

Abstract
Even in the absence of a template, glycosyltransferases can catalyze the synthesis of carbohydrate polymers of specific sequence. The paradigm has been that one enzyme catalyzes the formation of one type of glycosidic linkage, yet certain glycosyltransferases generate polysaccharide sequences composed of two distinct linkage types. In principle, bifunctional glycosyltransferases can possess separate active sites for each catalytic activity or one active site with dual activities. We encountered the fundamental question of one or two distinct active sites in our investigation of the galactosyltransferase GlfT2. GlfT2 catalyzes the formation of mycobacterial galactan, a critical cell-wall polymer composed of galactofuranose residues connected with alternating, regioisomeric linkages. We found that GlfT2 mediates galactan polymerization using only one active site that manifests dual regioselectivity. Structural modeling of the bifunctional glycosyltransferases hyaluronan synthase and cellulose synthase suggests that these enzymes also generate multiple glycosidic linkages using a single active site. These results highlight the versatility of glycosyltransferases for generating polysaccharides of specific sequence. We postulate that a hallmark of processive elongation of a carbohydrate polymer by a bifunctional enzyme is that one active site can give rise to two separate types of glycosidic bonds.
INTRODUCTION
Carbohydrate polymers function in critical biological processes. Polysaccharides form protective and fibrillar macromolecules, serve as stores of energy, and mediate many extra- and intra-cellular signaling events (1-6). These roles are dictated by the structural features of the polysaccharide, including the degree of polymerization, sequence, and connectivity. Because polysaccharide synthesis occurs without the aid of a template, polysaccharide function is governed by the specificity and selectivity of glycosyltransferases. Glycosyltransferases catalyze the addition of a glycosyl moiety from an activated donor sugar to an acceptor molecule. In glycosyltransferase-catalyzed reactions, a nucleophile (typically a hydroxyl group) on the acceptor attacks the electrophilic anomeric position of the donor to form a specific glycosidic linkage (7). To regulate polysaccharide structure precisely, glycosyltransferases select a donor residue from a multitude of cellular monosaccharides, prescribe which hydroxyl group is used as a nucleophile in glycosidic bond formation, and dictate the stereochemistry of bond formation (8, 9). Thus, the structural diversity of polysaccharides and their corresponding biological activities stem from the substrate specificity, stereoselectivity, and regioselectivity of glycosyltransferases.
A long-standing dictum of glycobiology is that one enzyme catalyzes the formation of one specific glycosidic linkage (10). Nevertheless, a number of polysaccharides contain a disaccharide as the repeating unit. This repeat can consist of either two distinct monosaccharides or of a single monosaccharide linked with alternating, regioisomeric linkages. Examples of the former include the vertebrate glycosaminoglycan (GAG) polymers such as hyaluronic acid, (Figure 1A), chondroitin sulfate, and heparan sulfate (11-13) as well as extracellular capsules in pathogenic bacteria (14, 15). An example of a polymer of alternating regioisomeric linkages is found in the polysialic acid capsules of certain bacteria (Figure 1A) (16, 17). Polymers possessing a disaccharide repeat are generated by glycosyltransferases that catalyze either the addition of both monosaccharides to the growing chain (12, 14, 15, 18, 19) or the formation of two distinct linkages between the same monosaccharide (16, 17). The bifunctionality of these enzymes raises questions about the number of active sites used to achieve dual activity. Some bifunctional glycosyltransferases contain two tandem glycosyltransferase domains in the primary structure and accordingly are thought to possess two distinct active sites. Such enzymes include certain glycosyltransferase family 2 (GT-2) enzymes (18, 20-23) and the enzymes that mediate heparan sulfate polymerization (24). In most cases, however, it is not known whether a bifunctional enzyme catalyzing these polymerization reactions uses one or two active sites.
Figure 1.
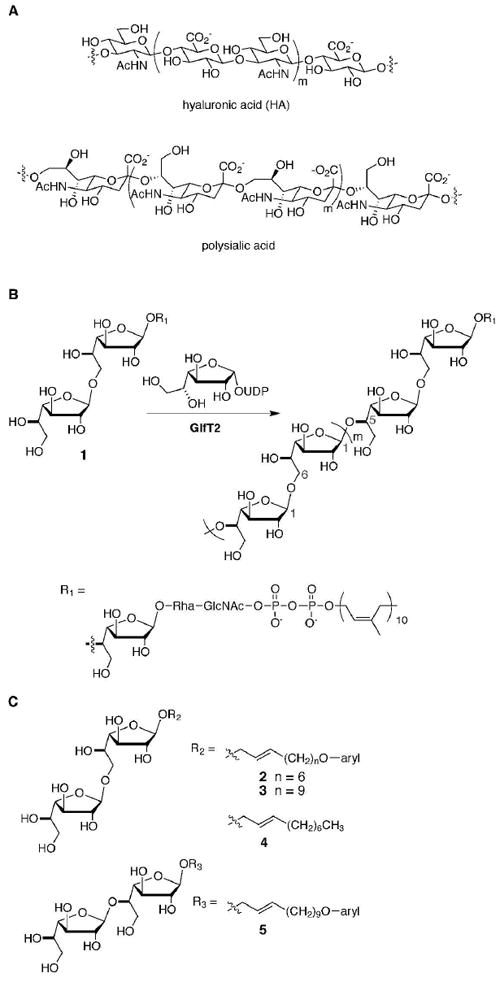
GlfT2 generates a heteropolysaccharide composed of alternating, regioisomeric linkages. (A) Disaccharide repeating units in heteropolysaccharides can consist of two distinct monosaccharides, as found in hyaluronic acid, or of a single monosaccharide linked with alternating, regioisomeric linkages, as found in polysialic acid or mycobacterial galactan (shown in part B). (B) GlfT2 mediates mycobacterial galactan formation by catalyzing the addition of Galf residues from UDP-Galf to an oligosaccharide–lipid conjugate (compound 1). GlfT2 has dual activities as it can generate alternating Galf-β-(1→5)-Galf and Galf-β-(1→6)-Galf linkages. (C) The synthetic acceptor substrates used in this study possess a disaccharide of Galf-β-(1→6)-Galf (compounds 2–4) or Galf-β-(1→5)-Galf (compound 5) linked to an aryl-terminated (2, 3, 5) or aliphatic (4) lipid.
Our interests in bifunctional glycosyltransferase activity originate from our investigations of the enzyme GlfT2, which mediates biosynthesis of a polysaccharide component of the mycobacterial cell wall. This polysaccharide, known as galactan, serves as a covalent connector between the peptidoglycan and mycolic acid-arabinan layers (25, 26). Galactan is a linear polysaccharide composed of galactofuranose (Galf) residues (26) (Figure 1B). GlfT2, encoded by the essential mycobacterial gene glfT (also known as Rv3808c), promotes galactan synthesis by catalyzing the transfer of Galf residues from the donor UDP-Galf to the nonreducing end of a lipid-linked oligosaccharide acceptor (27-30). Our previous investigations suggest that GlfT2 controls polymer length by tethering the acceptor at both the lipid and polysaccharide ends (30). Intriguingly, GlfT2 can catalyze the formation of alternating regioisomeric β-(1→5) and β-(1→6) linkages (27, 28), but it is unknown how the enzyme achieves this feat. Furthermore, GlfT2 is a processive enzyme (30, 31), which raises the question of how it can control regiochemistry without releasing the bound substrate during elongation. A key issue is whether GlfT2 uses one or two active sites for processive synthesis of an alternating polymer. A recent report demonstrated that trisaccharide acceptors composed of Galf residues that differ in their terminal linkage bind competitively to GlfT2 (32). These binding studies suggest that the two acceptors can bind the same site of GlfT2, but they did not address how many sites are required for bifunctional catalysis. Therefore, we were interested in elucidating the mechanistic attributes by which GlfT2 achieves bifunctionality and in determining whether other polymerizing glycosyltransferases share these attributes.
We sought to distinguish between the use of one or two active sites by using chemical biology and site-directed mutagenesis. We focused on identifying an active site in GlfT2 through sequence analysis and found that two motifs of invariant aspartic acid residues constitute one putative glycosyltransferase active site. Our results indicate that GlfT2 contains a single active site that exhibits dual regioselectivity. Further sequence analysis suggests that a similar one-site architecture occurs in additional glycosyltransferases that catalyze carbohydrate polymerization reactions. Lastly, we postulate that one-site bifunctionality is an intrinsic feature of processive enzymes.
