


Abstract
The Differential Radial Capillary Action of Ligand Assay (DRaCALA) allows detection of protein interactions with low-molecular weight ligands based on separation of the protein–ligand complex by differential capillary action. Here, we present an application of DRaCALA to the study of nucleic acid–protein interactions using the Escherichia coli cyclic AMP receptor protein (CRP). CRP bound in DRaCALA specifically to 32P-labeled oligonucleotides containing the consensus CRP binding site, but not to oligonucleotides with point mutations known to abrogate binding. Affinity and kinetic studies using DRaCALA yielded a dissociation constant and dissociation rate similar to previously reported values. Because DRaCALA is not subject to ligand size restrictions, whole plasmids with a single CRP-binding site were used as probes, yielding similar results. DNA can also function as an easily labeled carrier molecule for a conjugated ligand. Sequestration of biotinylated nucleic acids by streptavidin allowed nucleic acids to take the place of the protein as the immobile binding partner. Therefore, any molecular interactions involving nucleic acids can be tested. We demonstrate this principle utilizing a bacterial riboswitch that binds cyclic-di-guanosine monophosphate. DRaCALA is a flexible and complementary approach to other biochemical methods for rapid and accurate measurements of affinity and kinetics at near-equilibrium conditions.
INTRODUCTION
The Differential Radial Capillary Action of Ligand Assay (DRaCALA) was recently shown to allow rapid and quantitative analysis of protein–ligand interactions and permit high-throughput identification of receptors for the bacterial second messenger cyclic-di-guanosine monophosphate (cdiGMP) (1). In this assay, mixtures of protein and labeled ligand at equilibrium are spotted on dry nitrocellulose. By capillary action, free ligand moves radially outward from the initial spot while the proteins and bound ligands are immobilized by hydrophobic interactions with the nitrocellulose membrane. This allows differentiation of bound and unbound ligand based on its mobility. The advantage of DRaCALA over the traditional filter-binding assay is that the total amount of ligand in every sample is measured because there is no wash step. The speed of DRaCALA allows kinetic measurements at near-equilibrium conditions. The visual output of the method allows rapid assessment of molecular interactions. Quantitative measurements of protein–ligand interaction, such as fraction bound, can be readily calculated from measurements of four parameters: the total area, the total intensity, the sequestered area and the sequestered intensity (1). Previous studies have shown that DRaCALA can accurately measure protein–ligand interactions for purified proteins and whole-cell extracts expressing recombinant proteins. The simplicity of DRaCALA gives it potential for general applicability. Ligand mobility in DRaCALA is a necessity, but the possibility that ligands partition out of the mobile liquid phase during capillary action, and are therefore not mobile, has not yet been investigated.
Because mononucleotides and dinucleotides have been shown to be mobile, it is reasonable to expect that double stranded DNA would be mobile as well. This led us to apply the method to DNA–protein interactions using the well-characterized interaction between E. coli cyclic AMP receptor protein (CRP) and its DNA binding site ICAP. CRP is a transcription factor that has regulatory function at approximately 200 sites on the E. coli genome (2–4). CRP binds cAMP and cGMP (5), but DNA binding and transcriptional activation by CRP is solely dependent on cAMP binding (6). A 28-bp symmetrical synthetic consensus sequence, called ICAP, binds CRP with the greatest affinity (7). Through filter-binding assays, the affinity of the CRP–ICAP interactions and the contributions of specific nucleotides (such as guanines at positions 8 and 10 and the cytosines at positions 19 and 21) have been defined (8).
In this study, DRaCALA is shown to allow quantification of CRP–ICAP interactions using 32P-end-labeled oligonucleotides. Specificity of binding and competition studies were performed to establish this proof of principle. Furthermore, the method was used to obtain measurements of both affinity and kinetics. Much larger DNA probes derived from whole plasmids were tested in the same way. DNA could function as a carrier molecule for studying interactions between a protein and a molecule covalently linked to DNA. This also allows easy indirect 32P-labelling of molecules that are more difficult to label than DNA. Finally, immobilization of nucleic acids with the biotin-streptavidin system is shown to allow study of small molecule interactions with RNA (riboswitches). We show here the different ways DRaCALA can be used to study molecular interactions with nucleic acids including protein–nucleic acid and riboswitch-small ligand interactions.
METHODS
Proteins, nucleic acids and chemicals
The Vc2* DNA template was ordered from Integrated DNA Technologies (IDTs). Other DNA oligonucleotides, NucAway size exclusion columns and Turbo DNase were from Invitrogen. RNase was from Fermentas. RNase inhibitor and enzymes for restriction digests, PCR and other nucleic acid manipulations were from New England Biolabs. Streptavidin MagneSphere Paramagnetic Particles, Wizard miniprep and PCR Purification kits for DNA purification were from Promega. Biotin hydrazide and streptavidin were from Sigma Aldrich.
CRP was purified as described with the addition of dithiothreitol (DTT) to the dialysis buffer (1). Briefly, His-CRP (CRP) was expressed from pBAD-CRP (a gift from Dr Sankar Adhya) and purified using a Ni-NTA column. Proteins were dialyzed in 10 mM Tris, pH 8.0 and 100 mM NaCl and 1 mM DTT. His-CRP was subsequently purified and concentrated using cation exchange to a concentration of 36 µM, frozen in liquid nitrogen and stored at −80°C until thawing for use. The fraction of active CRP molecules in sequence-specific DNA binding (0.61) was determined by the titration of DNA fragment ICAP under stoichiometric binding conditions. Specifically, serial dilutions of CRP were incubated with a concentration of 32P ICAP dsDNA in excess of the Kd (200 nM) for 10 min at room temperature prior to determination of binding by DRaCALA and electrophoretic mobility shift assay (EMSA). All data are reported in terms of molar concentrations of active CRP dimers.
