

Abstract
Control of iron homeostasis is essential for healthy central nervous system function: iron deficiency is associated with cognitive impairment, yet iron overload is thought to promote neurodegenerative diseases. Specific genetic markers have been previously identified that influence levels of transferrin, the protein that transports iron throughout the body, in the blood and brain. Here, we discovered that transferrin levels are related to detectable differences in the macro- and microstructure of the living brain. We collected brain MRI scans from 615 healthy young adult twins and siblings, of whom 574 were also scanned with diffusion tensor imaging at 4 Tesla. Fiber integrity was assessed by using the diffusion tensor imaging-based measure of fractional anisotropy. In bivariate genetic models based on monozygotic and dizygotic twins, we discovered that partially overlapping additive genetic factors influenced transferrin levels and brain microstructure. We also examined common variants in genes associated with transferrin levels, TF and HFE, and found that a commonly carried polymorphism (H63D at rs1799945) in the hemochromatotic HFE gene was associated with white matter fiber integrity. This gene has a well documented association with iron overload. Our statistical maps reveal previously unknown influences of the same gene on brain microstructure and transferrin levels. This discovery may shed light on the neural mechanisms by which iron affects cognition, neurodevelopment, and neurodegeneration.
Keywords: neuroimaging genetics, twin modeling, pathway analysis, tensor-based morphometry, voxel based analysis
Iron and the proteins that transport it are critically important for brain function. Iron deficiency (ID) is the most common nutritional deficiency worldwide (1). Iron-deficient diets lead to poorer cognitive achievement in school-aged children (2). In rural areas where ID anemia is prevalent, iron supplements can increase motor and language capabilities in children (3). ID also impairs dopamine metabolism in the brain, particularly in the caudate and putamen regions (4).
ID clearly has adverse effects on cognitive development, but iron overload in later life is also associated with damage to the brain. Brain iron regulation is disrupted in several neurodegenerative diseases. Neuroimaging methods reveal abnormally high brain iron concentrations in Alzheimer's disease (5), Parkinson disease (6), and Huntington disease (7). High iron concentrations may even cause neuronal death (8, 9).
As deficiency and excess of iron can negatively impact brain function, the regulation of iron transport to the brain is crucial for cognition. Iron is transported throughout the body by the iron-binding protein transferrin. The interaction between transferrin and the transferrin receptors appears to regulate iron transport (10). When iron levels are low, the liver produces more transferrin for increased iron transport. In humans, transferrin can increase in iron-deficient states, which may help to distinguish ID anemia from anemia of chronic disease (11). Dietary ID has also been shown in rats to elevate the concentration of transferrin in the brain (12), specifically in the hippocampus and striatum (13). Transferrin is also decreased in cases of iron overload (14).
The gold standard for determining accurate iron measures is obtained from invasive bone marrow or liver tests, which are impractical for general applications. Serum levels of iron fluctuate greatly (15) and depend on dietary factors such as vitamin C intake (16) and the time of blood collection (17). Transferrin is arguably a more reliable and reproducible index of the long-term availability of iron to the brain (18, 19). In fact, in a 2-y study of postmenopausal women (20), total iron-binding capacity (equivalent to transferrin concentration) was a more reliable measure of iron status [(0.60; 95% confidence interval (CI), 0.44–0.76)], whereas serum iron measures varied more (0.50; 95% CI, 0.22–0.65). Transferrin is therefore used as a more reproducible measure to infer iron availability to the neural pathways.
As iron is a key determinant of neural development and degeneration, we set out to investigate whether brain structure in healthy adults depends on serum transferrin levels. We scanned 615 young adult twins and siblings with standard MRI. A total of 574 of them were also scanned with diffusion tensor imaging (DTI) to assess volumetric and microstructural white matter differences potentially associated with variations in serum transferrin levels measured during adolescence.
The participants in our study were healthy young adults, in whom iron overload is unlikely. We instead expected that iron levels toward the lower end of the normal range might lead to a poorer developmental phenotype in the brain of these young adults.
The brain synthesizes transferrin itself, so serum transferrin is not necessarily indicative of the levels of brain transferrin. However, in healthy populations without iron overload or hemochromatosis, all iron in the plasma is bound to transferrin (10). Iron enters the brain primarily by transport through the blood–brain barrier (21), yet transport through the blood–cerebrospinal fluid (CSF) and cellular–plasmalemma barriers have also been described (10). Upon binding to its receptor, transferrin is thought to be mostly released back into the blood stream (although some transcytosis of transferrin may occur). The iron can then bind to transferrin synthesized in the oligodendrocytes of white matter (22, 23).
Most of the brain's iron is found in oligodendrocytes, where it supports myelination (24). Oligodendrocytes also maintain iron homeostasis in the brain. Our primary hypothesis was that we might find poorer white matter integrity in adulthood in those who had lower iron levels available during development, as high transferrin levels are often a sign of the liver reacting to lower iron availability. We therefore framed our hypothesis by testing if serum transferrin levels in adolescence were related to fractional anisotropy (FA; measured later in adulthood from DTI scans of the brain). Lower FA can be a sign of less mature or poorer myelination.
We further hypothesized that brain structure volumes in iron-rich regions might be lower in people with high serum transferrin levels. Iron levels are highest in the basal ganglia and substantia nigra (25). By measuring brain volumes regionally with tensor-based morphometry (TBM), we predicted that we might find insufficiently developed (i.e., smaller) subcortical structures in those with higher transferrin levels. ID additionally alters dopamine metabolism in the caudate and putamen (4), so we predicted that people with high transferrin (and, by implication, lower brain iron) might have lower volumes for dopamine-containing structures, such as the caudate. Finally, we expected lower hippocampal volumes, as iron-deficient rats have lower iron concentrations in the hippocampus (13), a region vulnerable to neuronal loss in neurodegenerative disease (26).
Genetic factors explain 66% and 49% of the variance in serum transferrin levels in men and women, respectively (17). As such, if transferrin is found to be associated with neuroanatomical differences, we might expect that common genes influence both brain structure and transferrin levels. To understand such shared genetic contributions to brain variations and transferrin, we used a twin design. Many neuroimaging studies of identical and fraternal twins reveal substantial genetic contributions to brain structure (27–30) and function (31, 32). Cross-twin cross-trait designs can also discover overlapping (i.e., pleiotropic) genetic influences on very different biological traits, such as brain volume (33) or fiber integrity (34) with IQ.
After discovering a common genetic basis for transferrin levels and brain fiber integrity, we hypothesized that genes modulating transferrin also play a role in brain structure within the same regions. We performed exploratory tests on all SNPs within the two major transferrin related genes: the transferrin gene, TF, on chromosome 3, and the HFE gene on chromosome 6, where a handful of SNPs have been found to explain a remarkable 40% of the genetic variance in serum transferrin levels (35). We performed exploratory tests on all these SNPs and additional imputed ones (to HapMap2) within the same genes.*
Genes influencing transferrin are not the only cause of variation in iron levels measured in the blood serum. However, they do influence the limited amount of serum iron that becomes transported into the brain. Therefore, we expected genes that influence transferrin levels to show associations with brain structure. Some variants increase the risk for iron overload late in life, and these may also increase the availability of brain iron for developmental processes such as myelination. If high iron levels improve myelination, we might expect to see increased fiber integrity as measured through DTI.