MATERIALS AND METHODS
Sequence analysis and homology modeling
The full-length primary amino acid sequence for M. tuberculosis H37Rv GlfT2 (NCBI accession number: NP_218325.1) was used as the query sequence for a BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) search for homologs of GlfT2 using default parameters. The top 50 most similar sequences (including the query) were used as the input for a multiple sequence alignment by ClustalW2 (33). Alignments were also generated using the programs MAFFT (34), MUSCLE (35, 36), and T-Coffee (37) to check for overall precision in the alignment. The ClustalW2-generated alignment was analyzed using the program JalView (38), which was also used to generate the heat-map image shown in Figure 2A through coloring the alignment by conservation, with blue representing low conservation and red representing high conservation. The homology model of M. tuberculosis GlfT2 was computed using the “First Approach/Automatic Modeling” mode of SWISS-MODEL (39, 40) with default parameters. The full-length amino acid sequence was used as the input sequence. The model (of residues 160–398) was viewed and analyzed using PyMOL. Homology models for SpHasA (GenBank accession number AAA17984.1) and CesA (GenBank accession number P21877.3) were generated with this same process using the appropriate sequence as the query for the SWISS-MODEL server.
Figure 2.
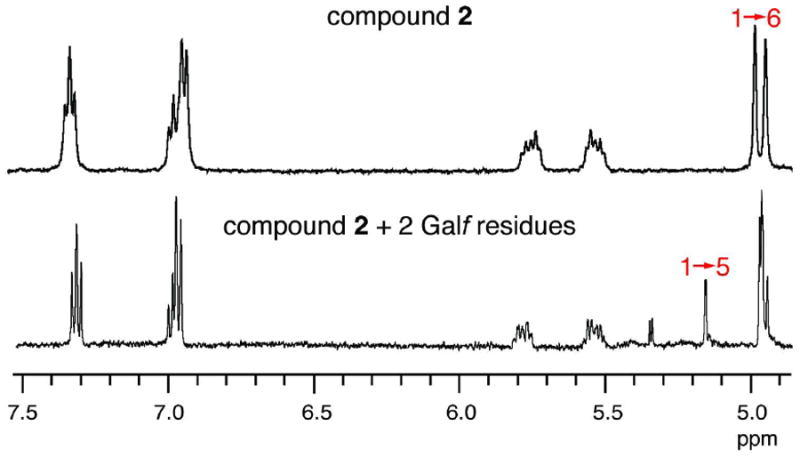
Partial 1H NMR spectra of disaccharide acceptor 2 (top) and isolated tetrasaccharide GlfT2 reaction product (bottom). The peaks at ~4.9 ppm are due to the anomeric protons of acceptor 2. The reaction product gives rise to an additional peak at 5.18 ppm, corresponding to the expected chemical shift for the anomeric proton in a Galf-β-(1→5)-Galf linkage (28).
Site-directed mutagenesis
Quik-Change mutagenesis (Stratagene) was used to introduce mutations in the glfT gene at specific sites. The sequences of oligonucleotide primers for each mutant can be found in Table S2 of the Supporting Information. The previously reported pET-24a:his6-glfT construct was used as template DNA for mutagenesis reactions (30). DpnI (New England Biolabs) was used to digest the template strand after PCR-amplification. DpnI-digested reaction mixtures were transformed by electroporation into DH5α E. coli and plated onto selective media (LBKan). Plasmid DNA was prepared from isolated transformants using a Spin Miniprep kit (Qiagen). The presence of the desired mutation was confirmed by DNA sequence analysis at the University of Wisconsin DNA Sequencing Facility.
Protein production and purification
Plasmid DNA containing either wild-type glfT or site-directed mutants was transformed by electroporation into Tuner® (DE3) E. coli. From an isolated transformant, wild-type or variant protein was produced in 1 L cultures and purified from the cultures as described previously (30). The GlfT2 variants yielded similar amounts of soluble, purified protein as wild-type GlfT2.
GlfT2 activity assays by HPLC
Production of UDP by GlfT2 was monitored using anion-exchange HPLC on a CarboPac PA-100 column (Dionex). Reactions were conducted as previously described (30) with the following changes: the total volume was 60 μL, and either MnCl2 or MgCl2 was used as a source of divalent cation. Mixtures were incubated for 45 min at room temperature and then quenched by the addition of 60 μL of 1:1 MeOH:CHCl3. The aqueous phase was injected onto the anion-exchange column, and UDP was separated from UDP-Galf using the following elution profile with a total flow rate of 0.6 mL/min: linear gradient from 95% ddH2O, 5% buffer (1M NH4OAc pH 8.0) to 20% ddH2O, 80% buffer over 10 min, then isocratic at 20% ddH2O, 80% buffer for 5 min, then linear gradient to 95% ddH2O, 5% buffer over 1 min, then isocratic at 95% ddH2O, 5% buffer for 15 min.
Analysis of GlfT2 reactions by MALDI-TOF MS
Reactions containing UDP-Galf, acceptor, and His6-GlfT2 (wild-type or variant) were carried out as described (30). Typical reaction conditions: 1.25 mM UDP-Galf, 200 μM acceptor, 0.2 μM His6-GlfT2 (wild-type or variant), 50 mM HEPES pH 7.0, 25 mM MgCl2, and 100 mM NaCl in a total volume of 120 μL. Mixtures were incubated for 20 h at room temperature and analyzed as previously described using MALDI-TOF MS (30).
Isolation and characterization of GlfT2 reaction products
To generate short polysaccharides for isolation, conditions for GlfT2-catalyzed polymerization were optimized to exclude the formation of longer polymeric products. Reaction mixtures containing 0.2 μM His6-GlfT2, 1.0 mM UDP-Galf, 1.0 mM acceptor 2, 50 mM HEPES, 25 mM MgCl2, and 100 mM NaCl in a total volume of 120 μL were incubated at room temperature for 1 hour. An equal volume of 1:1 MeOH:CHCl3 was added to quench the reactions, and the mixtures were evaporated to dryness. Reaction progress was assessed using MALDI-TOF MS. The resultant short polysaccharide products (ca. 2 to 7 Galf additions) were then separated by analytical C18 HPLC (Thermo Scientific Hypersil Gold column, 5 μm, 250 × 4.6 mm ID). Gradient elution from 20–80 % MeCN (v/v), 0.1 % AcOH (v/v) was employed for product separation at a flow rate of 1.0 mL/min. Due to low abundance of reaction products, the chromatograph was monitored at 200 nm. The identity of each collected fraction was analyzed by MALDI-TOF MS.
A peak eluting at 8.5 min contained a species with a mass corresponding to the addition of 2 Galf residues to acceptor 2 (m/z observed = 905.6, expected [M + Na]+ = 905.4). This fraction was concentrated under vacuum then resuspended in 99.9 % D2O for analysis by NMR. One-dimensional 1H NMR spectra were recorded on a Bruker Avance III 500i MHz spectrometer. A presaturation pulse was applied to suppress HOD signal. A total of 2048 scans were collected for the tetrasaccharide product. Data processing was performed using XWINNMR (Bruker) and TopSpin (Bruker).
Substrate binding assays using STD-NMR
His6-GlfT2 and variants were expressed and purified using a modification of a reported procedure (41). Purified fractions were concentrated with an Amicon Ultra centrifugal filter device (30,000 molecular weight cut-off) through multiple spins at 5000 × g. The concentrated protein was diluted to ca. 12 mL volume in 20 mM K2HPO4–KH2PO4 buffer, pH 7.6 and concentrated to < 1.5 mL. This process was repeated three times, at which time the concentrated protein sample was diluted in 20 mM K2HPO4–KH2PO4 buffer, pH 7.6 in 99.9 % D2O. The same dilution-concentration sequence was repeated three times until His6-GlfT2 was concentrated to ca. 1 mL (1.0-1.5 mg/mL). UDP-Galf and acceptor 4 were synthesized and purified as previously reported (30).
