


Abstract
Initiation of DNA replication in eukaryotes is exquisitely regulated to ensure that DNA replication occurs exactly once in each cell division. A conserved and essential step for the initiation of eukaryotic DNA replication is the loading of the mini-chromosome maintenance 2–7 (MCM2–7) helicase onto chromatin at replication origins by Cdt1. To elucidate the molecular mechanism of this event, we determined the structure of the human Cdt1-Mcm6 binding domains, the Cdt1(410–440)/MCM6(708–821) complex by NMR. Our structural and site-directed mutagenesis studies showed that charge complementarity is a key determinant for the specific interaction between Cdt1 and Mcm2–7. When this interaction was interrupted by alanine substitutions of the conserved interacting residues, the corresponding yeast Cdt1 and Mcm6 mutants were defective in DNA replication and the chromatin loading of Mcm2, resulting in cell death. Having shown that Cdt1 and Mcm6 interact through their C-termini, and knowing that Cdt1 is tethered to Orc6 during the loading of MCM2–7, our results suggest that the MCM2–7 hexamer is loaded with its C terminal end facing the ORC complex. These results provide a structural basis for the Cdt1-mediated MCM2–7 chromatin loading.
INTRODUCTION
To maintain genome integrity, DNA replication in eukaryotic cells is tightly regulated to ensure that the genome is replicated exactly once per cell cycle. This regulation is achieved through a two-step mechanism, the loading of the replicative DNA helicase, which includes the MCM2-7 complex as a major component and activation of this helicase (1).
The loading of the MCM2–7 complex requires the coordinated action of several proteins, most notably the six-subunit origin recognition complex (ORC), the cell division cycle 6 homolog (Cdc6), the chromatin licensing and DNA replication factor 1 (Cdt1) (2,3). The first step of this process is the binding of ORC to the replication origin on newly synthesized chromatin followed by the recruitment of Cdc6 and Cdt1. These two factors then recruit the MCM2–7 complex to form a prereplicative complex (pre-RC) during the late M and G1 phases (4,5). Activation of the pre-RC requires the sequential assembly of additional factors including Cdc45 and the GINS complex in a DDK- and S-CDK-dependent manner, culminating in the initiation of DNA replication in S-phase (6,7).
The MCM2–7 complex was first identified as a family of genes required for minichromosome maintenance in Saccharomyces cerevisiae (8). All six paralogous MCM proteins belong to the highly diversified AAA+ (ATPases associated with a variety of cellular activities) protein family (9). Structural analysis showed that the six MCM proteins form a double hexameric ring with head-to-head configuration, and DNA passes through the central channel of the double hexamer (10–12). The hexamer shows weak helicase activity in vitro (13–15).
The licensing factor Cdt1 is a critical component of the pre-RC, and its primary function is to recruit the MCM2–7 complex to the replication origin (16). Overexpression of Cdt1 alone in many types of mammalian cell lines causes rereplication of DNA (17–19).
Recently results showed that the Cdt1•MCM2–7 hepatmer is loaded onto DNA cooperatively to form a double hexamer (12). Previous studies on the interactions between Cdt1 and individual members of the MCM2–7 complex showed that Cdt1 interacts with Mcm2 and Mcm6 (16,20–22). The region of Cdt1 involved in the MCM2–7 interaction has been defined. Cdt1 binds to MCM2–7 complex through the region spanning residues 447–620 in Xenopus (16) and residues 407–477 of Cdt1 bind to Mcm6 in mouse (20). On the basis of the yeast two-hybrid assay we found that the interaction between human Mcm6 and Cdt1 is much stronger than that between Mcm2 and Cdt1. We further demonstrated that the conserved C-terminal domain of the human Mcm6 (residues 707–821) physically interacts with Cdt1 (residues 410–445) (23). However, the detailed molecular mechanism underlying the chromatin loading of the MCM2–7 complex through Cdt1 remains elusive.
In the present study, we determined the solution complex structure of the Cdt1-Mcm6 binding domains, the C-terminal helix (411–440) of Cdt1 binds to the C-terminal region (708–821) of Mcm6. In vivo studies in S. cerevisiae showed that interruption of this interaction prevented the loading of Mcm2 onto chromatin, inhibited DNA replication, and prevented cell proliferation. Our results reported here provided a structural basis for the Cdt1 mediated MCM2–7 chromatin loading.
MATERIALS AND METHODS
Sample preparation
Human Mcm6 C-terminal Cdt1-binding domain (hCBD) and the Mcm6-binding domain (hMBD) of human Cdt1 were expressed, enriched with 13C and 15N stable isotopes in Escherichia coli and purified essentially as described previously (23).