Preparation and activity of DNA oligonucleotides and plasmid probes
Reverse complementary oligonucleotides gd126-133 and vl1427-1428 (Supplementary Table S1) were used to generate probes by labeling 5 pmol of the forward primer with T4 Polynucleotide Kinase (PNK) and 15 pmol/5 mCi of γ-32P-labeled ATP. Five pmol of the reverse complementary primer were added and the PNK was heat inactivated during primer annealing in 80°C water bath for 10 min, which was then allowed to cool to room temperature >1 h. The annealed product was separated from free 32P-ATP using a NucAway column and diluted 1 : 10 for binding and competitions studies and 1 : 1000 for affinity and kinetics studies. Plasmids with binding sites were generated by cloning annealed, PNK-treated primers pairs (kr122-129 of Supplementary Table S1) into StuI-cut pVL-Blunt and sequencing for verification (Supplementary Table S2). Plasmids were 5′-end-labeled by sequential digestion with the single cutter BamHI, dephosphorylation of the 5′ overhang with Calf intestinal alkaline phosphatase, separation from enzymes by a Wizard PCR Purification column, and treatment with PNK in the presence of γ-32P-labeled ATP. The labeled product was purified by the Wizard column and a NucAway column and diluted 1 : 10 for affinity and kinetic study. The near 5′-end of these labeled plasmids is ∼40 bp from the cloned binding sites. Competitors for plasmid binding were PCR amplified from these plasmids using primers vl880–vl881, which amplify the cloned binding sites and 250-bp flanking on each side (Supplementary Table S1). The concentration of the ICAP probes was determined by NanoDrop 1000. The fraction of ICAP that is active for binding is determined by measuring the maximum fraction of 32P-labeled ICAP in excess of Kd (200 nM) that could be specifically bound by excess CRP by both EMSA and DRaCALA. The fraction was multiplied by concentration of ICAP to yield the concentration of ICAP that is active.
DRaCALA
Protein, 32P-labeled DNA and 200 µM cAMP (unless otherwise noted) were mixed in CRP buffer (10 mM Tris, pH 7.9, 200 mM NaCl, 0.1 mM DTT and 50 µg/ml bovine serum albumin) (7) and incubated at room temperature for 10 min. Five microliters of the mix was spotted on nitrocellulose by first pipetting the liquid out onto the tip of the pipette and then touching the drop to the membrane. Spots were allowed to dry completely (∼20 min) before exposing a phosphorimager screen and capturing with a Fujifilm FLA-7000. Photostimulated luminescence (PSL) from the inner spot and total PSL of the spot were quantitated with Fuji Image Gauge software. The fraction bound (FB) (1) was calculated using measurements of the total area (Aouter), the area of the inner circle (Ainner), the total PSL intensity (Itotal) and the inner intensity (Iinner) as follows:
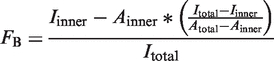 |
EMSA for CRP binding to ICAP oligonucleotide
A fraction of the same mix described for the DRaCALA assay is supplemented with 10% glycerol and 0.001% bromophenol blue and 5 µl was loaded onto an 8% 70 : 1 0.5 × TBE (90 mM Tris, pH 8.0, 90 mM H3BO3, 2.5 mM Na2 ethylenediaminetetraacetic acid (EDTA)) polyacrylamide gel (9). EMSA was preformed with 0.5× TBE buffer at 4°C. Gels were exposed to a phosphorimager screen and the image was captured with a Fujifilm FLA-7000. The fraction bound was calculated as the intensity of the bound ICAP probe as a fraction of the total intensity of the bound and free ICAP probe.
Non-radioactive ligands and detection
Fluorescent dyes were imaged with a GE Typhoon Trio. TNP was detected with electrochemiluminescence excitation at 555 nm emission. Fluorescein isothiocyanate was detected with 488 nM excitation and 526 nM emission. Ethidium bromide was imaged under a UV light source. Tetramethylrhodamine isothiocyanate, propidium iodide, crystal violet and coomassie brilliant blue were imaged in visible light.
Bioconjugate PCR
Biotinylated probes were generated by PCR using 5′-biotinylated primer vl881 for amplification of a ∼600-bp region of plasmids pGD9 and pGD13 (Supplementary Table S2). PCR products were extracted from an agarose gel and purified with a Wizard column. These were then γ-32P-labeled as described for the whole plasmids.
Preparation and purification of Vc2* RNA
The Vc2* template sequence (10) including T7 promoter sequence and complimentary T7 promoter sequence 5′-CTA ATA CGA CTC ACT ATA G-3′ were purchased from IDTs. Transcription was performed using 1.5 μg of template, 10 μl of 4 mg/ml T7 polymerase per 200 μl of transcription volume, 15 mM total NTP (A/C/G/UTPs), 15 mM MgCl2 in a transcription buffer of 40 mM Tris–HCl (pH 8.1), 1 mM spermidine, 5 mM DTT, 0.01% Trixon X-100, 2 U of RNase inhibitor, 2 U of inorganic pyrophosphatase. After 3 h, 0.4 U of Turbo DNase were added and incubated for another 15 min. The crude RNA was purified using a 12% denaturing PAGE with 1× TBE buffer. The product band was detected via UV shadowing the gel, excised and electroeluted in a Schleicher and Schuell Elutrap eletro-separation system. The purified RNA was precipitated with three volumes of absolute ethanol and 10% volumes of 0.3 M sodium acetate. The RNA pellet was then resuspended in water and dialyzed in a Nestroup Biodialyzer with a 500 MWCO membrane for 24 h against 100 mM potassium phosphate buffer (pH 6.4), 0.5 M KCl, 10 mM EDTA and then 1 and 0.1 mM EDTA, and finally against two changes of double-distilled H2O water before it was lyophilized.
Biotin labeling of RNA with biotin hydrazide at 3′-end
Seven microliters of freshly prepared 0.5 M NaIO4 was added to Vc2* RNA (210 μg) in 100 μl of water and the solution incubated at room temperature for 1 h. The excess NaIO4 was removed by filtration, using an Amicon ultra 0.5 ml centrifugal filter with 10 K cutoff membrane. The RNA was washed with 3 × 0.5 ml of water and then recovered by reverse spin. After that, 5μl of 1 M sodium acetate, pH 4.95 and 7 ml of 35 mM biotin hydrazide in dimethyl sulfoxide were added to the RNA. Coupling was carried out at 37°C for 1.5 h, then 3 μl of 1 M NaCNBH3 in acetonitrile was added and the reduction was carried out at room temperature for 1 h. The unused biotin hydrazide and NaCNBH3 were removed by centrifugal filter as above.