All STD NMR spectra were recorded on a Bruker DMX-600 spectrometer at 286K with 1024 scans. Samples for STD experiments with UDP-Galf contained 2.0 mM UDP-Galf, 2.0 mM MgCl2 and 6.0 μM His6-GlfT2 in 20 mM K2HPO4–KH2PO4 buffer, at pH 7.6 in 99.9 % D2O. Continuous-wave saturation of protein resonances was performed at -1.0 ppm with a saturation time of 2.0 s (-30 ppm for off resonance spectra). Phase cycling was used to subtract the saturation spectra from off resonance spectra. No ligand STD signal was observed in the absence of protein. Measurement of saturation transfer intensities was performed by direct comparison to a reference one-dimensional 1H spectra (256 scans) of the same sample that was obtained immediately prior to acquisition of the STD spectra. Samples for STD experiments with acceptor 4 contained 1.0 mM acceptor 4, 1.0 mM MgCl2, and 5.0 μM His6-GlfT2. Due to the presence of aliphatic protons in acceptor 4, saturation of protein resonances was performed at 10 ppm. Again, no ligand STD signal was observed in the absence of protein. All other conditions for the STD and reference 1H spectra were identical to spectra obtained with samples containing UDP-Galf. Data processing was performed using XWINNMR (Bruker). For all protein preparations, an unidentified lipid impurity could not be removed, which was observed in all reference 1H and STD NMR spectra in the aliphatic region.
RESULTS
GlfT2 mediates polysaccharide assembly using bifunctional catalytic activity
Purified, recombinant GlfT2 can catalyze the formation of both Galf-β-(1→5)-Galf and Galf-β-(1→6)-Galf linkages using synthetic acceptors (28). The substrates used in these studies were elongated by only a few Galf residues, and we wanted to ensure that GlfT2 behaved similarly when exposed to acceptors that could give rise to polysaccharides. We therefore examined whether our synthetic acceptors (30) could be transformed by GlfT2 into products composed of both β-(1→5) and β-(1→6) linkages. We used reversed phase HPLC to isolate products from exposure of compound 2 (Figure 1C) to His6-GlfT2. A glycoconjugate that had been elongated by 2 Galf residues was analyzed by 1H NMR spectroscopy to determine which glycosidic linkages had been formed. The resulting spectrum contained a new peak, and its chemical shift (5.18 ppm) corresponds to that expected for the anomeric proton of a 1→5 linked Galf residue (Figure 2) (28). Furthermore, the increase in area under the peak that corresponds to the anomeric proton of a 1→6 linked Galf residue (~4.9 ppm) suggested the presence of an additional 1→6 linkage in the +2 Galf product (Figure 2). These studies establish that synthetic acceptor substrates, such as 2, that are processively elongated can be used to investigate the bifunctional catalytic activity of the polymerase GlfT2.
Sequence analysis of GlfT2 suggests one putative active site
To identify potential active sites in GlfT2, we analyzed its amino acid sequence for conserved, functional motifs. Although glycosyltransferases generally have very divergent sequences (42, 43), a key identifying feature of their active sites is the DXD motif (7, 42). Within the primary sequence of GlfT2, the sequence DXD (or DDX) occurs six distinct times out of the 637 amino acids. Presuming that the most critical DXD motifs would be conserved, we examined sequence similarity between GlfT2 and homologous glycosyltransferases. A search using BLAST revealed numerous glycosyltransferases with significant similarity to GlfT2, most of which were from mycobacteria or closely related bacterial species. Multiple sequence alignment of the 50 most similar sequences to GlfT2 showed that most of the highly conserved residues occur within its only predicted domain, the GT-2 domain (Figure 3A, Table S1 and Figures S1 and S2 of the Supporting Information). Furthermore, two DXD motifs were strictly conserved among all 50 homologs: DDD (Asp256–Asp258) and DDA (Asp371–Ala373) (Figures S1 and S2 of the Supporting Information).
Figure 3.
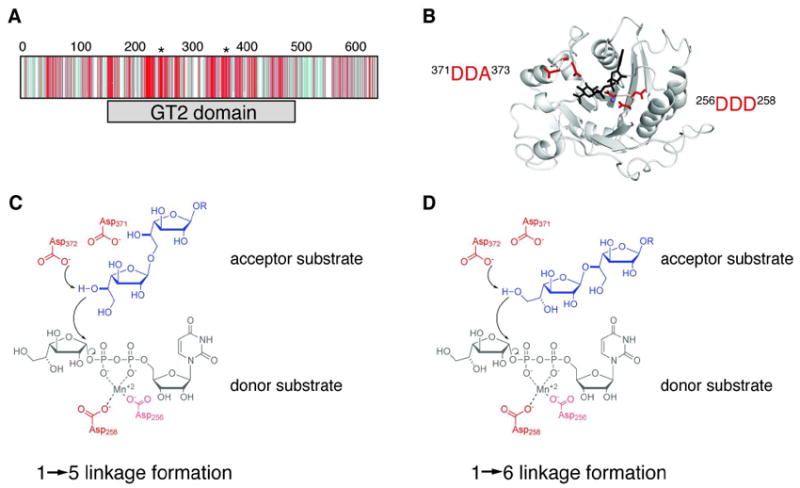
Identification of one putative active site in GlfT2. (A) Depiction of the CLUSTALW2-generated multiple sequence alignment of 50 homologs to GlfT2 shows that the conserved residues (shades of red) occur mostly within the GT-2 domain (gray bar), while divergent residues (shades of blue) occur mostly in the other regions of the protein. The positions of the two conserved DXD motifs are indicated with asterisks above the sequence alignment. (B) A homology model of the GT-2 domain of GlfT2 (residues 160-398) was computed from the structure of chondroitin polymerase from E. coli K4 (K4CP: PDB ID: 2Z86, subunit A). The DDA motif (red, left) and the DDD motif (red, right) are located in a putative active site near the donor substrate from K4CP, UDP-glucuronic acid (black) and the Mn2+ ion (purple sphere). (C, D) Chemical view of the hypothesized roles of the conserved motifs. The DDA motif is predicted to contain the catalytic base that deprotonates the nucelophilic hydroxyl on the acceptor to produce the Galf-β-(1→5)-Galf (C) or Galf-β-(1→6)-Galf (D) glycosidic linkage. The DDD motif is predicted to assist in binding the divalent cation.
To predict roles for these two invariant motifs, we built a homology model for GlfT2. Its full amino acid sequence was entered into the SWISS-MODEL server (http://swissmodel.expasy.org/), and the algorithm generated a structural model of residues 160–398, the region of the GT-2 domain (residues 158–487) (Figure 3A,B; Figure S3 of the Supporting Information). The homology model was based on the structure of E. coli K4 chondroitin polymerase (K4CP; pdb 2z86), a GT-2 family enzyme (23, 44). The modeling results predict that GlfT2, like other transferases in the GT-2 family, adopts the GT-A fold. Comparison of the modeled GlfT2 structure with the K4CP structure enabled identification of a putative donor nucleotide sugar binding site in GlfT2 and suggested functions for the conserved residues of GlfT2. Specifically, the DDD motif is found near the glycosyl donor, where it is positioned to bind a divalent cation that is chelated by the pyrophosphate group of the nucleotide (Figure 3B). The DDA motif is located near the anomeric position of the sugar-nucleotide donor substrate, the expected position for the catalytic base. Indeed, an Asp or Glu residue is found in a similar structural position in other glycosyltransferases of the GT-A superfamily (7). The homology model suggests that both the DDD and the DDA sequences play important roles in catalysis, and this prediction is consistent with the sequence conservation observed for both motifs. We therefore hypothesized that the conserved residues of GlfT2 form one active site in which the DDD and DDA motifs contribute distinct functions to catalysis (Figure 3C).