Results
Serum transferrin levels for the 615 individuals in our study ranged from 1.89 to 5.18 g/L of serum (mean, 2.99 ± 0.37 g/L; median, 2.96 g/L). Regressions fitted to data at each voxel in the brain DTI and MRI scans revealed significant associations with microstructural variations in diffusion anisotropy (Fig. 1). There were also strong associations between transferrin levels and gross anatomical volume differences (Fig. 2), even after controlling for age and sex. As ID is known to reduce myelination, we had expected in advance, to find a negative association between transferrin and FA (36); this was in fact observed.
Fig. 1.

Voxel-wise associations, between FA, a measure of white matter fiber integrity derived from the DW images, and serum transferrin levels in 574 subjects (five of whom had repeated scans). There are significant associations in the external capsule, superior longitudinal fasciculus, and the cingulum bilaterally. As transferrin levels increase, the diffusivity across the axons also tends to decrease by approximately 0.025 units for every g/L unit increase in the serum transferrin level. Significance was confirmed by enforcing a regional control over the FDR as described by Langers et al. (70) at the 5% level. Corrected P values of association are shown. Maps are adjusted for effects of age and sex; random-effects regression accounted for familial relatedness and the use of repeated scans. β-values shown represent the regression coefficient (or slope) of the transferrin level term, after accounting for covariates.
Fig. 2.

Brain regions where there are detectable associations between serum transferrin levels and patterns of brain morphometry. Higher blood transferrin levels were associated with greater regional brain volumes in the hippocampus and basal ganglia, including the globus pallidus bilaterally and midbrain regions appearing to contain the substantia nigra. Shrinkage in structure volume is seen as transferrin levels increase bilaterally in the caudates, the third ventricle, as well as temporoparietal regions of white matter. Lower regional volumes are also observed in frontal gray matter in those with higher serum transferrin levels. The greatest regional brain volume deficit, per unit difference in transferrin levels, is seen in the caudate, whereas the greatest expansion is detected in the hippocampus and basal ganglia. All highlighted regions were significant after a multiple comparisons correction that enforces a regional control over the FDR at the 5% level as described by Langers et al. (70). Maps are adjusted for effects of age and sex; random-effects regression accounted for familial relatedness and the use of repeated scans (N = 652 scans, N = 615 subjects). All images are in radiological convention: the left side shown is the right hemisphere. The β-value corresponds to the unnormalized slope of the regression. Corrected P values range from 0.001 to 0.05; uncorrected values range from 2.6 × 10−6 to 0.04 for the thresholded regions shown.
Transferrin Levels Relate to Neuroanatomical Structure.
As noted, cross-twin cross-trait models can determine whether a partially overlapping set of genes contributes to two traits of interest, such as fiber integrity (assessed by using FA) and transferrin levels. If this is the case, common genetic influences mediate the observed correlation between the two measures. Before examining the full cross-twin cross-trait model, we independently assessed these correlations within each group of twins; if the variables correlate more strongly for monozygotic (MZ) than for dizygotic (DZ) twin pairs, then we can infer the greater difference is a result of additive genetic factors. Fig. 3 shows the correlations between FA and transferrin levels in MZ twins and DZ twins separately, highlighting the higher magnitude of the correlations for MZ than DZ pairs.
Fig. 3.

The magnitude of the observed cross-twin cross-trait (FA and transferrin) correlations are higher in identical than fraternal twin pairs, supporting our hypothesis that partially overlapping sets of genes may explain some of the shared variance in brain structure and transferrin levels. This motivates the use of bivariate ACE modeling to estimate the degree of shared genetic influence.
Cross-Twin Cross-Trait Analysis of Shared Genetic Determination.
We performed cross-twin cross-trait heritability analysis starting from the full bivariate model as described in Methods, where the ACE structural equation model was used to fit the additive genetic (A), shared environmental (C), and unique environmental (E) components of variance for the brain measures and transferrin levels. We removed individual components one by one to determine the best fitting model. For both MRI- and DTI-based bivariate ACE models, the AE model fitted the best for transferrin and the full ACE model fitted best for the imaging measures. This means that genetic effects were detected in both cases, and the effects of common rearing environment were also detectable for the imaging measures. The path diagram for the best fitting model is shown in Fig. 4.
Fig. 4.

Path diagram for the best-fitting model of the bivariate association. The models that best fitted the data were the AE model for transferrin and ACE model for the imaging measures. The measures we examined included regional brain volumes and measures of microstructural white matter fiber integrity.
The cross-twin cross-trait correlation was then computed from the best fitting model. Significance of the correlation was determined by removing the rA component of the path model as described in Methods. The additive genetic determinants of voxel-wise FA measures (Fig. 5) and transferrin showed significant overlap after multiple comparisons correction using the false discovery rate (FDR) procedure (37). Although suggestive, no significant overlap was detected between the additive genetic determinants of transferrin levels and macroscopic structural morphometry as assessed through TBM.
Fig. 5.

Significant cross-twin cross-trait correlations for transferrin levels and brain FA. The P value controlling the FDR at the 5% level in regions of significant FA-transferrin associations was 0.032. The significant cross-twin cross-trait correlations presented here indicate that partially overlapping sets of genes are associated with transferrin levels and brain FA values in bilateral white matter regions, including the cingulum, external capsule, and superior longitudinal fasciculus. Negative correlations indicate lower anisotropy, perhaps indicating lower levels of myelination with increases in transferrin levels. Positive correlations were not significant.
Genetic Associations.
After filtering the SNPs in TF and HFE available in our imputed sample by minor allele frequency (MAF) greater than 0.05, 42 SNPs remained. SNPs chosen for analysis are listed in Table S1, along with their MAF according to the CEU population: Utah residents with Northern and Western European ancestry from the CEPH collection from HapMap. As a result of linkage disequilibrium, the effective number of SNPs tested (38, 39) was 20. When the significant voxels of the cross-twin cross-trait associations were clustered into regions of interest (ROIs), six survived a cluster threshold size of 27 voxels, corresponding to the size of a voxel with all its surrounding neighbors, or a 3 × 3 × 3 cube. These ROIs are shown in Fig. S1. Genetic associations of the 42 (effectively 20) SNPs assessed in these six regions revealed a significant association of the HFE rs1799945 SNP (also known as the H63D polymorphism) with the mean FA in the cluster along the left external capsule (P = 0.00017). The results of all of the genetic associations per ROI are also listed in Table S1.
Additionally, in the full sample of 565 genotyped subjects with serum transferrin levels available, we found that the H63D minor allele was associated with decreased transferrin levels as expected (t-statistic = 1.801, one-tailed P = 0.0361).
Post Hoc Voxel-Wise Analysis of HFE H63D Missense Polymorphism.
In our post hoc analysis, we performed a voxel-wise association of FA with the H63D polymorphism across the entirety of the white matter region. The SNP frequency information for this polymorphism in our sample is available in SI Methods. The map of voxel-wise associations of H63D to FA values was found to be significant in regions including the external capsule, and portions of the genu of the corpus callosum not initially found to have significant transferrin related associations (Fig. 6).
Fig. 6.