NMR spectroscopy
NMR spectra were acquired at 37°C on 750- and 500-MHz Varian NMR spectrometers with self-shielded z-axis gradients. All spectra were processed using NMRPipe (24,25) and analyzed using SPARKY 3 (Goddard and Kneller, University of California, San Francisco, CA, USA). The 1H, 15N and 13C resonances of backbone and side-chain atoms were assigned by using a standard set of triple resonance experiments on either uniformly 15N, 13C-labeled hCBD with/without unlabeled MBD or uniformly 15N, 13C-labeled hMBD with/without unlabeled hCBD, at protein concentrations of ∼0.6 mM (26). The hMBD–hCBD complex was prepared at a 1:2 ratio between 15N, 13C-labeled and unlabeled components. NOE-derived distance restraints were obtained from 15N- or 13C-edited 3D NOESY spectra each with a mixing time of 120 ms, complemented by 13C-edited, 13C/15N-filtered 3D NOESY spectra for the intermolecular contact (mixing time: 150 ms) (27).
NOE analysis and structure calculations
NOE assignment and structure calculations were performed using the program CYANA2.1 (28) for either hMBD or hCBD in the complex form. The initial 250 complex structures of hCBD and hMBD were generated with unambiguous intermolecular NOE restraints by CNS 1.1 (29). The 100 top scoring complex structures from CNS were further refined with additional chemical shift perturbation data using HADDOCK 1.3 (30). The quality of the structures was assessed using PROCHECK (31).
All of the figures representing the structures were generated by Pymol (http://www.pymol.org). The statistics of the structure refinement and the quality of the final structures are summarized in Table 1 for the hMBD/ hCBD complex, Supplementary Table S1 for hMBD.
Table 1.
Statistics of the NMR structure of the hCBD–hMBD complex
| NMR restraints | |
| Total experimental restraints | 1780 |
| Total NOE distance restraints | 1460 |
| Short-range, |i − j|≤1 | 773 |
| Medium-range, 1<|i − j|<5 | 352 |
| Long-range, |i − j|≥5 | 235 |
| Intra MBD | 90 |
| Inter CBD-MBD | 10 |
| Dihedral angle restraints(CBD + MBD) | |
| Phi | 160 |
| Psi | 160 |
| Statistics for structures | |
| Final Energies (kcal/mol) | |
| van der Waals (kcal/mol) | −447.129 ± 20.737 |
| NOE (kcal/mol) | 1.61991 ± 0.8470 |
| Violations | |
| Number of NOE violations > 0.5Å | 0 ± 0 |
| R.m.s. deviation (Å) from experimental distance restraints | 0.0323 ± 0.0094 |
| Number of dihedral angle constraint violations > 5° | 0 ± 0 |
| R.m.s. deviation (°) from experimental torsion restraints | 0.3751 ± 0.0795 |
| Deviations from idealized geometry | |
| Bonds (Å) | 0.00344 ± 0.0001 |
| Angles (°) | 0.40165 ± 0.0200 |
| Improper (°) | 0.47657 ± 0.0223 |
| Structural RMSD to the mean coordinate | |
| region (residue number) | bb/heavy (Å) |
| 708–821,411–440 | 3.922/4.171 |
| 718–737,745–756,763–780,787–789,811–813,421–433 | 0.764/1.345 |
| Ramachandran plot (% residues) | |
| Residues in most favored regions | 80.5 |
| Residues in additional allowed regions | 16.10 |
| Residues in generously allowed regions | 2.10 |
| Residues in disallowed regions | 1.30 |
Binding studies
To investigate the ligand binding, the 2D 1H-15N-HSQC spectra were recorded on uniformly 15N-labeled hMBD (∼0.2 mM) in the presence of different concentrations of hCBD ranging from 0 to 1.5 mM. Both the hMBD sample and the stock solutions of hCBD were prepared in the NMR buffer (50 mM sodium phosphate, 50 mM NaCl, 5% glycerol and 1 mM EDTA, pH 7.5). The chemical shift perturbation between the free-form and hCBD-bound hMBD was normalized by the following formula and expressed in ppm:
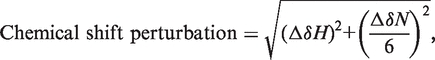 |
where ΔδH and ΔδN are the differences in chemical shifts of amide protons and nitrogen between the initial and final data points of the titration, respectively.
Site-directed mutagenesis study of the interaction between hCBD–Mcm6 and hMBD–Cdt1
The binding residues were confirmed by site-directed mutagenesis studies based on the information from the chemical shift perturbation experiment. The point mutants A414G, R425A, I426A, R427A, K429A, K433A, Q434A, L435A and Q437A of hCdt1 were expressed in E. coli and purified to homogeneity.
Strains, plasmids and antibodies
The strains and plasmids used in this study were described in Tables 2 and 3. Anti-Orc3 and anti-Mcm2 antibodies were kind gifts from B. Stillman. Anti-HA antibodies were purchased from Roche.
Table 2.