Altered activities of DDD motif variants support hypothesized active site
If GlfT2 contains only one active site, each distinct linkage should be formed through a similar catalytic mechanism. In general, GT-A glycosyltransferases require divalent cations such as Mg2+ or Mn2+ for catalysis, whereas glycosyltransferases in the GT-B superfamily exhibit activity in the absence of divalent cations (7, 42). It is unknown whether GlfT2 requires divalent cations for activity. Compounds 3 and 5 (Figure 1C) were employed as probes for GlfT2 bifunctionality, as they differ only in the terminal linkage between the Galf residues. Both compounds can be elongated by GlfT2 to give products with degrees of polymerization (20–40 Galf residues) similar to those of endogenous galactan (30, 45). We therefore examined the cation dependence on GlfT2 catalysis Although a coupled, continuous assay for UDP formation has been reported (46), the metal dependence of pyruvate kinase, one of the coupling enzymes, necessitated the development of an alternative approach. To this end, we devised a discontinuous assay in which the UDP product is separated from UDP-Galf substrate by anion-exchange HPLC. When EDTA was present, no UDP was detected. This observation suggests that a divalent metal is essential for catalysis. We next probed the role of the metal ion in polymer formation by exposing GlfT2 to saturating concentrations of donor and acceptor substrate in the presence of EDTA or divalent cation. The addition of EDTA abolished the transfer of Galf residues to either (1→6)-linked acceptor 3 or (1→5)-linked acceptor 5 (Figure S4 of the Supporting Information). These results are consistent with the placement of GlfT2 in the GT-A family, and they highlight the critical role of divalent cations for GlfT2 activity.
Substitution of residues involved in divalent cation binding can alter an enzyme’s cation preference (47). To further examine our prediction that the DDD motif is involved in metal binding, we used site-directed mutagenesis to engineer single point mutations of Asp256 or Asp258 to Ala or Glu in GlfT2. Rates of UDP production by these variants were slower than those obtained with wild-type GlfT2 (Figure 4A). We next tested the metal dependence of these variants. Whereas wild-type GlfT2 generates product at similar reaction rates when either Mg2+ or Mn2+ is present, the Asp256 and Asp258 variants were faster in the presence of Mn2+ (Figure 4A,B). These results provide additional evidence that the invariant DDD motif binds a metal ion required for GlfT2 catalysis. Furthermore, the ability of the DDD variants to form polymers is perturbed for both substrates. When reaction mixtures contained saturating concentrations of acceptor and donor the DDD variants afforded products with at most 15 Galf residues added to 3 or 1 Galf residue added to 5. These degrees of polymerization are lower than those (20–40 Galf residues) attained using wild-type GlfT2 (Figure 5). The DDD variants fail to generate polymers with compound 5, which has a terminal β-(1→5) linkage, and this observation is consistent with the reported preference of wild-type GlfT2 for disaccharide acceptors like 3 that terminate with a β-(1→6) residue (27, 28). Nevertheless, the ability of the DDD variants to utilize either 3 or 5 as an acceptor indicates that even these compromised enzymes can promote the formation of both types of linkage.
Figure 4.
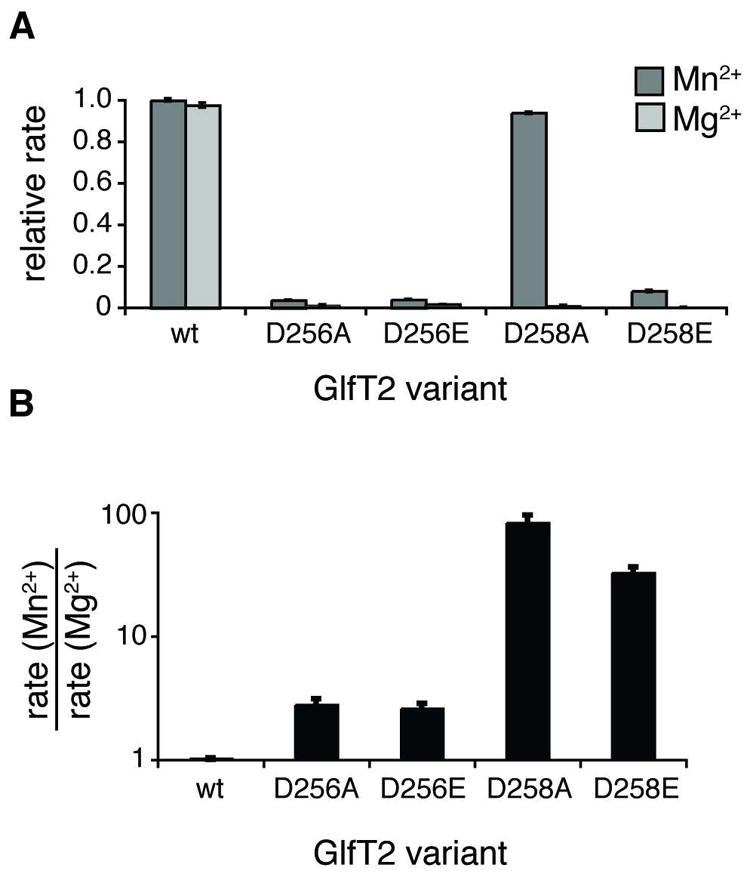
DDD motif variants show altered metal dependence. (A) Rates shown were obtained after 45 min from reaction mixtures containing UDP-Galf, compound 3 (aryl = naphthyl), and the indicated His6-GlfT2 variant in the presence of either Mg2+ (light grey) or Mn2+ (dark grey). The rates are normalized to those obtained with wild-type GlfT2 with added Mn2+. In the presence of Mg2+, the DDD motif variants exhibit significantly decreased rates. In the presence of Mn2+, the D258A variant has rate similar to wild-type GlfT2, and the rates obtained with the other variants are slower. (B) Reaction rates from part A are plotted as rate in the presence of Mn2+ divided by the rate in the presence of Mg2+ for each variant. Whereas wild-type GlfT2 shows no difference in kinetics with either metal, the DDD variants exhibit faster kinetics with Mn2+ than with Mg2+.
Figure 5.
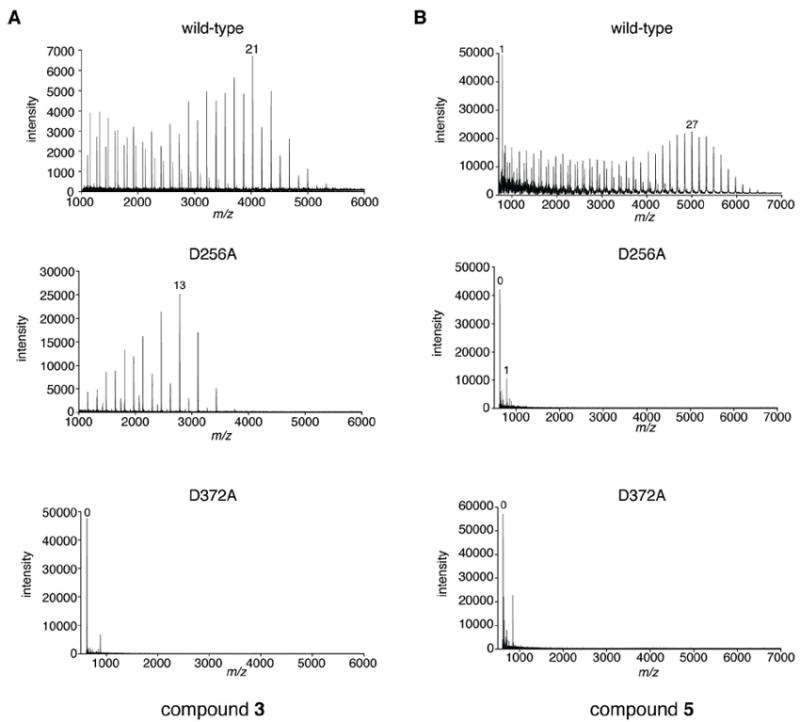
The DDA motif is critical for bifunctionality of GlfT2. Mass spectra are shown from analysis of 20 h incubations of (A) compound 3 or (B) compound 5, UDP-Galf, and the indicated His6-GlfT2 variant. The highest-intensity peak that corresponds to a product with m/z = [M+n*Galf+Na]+, where M is the mass of the unelongated acceptor, is labeled with the value of n. Results from the other DDD motif variants (D256E, D258A, D258E) were similar to those of the D256A variant. The activities of both the D371A and the D372A variants were diminished by approximately the same amount. The m/z value for the unlabeled peak in either spectrum of the D372A variant does not correspond to that of a reaction product.