Corrected p-map shows the HFE H63D associations with FA voxel-wise throughout the white matter. When regressing on the minor allele, there is a positive association between the number of minor alleles and the FA values. Significance was confirmed by enforcing a regional control over the FDR as described by Langers et al. (70) at the 5% level. We adjusted for effects of age and sex to be consistent with the previous tests. Positive correlations were not significant.
Discussion
ID, iron overload, and abnormalities in iron concentrations localized to particular structures in the brain have been linked to neurodevelopmental and neurodegenerative disorders.
Fig. 7 shows a schematic illustration relating several biological processes that motivated this study. Both brain structure and the iron transport protein, transferrin, are under strong genetic control, so we used a twin design to find brain regions with genetic determinants in common with transferrin. We were able to establish some previously unknown links between transferrin levels (and associated genes) and brain structure in 615 healthy young adults.
Fig. 7.

Several known relationships motivated our study (solid black lines); dashed lines show relationships we wanted to test. Genetic and environmental factors (e.g., diet) affect iron stores in the body; the liver synthesizes more transferrin in response to low iron stores. Our first goal was to relate transferrin levels to brain structure in healthy young adults. Our twin design determined if overlapping sets of genes influence transferrin levels and brain structure, as both are highly heritable. Transferrin levels are genetically modulated mainly through two genes (HFE and TF); to relate specific variants in transferrin-related genes to brain structure, we determined the additive effect of all variants within these two genes on brain structures that had shown genetic influences in common with transferrin.
Our analysis had three main findings. First, serum transferrin levels, measured during adolescence, were associated with both macro- and microneuroanatomical variations in a regionally selective pattern later, in early adulthood (approximately 9 y after blood was drawn). Second, these associations with white matter integrity were mediated by overlapping sets of genes. This was evident from the cross-twin cross-trait correlations between transferrin levels and white matter anisotropy. Third, we found that the HFE H63D polymorphism, well known for its association to iron overload (40, 41), influences both serum transferrin levels and white matter microstructure in the external capsule. This points to a direct link between blood serum related genomic variation and brain structure (Fig. 7, dashed lines).
Iron is important for neural development early in life. In rat brains, iron and transferrin are at extremely high levels, despite low brain transferrin mRNA levels before closure of the blood–brain barrier (42). Even after the barrier develops, serum transferrin levels, which are under high genetic control, influence how much iron is transported to the brain for crucial processes of development, such as myelination. Here we uncovered an association between brain structure in young adults and serum transferrin levels measured during their adolescent years.
By measuring transferrin levels 8 to 12 y before the imaging study, we were interested in knowing whether iron availability in this developmentally crucial period might impact the organization of the brain later in life. Adolescence is a period of high vulnerability to brain insults, and the brain is still very actively developing (43). Transferrin levels, measured before the brain is fully mature, may be especially relevant for the adult brain. Transferrin levels fall with age in children and adolescents, and older adolescents show similar ranges to adults (15, 44). Children have higher transferrin levels than adults, perhaps in response to physiologically low iron stores. By averaging transferrin levels assessed repeatedly at various ages (12, 14, and 16 y of age), we estimated the iron availability to the brain during adolescence. We relied on previous work showing that transferrin measures are stable and can be reliably collected (19), with high sensitivity and specificity, which makes associations easier to detect.
As key components of white matter, oligodendrocytes—the glial cells that produce myelin to insulate axons—stain for iron more than any other cell in the brain (24); these cells are the primary location for iron in the central nervous system (45).
Fig. 1 shows the negative association between serum transferrin levels and the diffusion-based measure of integrity, FA, in various brain regions, including the external capsule, cingulum, and superior longitudinal fasciculus. As FA can represent the degree of fiber integrity, myelination, or coherence in white matter fibers, the direction of this association is in line with previous reports indicating hypomyelination in cases of ID (36). Our findings of transferrin associations in human white matter tracts are consistent with previous studies of brain iron levels in rats. In a histochemical study of iron staining in the developing rat brain, Connor et al. (46) found major foci of iron staining in the cingulum, superior portions of the internal capsule, and the base of the external capsule.
As shown in Fig. 2, regional brain volume deficits are seen bilaterally in the caudates, the third ventricle, and in the temporoparietal white matter as transferrin levels increase. The caudate is particularly important in cognition, learning, and memory (47); as increased transferrin levels have been implicated in ID, the inverse relation between transferrin levels and caudate volume may indicate underdevelopment of caudates in ID, in line with evidence of poorer cognition in children with ID. If severe enough, a caudate volume reduction related to elevated transferrin levels may explain why certain developmental cognitive deficits are associated with ID. This finding fits with our previous hypothesis that insufficient iron transport to the brain may hinder development of subcortical structures.
Although transferrin levels are low, transferrin in the CSF is fully saturated with iron (48); the regional deficit in the volume of the third ventricle is therefore intriguing, as it may indicate an altered pattern and/or rate of transport for transferrin-bound iron.
Some brain regions were smaller and some larger in people with lower transferrin levels; in fact, there was statistically significant evidence in favor of both effects occurring in different parts of the brain. This pattern of anomalies was somewhat surprising: we expected smaller volumes, not larger ones, in people with low iron (and elevated transferrin) levels. Opposing this, some brain regions involved in neurodegeneration did show lower volumes in those with high iron and low transferrin levels, so iron overload may promote neuronal atrophy in iron-containing structures. The direct association of the volume of these regions to transferrin levels may therefore indicate a future susceptibility to the effects of iron overload and altered transport in these disorders.
Our DTI-based analyses supported a model wherein signs of a less mature or well myelinated brain were found in those with high transferrin levels during adolescence; this may reflect the liver's reaction to sustained periods of lower iron availability. The analysis of brain volumes with TBM gives a more complex picture: in segregated comparisons, there were some brain regions that were larger, and some were smaller in those with high transferrin. This imbalance of structure volumes is similar to that seen in some neurogenetic disorders, in which patterns of abnormally high and low volumes are seen (49, 50). As this was not hypothesized, future independent studies are needed to confirm the localization and direction of these effects.
As indicated by a dashed line in Fig. 7, a way to study the iron pathway's association to brain structure is to determine whether genes influencing transferrin levels also modulate structural variation. Our cross-twin cross-trait genetic analysis revealed that common additive genetic factors influence transferrin concentrations and white matter fiber integrity. Finding neuroanatomical regions whose underlying structure is partially under the same genetic control as transferrin levels can help shed light on the inherited properties of these regions as they develop. Discovering specific iron-associated genetic variants that influence the underlying microstructure in these brain regions could potentially help uncover the neural mechanisms affected by iron transport in the brain. These may lead to downstream genetically mediated impairments.
Specific variants associated with iron mediating proteins in healthy young adults have also been discovered; 40% of the genetic variance in serum transferrin levels is explainable by just a few genetic variants in the TF gene (rs3811647, rs1799852, and rs2280673) and the C282Y mutation in the HFE gene (35). Additionally, interaction between variants in these two genes has been linked to an increased risk of Alzheimer's disease (51), so the TF and HFE genes are neurobiologically linked. To comprehensively explore these two genes further and determine any coexisting associations to brain structure, we examined all available variants within these genes in regions where the shared additive genetic component between the two traits, transferrin levels and brain microstructure, was statistically significant. We found the H63D polymorphism within the HFE gene is significantly associated with the mean FA of the left external capsule, one of the regions shown to have significant cross-twin cross-trait correlations. The FA of the external capsule has also been shown to be highly heritable (∼60%), however, a sex-by-heritability analysis also shows this region is much more heritable in male subjects (28). Intriguingly, genetic factors also explain a higher proportion of the variance for transferrin levels in men than in women (17).