Yeast strains
| Strain | Genotype |
|---|---|
| YL135 | W303-1a mcm6-td::URA3 ubr1Δ::GAL-UBR1::HIS3 |
| YL1208 | W303-1a cdt1-td ubr1Δ::GAL-UBR1::HIS3 leu2::pCM244x3 cdc54Δmcm4-GFP::TRP1 |
Table 3.
Plasmids
| Plasmid | Description |
|---|---|
| PL1808 | pGBKT7-Cdt1 |
| PL1843 | pGBKT7- Cdt1-3A(R486A, L487A, R490A) |
| PL1815 | pGADT7-Mcm6 |
| PL1850 | pGADT7-Mcm6-5A(E945A, D947A, L951A, E953A, Y954A) |
Cell cycle synchronization and flow cytometry
Cell cycle block and release with α-factor or nocodazole were carried out as described previously (32). Flow cytometry was performed as previously described (33). mcm6-td cells (S. cerevisiae) expressing yeast AD-Mcm6 or AD-Mcm6-5A and cdt1-td cells (S. cerevisiae) expressing yeast BD-Cdt1 or BD-Cdt1-3A were synchronized in M phase before being shifted to 37°C for 1 h to degrade the Mcm6-td or Cdt1-td protein. Cells were then released into fresh medium containing α-factor (α-F) at 37°C for 2 h and then released into fresh medium at 37°C.
Chromatin binding assay
The chromatin-binding assay to examine chromatin-associated proteins was performed as previously described (33). mcm6-td cells (S. cerevisiae) expressing yeast AD-Mcm6 or AD-Mcm6-5A were synchronized in M phase by nocodazole (Noc) and then shifted to 37°C for 1 h to deplete the Mcm6-td protein before being released into G1 phase in fresh medium containing α-factor at 37°C for 2 h. cdt1-td cells containing the plasmid expressing BD-Cdt1 or BD-Cdt1-3A were synchronized in M phase and then shifted to 37°C for 1.5 h before being released into fresh medium containing α-factor at 37°C for 2 h. Samples were taken at 30-min intervals. The chromatin fraction (Chr.) and whole cell extracts (WCE) from the cells were immunoblotted.
Data deposition
Atomic coordinate has been deposited at Protein Data Bank for the Cdt1–Mcm6 complex (code-2LE8).
RESULTS
Mapping the hMcm6 binding sites and determining the structure of hMBD
To circumvent the challenge of studying the large MCM2–7 and Cdt1 protein assembly by X-ray and NMR techniques, we conducted an extensive mapping of the interaction domains of the human MCM–Cdt1 complex. We showed that Cdt1 specifically interacts with Mcm6 (also confirmed in yeast) through Cdt1 410–445, the human Mcm6-binding domain of Cdt1 (hMBD) and Mcm6 708–821, the human Cdt1-binding domain of Mcm6 (hCBD) (23). To determine the structure of the interacting domains of the human MCM6 and Cdt1 complex (hCBD/hMBD), we first determined the individual structures of hCBD and hMBD by NMR spectroscopy. We previously reported the structure of hCBD (23). In this study, we determined the hMBD structure, which adopts an amphipathic α-helical conformation (Figure 1a and b) containing residues 421–432 (Supplementary Table S1). NMR titration experiments (Figure 2) showed that residues of hMBD that displayed large chemical shift changes (>0.08 ppm) corresponded to amino acids on one side of the amphipathic helix formed by residues Asp421, Glu424, Ala428, Lys429, Glu430, Gln432, Lys433 (Figures 1a and b and 2), while residues Leu422, Leu423, Ile426 and Arg427 completely disappeared in the titrated HSQC spectra.
Figure 1.

Solution structure of hMBD of human Cdt1 and the Mcm6-Cdt1 complex. (a) The left panel shows the backbone superposition of the 20 lowest-energy NMR structures of hMBD. The α-helix is colored red. N-terminal and C-terminal ends are indicated as N and C. The right panel shows a ribbon representation of the same structure of hMBD using the coordinates of the lowest energy structure. (b) Chemical shift perturbations in the presence of hCBD are colored onto the structure of hMBD in the surface representation. Residues with chemical shift perturbations ranging from 0.00 to 0.08 ppm are colored in green while residues with chemical shift perturbations larger than 0.08 ppm are shown in red and residues disappeared in HSQC spectrum are in blue. (c) Backbone superposition of the 19 lowest-energy NMR structures. Secondary structural elements of hCBD are color-coded: α-helices (red), β-strands (green), and loops (gray). hMBD is shown in blue. N-terminal and C-terminal ends are indicated as N and C. (d) Ribbon diagram of the complex using the coordinates of the lowest energy structure. (e) Expanded view of the complex binding surface. Residues having intermolecular NOEs are shown in sticks, yellow for hCBD and green for hMBD. The backbones of hCBD and hMBD are colored gray and blue respectively. (f) Surface representation of hCBD colored by residue type: red, acidic; blue, basic; yellow, hydrophobic; gray, non-interacting. hMBD is indicated as in (e) except that the backbone is in purple.