To examine further the molecular interactions of the DDD motif variants with donor substrate, we used saturation transfer distance NMR (STD-NMR) spectroscopy. This technique can report on the interaction of small molecule ligands with a protein through transfer of magnetization from the protein to the bound ligand (48). STD-NMR has been used to monitor acceptor and donor binding to GlfT2 (32, 49). Upon addition of wild-type GlfT2 to a solution of UDP-Galf, we observed STD signals that correspond to the sugar and nucleotide portion of the donor substrate. These data indicate that complex formation occurs. (Figure 6A, Figure S5 of the Supporting Information). The D256A GlfT2 and D258A GlfT2 variants still bind UDP-Galf but give rise to weaker signals corresponding to protons of the uracil and ribose moieties (Figure 6A, Figure S5 of the Supporting Information). These differences suggest that the DDD motif variants have fewer specific interactions with UDP-Galf. Regardless of the underlying basis for the differences, the STD-NMR spectra provide additional evidence that the DDD motif contributes to donor binding. Taken together, the data support the predicted importance of the DDD motif for catalysis and corroborate the homology model prediction that GlfT2 possesses a single active site.
Figure 6.
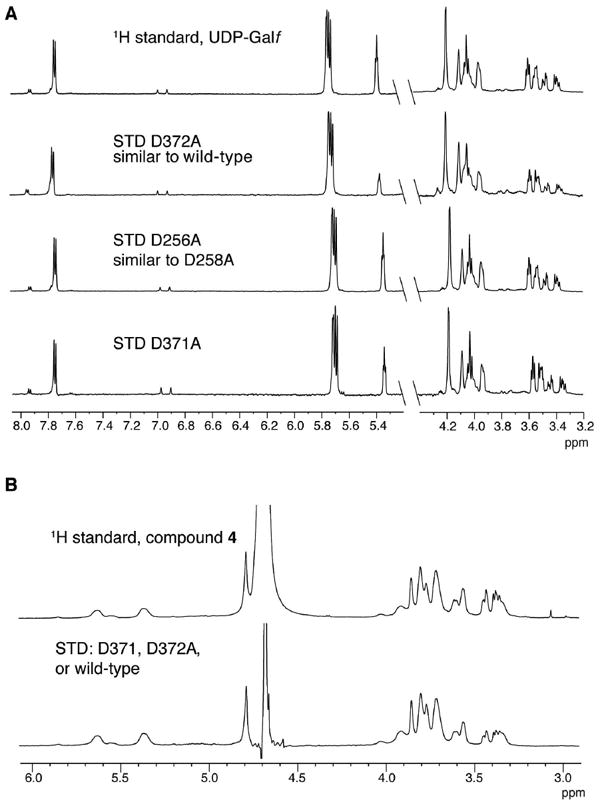
Binding of donor and acceptor substrates by GlfT2 variants. (A) The 1H NMR spectrum of UDP-Galf (in the absence of protein; top) is overlaid with the STD NMR spectra observed upon addition of GlfT2 (wild-type or variant as indicated) to a solution of UDP-Galf. (B) The 1H NMR spectrum of compound 4 (in absence of protein; top) is overlaid with the STD NMR spectra observed upon addition of the indicated GlfT2 variant to a solution of compound 4 (bottom spectrum). The STD spectra from wild-type, D371A, and D372A GlfT2 were nearly identical and are plotted as one spectrum for clarity.
Substitution of the DDA motif impairs GlfT2 activity
We postulated the invariant DDA motif of GlfT2 would contain residues critical for catalysis. Identification of such residues would allow us to distinguish between two mechanistic hypotheses for the bifunctionality of GlfT2. Specifically, if GlfT2 contains two active sites, then alteration of the DDA motif should only disrupt the ability of GlfT2 to catalyze the formation of either the β-(1→6) linkage or the β-(1→5) linkage but not both. In this scenario, if DDA variants were unable to catalyze Galf addition to one acceptor (for example, 3), we would still expect to observe significant reaction with an acceptor possessing the differing terminal linkage (for example, 5). A similar mutagenesis strategy has uncovered the two-active-site bifunctionality of other GT-2 enzymes such as PmHAS (20-22) and K4 chondroitin polymerase (18, 23). In contrast, if GlfT2 has one active site, then variation of the DDA motif should disrupt both catalytic activities and thereby impair Galf addition to both 3 and 5.
We engineered variants of Asp371 and Asp372 to Ala in GlfT2 and assessed their activity. Using a coupled, continuous assay (UDP detection limit of approximately 0.04 μM/min), no UDP formation was observed in reaction mixtures containing D371A or D372A GlfT2 variants and either acceptor 3 or 5. These protein variants are at least 300-fold slower than wild-type GlfT2 (steady-state rate of 12 μM/min). To test whether the DDA variants can promote efficient elongation, we exposed D371A or D372A GlfT2 to saturating concentrations of donor and acceptor substrates. Analysis of reaction mixtures by MALDI-TOF MS showed very low intensity peaks consistent with the presence of +1 and +2 Galf products. The reaction did not proceed efficiently to afford the +1 product and no polymeric products were observed (Figure 5). The inability of D371A or D372A GlfT2 to elongate either 3 or 5 indicates that the DDA motif is required for both catalytic activities of GlfT2.
To test whether the severely reduced activity of the DDA motif variants is due to impaired binding of the donor and acceptor substrates, we again used STD-NMR. These investgations revealed that both the D371A or D372A GlfT2 variants bind UDP-Galf (Figure 6A, Figure S5 of the Supporting Information). The STD signals obtained with D372A and wild-type GlfT2 are similar, indicating the enzymes bind their donor substrate in a similar orientation, whereas the distinct STD signals for D371A suggest some flexibility in the donor binding site. To test whether the D371A and D372A variants can bind the acceptor, we used acceptor 4 instead of the aryl-containing acceptors. The latter were not amenable to STD-NMR, because resonances from the aryl group overlap with the frequency used to saturate the protein (10 ppm). When a solution of compound 4 was exposed to either wild-type GlfT2 or a GlfT2 variant (D371A or D372A), nearly identical STD signals were obtained (Figure 6B). These results indicate that the DDA variants bind acceptor and that they bind acceptor in the same orientation as wild-type GlfT2 (Figure 6B). Thus, the decreased activity of the DDA motif variants is not due to loss of substrate binding.
We tested the possibility that the two Asp residues within the DDA motif could have different. For an Asp serving as a catalytic base, substitution with Glu should be deleterious, as a change in side-chain length should alter the orientation of the residue in the active site and disrupt the geometric requirements for catalysis. In several sugar-modifying enzymes, including GT-2 family glycosyltransferases, replacement of the proposed catalytic Asp or Glu with Glu or Asp, respectively, greatly diminishes activity (50-52). Therefore, we tested the activities of D371E and D372E GlfT2 with substrates 3 and 5. Reaction mixtures containing D371E GlfT2 yielded polymeric products (Figure S6 of the Supporting Information), but those with D372E GlfT2 did not. The results suggest that Asp372 acts as the catalytic base that participates in forming β-(1→5) and β-(1→6) linkages.
GlfT2 exhibits greater processivity with a tetrasaccharide acceptor (31); therefore, we tested whether the DDA motif variants elongate a substrate that more readily gives rise to polymers. We incubated GlfT2 and DDA motif variants with the purified +2 glycoconjugate generated from acceptor 2 and analyzed reaction mixtures by MALDI-TOF MS. The variants D371A, D372A, or D372E GlfT2 did not promote addition of Galf residues to the tetrasaccharide, whereas D371E and wild-type GlfT2 did catalyze polymer formation (Figure S7 of the Supporting Information). This observation of impaired activity with the best available synthetic acceptor further supports the critical role for the DDA motif in bifunctional catalysis. Overall, the data indicate that both catalytic activities of GlfT2 reside in a single active site.