In a recent study of HFE and TF variants on iron levels and risk for AD, Giambattistelli et al. (52) found that patients with AD with the H63D polymorphism had increased plasma iron and transferrin levels, but this pattern was not found in healthy control subjects with the variant; in fact, a meta-analysis found that the H63D polymorphism may be protective against AD (53). As shown in Table S1, the minor allele at rs1799945 (H63D) showed a positive effect on FA. This is the expected direction of association; as mentioned previously, ID can cause deficits in myelin formation, so it is reasonable that an iron overload allele may play a protective role for myelination during neuronal development of these healthy controls.
Our work here is one of the largest bimodal neuroimaging genetics studies of healthy humans to date. It describes a three-step top-down method to analyze gene effects on the brain. First, we related a heritable serum measure—with known cognitive associations—to specific locations in the brain; second, we used a genetic correlation model to home in on brain regions with evidence of joint genetic determination; and finally, we searched these neuroanatomical locations for variants within genes known to associate with the highly heritable phenotype, serum transferrin. We found localized regions of transferrin association on both the macro- and micro-anatomical scale within the brain. These regions showed additional associations with HFE variants, in a direction that is consistent with several previous studies of brain iron and associated proteins. Future analyses may include studying the mechanistic process of transferrin and HFE in children with cognitive impairment and in elderly subjects with neurodegenerative diseases.
Several conclusions can be drawn about genetic variations that affect transferrin levels and their effect on brain microstructure. Transferrin levels are influenced by at least two known factors: a shortage of iron, which drives them up, and polymorphisms in the two major transferrin-related genes, HFE and TF, as shown in Fig. 7. According to a principle known as Mendelian randomization (54), one can examine genes that are known to affect a measure, such as transferrin, to get a sense of the downstream biological effects of other factors that affect transferrin, such as a shortage of dietary iron. We did find that the H63D mutation within the HFE gene was related to brain FA, but we did not find an effect on FA of the other common (i.e., MAF > 0.05) SNPs in the HFE or TF genes, which are known to explain 40% of variation in transferrin levels. A larger sample may be needed to uncover effects of these SNPs, but a more skeptical alternative interpretation may be that some variants known to powerfully affect transferrin may not affect DTI measures at all. At this point, we cannot say conclusively whether polymorphisms in the TF gene have any causal role on white matter, although the H63D mutation within the other major transferrin-related gene—HFE—was related to brain FA. A qualified interpretation of the available data would suggest that transferrin levels do relate to brain structure, but further work is needed to clarify which of the several known transferrin-related SNPs, other than HFE H63D, are contributing to the effect.
Methods
Subject Information.
A total of 615 subjects (mean age ± SD, 23.5 ± 2.1 y; 375 women) were included in this study; all subjects had standard structural T1-weighted brain MRI scans and serum iron and transferrin levels measured; 574 also underwent DTI. As part of a reliability analysis, 37 subjects had a duplicate MRI scan taken 3 mo later, and five had a second DTI scan. All subjects were of European ancestry from 350 families. Subjects were recruited as part of a 5-y research project examining healthy young adult Australian twins using structural and functional MRI and DTI with a projected sample size of approximately 1,150 at completion (55). Subjects were screened to exclude cases of pathology known to affect brain structure. No subjects reported a history of significant head injury, a neurological or psychiatric illness, substance abuse or dependence, or had a first-degree relative with a psychiatric disorder. All subjects were right-handed as determined using 12 items from the Annett handedness questionnaire (56). We selected only the paired MZ (T1, n = 107; DTI, n = 95 pairs) and same-sex DZ (T1, n = 65; DTI, n = 59 pairs) twins for the cross-twin cross-trait genetic analysis. The rest of the subjects included 52 (n = 43 DTI) pairs of mixed-sex DZ twins, two sets of fraternal triplets (n = 6 individuals), and 112 (n = 95 DTI) individuals unrelated to anyone else in the study; additional subjects included non-twin siblings or unpaired twins with siblings also in the study in which kinship existed between members. A total of 544 of the subjects with standard MRI scans were genotyped, of whom 509 also had DTI scans available. Study participants gave informed consent; the studies were approved by the institutional ethics committees at the University of Queensland and the University of California, Los Angeles. All images used in this analysis went through, and passed, a rigorous quality control; subjects with anatomical abnormalities, imaging artifacts, and misregistered images were removed from analysis and not included in the subject counts.
Blood was collected from subjects at ages 12, 14, and 16 y. Serum was separated from blood samples and stored at −70 °C until assayed; iron, transferrin, and ferritin were measured by using standard clinical chemistry methods (Roche Diagnostics) on a 917 or Modular P analyzer. Data on serum iron and transferrin levels were extracted from these time points and averaged for use in this analysis.
Establishing Zygosity, Genotyping, and Imputation.
Zygosity was established objectively by typing nine independent DNA microsatellite polymorphisms (polymorphism information content > 0.7), by using standard PCR methods and genotyping. Results were cross-checked with blood group (ABO, MNS, and Rh) and phenotypic data (hair, skin, and eye color), giving an overall probability of correct zygosity assignment greater than 99.99%. Genomic DNA samples were analyzed on the Human610-Quad BeadChip (Illumina) according to the manufacturer's protocols (Infinium HD Assay; Super Protocol Guide; Revision A, May 2008). Imputation was performed by mapping the genotyped information to HapMap (release 22, build 36) with Mach software (http://www.sph.umich.edu/csg/abecasis/MACH/index.html).
Image Acquisition.
Structural and diffusion-weighted (DW) whole-brain MRI scans were acquired for each subject (4 Tesla Medspec; Bruker). T1-weighted images were acquired with an inversion recovery rapid gradient-echo sequence (inversion/repetition/echo times, 700/1500/3.35 ms; flip angle, 8°; slice thickness, 0.9 mm; 256 × 256 acquisition matrix). DW images were acquired using single-shot echo-planar imaging with a twice-refocused spin echo sequence to reduce eddy current-induced distortions. A 3-min, 30-gradient acquisition was designed to optimize signal-to-noise ratio for diffusion tensor estimation (57). Imaging parameters were repetition/echo times of 6,090/91.7 ms, field of view of 23 cm, and 128 × 128 acquisition matrix. Each 3D volume consisted of 21 axial slices 5 mm thick with a 0.5-mm gap and 1.8 × 1.8 mm2 in-plane resolution. Thirty images were acquired per subject: three with no diffusion sensitization (i.e., T2-weighted b0 images) and 27 DW images (b = 1,146 s/mm2) with gradient directions uniformly distributed on the hemisphere.
Image Preprocessing.
Nonbrain regions were automatically removed from each T1-weighted MR image and from a T2-weighted image from the DW image set using FSL software brain extraction tool (58) to enhance coregistration between subjects. All T1-weighted images were corrected for field nonuniformities using FreeSurfer (http://surfer.nmr.mgh.harvard.edu/), linearly aligned [with 9 degrees of freedom (df)] to a common space (59). The raw DW images were corrected for eddy current distortions by using the FSL tool “eddy_correct” (http://fsl.fmrib.ox.ac.uk/fsl/). For each subject, the three eddy-corrected images with no diffusion sensitization were averaged, linearly aligned, and resampled to the subject's corresponding down-sampled T1 image. The average b0 maps were then elastically registered to the subject's aligned T1-weighted structural scan by using an inverse consistent registration with a mutual information cost function (60) to adjust for any echo-planar–induced susceptibility artifacts.