Testing the biotinylation efficiency with magnetic streptavidin beads
Four hundred microliters of streptavidin MagneSphere Paramagnetic Particle solution [Promega; Binding capacity: >0.75 nmol of biotinylated oligonucleotide (dT) bind per ml of particles] was taken and washed three times with 500 μL saline–sodium citrate (SSC) buffer (0.5 times). The washing step was facilitated by applying a magnet to the side of the tube and the supernatant discarded during each wash. SSC buffer with 100 µl of dissolved biotinylated RNA (2 μM) was added to streptavidin-coated magnetic particles and the tube was gently tapped to suspend the beads. The suspended beads were incubated at room temperature for 30 min, with occasional agitation by hand. A magnet was applied to the side of the tube and the supernatant was collected. The beads were washed with 100 μl SSC buffer (0.5×) two more times and the supernatant was collected and combined and UV260 nm measurement was made (OD260 = 0.123; 300 μl of supernatant wash). Because the supernatant was diluted three times, the OD of the original supernatant must be 0.531. This OD value was compared to the OD of the biotinylated RNA before incubation with streptavidin-coated beads. The yield of the biotinylated RNA was calculated to be 76.8%.
To confirm that the biotinylated RNA was bound to the streptavidin magnetic beads, 0.5 μl of RNAse A/T1 Mix was added to the washed beads in 100 μl of SSC buffer (0.5×). The beads were incubated at 37°C for 30 min before the supernatant was collected by applying a magnet. The OD260 for the eluted nucleotides was 0.560. The slight increase in absorbance at 260 nm (compare OD of 0.531 for the RNA with an OD of 0.56 for the nucleotides generated from the RNA hydrolysis) is expected as free nucleotides have higher absorption than that in a polynucleotide (hypochromic effect).
EMSA for RNA binding to cdiGMP
Gel shift assays were performed using 8% acrylamide gels with 100 mM Tris/HEPES, pH 7.5, 10 mM MgCl2 and 0.1 mM EDTA in the gel and running buffer. Gels were run at 4°C at 100 V for 2 h. Gels were imaged with a phosphorimager and fraction bound quantified with Fuji Image Guage software. The 32P cdiGMP probe was synthesized from α-32P-GTP by incubating overnight with purified diguanylate cyclase WspR (PA3702 from Pseudomonas aeruginosa) in 10 mM Tris, pH 8, 100 mM NaCl and 5 mM MgCl2 at 37°C.
RESULTS
DNA oligonucleotides are mobile in DRaCALA and sequestered by protein binding
Because radiolabeled mononucleotides and dinucleotides are mobile on nitrocellulose by capillary action (1), we reasoned this would be a property of double-stranded DNA as well. If confirmed, this would allow study of DNA using the DRaCALA technique. Double-stranded mobility on nitrocellulose was tested using 5′-end-labeled duplex DNA formed by annealing a pair of 40-bp oligonucleotides that generate the CRP consensus binding site, ICAP (gd126 and gd127 in Supplementary Table S1). When the 32P-labeled DNA was spotted on dry nitrocellulose, the 32P radiolabel was mobilized by radial capillary action resulting in a homogenous signal across the total sample area (Figure 1A) similar to results previously obtained for cAMP and ATP (1). Addition of 100 nM CRP and 200 µM cAMP to the ICAP probe is known to promote DNA–protein complexes (8). Spotting of the CRP–ICAP mixture at equilibrium resulted in sequestration of the soluble probe by the immobilized protein. Maltose binding protein (MBP), which does not bind DNA, did not sequester the probe, resulting in a uniform distribution of the radiolabel as in the control without any protein. This shows that specific molecular interaction is required for probe sequestration. Quantification of the fraction bound revealed that probe alone and probe mixed with non-specific protein have no fraction bound (Figure 1B). These results demonstrate the ability of DRaCALA to detect interactions between proteins and double-stranded DNA.
Figure 1.

Detection of protein–DNA interaction by DRaCALA. (A) Phosphorimager visualization of DRaCALA spots of indicated proteins at 100 nM mixed with 4 nM 32P-labeled ICAP fragments and 200 µM cAMP show distributions of the radioligand, which are diffused and homogenous (no protein, MBP) or sequestered (CRP). (B) The fraction bound was quantified using the formula in the ‘Methods’ section and error bars indicate the SD for three spots.
Oligonucleotide–protein interactions are specific in DRaCALA
CRP interaction with ICAP requires sequence-specific inverted repeats (8). To test if DRaCALA can detect changes in DNA–protein interaction with single base pair changes, point mutants were generated in the ICAP site at positions that are known to abolish binding (8). Specifically, the guanosines at position 8 and position 10 were changed to cytosines. Because the site is symmetrical, the corresponding cytosines at positions 19 and 21 were changed to guanosines (Figure 2A). These various probes were tested, at 4 nM, for binding to CRP by DRaCALA. The wild-type ICAP was sequestered by 100 nM CRP as before. The 8:GC mutant (G to C at position 8 and C to G at position 21) showed a very low level of binding to CRP while the 10:GC mutant (G to C at 10 and C to G at 19) and the 8, 10:GC double mutant exhibited no binding (Figure 2B). To confirm specificity, the binding between wild-type ICAP and 100 nM CRP was subjected to competition by wild-type and mutant-unlabeled DNA at 10, 100, or 1000 times the concentration of the labeled DNA. The wild-type competitor partially competed at 10-fold excess and competed more significantly with increased amount of competitor (Figure 2C). The 8:GC competitor showed no competition at 10- or 100-fold excess but did display some minor competition at 1000-fold. The 10:GC and 8, 10:GC failed to compete regardless of their concentration. These results collectively show that DRaCALA measures sequence-specific DNA binding.
Figure 2.

CRP binding to specific DNA sequences detected by DRaCALA. (A) The sequence of the 28-bp ICAP site. The positions perturbed in this study are marked in red. Names of mutant versions are listed next to the point mutations that define them. Equivalent nomenclature for mutants from Gunasekera et al. (8) is indicated in parentheses. (B) DRaCALA spots for direct binding of 100 nM CRP to 4 nM of ICAP, 8:G-C, 10:G-C and 8, 10:G-C probes with 200 µM cAMP are shown above the graphed quantification of fraction bound. (C) Binding of the ICAP probe to CRP was subjected to competition by unlabeled probes at 10, 100 or 1000 times the concentration of the radioligand. All error bars represent SD of three spots. DRaCALA spots shown above their respective conditions are separate images consolidated to fit the graph.