Generality of single active site bifunctionality in carbohydrate polymerization
Because the GT-2 family of glycosyltransferases includes a number of bifunctional carbohydrate polymerases, we analyzed other family members to ascertain whether they mediate bifunctional catalysis using a single active site. First, we computed homology models for hyaluronan synthases that contain only one GT-2 domain, such as those from Gram-positive bacteria and from animals (18) (Figure 7A, Figure S8A of the Supporting Information). Superposition of the model for GlfT2 with that postulated for the bacterial hyaluronan synthase SpHasA reveals that the conserved Asp residues in SpHasA align with the DDD and DDA motifs of GlfT2, reflecting the predicted structural homology between these proteins. Moreover, the putative catalytic base in GlfT2 corresponds to an Asp residue in SpHasA that is conserved in mammalian hyaluronan synthases (53). Interestingly, replacement of this Asp residue by Glu knocks out both GlcUA-β-(1→3)-GlcNAc and GlcNAc-β-(1→4)-GlcUA transferase activities for mouse hyaluronan synthase (53). Although the precise role of that residue was not determined, our results suggest it is the catalytic base and that these hyaluronan synthases contain only one active site. Another bifunctional GT-2-family polymerase is the enzyme that catalyzes the synthesis of a repeating disaccharide sequence in the type 3 streptococcal capsular polysaccharide (53). It is unclear whether this enzyme has one or two separate catalytic sites (15, 54). Nevertheless, the presence of a conserved Asp residue at the position corresponding to critical Asp residues of hyaluronan synthase and GlfT2 suggests that this streptococcal polymerase also functions via using a single bifunctional catalytic site.
Figure 7.
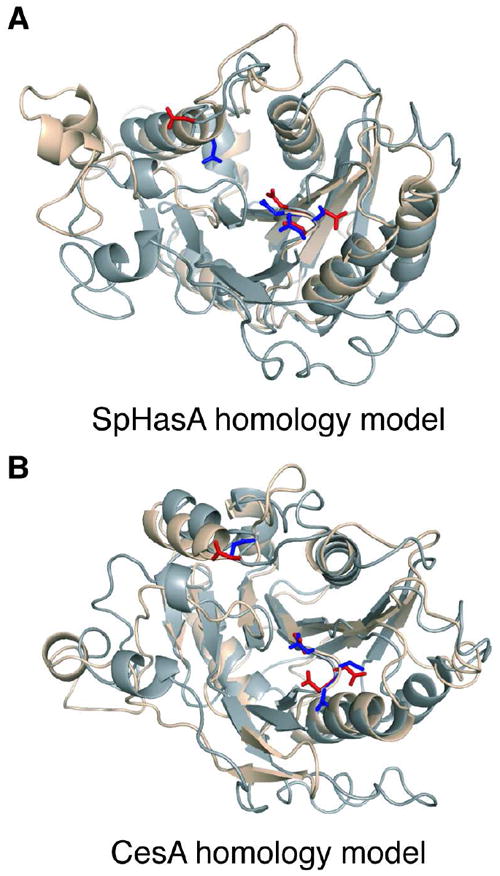
Conservation of single active site architecture in other GT-2 carbohydrate polymerases. (A) Superposition of SpHasA homology model (wheat) with GlfT2 homology model (grey) shows that D372 (red, left) in GlfT2 overlaps with D235 (blue, left) in SpHasA and that the DDD motif (red, right) in GlfT2 overlaps with the DSD motif of SpHasA (blue, right). (B) Superposition of CesA homology model (wheat) with GlfT2 homology model (grey) reveals that D372 (red, left) in GlfT2 is in similar position as D333 (blue, left) in CesA. Furthermore, the DDD motif of GlfT2 (red, right) overlaps with the DCD motif in CesA (blue, right).
The GT-2 family encompasses enzymes that catalyze cellulose polymerization. Cellulose is a linear polymer consisting of β-(1→4) linked glucose residues. The repeating unit can be thought of as a disaccharide because each β-(1→4) linked glucose is inverted with respect to the next (1). It has been noted that this arrangement of glucose residues within the acceptor could raise torsional challenges for catalysis, leading to the proposal that cellulose synthase uses two active sites (55, 56). Still, bacterial cellulose synthase contains one GT-2 domain that has been postulated to possess one active site (57). Our homology model of bacterial cellulose synthase (Figure 7B, Figure S8B of the Supporting Information) indicates that the putative catalytic base in GlfT2 corresponds to a cellulose synthase Asp residue that is conserved in bacterial (55) and plant (58, 59) enzymes. Indeed, mutagenesis studies have shown this residue is critical for cellulose formation in bacteria (55). Notably, a strain of Arabidopsis thaliana that showed severely reduced cellulose content and collapsed vasculature contained a single missense mutation in the codon for the analogous conserved Asp residue (Asp683) in one of its cellulose synthase genes (59). Our studies with GlfT2 suggest a molecular mechanism for the phenotypes of cellulose synthase mutants. They also support the proposal that cellulose synthases, like other bifunctional GT-2 polymerases including GlfT2, have a single active site. Together, our results lead us to postulate that many GT-2 family bifunctional carbohydrate polymerases share the ability to catalyze polysaccharide synthesis using a single active site.
DISCUSSION
Single-active site bifunctionality in carbohydrate polymerization
A significant question in carbohydrate polymerization is how glycosyltransferases direct polysaccharide sequence and length in the absence of a template. The one-enzyme-one-linkage paradigm predicts that one enzyme would be sufficient to synthesize polysaccharides containing one repeating monomer unit (homopolysaccharides), and that multiple enzymes would be necessary to synthesize polysaccharides containing a disaccharide repeating unit (heteropolysaccharides). Our results highlight the importance of an alternative strategy for control of polysaccharide structure in the absence of a template: some carbohydrate polymerases employ one active site to mediate formation of a heteropolysaccharide.
Our results address questions about the mechanism for carbohydrate polymerization by GT-2 family polymerases. The use of a single active site for production of an alternating polysaccharide has been previously suggested for hyaluronan synthase, cellulose synthase, and the enzyme that catalyzes formation of type 3 streptococcal capsular polysaccharide (18, 53, 54, 57). These conclusions, however, were based on in vitro assays using crude membrane preparations or solely on the structure of a homologous glycosyltransferase (SpsA) whose particular substrates and products are unknown. The finding that one active site contains dual catalytic activity for the sialyltransferase CstII (60) is intriguing but does not provide insight into general mechanisms for bifunctional polymerization; CstII does not form a polymer, and its catalytic mechanism is expected to be distinct from GlfT2 and other bifunctional GT-2 enzymes (60). Our investigations using purified enzyme and substrates provide strong evidence for single-active site bifunctionality in a carbohydrate polymerization.
Intriguingly, analysis of data from carbohydrate polymerases that mediate bifunctional polymerization suggests that the mechanistic features that allow for bifunctional catalysis appear to be intertwined with those underlying elongation. Polymerases can carry out catalysis using either processive or distributive mechanisms. In processive elongation, the enzyme retains the growing polymer through multiple rounds of monomer addition; in distributive elongation, the enzyme releases the growing polymer after each addition of monomer unit (61). HA polymerization by PmHAS, a two-active site polymerase, is distributive (18), and a similar catalytic mode is likely used by the homologous K4CP, another two-active site polymerase (23). In contrast, those bifunctional polymerases that use a processive mechanism for polymerization likely use one active site. For example, only a single active site is apparent in the processive bacterial and mammalian HA synthases (18). Similarly, the polymerase that catalyzes processive elongation of streptococcal type 3 capsular polysaccharide is thought to have a single active site (54). Finally, our results with GlfT2, a processive polymerase (30, 31), place it in the category of bifunctional enzymes with single active sites.
These examples suggest that the number of active sites in a bifunctional carbohydrate polymerase influences the mechanism for chain elongation. Two active sites would suffice for a distributive, bifunctional polymerization, as release of product from one site would allow that product to bind as the acceptor substrate in the second active site. Two active sites may be unfavorable, however, for a processive bifunctional polymerization, as a processive polymerase retains the polymer through multiple rounds of monomer addition. Thus, one active site with dual activity may be a requirement for bifunctional processive carbohydrate polymerases (Figure 8). The processivity of GlfT2 is therefore mechanistically consistent with its usage of a single active site for bifunctional catalysis. We note, however, that our data do not address whether the polymerizing glycosyltransferase functions as a monomer or as an oligomer. For monomeric or multimeric bifunctional glycosyltransferases, we anticipate that any/each active site that catalyzes processive chain elongation will be capable of catalyzing the formation of two distinct types of glycosidic bonds.
Figure 8.
