secretion from reactive astrocytes: relevance to multiple sclerosis pathogenesis. Brain. 2011;134:555–570. doi: 10.1093/brain/awq330. [DOI] [PubMed] [Google Scholar]
- Kuang Y, Lackay SN, Zhao L, Fu ZF. Role of chemokines in the enhancement of BBB permeability and inflammatory infiltration after rabies virus infection. Virus Res. 2009;144:18–26. doi: 10.1016/j.virusres.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortat-Jacob H, Grosdidier A, Imberty A. Structural diversity of heparan sulfate binding domains in chemokines. Proc Natl Acad Sci U S A. 2002;99:1229–1234. doi: 10.1073/pnas.032497699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Manaenko A, Khatibi NH, Chen W, Zhang JH, Tang J. Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 2011;31:881–893. doi: 10.1038/jcbfm.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Meng W, Wang X, Asahi M, Asahi K, Moskowitz MA, Lo EH. Evidence for apoptosis after intercerebral hemorrhage in rat striatum. J Cereb Blood Flow Metab. 2000;20:396–404. doi: 10.1097/00004647-200002000-00022. [DOI] [PubMed] [Google Scholar]
- Moxon-Emre I, Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol. 2011;70:218–235. doi: 10.1097/NEN.0b013e31820d94a5. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Hao C, Manos J, Ransohoff RM, Gruol DL. Altered hippocampal synaptic transmission in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Brain Behav Immun. 2011;25(Suppl 1):S106–119. doi: 10.1016/j.bbi.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeling J, Yan HJ, Corbett D, Xue M, Del Bigio MR. Effect of FK-506 on inflammation and behavioral outcome following intracerebral hemorrhage in rat. Exp Neurol. 2001;167:341–347. doi: 10.1006/exnr.2000.7564. [DOI] [PubMed] [Google Scholar]
- Provencio JJ, Altay T, Smithason S, Moore SK, Ransohoff RM. Depletion of Ly6G/C(+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J Neuroimmunol. 2011;232:94–100. doi: 10.1016/j.jneuroim.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi JA, Notta NJ, Salahuddin N, Zaman V, Khan JA. An epidemic of Dengue fever in Karachi--associated clinical manifestations. J Pak Med Assoc. 1997;47:178–181. [PubMed] [Google Scholar]
- Reijerkerk A, Kooij G, van der Pol SM, Leyen T, van Het Hof B, Couraud PO, Vivien D, Dijkstra CD, de Vries HE. Tissue-type plasminogen activator is a regulator of monocyte diapedesis through the brain endothelial barrier. J Immunol. 2008;181:3567–3574. doi: 10.4049/jimmunol.181.5.3567. [DOI] [PubMed] [Google Scholar]
- Ribo M, Grotta JC. Latest advances in intracerebral hemorrhage. Curr Neurol Neurosci Rep. 2006;6:17–22. doi: 10.1007/s11910-996-0004-0. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Semple BD, Frugier T, Morganti-Kossmann MC. CCL2 modulates cytokine production in cultured mouse astrocytes. J Neuroinflammation. 2010;7:67. doi: 10.1186/1742-2094-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan JJ, Zhou C, Gravanis I, Rogove AD, Wu YP, Bogenhagen DF, Tsirka SE. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci. 2007;27:1738–1745. doi: 10.1523/JNEUROSCI.4987-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281:8379–8388. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, Andjelkovic AV. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005;25:593–606. doi: 10.1038/sj.jcbfm.9600055. [DOI] [PubMed] [Google Scholar]
- Thiex R, Mayfrank L, Rohde V, Gilsbach JM, Tsirka SA. The role of endogenous versus exogenous tPA on edema formation in murine ICH. Exp Neurol. 2004;189:25–32. doi: 10.1016/j.expneurol.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes [corrected] Brain Res. 2009;1287:47–57. doi: 10.1016/j.brainres.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukuda K, Mogi M, Iwanami J, Min LJ, Jing F, Oshima K, Horiuchi M. Irbesartan attenuates ischemic brain damage by inhibition of MCP-1/CCR2 signaling pathway beyond AT receptor blockade. Biochem Biophys Res Commun. 2011;409:275–279. doi: 10.1016/j.bbrc.2011.04.142. [DOI] [PubMed] [Google Scholar]
- Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- Wang J, Rogove AD, Tsirka AE, Tsirka SE. Protective role of tuftsin fragment 1–3 in an animal model of intracerebral hemorrhage. Ann Neurol. 2003;54:655–664. doi: 10.1002/ana.10750. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005a;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Tuftsin fragment 1–3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005b;36:613–618. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Xue M, Del Bigio MR. Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett. 2000a;283:230–232. doi: 10.1016/s0304-3940(00)00971-x. [DOI] [PubMed] [Google Scholar]
- Xue M, Del Bigio MR. Intracortical hemorrhage injury in rats : relationship between blood fractions and brain cell death. Stroke. 2000b;31:1721–1727. doi: 10.1161/01.str.31.7.1721. [DOI] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. The C terminus of mouse monocyte chemoattractant protein 1 (MCP1) mediates MCP1 dimerization while blocking its chemotactic potency. J Biol Chem. 2010;285:31509–31516. doi: 10.1074/jbc.M110.124891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. Mouse MCP1 C-terminus inhibits human MCP1-induced chemotaxis and BBB compromise. J Neurochem. 2011a;118:215–223. doi: 10.1111/j.1471-4159.2011.07319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsirka SE. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood-brain barrier disruption. J Cell Sci. 2011b;124:1486–1495. doi: 10.1242/jcs.082834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of CCL2 and CCR2 on hematoma size. Luxol Fast Blue/Cresyl Violet staining was used to visualize the hematoma.
ICH induces truncation of CCL2. Six hours after ICH, the contralateral and ipsilateral hemispheres were collected and subjected to western blot using anti-CCL2 antibody (Abcam). Two bands corresponding to intact and truncated CCL2 were detected in the ipsilateral but not the contralateral hemisphere. Actin was used as a loading control.
Collagenase does not cleave mouse CCL2. Recombinant FL-CCL2 was incubated with collagenase for 24hrs at 37° C, resolved on 15% SDS-PAGE and visualized using anti-6His and anti-CCL2 antibodies.
Individual neurological scores for the animals subjected to ICH. The neurological scores for each category and each animal are presented.