DNA-binding affinity and kinetics can be measured by DRaCALA
In order to accurately describe the activity of a transcription factor or other protein on a DNA binding site, it is desirable to determine the affinity and kinetics of the DNA–protein interaction. Because radionuclides can be detected with high sensitivity, DRaCALA can be used to make such measurements for high-affinity interactions. Serial 2-fold dilutions of CRP were mixed with limiting 32P-labeled ICAP probe (5 pM) to find the affinity of CRP for ICAP. CRP bound ICAP with maximum affinity when it was saturated with 200 µM cAMP. Analysis of these results indicated a dissociation constant (Kd) of 3.6 ± 0.4 × 10−11 M (SE) (Figure 3A). This is consistent with previously reported values for ICAP (5) (Table 1). In the absence of cAMP, the affinity of CRP for ICAP was at least 10 000-fold lower (Kd > 1.0 × 10−6 M) (Figure 3C). To confirm our DRaCALA results, we applied the same sample to an EMSA to determine the fraction bound (Figure 3B). The autoradiogram of the EMSA shows that the mobility of the free annealed oligonucleotide is retarded in the presence of CRP while the free the single-stranded oligonucleotide is not (Figure 3 and Supplementary Figure S1). The results from the EMSA assay yielded a similar dissociation constant Kd of 8.1 ± 0.8 × 10−11 M (Figure 3C).
Figure 3.

DRaCALA allows determination of affinity and kinetics of protein–DNA interaction. The affinity of CRP to the ICAP binding site reconstituted from annealed oligonucleotides (vl1427 and vl1428) was determined by the ability of serially diluted CRP to sequester 4 pM 32P-labeled ICAP probe in the presence of 200 µM cAMP by (A) DRaCALA and (B) EMSA. (C) The fraction bound is plotted against each concentration of CRP as detected by EMSA in the presence of 200 µM cAMP or DRaCALA at 0 or 200 µM cAMP. Plot represents a single replicate for EMSA and three replicates for DRaCALA. All Kd values are reported in Table 1. (D) The observed off-rate, koff = 2.6 ± 0.40 × 10−3 s−1 (SD), was measured by adding 1000-fold unlabeled competitor to 5 nM CRP with 5 pM ICAP oligonucleotide probe and spotting at different time points. All error bars represent the SD of three spots.
Table 1.
Observed affinity of CRP to various ICAP probes from this and previous studies with indicated amounts of cAMP
| Source | Method | Probe | [cAMP] (M) | Kd (M) (± SEM) |
|---|---|---|---|---|
| (8) | Filter binding | ICAP oligo 32P | 2 × 10−4 | 1.4 ± 0.3 × 10−11a |
| (8) | Filter binding | ICAP oligo 32P | 0 | >1.0 × 10−7a |
| This study | DRaCALA | ICAP oligo 32P | 2 × 10−4 | 3.6 ± 0.4 × 10−11 |
| This study | DRaCALA | ICAP oligo 32P | 0 | > 1.0 × 10−6 |
| This study | DRaCALA | ICAP* plasmid 32P | 2 × 10−4 | 4.1 ± 1.0 × 10−11 |
| This study | DRaCALA | ICAP* plasmid 32P | 0 | >1.0 × 10−6 |
| This study | EMSA | ICAP oligo 32P | 2 × 10−4 | 8.1 ± 0.8 × 10−11 |
All reported Kd values from this study were determined by DRaCALA and the standard error of three trials is reported.
aThe affinity reported in the Gunasekera et al. paper is the binding constant or association constant Ka. We have taken the inverse of those values to give the Kd reported in Table 1 to match our Kd measurements.
We also used the CRP–ICAP binding interaction to test whether DRaCALA can be used to easily monitor the dissociation kinetics for protein–DNA complexes. A limiting amount of 32P-labeled ICAP (5 pM) was mixed with a protein concentration just above the Kd (5 nM). Then, unlabeled competitor ICAP was added in 1000-fold excess of radiolabeled ligand and spots were made over time, and these spots were analyzed to monitor the fraction of ICAP bound as a function of time. Our analysis indicated a dissociation rate (koff) of 2.6 ± 0.40 × 10−3 s−1 (SD) for the CRP–cAMP complex, corresponding to a half-life of 4.42 min (Figure 3D). Using the DRaCALA-observed off-rate and affinity, the calculated on-rate is kon = 7.2 × 107 M−1 s−1. These results show that DRaCALA is a rapid method for determining affinity and kinetics of protein–DNA interactions.
Protein binding of whole-plasmid ligands is detected specifically by DRaCALA
The mobility of both nucleotides and double-stranded oligonucleotides on nitrocellulose suggests that molecular weight is not a critical limiting factor for what types of molecules can be used as the mobile, detectable ligand. The size limit of DNA ligands in DRaCALA was tested by cloning the same ICAP binding site and mutant sites onto a 3.5 kb pVL-Blunt plasmid, and using the entire linearized vector as a ligand. Each of the linearized plasmids was labeled with 32P and shown to be mobile in DRaCALA (plasmids listed in Supplementary Table S2). Plasmids (50 pM) with ICAP sites bound 100 nM CRP. In contrast, plasmids with 8:GC bound weakly and 10:GC or 8, 10:GC sites did not bind at all (Figure 4A).
Figure 4.

DRaCALA allows detection of specific interaction of CRP with plasmid carrying the ICAP site. (A) 50 pM individual plasmids with 1×, 3× or 5× wild-type binding sites or 3× mutant-binding sites cloned in series were tested for binding in the presence of 100 nM CRP and 200 µM cAMP. (B) Specificity was determined by competition of binding to 32P-labeled 1× wild-type plasmid with unlabeled PCR products. Competitors used were 1× ICAP, 3× 8:G-C, 3× 10:G-C, 3× 8, 10:G-C. All error bars represent SD of three spots with a representative spot (spot images consolidated to fit graph) shown above each column.
Binding of a single ICAP insert on a plasmid probe (50 pM) to 100 nM CRP was next subjected to competition. Competitors in this case were made by PCR amplification of a 600-bp region of the plasmids containing wild-type and mutant ICAP sites. The wild-type PCR competitor partially inhibited radiolabeled plasmid binding to CRP at 10-fold excess of the radiolabeled ligand and fully competed at 1000-fold excess (Figure 4B). PCR products containing 8:GC, 10:GC, or 8, 10:GC did not compete away binding even at 1000-fold excess concentration. Detected binding of CRP to whole-plasmid probes is therefore also site-specific in DRaCALA. These results show that the critical parameter for detection of protein–DNA interaction by DRaCALA is the mobility of the ligand on the solid support and not the molecular weight of the ligand.