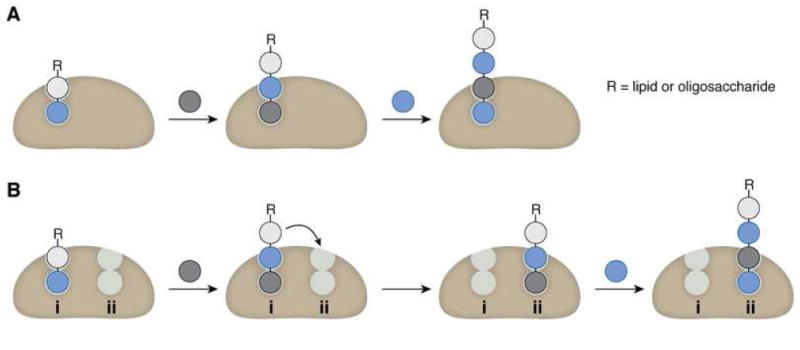
Comparison of one active site versus two active sites for processive synthesis of an alternating polymer. (A) The use of one multifunctional active site facilitates retention of the growing polymer by the polymerase through multiple catalytic additions. (B) The use of two active sites for processive synthesis requires translocation of the growing chain between each site, which adds complexity to the mechanism of elongation and may be disfavored by the principle of least motion.
This correlation between bifunctionality and the mechanism of elongation has implications for understanding how single-enzyme bifunctionality could evolve. Fusion of two GT domains into a single polypetide to form a bifunctional enzyme may constrain such an enzyme to a distributive mechanism of polymerization. It is not easy to envision how an enzyme possessing two distinct GT domains might evolve to use a processive mechanism. In contrast, a single-active site GT that is initially processive (or initially bifunctional) could accumulate mutations to confer dual catalytic activity (or processivity).
Analysis of possible mechanisms for specificity in elongation of an alternating polymer
For single-active site, processive glycosyltransferases with dual catalytic activity, a key question is how one active site can form two distinct glycosidic linkages to assemble an alternating polymer. This question is particularly significant, as glycosyltransferases do not use a template so they cannot employ mismatch-based proofreading. Two types of polymerizations are prevalent in biosynthesis of heteropolysaccharides. In one, the polymerase connects two distinct donor residues to form a polymer of alternating sugar residues. In the second type, the glycosyltransferase uses a single donor sugar to generate a polymer composed of alternating, regioisomeric linkages. For either type of bifunctional polymerization to occur processively in a single active site, a means of maintaining fidelity is required.
Four parameters affect glycosyltransferase specificity: two involve selective binding of a donor substrate and an acceptor substrate and the other two include regioselectivity and stereoselectivity in glycosidic bond formation. Examples of glycosyltransferases in which one of these specificity parameters is relaxed have emerged recently (62-65). Synthesis of each of the prevalent types of heteropolysaccharides (see above) by one active site requires only some of these four parameters to have dual specificity. For formation of a polymer with alternating donor residues, a single-active site polymerase would need to exhibit relaxed donor specificity and relaxed acceptor specificity. For formation of a heteropolysaccharide composed of alternating, regioisomeric linkages, a single-active site polymerase would maintain donor specificity but exhibit relaxed acceptor specificity and relaxed regioselectivity.
These general considerations allow us to speculate on features that could confer bifunctional catalytic activity to the single active site in GlfT2. The bifunctional active site of GlfT2 must accommodate dual acceptor specificity and dual regioselectivity. Although GlfT2 exhibits dual acceptor specificity, its single active site still can discriminate against acceptor substrates that do not contain Galf residues, including ones with structurally similar sugars or a galactopyranose residue (28, 45). For glycosyltransferases, regioselectivity is determined by the relative orientation of the catalytic residue, the nucleophile of the acceptor, and the anomeric position of the donor (7). We therefore postulate that the dual activity of a bifunctional carbohydrate polymerase arises from the ability of the catalytic aspartic acid residue to form a catalytically productive orientiation with 2 distinct acceptor hydroxyl groups. Such dual positioning could be governed by the enzyme, the acceptor, or a combination of both (66-68). In the case of GlfT2, for example, differential binding of the two different terminal linkages (β-(1→6) or β-(1→5)) could orient the appropriate hydroxyl group such that it serves as the nucleophile (Figure 3c, 3D). Alternatively, the intrinsic conformation of the acceptor itself could dictate which linkage is formed (69). For the dual regioselectivity of GlfT2, the conformation of the growing polymer could present geometric constraints that ensure placement of the appropriate hydroxyl nucleophile of the terminal residue within the active site. Structural data on GlfT2 in complex with its acceptor substrates, computational modeling of conformations of polymeric products, and studies using additional synthetic acceptor analogs could illuminate the relative contributions of these different possible means of achieving bifunctional yet selective catalytic activity during polymer elongation.
In conclusion, our data indicate that GlfT2 uses one active site with dual regioselectivity to mediate polymerization of a carbohydrate containing a disaccharide repeating unit. Our findings and analysis suggest that one active site, bifunctional catalysis occurs in additional GT-2 family enzymes. Furthermore, we postulate that processive bifunctional glycosyltransferases are likely to employ one active site. Structural data on bifunctional carbohydrate polymerases would further illuminate how an enzyme with a single active site can form a heteropolymer in a selective manner.
Supplementary Material
Acknowledgments
The authors thank W.M. Westler and T.D. Gruber for assistance with NMR experiments and M.H. Foss for assistance in engineering some of the site-directed variants.
Funding: This research was supported by National Institutes of Health (R01-AI063596). JFM was supported by a National Science Foundation Graduate Research Fellowship and by the Molecular Biosciences Training Grant (NIH T32-GM007215) at the University of Wisconsin–Madison. RAS was supported by an American Chemical Society Division of Medicinal Chemistry Graduate Fellowship. MRL was supported by an NIH postdoctoral fellowship (F32-GM089219). MALDI-TOF MS data were obtained at the University of Wisconsin, Madison Biophysics Instrumentation Facility, which is supported by the University of Wisconsin, Madison; NSF Grant BIR-9512577; and the NIH Grant S10 RR13790. NMR experiments were performed at the National Magnetic Resonance Facility at Madison, supported by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS). This facility is also supported by the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
ABBREVIATIONS
- BLAST
basic local alignment search tool
- CesA
bacterial cellulose synthase
- GAG
glycosaminoglycan
- Galf
galactofuranose
- GlfT2
galactofuranosyl-transferase 2
- GT-2
glycosyltransferase family 2
- GT-A
glycosyltransferase superfamily A
- GT-B
glycosyltransferase superfamily B
- HA
hyaluronic acid
- K4CP
K4 chondroitin polymerase
- MALDI-TOF
matrix-assisted laser desorption/ionization time-of-flight
- PDB
protein data bank
- PmHAS
hyaluronic acid synthase from Pasteurella multocida
- SpHasA
hyaluronic acid synthase from Streptococcus pyogenes
- STD-NMR
saturation transfer difference nuclear magnetic resonance
Footnotes
SUPPORTING INFORMATION AVAILABLE. GlfT2 homologs used in the multiple sequence alignment (Table S1); Multiple sequence alignment showing conservation of DDD motif (Figure S1); Multiple sequence alignment showing conservation of DDA motif (Figure S2); Sequence alignment used for homology modeling of GlfT2 (Figure S3); MALDI-TOF spectra showing metal-dependence of GlfT2 catalysis (Figure S4); Analysis of STD NMR spectra for GlfT2 variants binding to UDP-Galf (Figure S5); Wild-type polymerization activity of GlfT2 D371E variant (Figure S6); GlfT2 DDA motif variants show impaired activity with tetrasaccharide acceptor substrate (Figure S7); Sequence alignments used for homology modeling of other GT-2 glycosyltransferases (Figure S8); and oligonucleotide primers used for site-directed mutagenesis (Table S2). This material is available free of charge at http://pubs.acs.org