TBM.
TBM is a technique that identifies regional structural differences from the gradients of the deformation fields that align brain images to a common anatomical template. After nonlinearly aligning the full brain of all subjects to their corresponding minimum deformation template (MDT), a separate Jacobian map (i.e., relative volume map) was created for each subject. These Jacobian maps, which share the common space defined by the MDT, help to characterize the local volume differences between one individual and the normal anatomical template. These maps explain the relative expansion and contraction of regions from each individual relative to the template.
Computing Anisotropy and Diffusivity.
Under a single-tensor model (61), diffusion of water molecules attenuates the MR signal in direction r, according to the Stejskal–Tanner equation:
Here, S0(r) is the non-DW baseline intensity in direction r, Dk(r) is the apparent diffusion coefficient, and bk is a constant depending on the gradient k. Diffusion tensors were computed from the 27-gradient DW images using FSL software (http://fsl.fmrib.ox.ac.uk/fsl/). The FA of diffusion was computed from the tensor eigenvalues (λ1, λ2, λ3) at each voxel. FA is influenced by both axial diffusivity (λ1; a measure of diffusion along the axonal fibers) and radial diffusivity (the average of λ2 and λ3; a measure of diffusion orthogonal to the axonal fibers).
Template Creation and Registration.
We created an MDT by using nonlinear fluid registration (62), with the method proposed by Kochunov and colleagues (63, 64). The N 3D vector fields fluidly registering a specific individual to all other N participants were averaged and applied to that subject. This geometrically adjusts the anatomy but preserves the intensities and anatomical features of the template subject.
To create a representative MDT for the TBM analysis, we randomly selected 32 (16 female/16 male) nonrelated participants’ T1-weighted images, after down-sampling to be in the same space as the DW imaging and aligning to the Colin template (59), and created two MDTs with 16 subjects (eight female) in each group. For one group, the target was male and the other female. The templates for each group were then averaged to create one representative anatomically centered target. Skull-stripped T1-weighted images for each subject were registered to the final population averaged FA-based MDT by using an inverse consistent 3D elastic warping technique using a mutual information cost function (60).
To create an MDT for DTI analysis, we selected the same 32 participants, yet we used their FA images (calculated after b0 susceptibility correction) to create the MDT in the exact same manner as the T1-weighted structural scans. FA maps for each of the susceptibility corrected subjects were registered to the final population averaged FA-based MDT by using a 3D elastic warping technique with a mutual information cost function (60). To further align white matter regions of interest, the FA-based MDT and all whole-brain registered FA maps were then thresholded at 0.25, as FA values lower than 0.25 in healthy-appearing white matter may reflect contributions from nonwhite matter. Individual thresholded FA maps were then reregistered to the thresholded MDT in the same way as the whole brain registration. After registration of the FA maps, the FA images were smoothed with a Gaussian filter with an isotropic full-width half maximum of 5 mm).
Random-Effects Regression.
The relationships of transferrin to measures of anisotropy and brain morphometry were assessed at each voxel in the brain by using a mixed-effects regression model to account for similarities within families while controlling for the effects of sex and age. To boost power of the association, and reduce random noise brought on by image acquisition, we included duplicate scans for the subjects who had them available. The variable of interest (transferrin), sex, and age were included as fixed effects. Random intercepts were included for each family and subjects to account for relatedness within families as well as the duplicate scans used. The analysis was implemented in the R statistical package (version 2.9.2; http://www.r-project.org/) using the ‘nlme’ library (65).
As noted earlier, extremely high and extremely low levels of iron can adversely affect the brain, but these observations do not completely imply in which direction the correlation would be in healthy people who maintain their iron levels for the most part in the normal range. As iron overload was not expected in this young, healthy population, mild insufficiencies in iron were considered more likely. This led to a directional hypothesis that poorer brain phenotypes might be found in those with lower chronic levels of iron (as inferred from transferrin measures). However, we considered it also possible, but less plausible, that there might be enough people with very high iron levels to drive the effect in the opposite direction. To allow for this alternative but less likely hypothesis, we ran our analyses with a more conservative searchlight FDR threshold of 0.025, to allow us to reject the null hypothesis in either direction, but distinguish between the alternative hypotheses in different directions. More information may be found in SI Methods.
Cross-Twin Cross-Trait Analysis.
We used a cross-twin cross-trait analysis (66) to detect common genetic or environmental factors influencing both brain structure (or microstructure) and serum transferrin levels at every voxel within the brain. Covariance matrices for the phenotypes, in this case the voxel-wise structural measure of interest (structural deformation, microstructural anisotropy, or diffusivity) and serum transferrin levels were computed between the MZ twins who share all the same genes, and the DZ twins who share, on average, half of their genetic polymorphisms. These covariance matrices were then entered into a multivariate structural equation model [SEM (67)], using OpenMx software (http://openmx.psyc.virginia.edu/) to fit the relative contributions of additive genetic (A), shared environmental (C), and unshared or unique environmental (E) components to the population variances and covariances of the observed variables. Experimental measurement error is also included in the E component, and is assumed to be independent between twins 1 and 2 (i.e., no correlation).
In multivariate SEM, it is assumed that there are common genetic and environmental factors that affect various phenotypes, as also described in (34). Here we consider bivariate models with two phenotypes, transferrin levels and the brain MRI- or DTI-derived value at each voxel. The common genetic and environmental components of the variance may be estimated from the total population variance by examining the difference between the covariances between the MZ and DZ twins within the same individual (cross-trait within individual) and also between one phenotype in one twin with the other phenotype in the second twin (cross-twin cross-trait). By using this multivariate SEM, we can also obtain the additive genetic and shared environmental influences on the correlations between the two phenotypes, denoted as rA and rC, respectively. A path diagram describing the SEM and the connections between the twins is shown in Fig. 8.
Fig. 8.

Path diagram for the full bivariate ACE model.
The cross-trait within-individual correlation [i.e., the correlation between the voxel value (V) and transferrin (T) in twin 1 or in twin 2] is divided into additive genetic and shared and unique environmental components (e.g., AV,i, CV,i, and EV,i for voxel value and AT,i, CT,i, and ET,i for transferrin; i = 1 or 2 for twin 1 or 2), and the correlation coefficients between AV,i and AT,i, CV,i and CT,i, and EV,i and ET,i, are denoted by ra, rc, and re, respectively. The cross-twin cross-trait correlation is shown as AV,i and AT,j, and CV,i and CT,j for the voxel value in twin i and the transferrin level in twin j, where i, j = 1 or 2, and i ≠ j. There is no re term for EV,i and ET,j because the unique environmental factors between subjects are independent. The covariance across the two phenotypes within the same subject, or separately in the two subjects, is then derived by multiplication of the path coefficients for the closed paths in the path diagram. For example, covariance between the voxel values in twin 1 and the transferrin level in twin 2 is equal to aV·ra·aT + cV·rc cT for MZ twins, and aV·1/2ra aT + cV·rc cT for DZ twins. This implies that any excess in cross-twin cross-trait correlation in MZ twins over that in DZ twins is attributed to common genetic factors that affect both voxel values and transferrin levels.