Figure 2.

NMR studies of the interaction between hMBD and hCBD. (a) Overlays of 1H-15N HSQC spectra of 15N-labeled hMBD in free form (black) titrated with hCBD at a molar ratio of 1:3 (blue), 1:6 (red). The arrows point to the shifted positions (black to red) of the amide proton resonance. (b) Chemical shift differences between the free-form and hCBD saturated hMBD. NMR titration experiments show an intermediate exchange kinetics indicating a relatively strong binding between hCBD and hMBD.
Three-dimensional structure of the hCBD–hMBD complex
To fully characterize this interaction at the atomic level, we determined the structure of the hCBD–hMBD complex which is well-defined by the NMR data (described in details under ‘Materials and Methods’ section). The 19 lowest energy structures are characterized by good backbone geometry, with no significant restraint violation, and low pairwise rmsd values (Table 1). The structure of hCBD in the complex presents a typical winged helix–turn–helix (HTH) fold as previously reported: the canonical three-helix bundle is packed against two short antiparallel β strands. The core region of the hMBD peptide (residues 421–432) adopts a well-defined amphipathic α-helical conformation, whereas the N- and C-terminal flanking residues are flexible and do not display a defined secondary structure (Figure 1c and d). The orientation of the peptide is defined by unambiguous intermolecular NOEs (Figure 3) and the structure of hCBD was validated by residual dipolar couplings (RDC) (Supplementary Figure S1). The size of the protein–peptide binding interface is ≈1260 Å2, indicating relative strong binding, which is consistent with the NMR titration experiment showing an intermediate binding.
Figure 3.

Intermolecular NOEs between hCBD and hMBD. The left two strips of a 3D F1 13C,15N-filtered, F2 13C-edited NOESY-HSQC spectrum (150 ms mixing time) of a sample containing 13C,15N-labeled hCBD and unlabeled hMBD showing NOEs from the hMBD (I426, L422, the ambiguous assignment is colored by blue) to the hCBD (K770, I767). The right strip of the same experiment of a sample containing 13C, 15N-labeled hMBD and unlabeled hCBD showing NOEs from the hCBD (L766, K770) to the hMBD (L422). CD1: the ε1 carbon atom of the Isoleucine methyl group; CD#: degenerate pairs of Leucine methyl carbon atoms; CE: the ε carbon of the Lysine; HA: the α proton attached to α carbon; QG: degenerate pairs of γ methylene protons; HG: the γ proton of Leucine; QG1: degenerate pairs of Isoleucine γ1 methylene protons; QD: degenerate pairs of δ methylene protons; QE: degenerate pairs of ε methylene protons; QD1: degenerate pairs of Isoleucine δ1 methyl protons; QQD: degenerate pairs of Leucine methyl protons.
Important interactions at the hCBD–hMBD interface
The binding interface of the hCBD–hMBD complex comprises the helix-turn-helix region formed by helix α2, loop2 and helix α3 of hCBD. The hMBD binding site on hCBD is characterized by a hydrophobic surface formed by Ile760, Leu766, Ile767 that engages the hydrophobic residues Leu423, Ile426 and makes van der Waals contacts with the side chains of Lys429 and Lys433 of hMBD. Additionally the basic residues Lys429 and Lys433 of hMBD make a close contact with the acidic residues E757 and E760 of hCBD, suggesting that charge complementarity is a key determinant of the observed hCBD–hMBD interactions (Figure 1e and f).
Mutagenesis studies of the interaction between hMBD and hCBD
To investigate the structural requirements for the binding of hMBD peptides by independent means, we identified critical residues in the respective interaction motifs by mutational analysis. Guided by the observation in the chemical shift perturbation experiment, we performed site-directed mutagenesis at several amino acid residues of hMBD which are expected to interfere with the binding between hCBD and hMBD. These point mutant peptides A414G, R425A, I426A, R427A, K429A, K433A, Q434A, L435A and Q437A were expressed and purified to homogeneity. However, L422A could not be expressed in E. coli. Pulldown assay (Figure 4a, Supplementary Figure S2A) showed that changing residues R425, I426, K429 and K433 to alanines significantly decreased the binding affinity as predicted from the structure. We noted that the interaction is disrupted for the R425A mutant although R425 does not have direct contact with the hMBD in the hCBD/hMBD tertiary structure. We believe that there is a conformational change in the R425A mutant, disrupts the interaction between hCBD and hMBD. On the basis of our previous mutational studies, the residues of hCBD E757, E763 and L766 are crucial for the interaction at the binding interface of the complex. Pulldown assays were also performed using the hMBD mutants. The charged residues R425, R427, K429 and K433 were replaced with neutral (M) and opposite-charged (E) amino acids of similar side chains. The similar results obtained further support that charge complementarity is the key determinant (Supplementary Figure S2B and C) of the Cdt1–Mcm6 interaction. Together, these data demonstrate that the interaction between hCBD and hMBD is highly specific.