Affinity and kinetics determined for whole plasmid ligand
Whole plasmids can also be used in affinity and kinetic studies. With 200 µM cAMP, the observed Kd of CRP and a plasmid with a single ICAP site was 4.1 ± 0.3 × 10−11 M (SD) (Figure 5A). Without cAMP the Kd was >1.0 × 10−6 M. These values are similar to those obtained for the labeled oligonucleotides and those from previous studies (Table 1). The off-rate for the plasmid was observed at koff = 4.8 ± 0.17 × 10−4 s−1, corresponding to a half-life of 23.9 min (Figure 5B). The calculated on-rate for the plasmid was kon = 1.2 × 107 M−1 s−1, which is 6-fold lower than that of the annealed oligonucleotides, likely due to the large excess of non-specific DNA in the plasmid probe. Affinity and kinetics can thus also be measured for sites contained on a plasmid.
Figure 5.

Affinity and kinetics of DNA-binding determined using 5 pM whole-plasmid probe with a single ICAP site. (A) Graphs of fraction of ICAP plasmid bound by various concentrations of CRP with indicated levels of cAMP (Kd reported in Table 1). (B) Graph of observed off-rate of koff = 4.8 ± 0.17 × 10−4 s−1 (SD) for ICAP plasmid generated by adding 1000-fold unlabeled PCR product (1× ICAP) competitor to 5 nM CRP with plasmid probe and spotting at time points >3 h. All error bars represent SDs of three spots.
Use of DNA as a carrier/label molecule
Because such large pieces of DNA can be used in DRaCALA without altering specificity, we hypothesized that DNA could be used as a label and carrier for molecules that are not ordinarily mobile in DRaCALA and/or not easily labeled. Because ligand mobility and ligand detection are the only requirements for the mobile binding partner, DNA-conjugation could potentially make any molecule adaptable for use as a DRaCALA probe. A DNA component to the probe allows for easy labeling with 32P. Many small, soluble molecules are not mobile in DRaCALA suggesting that fluorescently labeled low molecular weight ligands are not suitable for DRaCALA technique (Supplementary Figure S2). However, addition of DNA to immobile ethidium bromide conferred mobility to the interacting dye (Supplementary Figure S2) implying that conjugation to DNA can overcome the immobility of some dye molecules. DNA can also be covalently linked to molecules through bioconjugate PCR with modified primers. This technique was tested using the biotin–streptavidin system. PCR products including the binding sites of the 3× ICAP plasmid and 3 × 8, 10:GC plasmid were generated with a 5′-biotinylated primer and labeled with 32P on the free 5′-end. These bioconjugate probes were tested with DRaCALA for binding to CRP, streptavidin and MBP. The wild-type probe without biotin-bound CRP but not streptavidin or MBP (Figure 6A). The biotinylated wild-type probe bound both CRP and streptavidin but not MBP. The 8, 10:GC probe without biotin bound none of the proteins, whereas the biotinylated version bound only streptavidin.
Figure 6.

Bioconjugate DNA probes. Bioconjugate probes were generated by PCR with 5′-biotinylated primers. (A) Four probes (ICAP and 8, 10:G-C with and without biotin) were tested for binding to CRP, streptavidin and MBP. A mix of 50 pM 32P-labeled probe, 100 nM protein and 200 µM cAMP was spotted on 0.8 µ nitrocellulose and phosphor images of the spots are shown. (B) The ICAP–biotin probe affinity for streptavidin in PBS was determined by DRaCALA with 100 pM probe (Kd = 4.0 ± 0.6 × 10−10 M). (C) Binding of 10 nM streptavidin to the ICAP–biotin probe was competed with serial dilutions of free biotin (IC50 = 3.3 × 10−8 M).
The affinity of the biotinylated ICAP probe was determined using DRaCALA by diluting streptavidin (Figure 6B). The affinity was limited by the concentration of the probe that could not be diluted below tens of pM without loss of signal. The limit of DRaCALA detecting binding seems to be therefore the limit of detection of the probe. The IC50 of free biotin was determined by competing against the probe with different concentrations of free biotin (Figure 6C). Here the IC50 of 33 nM is approximately enough to occupy the four sites of the 10 nM streptavidin. The observed affinity is lower than the previous published values for free biotin probably because the biotin molecule was conjugated to DNA (10). We were also able to measure the off-rate of the conjugated biotin by observing the exchange with excess free biotin (Supplementary Figure S3). The exchange occurred in two steps, with an initial rapid off-rate and then a second slower rate corresponding to a half-life of 112 h and exchange-rate of koff = 1.7 × 10−6 s−1. The two-step rate has been previously reported in a study of avidin and unconjugated biotin and is likely due to the tetramer protein having different affinites for biotin depending on the number of occupied sites (10). These results demonstrate that PCR conjugation can be used to link a molecule/ligand of interest to DNA, which allows facile 32P-labeling and can confer mobility (in DRaCALA), allowing rapid determination of affinity and kinetics of the protein–ligand interaction.
Riboswitch-binding cdiGMP
We have shown that protein interaction with DNA can be detected by DRaCALA. We wondered if the principle of DRaCALA would also apply to ribonucleic acids. In particular, can the DRaCALA technology be used to detect the interaction of riboswitches with their small molecule ligands? One example of such an interaction that has been of recent interest is the cdiGMP responsive Vc2 riboswitch identified in bacteria (11). To study such an interaction with DRaCALA, one of the binding partners must be immobilized. We achieved this through biotinylation of Vc2* riboswitch RNA (with a modified tetraloop and shortened 5′- and 3′-ends compared to the original Vc2) at the 3′-end by periodate cleavage of the terminal ribose and reductive amination to conjugate the biotin moiety. The biotinylated riboswitch was sequestered by streptavidin, allowing the nucleic acid to take the place of protein as the immobile partner in the binding assay. Vc2* was tested directly for sequestration of cdiGMP and also biotinylated and tested for binding to cdiGMP in the presence or absence of streptavidin. The 4 nM radiolabeled cdiGMP was mobile alone and in the presence of the Vc2* or biotinylated Vc2* RNA (Figure 7A, lanes 1–3). This suggests that RNA, like DNA, is mobile in this system, and therefore could be used as a labeled probe as well. Streptavidin did not sequester radiolabeled cdiGMP alone or with Vc2* RNA, so there is no detectable interaction between streptavidin and Vc2* RNA (lanes 4–5). Biotinylated Vc2* RNA and bound cdiGMP were immobilized by streptavidin as expected (lane 6). The affinity of Vc2* for cdiGMP was tested using both DRaCALA and an EMSA (or gel shift). These measurements were made in a Vc2 binding buffer (10 mM sodium cacodylate, 10 mM MgCl2, 10 mM KCl) by heating the binding reaction to 70°C for 3 min, slowly cooling to room temperature, and then incubating at room temperature for 48 h (12). Remarkably similar results were obtained using DRaCALA and gel shift (Figure 7B). The affinity of the Vc2* RNA for cdiGMP was observed to be Kd = 7.8 ± 1.9 × 10−9 M with DRaCALA and Kd = 9.8 ± 1.6 × 10−9 M with EMSA. These results show that DRaCALA works as well as EMSA for studying the molecular interactions of riboswitches. This strategy can be adapted to study interactions between the biotinylated nucleic acids and a mobile ligand (another nucleic acid or nucleotide).