Paths drawn between the same phenotype would be identical to considering a univariate voxel-wise SEM model (27). For A1 and A2, the correlation coefficient is equal to 1 for MZ and 0.5 for DZ twin pairs. The correlation coefficient between C1 and C2 is always 1 from the definition of the shared environment, and E1 and E2 are assumed to be independent and there is no correlation.
In twin studies, it is common to examine whether the observed measures are best modeled by using a combination of additive genetic and shared and unshared environmental factors, or whether only one or two of these factors is sufficient to explain the observed pattern of inter-twin correlations.
If the correlation between the voxel value of the image in one twin and the level of transferrin in the other twin is greater in MZ pairs than in DZ pairs, then, under standard assumptions, the greater correlation may be assumed to be caused by common genetic factors controlling both factors. In the univariate model with a single phenotype, which we denote x, the genetic and environmental contributions in twin j (j = 1 or 2) is modeled by defining the following:
A, C, and E, respectively, denote the additive genetic and shared and unshared environmental components. Cross-trait correlations between voxel values (v) and serum transferrin (t) level are then derived from the covariance matrix of the following vector:
given by the following 4 × 4 matrix:
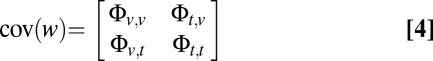 |
where Φvv and Φtt are the 2 × 2 covariance matrices for phenotype v or t between twins 1 and 2, as performed in univariate SEM. Φvt is the cross-trait covariance matrix, composed of the covariance between the two traits within the different unrelated individuals [cov(v1, t1) and cov(v2, t2)] and the cross-twin cross-trait covariance between the pairs [cov(v1, t2) and cov(v2, t1)], as detailed below:
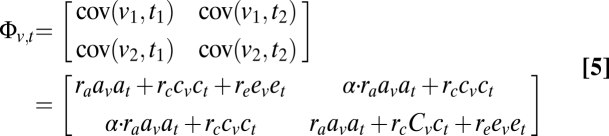 |
where α is 1 for MZ twins, and 0.5 for DZ twins. ra, rc, and re are the cross-trait correlation coefficients for Av and AT, CV and CT, and EV and ET, respectively. A higher value of ra indicates that the two phenotypes are more likely mediated by a common set of genes (34, 68). The path coefficients were estimated by comparing the covariance matrix implied by the model and the sample covariance matrix of the observed variables, using maximum-likelihood fitting to give a χ2 value. We started from the full set of path coefficients (av, cv, ev, at, ct, et, ra, rc, and re) and removed one of av, cv, at, and ct from the model step by step. Removing av or at/cv or ct also removed ra/rc. e1, e2, and re were always kept in the model to include random noise. A model was considered to better fit the data if the difference in χ2 values between it and the more comprehensive model at the previous step was not significant. If two models contained the same number of parameters, the model with a smaller χ2 value was considered better. Model selection ended when the best model was achieved, i.e., when either (i) all possible more restricted models were not better than the current model or (ii) the current model was the most restricted and contained ev, et, and re only. If ra was included in the best model, the significance of ra was then determined by comparing the χ2 values of the best model and its submodel where ra is 0. To determine the significance of the submodels, or the restricted models, with respect to the full model, we obtain the log-likelihood for the full and the restricted models, denoted by log(Lf) and log(Lr), respectively. Minus two times this difference, or −2[log(Lr) − log(Lf)], is asymptotically distributed approximately as a χ2 distribution with the df equal to the difference between the df of the two models, and therefore the inverse χ2 distribution is estimated with these parameters.
SNP Selection.
We examined all SNPs within two genes previously shown (35) to affect serum transferrin levels in healthy adults: TF and HFE. By using HapMap, we searched for SNPs that met our criterion of having an MAF greater than 5%; when matching these SNPs to those with available genotype information in our imputed data, 42 valid SNPs from these genes were available for analysis. We determined significance levels for association tests by first examining the total number of independent tests performed. Linkage disequilibrium among SNPs tested corresponds to correlation between the SNPs, and therefore each test is not completely independent. By first estimating the effective number of independent tests, we can avoid using a significance level too conservative for the number of tests we performed. As a result of linkage disequilibrium, the effective number of SNPs tested (38, 39) was 20.
ROI SNP Association in Significantly Correlated Clusters.
In each cluster (>27 voxels to represent a size equivalent to one voxel and all its surrounding neighbors) that was found to have significant cross-twin cross-trait additive genetic associations, we found the average value across all of the voxels in that region and performed univariate associations with all 42 SNPs by using a mixed-model approach controlling for age and sex (emmaX; http://genetics.cs.ucla.edu/emmax/news.html) (69) to account for the familial relatedness between subjects through the use of a kinship matrix describing the approximate proportion of genetic similarities between subjects. A 0 in the kinship matrix represents the relation between unrelated individuals, MZ twins are related by 1 (with identical genomes), and DZ twins and non-twin siblings within the same family by 0.5 (as they share approximately half). Duplicate scans were not used for genetic associations.
Multiple Comparisons Correction.
Computing thousands of tests of associations on a voxel-wise level can introduce a high Type I (i.e., false-positive) error rate in neuroimaging studies. To control these errors, we used a searchlight method for FDR correction as described by Langers et al. (70), which ensures a regional control over the FDR in any reported findings. To ensure adequate regionally selective associations with the transferrin levels, we use this searchlight method to correct the associations between the image phenotypes (morphometry or anisotropy) or transferrin. All maps shown are thresholded at the appropriate corrected P value after performing searchlight FDR (q = 0.05) to show only regions of significance; uncorrected P values are then shown only within these significant regions. To determine the best overall model for the SEM cross-twin cross-trait analysis, we use the standard FDR (37, 71) procedure as opposed to searchlight FDR, as we would like to determine the best overall fit of the SEM model, and not necessarily examine any localized or clustering effects. When examining the significance of the effects of the SNPs regressed on the mean FA value within ROIs with significant cross-twin cross-trait associations, we corrected for multiple comparisons by using the strict Bonferroni correction controlled at the q level of 0.05, at which a threshold for significance was determined by dividing 0.05 by the effective number of SNPs tested (20), and the number of ROIs where these SNPs were each tested previously (5). The Bonferroni threshold for significance was therefore set as follows:
Post Hoc Analysis: Voxel-Wise Effect of HFE H63D Polymorphism on Fiber Integrity.
The number of minor alleles for each subject at HFE H63D (rs1799945) was regressed against the FA at each voxel within the white matter, after adjusting for sex and age as before. Family structure was taken into account with mixed-effects modeling (72). To correct for multiple comparisons across voxels, we used a searchlight method to control the FDR regionally (70).