Figure 4.

Disruption of the Cdt1–Mcm6 interaction by mutations on their interacting surfaces impaired pre-RC formation and DNA replication. (a) Wild-type and mutants of hMBD were pulled down by GST-tagged wild-type hCBD and visualized by Coomassie Blue staining. (b)Chromatin loading of Mcm2 (S. cerevisiae) is interrupted by the Mcm6-5A mutant (Supplementary Material). (c) Chromatin loading of Mcm2 (S. cerevisiae) is interrupted by the Cdt1-3A mutant (Supplementary Data). (d) Mcm6-5A cells arrest in S-phase with large buds (S. cerevisiae) (Supplementary Data). Flow cytometry was performed for the cell samples taken at the indicated time points. % Bud. percentage of budding cells. (e) Cdt1-3A cells arrest in S-phase as in (d).
The interaction between hMBD and hCBD is crucial for pre-RC assembly and DNA replication
To investigate the biological significance of the Cdt1–Mcm6 interaction through the residues identified by NMR, we disrupted the Cdt1–Mcm6 interaction by introducing the corresponding mutations on the highly conserved MBD–CBD interacting surface in the budding yeast (Figure 5). We constructed yeast mutants bearing the combined E945A, D947A, L951A, E953A and Y954A mutations in the CBD (Mcm6-5A) and the combined R486A, L487A and R490A, alleles in the MBD (Cdt1-3A) (Figure 5). GST retention assay showed that each of the interface surface mutant impaired the interaction between hCdt1 and hMcm6 as predicted by the structure (Figures 4a and 5, Supplementary Figure S2A and B) and failed to support cell proliferation (Figure 4d and e). Yeast two-hybrid analysis also showed that Mcm6-5A and Cdt1-3A did not interact with their corresponding wild-type partners (data not shown).
Figure 5.

Alignments of the Cdt1 and Mcm6 sequences from different organisms. (a) Multiple sequence alignment of a representative set of Mcm6 proteins: Q14566, H. sapiens; P97311, Mouse; BAF94254, Rattus norvegicus; Q5FWY4, XENLA; Q9V461, D. melanogaster; P4973, S. pombe; P53091, S. cerevisiae. The dashes indicate the positions of gaps in eukaryotic sequences. Secondary structural elements at the top of the alignment are indicated with color coding as in Figure 1a and b. (b) Multiple sequence alignment of a representative set of the C-terminal regions of Cdt1: Q9H211, H. sapiens; Q8R4E9, Mouse; D3ZKD4, Rattus norvegicus; Q9I9A7, XENLA; Q7JVY2; D. melanogaster; P40382, S. pombe; P47112, S. cerevisiae. The sequence alignment was produced with ClustalX(34). Blue arrows indicate the amino acids that greatly reduce the interaction between Mcm6 and Cdt1 upon Ala substitution. Residues are gray-scaled based on percentage identity.
The activation domain-Mcm6 fusion protein (AD-Mcm6) and the DNA binding-domain-Cdt1 fusion protein (BD-Cdt1) expressed from the yeast two-hybrid plasmids are biologically functional. We expressed AD-Mcm6 and AD-MCM6-5A in the mcm6-td (td, temperature-inducible degron) cells, and BD-Cdt1 and BD-Cdt1-3A in cdt1-td cells to examine the effects of Mcm6-5A and Cdt1-3A on cell viability, pre-RC formation and DNA replication. To investigate if the Cdt1–Mcm6 interaction is essential for the assembly of pre-RC, we examined the chromatin association of pre-RC proteins during the M-to-G1 transition in cells expressing the AD-Mcm6-5A mutant at 37°C when the Mcm6-td protein is depleted. The results showed that in cells expressing the wild-type AD-Mcm6, Mcm2 was loaded onto chromatin at 30 min after release from the M-phase block (Figure 4b). In contrast, AD-Mcm6-5A did not support pre-RC formation as shown by the absence of Mcm2 on chromatin in G1 phase cells (Figure 4b). Similarly, BD-Cdt1 but not BD-Cdt1-3A supported pre-RC formation (Figure 4c).