Figure 7.

Vc2* RNA binding to 32P-cdiGMP is detected by DRaCALA. (A) Spots visualized by phosphorimager with streptavidin used to immobilize biotinylated RNA. The binding reaction contained 4 nM 32P-cdiGMP, 1 µM RNA and 200 nM streptavidin in buffer (10 mM KCl, 10 mM sodium cacodylate, 3 mM MgCl2). (B) The affinity of Vc2*-biotin RNA for cdiGMP was determined with both EMSA and DRaCALA by diluting RNA in the binding reaction. The fraction bound is normalized such that 1.0 represents maximal binding. The DRaCALA-obtained affinity was Kd = 7.8 ± 1.9 × 10−9 M and the apparent affinity in EMSA was Kd = 9.8 ± 1.6 × 10−9 M.
DISCUSSION
Nucleic acid interactions with proteins and low molecular weight ligands are fundamental for biological function. This has inspired the development of a number of assays to measure these interactions. We will compare and contrast the merits of DRaCALA with these various assay systems using CRP–cAMP as the classic example of a signal-responsive transcription factor and a model system for protein interaction with both a large nucleic acid and a mononucleotide. Study of the CRP system led to the establishment of some fundamental concepts related to transcription, such as regulation by second messengers (13), multiple promoter control of a single operon (14–16) and promoter control of RNA polymerase binding (17). Because of this foundational work, CRP and the lac repressor have been traditionally used to demonstrate proof of principal for various methods to detect DNA–protein interactions.
Nucleic acid–protein DRaCALA utilizes the differential mobility of nucleic acids through nitrocellulose to separate DNA that is bound to a protein from that which is unbound. The interactions measured in this way were specific to the nucleic acid sequences because point mutations at previously identified critical nucleic acids abolished specific binding of CRP to ICAP in both annealed oligonucleotides and plasmids (18). The affinity of the interaction was measured by diluting protein with limiting amounts of probe. Remarkably, the Kd measured for the annealed oligonucleotide and plasmid closely matched what was reported in a previous study that used a filter-binding assay (8) as well as a study that used gel shift (19) (Table 1). The off-rate determined with DRaCALA was slower for the plasmid than for the oligonucleotide probe, which is consistent with the finding that non-specific DNA concentration can affect the kinetics of specific DNA binding with protein (20). The off-rate for the plasmid (koff = 4.84 ± 0.17 × 10−4 s−1) was similar to that reported in a gel shift study (koff = 1.2 × 10−4 s−1) (20). This corresponds to an observed half lives of 23.9 min for DRaCALA and about an hour for gel shift. This difference may be explained by the amount of unlabeled competitor used to chase off the probe, which was at 25 times molar excess for the gel shift and 1000 times for DRaCALA. For DRaCALA with plasmid probes, another advantage is that high concentrations of competitor can easily be obtained by PCR amplification. The on-rate cannot be measured using DRaCALA, but it can be approximated with a calculation based on the affinity and off-rate. Using DRaCALA with plasmid probes allows for easy testing of direct binding and specific competition of any potential DNA-binding site simply by cloning into a plasmid that can be labeled for detection. Studying kinetics in this system is more analogous to DNA-binding activity in a cell because there is a great excess of DNA to which the protein can bind non-specifically.
Comparing DRaCALA to the traditional separation-based methods reveals the advantages of the new technique. The filter-binding assay was the first popular method that depended on separation of bound and unbound ligands based on differential mobility through a support (21). This technique was used for the first study of the interaction of CRP with DNA (18). One key difference between DRaCALA and filter-binding assays is that for DRaCALA, both the bound ligand and the total amount of ligand are always measured. In contrast, the traditional filter-binding assay typically only measures the bound ligand. Thus, results of filter-binding assays are typically normalized to 1.0 fraction bound for the highest concentration of protein or ligand. In contrast, results for DRaCALA for the highest concentration of protein is often <1.0. There are three potential reasons for the fraction bound detected by DRaCALA to be less than the theoretical 1.0. First, the off-rate of the protein–ligand interaction dictate that, during the assay time, a population of the bound ligand is dissociated, mobilized and can not rebind the protein. Second, for all 5′-end-labeled nucleotide, a small fraction of labeled free phosphate can be hydrolyzed and appear as free ligand. Third, oligonucleotide probes can fold into non-native conformations. For example, the inverted repeat of the ICAP oligo can lead to the folding into hairpins that cannot be bound by CRP. Because DRaCALA measures both free and bound ligand, the determined fraction bound is far more accurate despite the detection of fraction bound of <1.0. We do not think this is a concern as the Kd and koff that we measured for CRP–ICAP interactions are similar to previously reported results. A similarity of DRaCALA and filter-binding assays is the interaction of proteins with nitrocellulose may alter the behavior of proteins. For DRaCALA, this effect is likely protein specific since soluble and insoluble forms of Alg44 and PelD behave similarly when assayed for binding to cdiGMP by DRaCALA (1).