Supplementary Material
Acknowledgments
We thank the twins and siblings for their participation. In Brisbane, we thank Marlene Grace and Ann Eldridge for twin recruitment, Aiman Al Najjar and other radiographers for scanning, Kori Johnson for scanning and data transfer, Kerrie McAloney and Daniel Park for research support, and staff in the Molecular Epidemiology Laboratory for serum and DNA sample processing and preparation. This work was supported by National Institute of Child Health and Human Development Grant R01 HD050735, National Health and Medical Research Council (NHMRC; Australia) Grant 486682, Grant T15 LM07356 (to N.J.), the Achievement Rewards for College Scientists Foundation (J.L.S.), National Institute of Mental Health Grant 1F31MH087061 (to J.L.S.), and Australian Research Council Future Fellowship FT0991634 (to G.I.d.Z.). Genotyping was supported by NHMRC Grant 389875. Additional support for algorithm development was provided by National Institutes of Health Grants R01 EB008432, R01 EB008281, and R01 EB007813.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
*Our dataset included a genotype list that had been imputed (to HapMap2) whereas the Benyamin et al. (2008) paper (35) did not; the previous paper therefore did not analyze the H63D polymorphism at all.
See Author Summary on page 5162 (volume 109, number 14).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1105543109/-/DCSupplemental.
References
- 1.CDC From the Centers for Disease Control and Prevention. Iron deficiency—United States, 1999-2000. JAMA. 2002;288:2114–2116. [PubMed] [Google Scholar]
- 2.Halterman JS, Kaczorowski JM, Aligne CA, Auinger P, Szilagyi PG. Iron deficiency and cognitive achievement among school-aged children and adolescents in the United States. Pediatrics. 2001;107:1381–1386. doi: 10.1542/peds.107.6.1381. [DOI] [PubMed] [Google Scholar]
- 3.Stoltzfus RJ, et al. Effects of iron supplementation and anthelmintic treatment on motor and language development of preschool children in Zanzibar: Double blind, placebo controlled study. BMJ. 2001;323:1389–1393. doi: 10.1136/bmj.323.7326.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson C, Erikson K, Piñero DJ, Beard JL. In vivo dopamine metabolism is altered in iron-deficient anemic rats. J Nutr. 1997;127:2282–2288. doi: 10.1093/jn/127.12.2282. [DOI] [PubMed] [Google Scholar]
- 5.Bartzokis G, et al. In vivo evaluation of brain iron in Alzheimer's disease and normal subjects using MRI. Biol Psychiatry. 1994;35:480–487. doi: 10.1016/0006-3223(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 6.Bartzokis G, et al. MRI evaluation of brain iron in earlier- and later-onset Parkinson's disease and normal subjects. Magn Reson Imaging. 1999;17:213–222. doi: 10.1016/s0730-725x(98)00155-6. [DOI] [PubMed] [Google Scholar]
- 7.Jurgens CK, et al. MRI T2 hypointensities in basal ganglia of premanifest Huntington's disease. PLoS Curr. 2010;2:RRN1173. doi: 10.1371/currents.RRN1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ke Y, Ming Qian Z. Iron misregulation in the brain: A primary cause of neurodegenerative disorders. Lancet Neurol. 2003;2:246–253. doi: 10.1016/s1474-4422(03)00353-3. [DOI] [PubMed] [Google Scholar]
- 9.Benarroch EE. Brain iron homeostasis and neurodegenerative disease. Neurology. 2009;72:1436–1440. doi: 10.1212/WNL.0b013e3181a26b30. [DOI] [PubMed] [Google Scholar]
- 10.Moos T, Morgan EH. Transferrin and transferrin receptor function in brain barrier systems. Cell Mol Neurobiol. 2000;20:77–95. doi: 10.1023/a:1006948027674. [DOI] [PubMed] [Google Scholar]
- 11.Das Gupta A, Abbi A. High serum transferrin receptor level in anemia of chronic disorders indicates coexistent iron deficiency. Am J Hematol. 2003;72:158–161. doi: 10.1002/ajh.10260. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Connor JR, Beard JL. Brain iron, transferrin and ferritin concentrations are altered in developing iron-deficient rats. J Nutr. 1995;125:1529–1535. doi: 10.1093/jn/125.6.1529. [DOI] [PubMed] [Google Scholar]
- 13.Erikson KM, Pinero DJ, Connor JR, Beard JL. Regional brain iron, ferritin and transferrin concentrations during iron deficiency and iron repletion in developing rats. J Nutr. 1997;127:2030–2038. doi: 10.1093/jn/127.10.2030. [DOI] [PubMed] [Google Scholar]
- 14.Khumalo H, et al. Serum transferrin receptors are decreased in the presence of iron overload. Clin Chem. 1998;44:40–44. [PubMed] [Google Scholar]
- 15.Suominen P, et al. Regression-based reference limits for serum transferrin receptor in children 6 months to 16 years of age. Clin Chem. 2001;47:935–937. [PubMed] [Google Scholar]
- 16.Lynch SR, Cook JD. Interaction of vitamin C and iron. Ann N Y Acad Sci. 1980;355:32–44. doi: 10.1111/j.1749-6632.1980.tb21325.x. [DOI] [PubMed] [Google Scholar]
- 17.Whitfield JB, et al. Effects of HFE C282Y and H63D polymorphisms and polygenic background on iron stores in a large community sample of twins. Am J Hum Genet. 2000;66:1246–1258. doi: 10.1086/302862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baynes RD. Assessment of iron status. Clin Biochem. 1996;29:209–215. doi: 10.1016/0009-9120(96)00010-k. [DOI] [PubMed] [Google Scholar]
- 19.Ahluwalia N. Diagnostic utility of serum transferrin receptors measurement in assessing iron status. Nutr Rev. 1998;56:133–141. doi: 10.1111/j.1753-4887.1998.tb01738.x. [DOI] [PubMed] [Google Scholar]
- 20.Zeleniuch-Jacquotte A, et al. Reliability of serum assays of iron status in postmenopausal women. Ann Epidemiol. 2007;17:354–358. doi: 10.1016/j.annepidem.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pardridge WM, Eisenberg J, Yang J. Human blood-brain barrier transferrin receptor. Metabolism. 1987;36:892–895. doi: 10.1016/0026-0495(87)90099-0. [DOI] [PubMed] [Google Scholar]
- 22.Rouault TA, Zhang DL, Jeong SY. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab Brain Dis. 2009;24:673–684. doi: 10.1007/s11011-009-9169-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 24.Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: The role of iron. Glia. 2009;57:467–478. doi: 10.1002/glia.20784. [DOI] [PubMed] [Google Scholar]
- 25.Aoki S, et al. Normal deposition of brain iron in childhood and adolescence: MR imaging at 1.5 T. Radiology. 1989;172:381–385. doi: 10.1148/radiology.172.2.2748819. [DOI] [PubMed] [Google Scholar]
- 26.Frisoni GB, Fox NC, Jack CR, Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jahanshad N, et al. Genetic influences on brain asymmetry: A DTI study of 374 twins and siblings. Neuroimage. 2010;52:455–469. doi: 10.1016/j.neuroimage.2010.04.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiang MC, et al. Genetics of white matter development: A DTI study of 705 twins and their siblings aged 12 to 29. Neuroimage. 2011;54:2308–2317. doi: 10.1016/j.neuroimage.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou YY, et al. Mapping genetic influences on ventricular structure in twins. Neuroimage. 2009;44:1312–1323. doi: 10.1016/j.neuroimage.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson PM, et al. Genetic influences on brain structure. Nat Neurosci. 2001;4:1253–1258. doi: 10.1038/nn758. [DOI] [PubMed] [Google Scholar]