To investigate if Mcm6-5A is defective for DNA replication, the DNA content of mcm6-td cells expressing AD-Mcm6 or AD-Mcm6-5A after G1 arrest-and-release was studied. The cells expressing AD-Mcm6 replicated their DNA normally after the Mcm6-td protein was depleted (Figure 4d). In contrast, cells expressing the AD-Mcm6-5A mutant did not replicate their DNA as evident from their failure to traverse S phase, even though budding progressed normally (Figure 4d). Similar results were obtained in Cdt1-depleted cells expressing BD-Cdt1-3A (Figure 4e). Therefore, mutations on the interface surface that disrupted the Cdt1–Mcm6 interaction prevented pre-RC formation, DNA replication and cell proliferation. Together, these results demonstrated that the residues identified from the hMBD–hCBD complex structure indeed mediate MCM2–7 chromatin loading through the C-terminal interaction of Cdt1 and Mcm6.
DISCUSSION
In this study, we determined the atomic structure of the conserved C-terminal domain of human Mcm6 in complex with the C-terminal region of Cdt1 by NMR spectroscopy and identified the residues which are crucial for pre-RC assembly in DNA replication. Our structural and functional analyses in conjunction with previous studies showed that Mcm6–Cdt1 interaction played a major role in Cdt1-mediated MCM2–7 chromatin loading, a critical and precisely regulated event during the initiation of replication in eukaryotes.
Structural studies showed that the C-terminus of human Cdt1, the human Mcm6 binding domain (hMBD), adopts an amphipathic α-helical conformation in the hCBD–hMBD complex. The hCBD bears a winged-helix fold that consists of a helix-turn-helix (HTH) motif followed by the wing (β–loop–β) motif. The hCBD binds to one side of hMBD through the HTH region (helix α2—loop2—helix α3). Structural analysis on the binding surface show that residues Ile760, Leu766 and Ile767 of hCBD make hydrophobic contributions to the complex through interaction with residues Leu423, and Ile426 of hMBD. In addition, the acidic residues of E757 and E760 of hCBD engage in close contacts with the side chains of Lys429 and Lys433 of hMBD, suggesting that charge complementarity is essential for the hCBD–hMBD interactions.
To verify the binding mode for the observed interactions in hCBD–hMBD complex, the structure-based site-directed mutagenesis study was performed on the residues located on the binding surface. The GST pull-down assay indicates that the point mutant R425A, I426A, K429A and K433A significantly decreased the binding affinity. Previous studies showed that point mutations of residues Leu766, E757 and E760 on hCBD significantly reduced the binding between hCBD and hMBD. In our previous work we found that the interaction between human Mcm6 and Cdt1 is much stronger than that between Mcm2 and Cdt1 (23). Based on the alignment of the WH domains of the MCM proteins (Supplementary Figure S3), the charged residues of Mcm6 required for Cdt1 binding (E757 and E760) are not conserved among the MCM proteins, consistent with the observation that Cdt1 only interacts strongly with Mcm6. These results together confirmed the strong and specific interactions in hCBD–hMBD complex.
Disrupting the Cdt1–Mcm6 interaction in budding yeast by the Cdt1-3A or Mcm6-5A mutants prevented pre-RC formation and resulted in DNA replication defect. The sequence conservation of these two domains suggests that the mutated residues play an important and evolutionarily conserved role in the loading of the MCM2–7 complex during DNA replication licensing in eukaryotes. The conserved binding residues among different species underscore the biological significance of this interaction.
Current information suggests that the MCM2–7 helicase is recruited to ORC by tethering Cdt1 to Orc6 during pre-RC formation (35). The spacing and orientation of the Cdt1–Mcm6 interaction domains determined here provide further insight into the loading mechanism of the MCM2–7 helicase. Because Cdt1 interacts with Mcm6 via their C-terminal regions, the MCM2–7 complex must be loaded with its C-terminal end facing ORC (Figure 6). Furthermore, the position of the Cdt1 binding site on the extreme C-terminal region of the MCM2–7 hexamer suggests that the two Cdt1-binding sites on a single Orc6 are too close to accommodate the independent loading of two MCM2–7 hexamers by two Orc6-bound Cdt1 (36). The spatial constraint suggested by our structure supports alternative loading mechanisms such as the loading of the head-to-head MCM2–7 double hexamer by a single Cdt1 or the independent loading of the MCM2–7 hexamer by two Cdt1 associated with a dimeric ORC.
Figure 6.

Proposed model for the Cdt1/MCM2-7 recruitment and MCM hexmer loading events. The Cdt1 functions as the bridge to bring Orc6 and MCM2–7 together to form pre-RC. The C-terminal Mcm6-binding domain of Cdt1 tethers MCM2–7, while the N-terminal Orc6-binding domain recruits Cdt1/MCM2-7 to ORC complex.