The EMSA (or gel shift), which detects interactions because they cause retardation in DNA mobility through a gel, was first introduced as an alternative to the filter-binding assay using the lac repressor as an example (22,23). Later it was used to study CRP in greater detail (19,20). The major strengths of the gel shift are that both bound and unbound ligands are measured and supershifts provide information about binding structure. A potential issue is the length of time required to run the gel, during which time the protein and DNA can dissociate, which is a particular concern for lower affinity interactions (9). DRaCALA does not have a wash step and it measures total signal in every sample with a visual readout, making it preferable to the filter-binding assay. EMSA also measures total signal with a visual readout, but requires a much greater assay time than DRaCALA. Although DRaCALA is more rapid, EMSA still retains an advantage in the detection of supershifts that result from an antibody binding to a DNA-bound protein or multiple proteins binding to DNA. The ability of DRaCALA to detect interactions on plasmid DNA is a significant improvement over EMSA, which is most sensitive with probes <300-bp long (24).
More modern techniques include chromatin immunoprecipitation on a microarray chip (ChIP-chip) and sequencing of chromatin immunoprecipitated DNA (ChIP-Seq). These assays allow for a high-throughput approach to identify binding sites on the chromosome but provide no measure of affinity and cannot rule out indirect interactions (25,26). Because the readout of ChIP-chip is precipitation or a lack thereof, studies of transcription factors such as CRP often have false negatives and include a lot of background noise attributable to low-affinity binding sites (27). The most accurate analytical assays include isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR). ITC uses a controlled chamber to assess heat changes as DNA binds protein, allowing for thermodynamic and kinetic measurements (28). SPR detects molecular weight changes on a metal surface in real time and can determine affinity and kinetics with remarkable sensitivity (29). The proof of principle for SPR studies of DNA–protein interaction was first demonstrated using the lac repressor (30). ITC and SPR have the advantage over DRaCALA in that neither technique requires labeling of the ligand of interest. However, the common drawbacks of ChIP-chip, ITC and SPR are the relatively high associated costs and need for specialized equipment. DRaCALA uses small amounts of inexpensive materials and requires no special equipment, making biochemistry accessible to molecular biologists. DRaCALA is precise, with standard deviations (SDs) of measurements that are typically <5% of the mean. The value of DRaCALA lies in the simplicity of the technique. The only special tool required is a detector of the label on the probe. Only a small amount of sample and nitrocellulose are needed, making it inexpensive and easy to scale up. Capillary action of small volumes is fast, so separation of bound and unbound ligand takes only seconds. Together, these traits make DRaCALA especially cost- and time efficient in comparison to established methods.
The simplicity of DRaCALA allows adaptation of the technique to study other molecular interactions. PCR conjugation of DNA to a variety of molecules can be achieved using commercially available modified primers that can have 5′ reactive groups such as aldehydes, amines and thiols. This can serve the dual function of keeping the molecule mobile through nitrocellulose and providing a mechanism to label the probe in different ways. Radiolabelling small molecules directly is often impractical due to costs associated with chemical synthesis with radiolabeled chemicals, so DNA conjugation could be a good alternative. The free 5′-end of the DNA can be 32P-labeled as in this study or occupied with a fluorescent dye from a second modified primer in the original PCR reaction. While fluorescence may be desirable for its ease of use, it cannot match the sensitivity of 32P. Bioconjugate PCR was used in this study with the simple streptavidin–biotin system. Biotinylated PCR products were mobile, detectable and showed specific interactions with CRP and streptavidin. This also allows selective immobilization of biotinylated nucleic acids so that they can take the role of the immobile binding partner in DRaCALA.
As shown in this study, immobilization of RNA allowed detection of RNA interaction with a small ligand. This area has been of great interest since the discovery of riboswitches, cis-acting RNA sequences on mRNAs that directly interact with small molecules and consequently self-regulate their transcriptional termination and/or translation (31,32). Such RNAs have been found to bind a variety of small molecules, including amino acid derivatives, coenzyme B12 and the bacterial second messenger cdiGMP (10,33,34). Studies of riboswitches have primarily used in-line probing and equilibrium dialysis to analyze direct RNA binding to its target molecule. These methods require long incubations that limit their accuracy in determining biochemical parameters. Others have used gel shift assays to measure the affinity and kinetics for riboswitches (12). By comparing DRaCALA to gel shift assays using a Vc2* RNA to establish a proof of principle, we have demonstrated that DRaCALA is a powerful alternative to these methods, which is much faster with at least equal accuracy and precision (Figure 7). In this study, RNA was immobilized using biotinylation, but RNA could also be immobilized by other means such as with a known binding protein or an additional sequence on the RNA that specifically binds a protein. Another alternative strategy is to use a biotinylated DNA oligonucleotide that can hybridize with the RNA molecule (3′-end of riboswitch) to provide a method for immobilization. The same technique could also be used to study RNA–RNA interactions in the context of regulatory RNAs, which are ubiquitous in prokaryotes and eukaryotes and have therapeutic potential (35,36). As more research is done involving RNA interactions with a variety of other molecules, it is critical to have a rapid, quantitative and cost-effective method for directly testing these interactions.
DRaCALA requires one immobile binding partner and a ligand that is detectable and mobile by capillary action. This study provides a foundation for universal applicability of DRaCALA for studying any molecular interaction. There is evidence that DNA could serve as a label and carrier for any molecule that can be conjugated to it. Because bioconjugate PCR allows specific immobilization of biotinylated nucleic acids, the assay can be used with nucleic acids as the immobile and/or the mobile piece in binding studies. These manipulations of the mobility of molecules provide a window to the many potential uses of this assay. Additionally, the ease of running DRaCALA (little volume needed, no wash step, inexpensive materials and visual readout) makes it possibly amenable to usage as a portable rapid diagnostic tool in a ‘lab-on-paper’ design (37).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–3.
FUNDING
National Institutes of Health (NIH) (AI096083 to V.T.L.); University of Maryland Seed Grant (to V.T.L.); NSF-CHE0746446 (to H.O.S.). Funding for open access charge: NIH – NIAID.
Conflict of interest statement. Provisional patent has been filed by University of Maryland.
Supplementary Material
ACKNOWLEDGEMENTS
We thank R. Stewart, Z. Kelman and A. Records for critical reading of the article and S. Adhya for the CRP expression plasmid.