- 31.Blokland GA, et al. Quantifying the heritability of task-related brain activation and performance during the N-back working memory task: A twin fMRI study. Biol Psychol. 2008;79:70–79. doi: 10.1016/j.biopsycho.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koten JW, Jr, et al. Genetic contribution to variation in cognitive function: An FMRI study in twins. Science. 2009;323:1737–1740. doi: 10.1126/science.1167371. [DOI] [PubMed] [Google Scholar]
- 33.Posthuma D, et al. The association between brain volume and intelligence is of genetic origin. Nat Neurosci. 2002;5:83–84. doi: 10.1038/nn0202-83. [DOI] [PubMed] [Google Scholar]
- 34.Chiang MC, et al. Genetics of brain fiber architecture and intellectual performance. J Neurosci. 2009;29:2212–2224. doi: 10.1523/JNEUROSCI.4184-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benyamin B, et al. Variants in TF and HFE explain approximately 40% of genetic variation in serum-transferrin levels. Am J Hum Genet. 2009;84:60–65. doi: 10.1016/j.ajhg.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- 38.Gao X, Starmer J, Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol. 2008;32:361–369. doi: 10.1002/gepi.20310. [DOI] [PubMed] [Google Scholar]
- 39.Gao X, Becker LC, Becker DM, Starmer JD, Province MA. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol. 2010;34:100–105. doi: 10.1002/gepi.20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feder JN, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA. 1998;95:1472–1477. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aguilar-Martinez P, et al. Variable phenotypic presentation of iron overload in H63D homozygotes: are genetic modifiers the cause? Gut. 2001;48:836–842. doi: 10.1136/gut.48.6.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beard JL, Connor JR, Jones BC. Iron in the brain. Nutr Rev. 1993;51:157–170. doi: 10.1111/j.1753-4887.1993.tb03096.x. [DOI] [PubMed] [Google Scholar]
- 43.Gogtay N, et al. Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA. 2004;101:8174–8179. doi: 10.1073/pnas.0402680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virtanen MA, et al. Higher concentrations of serum transferrin receptor in children than in adults. Am J Clin Nutr. 1999;69:256–260. doi: 10.1093/ajcn/69.2.256. [DOI] [PubMed] [Google Scholar]
- 45.Gerber MR, Connor JR. Do oligodendrocytes mediate iron regulation in the human brain? Ann Neurol. 1989;26:95–98. doi: 10.1002/ana.410260115. [DOI] [PubMed] [Google Scholar]
- 46.Connor JR, Pavlick G, Karli D, Menzies SL, Palmer C. A histochemical study of iron-positive cells in the developing rat brain. J Comp Neurol. 1995;355:111–123. doi: 10.1002/cne.903550112. [DOI] [PubMed] [Google Scholar]
- 47.Packard MG, Knowlton BJ. Learning and memory functions of the basal ganglia. Annu Rev Neurosci. 2002;25:563–593. doi: 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- 48.Moos T, Rosengren Nielsen T, Skjørringe T, Morgan EH. Iron trafficking inside the brain. J Neurochem. 2007;103:1730–1740. doi: 10.1111/j.1471-4159.2007.04976.x. [DOI] [PubMed] [Google Scholar]
- 49.Thompson PM, et al. Abnormal cortical complexity and thickness profiles mapped in Williams syndrome. J Neurosci. 2005;25:4146–4158. doi: 10.1523/JNEUROSCI.0165-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee AD, et al. 3D pattern of brain abnormalities in Fragile X syndrome visualized using tensor-based morphometry. Neuroimage. 2007;34:924–938. doi: 10.1016/j.neuroimage.2006.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lehmann DJ, et al. Transferrin and HFE genes interact in Alzheimer's disease risk: The Epistasis Project. Neurobiol Aging. 2012;33:202.e1–202.e13. doi: 10.1016/j.neurobiolaging.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 52.Giambattistelli F, et al. Effects of hemochromatosis and transferrin gene mutations on iron dyshomeostasis, liver dysfunction and on the risk of Alzheimer's disease. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Lin M, et al. Association between HFE polymorphisms and susceptibility to Alzheimer's disease: A meta-analysis of 22 studies including 4,365 cases and 8,652 controls. Mol Biol Rep. 2011 doi: 10.1007/s11033-011-1072-z. [DOI] [PubMed] [Google Scholar]
- 54.Davey Smith G, Ebrahim S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 55.de Zubicaray GI, et al. Meeting the challenges of neuroimaging genetics. Brain Imaging Behav. 2008;2:258–263. doi: 10.1007/s11682-008-9029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Annett M. A classification of hand preference by association analysis. Br J Psychol. 1970;61:303–321. doi: 10.1111/j.2044-8295.1970.tb01248.x. [DOI] [PubMed] [Google Scholar]
- 57.Jones DK, Horsfield MA, Simmons A. Optimal strategies for measuring diffusion in anisotropic systems by magnetic resonance imaging. Magn Reson Med. 1999;42:515–525. [PubMed] [Google Scholar]
- 58.Smith SM. Fast robust automated brain extraction. Hum Brain Mapp. 2002;17:143–155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holmes CJ, et al. Enhancement of MR images using registration for signal averaging. J Comput Assist Tomogr. 1998;22:324–333. doi: 10.1097/00004728-199803000-00032. [DOI] [PubMed] [Google Scholar]
- 60.Leow A, et al. Inverse consistent mapping in 3D deformable image registration: its construction and statistical properties. Inf Process Med Imaging. 2005;19:493–503. doi: 10.1007/11505730_41. [DOI] [PubMed] [Google Scholar]
- 61.Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- 62.Lepore N, et al. Medical Imaging 2008: Physiology, Function, and Structure from Medical Images. Vol. 6916. San Diego: SPIE; 2008. Fast 3D fluid registration of brain magnetic resonance images; pp. 69160Z–69160Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kochunov P, et al. Regional spatial normalization: Toward an optimal target. J Comput Assist Tomogr. 2001;25:805–816. doi: 10.1097/00004728-200109000-00023. [DOI] [PubMed] [Google Scholar]
- 64.Kochunov P, et al. An optimized individual target brain in the Talairach coordinate system. Neuroimage. 2002;17:922–927. [PubMed] [Google Scholar]
- 65.Pinheiro JC, Bates DM. Mixed-Effects Models in S and S-PLUS. New York: Springer; 2000. [Google Scholar]
- 66.Neale MC, Cardon LR North Atlantic Treaty Organization. Scientific Affairs Division. Methodology for Genetic Studies of Twins and Families. Dordrecht, The Netherlands: Kluwer; 1992. [Google Scholar]
- 67.Rijsdijk FV, Sham PC. Analytic approaches to twin data using structural equation models. Brief Bioinform. 2002;3:119–133. doi: 10.1093/bib/3.2.119. [DOI] [PubMed] [Google Scholar]
- 68.Lange K, Boehnke M. Extensions to pedigree analysis. IV. Covariance components models for multivariate traits. Am J Med Genet. 1983;14:513–524. doi: 10.1002/ajmg.1320140315. [DOI] [PubMed] [Google Scholar]
- 69.Kang HM, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langers DR, Jansen JF, Backes WH. Enhanced signal detection in neuroimaging by means of regional control of the global false discovery rate. Neuroimage. 2007;38:43–56. doi: 10.1016/j.neuroimage.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 71.Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15:870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- 72.Kang HM, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