Structural information related to the MCM protein family is accumulating, including the crystal structures of the N-terminal domains of MthMCM (10) and SsoMCM (37), the low resolution three-dimensional electron microscopy (EM) reconstructions of the full-length MthMCM (38–40) and the near full length SsoMCM (41) as well as the EM structures of MCM2–7 complex of yeast proteins (11,12) and Drosophila melanogaster (7). Numerous structural studies on the key residues and the relevant region of Cdt1 that bind the MCM proteins have been reported (42,43). However, none of these studies provided detailed structural information on the Cdt1-mediated MCM2–7 chromatin loading. We believe that the complex structure presented here uniquely provides mechanistic insights for the Cdt1-mediated MCM2–7 chromatin loading.
In conclusion, we present the solution structure of hCBD/hMBD complex by NMR spectroscopy and identified the interacting residues of Mcm6 and Cdt1 essential for the chromatin loading of the MCM2–7 hexamer in the initiation of DNA replication. Our results provide in molecular detail a structural basis for the essential role of Cdt1 in loading the MCM2–7 complex onto chromatin during pre-RC assembly.
ACCESSION NUMBER
PDB code: 2LE8
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online: Supplementary Table 1, Supplementary Figures 1–3, and Supplementary references [44].
FUNDING
Funding for open access charge: Hong Kong Research Grants Council [RGC664109 and RGC663911], AoE/M-06/08, TUYF10SC03; and HKUST Research Fund.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Remus D, Diffley JF. Eukaryotic DNA replication control: lock and load, then fire. Curr. Opin. Cell. Biol. 2009;21:771–777. doi: 10.1016/j.ceb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 3.Diffley JFX. Regulation of early events in chromosome replication. Curr. Biol. 2004;14:778–786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 4.Maiorano D, Moreau J, Mechali M. XCDT1 is required for the assembly of pre-replicative complexes in X. laevis. Nature. 2000;404:622–625. doi: 10.1038/35007104. [DOI] [PubMed] [Google Scholar]
- 5.Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- 6.Masai H, Sato N, Takeda T, Arai K. CDC7 kinase complex as a molecular switch for DNA replication. Front Biosci. 1999;4:834–840. doi: 10.2741/masai. [DOI] [PubMed] [Google Scholar]
- 7.Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011;18:471–477. doi: 10.1038/nsmb.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maine GT, Shiha P, Tye BK. Mutants of S. Cerevisiae. defective in the maintenance of minichromsome. Genetics. 1984;106:356–385. doi: 10.1093/genetics/106.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 10.Fletcher RJ, Bishop BE, Leon RP, Sclafani RA, Ogata CM, Chen XS. The structure and function of MCM from archaeal M. thermoautotrophicum. Nat. Struct. Biol. 2003;10:160–167. doi: 10.1038/nsb893. [DOI] [PubMed] [Google Scholar]
- 11.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl Acad. Sci. 2009;106:20240–20245. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of MCM2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719–730. doi: 10.1016/j.cell.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davey MJ, Indiani C, O'Donnell M. Reconstitution of the MCM2–7p heterohexamer, subunit arrangement, and ATP site architecture. J. Biol. Chem. 2003;278:4491–4499. doi: 10.1074/jbc.M210511200. [DOI] [PubMed] [Google Scholar]
- 14.Schwacha A, Bell SP. Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol. Cell. 2001;8:1093–1104. doi: 10.1016/s1097-2765(01)00389-6. [DOI] [PubMed] [Google Scholar]
- 15.Bochman ML, Schwacha A. The MCM2-7 complex has in vitro helicase activity. Mol. Cell. 2008;31:287–293. doi: 10.1016/j.molcel.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 16.Ferenbach A, Li A, Brito-Martins M, Blow JJ. Functional domains of the Xenopus replication licensing factor Cdt1. Nucleic Acids Res. 2005;33:316–324. doi: 10.1093/nar/gki176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arentson E, Faloon P, Seo J, Moon E, Studts JM, Fremont DH, Choi K. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene. 2002;21:1150–1158. doi: 10.1038/sj.onc.1205175. [DOI] [PubMed] [Google Scholar]
- 18.Tatsumi Y, Sugimoto N, Yugawa T, Narisawa-Saito M, Kiyono T, Fujita M. Deregulation of Cdt1 induces chromosomal damage without rereplication and leads to chromosomal instability. J. Cell. Sci. 2006;119:3128–3140. doi: 10.1242/jcs.03031. [DOI] [PubMed] [Google Scholar]
- 19.Seo J, Chung YS, Sharma GG, Moon E, Burack WR, Pandita TK, Choi K. Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene. 2005;24:8176–8186. doi: 10.1038/sj.onc.1208881. [DOI] [PubMed] [Google Scholar]