REFERENCES
- 1.Roelofs KG, Wang J, Sintim HO, Lee VT. Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc. Natl Acad. Sci. USA. 2011;108:15528–15533. doi: 10.1073/pnas.1018949108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng D, Constantinidou C, Hobman JL, Minchin SD. Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 2004;32:5874–5893. doi: 10.1093/nar/gkh908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zubay G, Schwartz D, Beckwith J. Mechanism of activation of catabolite-sensitive genes: a positive control system. Proc. Natl Acad. Sci. USA. 1970;66:104–110. doi: 10.1073/pnas.66.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Crombrugghe B, Busby S, Buc H. Cyclic AMP receptor protein: role in transcription activation. Science. 1984;224:831–838. doi: 10.1126/science.6372090. [DOI] [PubMed] [Google Scholar]
- 5.Garges S, Adhya S. Sites of allosteric shift in the structure of the cyclic AMP receptor protein. Cell. 1985;41:745–751. doi: 10.1016/s0092-8674(85)80055-6. [DOI] [PubMed] [Google Scholar]
- 6.Emmer M, deCrombrugghe B, Pastan I, Perlman R. Cyclic AMP receptor protein of E. coli: its role in the synthesis of inducible enzymes. Proc. Natl Acad. Sci. USA. 1970;66:480–487. doi: 10.1073/pnas.66.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebright RH, Ebright YW, Gunasekera A. Consensus DNA site for the Escherichia coli catabolite gene activator protein (CAP): CAP exhibits a 450-fold higher affinity for the consensus DNA site than for the E. coli lac DNA site. Nucleic Acids Res. 1989;17:10295–10305. doi: 10.1093/nar/17.24.10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunasekera A, Ebright YW, Ebright RH. DNA sequence determinants for binding of the Escherichia coli catabolite gene activator protein. J. Biol. Chem. 1992;267:14713–14720. [PubMed] [Google Scholar]
- 9.Revzin A, Ceglarek JA, Garner MM. Comparison of nucleic acid-protein interactions in solution and in polyacrylamide gels. Anal. Biochem. 1986;153:172–177. doi: 10.1016/0003-2697(86)90077-1. [DOI] [PubMed] [Google Scholar]
- 10.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green NM. Avidin. 1. The use of (14-c)biotin for kinetic studies and for assay. Biochem. J. 1963;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith KD, Lipchock SV, Livingston AL, Shanahan CA, Strobel SA. Structural and biochemical determinants of ligand binding by the c-di-GMP riboswitch. Biochemistry. 2010;49:7351–7359. doi: 10.1021/bi100671e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pastan I, Perlman R. Cyclic adenosine monophosphate in bacteria. Science. 1970;169:339–344. doi: 10.1126/science.169.3943.339. [DOI] [PubMed] [Google Scholar]
- 14.Adhya S, Miller W. Modulation of the two promoters of the galactose operon of Escherichia coli. Nature. 1979;279:492–494. doi: 10.1038/279492a0. [DOI] [PubMed] [Google Scholar]
- 15.Busby S, Aiba H, de Crombrugghe B. Mutations in the Escherichia coli operon that define two promoters and the binding site of the cyclic AMP receptor protein. J. Mol. Biol. 1982;154:211–227. doi: 10.1016/0022-2836(82)90061-4. [DOI] [PubMed] [Google Scholar]
- 16.Irani MH, Orosz L, Adhya S. A control element within a structural gene: the gal operon of Escherichia coli. Cell. 1983;32:783–788. doi: 10.1016/0092-8674(83)90064-8. [DOI] [PubMed] [Google Scholar]
- 17.Meiklejohn AL, Gralla JD. Entry of RNA polymerase at the lac promoter. Cell. 1985;43:769–776. doi: 10.1016/0092-8674(85)90250-8. [DOI] [PubMed] [Google Scholar]
- 18.Riggs AD, Reiness G, Zubay G. Purification and DNA-binding properties of the catabolite gene activator protein. Proc. Natl Acad. Sci. USA. 1971;68:1222–1225. doi: 10.1073/pnas.68.6.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fried MG, Crothers DM. Equilibrium studies of the cyclic AMP receptor protein-DNA interaction. J. Mol. Biol. 1984;172:241–262. doi: 10.1016/s0022-2836(84)80025-x. [DOI] [PubMed] [Google Scholar]
- 20.Fried MG, Crothers DM. Kinetics and mechanism in the reaction of gene regulatory proteins with DNA. J. Mol. Biol. 1984;172:263–282. doi: 10.1016/s0022-2836(84)80026-1. [DOI] [PubMed] [Google Scholar]
- 21.Nirenberg M, Leder P. RNA codewords and protein synthesis. The effect of trinucleotides upon the binding of srna to ribosomes. Science. 1964;145:1399–1407. doi: 10.1126/science.145.3639.1399. [DOI] [PubMed] [Google Scholar]
- 22.Fried M, Crothers DM. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savery NJ, Busby SJW. In: Molecular Biomethods Handbook. Rapley R, Walker JM, editors. Humana Press; 1998. pp. 121–129. [Google Scholar]
- 25.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 26.Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 27.Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl Acad. Sci. USA. 2005;102:17693–17698. doi: 10.1073/pnas.0506687102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 29.Pockrand I, Swalen JD, Gordon JG, Philpott MR. Surface plasmon spectroscopy of organic monolayer assemblies. Surface Sci. 1978;74:237–244. [Google Scholar]
- 30.Bondeson K, Frostell-Karlsson A, Fägerstam L, Magnusson G. Lactose repressor-operator DNA interactions: kinetic analysis by a surface plasmon resonance biosensor. Anal. Biochem. 1993;214:245–251. doi: 10.1006/abio.1993.1484. [DOI] [PubMed] [Google Scholar]
- 31.Winkler WC, Cohen-Chalamish S, Breaker RR. An mRNA structure that controls gene expression by binding FMN. Proc. Natl Acad. Sci. USA. 2002;99:15908–15913. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winkler W, Nahvi A, Breaker RR. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419:952–956. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 33.Epshtein V, Mironov AS, Nudler E. The riboswitch-mediated control of sulfur metabolism in bacteria. Proc. Natl Acad. Sci. USA. 2003;100:5052–5056. doi: 10.1073/pnas.0531307100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nahvi A, Barrick JE, Breaker RR. Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res. 2004;32:143–150. doi: 10.1093/nar/gkh167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 37.Lu Y, Shi W, Qin J, Lin B. Fabrication and characterization of paper-based microfluidics prepared in nitrocellulose membrane by wax printing. Anal. Chem. 2010;82:329–335. doi: 10.1021/ac9020193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.