- 20.Yanagi K, Mizuno T, You Z, Hanaoka F. Mouse geminin inhibits not only Cdt1-MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J. Biol. Chem. 2002;277:40871–40880. doi: 10.1074/jbc.M206202200. [DOI] [PubMed] [Google Scholar]
- 21.Teer JK, Dutta A. Human Cdt1 lacking the evolutionarily conserved region that interacts with MCM2-7 is capable of inducing re-replication. J. Biol. Chem. 2008;283:6817–6825. doi: 10.1074/jbc.M708767200. [DOI] [PubMed] [Google Scholar]
- 22.You Z, Masai H. Cdt1 forms a complex with the minichromosome maintenance protein (MCM) and activates its helicase activity. J. Biol. Chem. 2008;283:24469–24477. doi: 10.1074/jbc.M803212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Z, Liu C, Wu X, Xu N, Zhou B, Liang C, Zhu G. Characterization and structure determination of the Cdt1 binding domain of human minichromosome maintenance (MCM) 6. J. Biol. Chem. 2010;285:12469–12473. doi: 10.1074/jbc.C109.094599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Zhu G, Bax A. Improved linear prediction of truncated damped sinusoids using modified backward-forward linear prediction. J. Magn. Reson. 1992;100:202–207. [Google Scholar]
- 26.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:93–158. [Google Scholar]
- 27.Zwahlen C, Legault P, Vincent SJF, Greenblat J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage λ N-peptide/boxB RNA complex. J. Am. Chem. Soc. 1997;119:6711–6721. [Google Scholar]
- 28.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography and NMR System (CNS), 1.1 edn. Yale University, New Haven, CT; 2001. [DOI] [PubMed] [Google Scholar]
- 30.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 32.Labib K, Kearsey SE, Diffley JF. MCM2-7 proteins are essential components of prereplicative complexes that accumulate cooperatively in the nucleus during G1-phase and are required to establish, but not maintain, the S-phase checkpoint. Mol. Biol. Cell. 2001;12:3658–3667. doi: 10.1091/mbc.12.11.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Yu L, Wu X, Zou L, Sou KK, Wei Z, Cheng X, Zhu G, Liang C. The interacting domains of hCdt1 and hMCM6 involved in the chromatin loading of the MCM complex in human cells. Cell Cycle. 2010;9:4848–4857. doi: 10.4161/cc.9.24.14136. [DOI] [PubMed] [Google Scholar]
- 34.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S, de Vries MA, Bell SP. Orc6 is required for dynamic recruitment of Cdt1 during repeated MCM2-7 loading. Genes. Dev. 2007;21:2897–2907. doi: 10.1101/gad.1596807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Bell SP. CDK prevents MCM2-7 helicase loading by inhibiting Cdt1 interaction with Orc6. Genes. Dev. 2011;25:363–372. doi: 10.1101/gad.2011511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu W, Pucci B, Rossi M, Pisani F, Ladenstein R. Structural analysis of the Sulfolobus solfataricus MCM protein N-terminal domain. Nucleic Acids Res. 2008;36:3235–3243. doi: 10.1093/nar/gkn183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pape T, Meka H, Chen S, Vicentini G, van Heel M, Onesti S. Hexameric ring structure of the full-length archaeal MCM protein complex. EMBO Rep. 2003;4:1079–1083. doi: 10.1038/sj.embor.7400010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez-Llorente Y, Fletcher RJ, Chen XS, Carazo JM, San Martin C. Polymorphism and double hexamer structure in the archaeal minichromosome maintenance (MCM) helicase from Methanobacterium thermoautotrophicum. J. Biol. Chem. 2005;280:40909–40915. doi: 10.1074/jbc.M509760200. [DOI] [PubMed] [Google Scholar]
- 40.Costa A, van Duinen G, Medagli B, Chong J, Sakakibara N, Kelman Z, Nair SK, Patwardhan A, Onesti S. Cryo-electron microscopyreveals a novel DNA-binding site on the MCM helicase. EMBO J. 2008;27:2250–2258. doi: 10.1038/emboj.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brewster AS, Wang G, Yu X, Greenleaf WB, Carazo JM, Tjajadia M, Klein MG, Chen XS. Crystal structure of a near-full-length archaeal MCM: functional insights for an AAA+ hexameric helicase. Proc. Natl Acad. Sci. 2008;105:20191–20196. doi: 10.1073/pnas.0808037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khayrutdinov BI, Bae WJ, Yun YM, Lee JH, Tsuyama T, Kim JJ, Hwang E, Ryu KS, Cheong HK, Cheong C, et al. Structure of the Cdt1 C-terminal domain: conservation of the winged helix fold in replication licensing factors. Protein Sci. 2009;18:2252–2264. doi: 10.1002/pro.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jee J, Mizuno T, Kamada K, Tochio H, Chiba Y, Yanagi K, Yasuda G, Hiroaki H, Hanaoka F, Shirakawa M. Structure and mutagenesis studies of the C-terminal region of licensing factor Cdt1 enable the identification of key residues for binding to replicative helicase MCM proteins. J. Biol. Chem. 2010;285:15931–15940. doi: 10.1074/jbc.M109.075333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in dilute liquid crystalline medium of outstanding interest. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.