References
- 1.Somerville C. Cellulose synthesis in higher plants. Annu Rev Cell Dev Biol. 2006;22:53–78. doi: 10.1146/annurev.cellbio.22.022206.160206. [DOI] [PubMed] [Google Scholar]
- 2.Ross P, Mayer R, Benziman M. Cellulose biosynthesis and function in bacteria. Microbiol Rev. 1991;55:35–58. doi: 10.1128/mr.55.1.35-58.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stubbe J, Tian J, He A, Sinskey AJ, Lawrence AG, Liu P. Nontemplate-dependent polymerization processes: polyhydroxyalkanoate synthases as a paradigm. Annu Rev Biochem. 2005;74:433–480. doi: 10.1146/annurev.biochem.74.082803.133013. [DOI] [PubMed] [Google Scholar]
- 4.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 5.Kjellén L, Lindahl U. Proteoglycans: structures and interactions. Annu Rev Biochem. 1991;60:443–475. doi: 10.1146/annurev.bi.60.070191.002303. [DOI] [PubMed] [Google Scholar]
- 6.Kiessling LL, Splain RA. Chemical approaches to glycobiology. Annu Rev Biochem. 2010;79:619–653. doi: 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]
- 7.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 8.Herget S, Toukach PV, Ranzinger R, Hull WE, Knirel YA, von der Lieth CW. Statistical analysis of the Bacterial Carbohydrate Structure Data Base (BCSDB): characteristics and diversity of bacterial carbohydrates in comparison with mammalian glycans. BMC Struct Biol. 2008;8:35–54. doi: 10.1186/1472-6807-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werz DB, Ranzinger R, Herget S, Adibekian A, von der Lieth CW, Seeberger PH. Exploring the structural diversity of mammalian carbohydrates (“glycospace”) by statistical databank analysis. ACS Chem Biol. 2007;2:685–691. doi: 10.1021/cb700178s. [DOI] [PubMed] [Google Scholar]
- 10.Hagopian A, Eylar EH. Glycoprotein biosynthesis: studies on the receptor specificity of the polypeptidyl:N-acetylgalactosaminyl transferase from bovine submaxillary glands. Arch Biochem Biophys. 1968;128:422–433. doi: 10.1016/0003-9861(68)90048-9. [DOI] [PubMed] [Google Scholar]
- 11.Lee JY, Spicer AP. Hyaluronan: a multifunctional, megaDalton, stealth molecule. Curr Opin Cell Biol. 2000;12:581–586. doi: 10.1016/s0955-0674(00)00135-6. [DOI] [PubMed] [Google Scholar]
- 12.Sugahara K, Kitagawa H. Heparin and heparan sulfate biosynthesis. IUBMB Life. 2002;54:163–175. doi: 10.1080/15216540214928. [DOI] [PubMed] [Google Scholar]
- 13.Silbert JE, Sugumaran G. Biosynthesis of chondroitin/dermatan sulfate. IUBMB Life. 2002;54:177–186. doi: 10.1080/15216540214923. [DOI] [PubMed] [Google Scholar]
- 14.DeAngelis PL. Microbial glycosaminoglycan glycosyltransferases. Glycobiology. 2002;12:9R–16R. doi: 10.1093/glycob/12.1.9r. [DOI] [PubMed] [Google Scholar]
- 15.Forsee WT, Cartee RT, Yother J. Biosynthesis of type 3 capsular polysaccharide in Streptococcus pneumoniae. Enzymatic chain release by an abortive translocation process. J Biol Chem. 2000;275:25972–25978. doi: 10.1074/jbc.M002613200. [DOI] [PubMed] [Google Scholar]
- 16.Shen GJ, Datta AK, Izumi M, Koeller KM, Wong CH. Expression of α2,8/2,9-polysialyltransferase from Escherichia coli K92. Characterization of the enzyme and its reaction products. J Biol Chem. 1999;274:35139–35146. doi: 10.1074/jbc.274.49.35139. [DOI] [PubMed] [Google Scholar]
- 17.McGowen MM, Vionnet J, Vann WF. Elongation of alternating α2,8/2,9 polysialic acid by the Escherichia coli K92 polysialyltransferase. Glycobiology. 2001;11:613–620. doi: 10.1093/glycob/11.8.613. [DOI] [PubMed] [Google Scholar]
- 18.Weigel PH, DeAngelis PL. Hyaluronan synthases: a decade-plus of novel glycosyltransferases. J Biol Chem. 2007;282:36777–36781. doi: 10.1074/jbc.R700036200. [DOI] [PubMed] [Google Scholar]
- 19.Busse M, Feta A, Presto J, Wilén M, Grønning M, Kjellén L, Kusche-Gullberg M. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J Biol Chem. 2007;282:32802–32810. doi: 10.1074/jbc.M703560200. [DOI] [PubMed] [Google Scholar]
- 20.Williams KJ, Halkes KM, Kamerling JP, DeAngelis PL. Critical elements of oligosaccharide acceptor substrates for the Pasteurella multocida hyaluronan synthase. J Biol Chem. 2006;281:5391–5397. doi: 10.1074/jbc.M510439200. [DOI] [PubMed] [Google Scholar]
- 21.Jing W, DeAngelis PL. Dissection of the two transferase activities of the Pasteurella multocida hyaluronan synthase: two active sites exist in one polypeptide. Glycobiology. 2000;10:883–889. doi: 10.1093/glycob/10.9.883. [DOI] [PubMed] [Google Scholar]
- 22.Jing W, DeAngelis PL. Analysis of the two active sites of the hyaluronan synthase and the chondroitin synthase of Pasteurella multocida. Glycobiology. 2003;13:661–671. doi: 10.1093/glycob/cwg085. [DOI] [PubMed] [Google Scholar]
- 23.Sobhany M, Kakuta Y, Sugiura N, Kimata K, Negishi M. The chondroitin polymerase K4CP and the molecular mechanism of selective bindings of donor substrates to two active sites. J Biol Chem. 2008;283:32328–32333. doi: 10.1074/jbc.M804332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zak BM, Crawford BE, Esko JD. Hereditary multiple exostoses and heparan sulfate polymerization. Biochim Biophys Acta. 2002;1573:346–355. doi: 10.1016/s0304-4165(02)00402-6. [DOI] [PubMed] [Google Scholar]
- 25.Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 26.Besra GS, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. A new interpretation of the structure of the mycolyl-arabinogalactan complex of Mycobacterium tuberculosis as revealed through characterization of oligoglycosylalditol fragments by fast-atom bombardment mass spectrometry and 1H nuclear magnetic resonance spectroscopy. Biochemistry. 1995;34:4257–4266. doi: 10.1021/bi00013a015. [DOI] [PubMed] [Google Scholar]
- 27.Kremer L, Dover LG, Morehouse C, Hitchin P, Everett M, Morris HR, Dell A, Brennan PJ, McNeil MR, Flaherty C, Duncan K, Besra GS. Galactan biosynthesis in Mycobacterium tuberculosis. Identification of a bifunctional UDP-galactofuranosyltransferase. J Biol Chem. 2001;276:26430–26440. doi: 10.1074/jbc.M102022200. [DOI] [PubMed] [Google Scholar]
- 28.Rose NL, Completo GC, Lin S, McNeil MR, Palcic MM, Lowary TL. Expression, purification, and characterization of a galactofuranosyltransferase involved in Mycobacterium tuberculosis arabinogalactan biosynthesis. J Am Chem Soc. 2006;128:6721–6729. doi: 10.1021/ja058254d. [DOI] [PubMed] [Google Scholar]
- 29.Belánová M, Dianisková P, Brennan PJ, Completo GC, Rose NL, Lowary TL, Mikusová K. Galactosyl transferases in mycobacterial cell wall synthesis. J Bacteriol. 2008;190:1141–1145. doi: 10.1128/JB.01326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.May JF, Splain RA, Brotschi C, Kiessling LL. A tethering mechanism for length control in a processive carbohydrate polymerization. Proc Natl Acad Sci USA. 2009;106:11851–11856. doi: 10.1073/pnas.0901407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levengood MR, Splain RA, Kiessling LL. Monitoring Processivity and Length Control of a Carbohydrate Polymerase. J Am Chem Soc. 2011 doi: 10.1021/ja204448t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szczepina MG, Zheng RB, Completo GC, Lowary TL, Pinto BM. STD-NMR studies suggest that two acceptor substrates for GlfT2, a bifunctional galactofuranosyltransferase required for the biosynthesis of Mycobacterium tuberculosis arabinogalactan, compete for the same binding site. ChemBioChem. 2009;10:2052–2059. doi: 10.1002/cbic.200900202. [DOI] [PubMed] [Google Scholar]
- 33.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 34.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 35.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 38.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 40.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alderwick LJ, Dover LG, Veerapen N, Gurcha SS, Kremer L, Roper DL, Pathak AK, Reynolds RC, Besra GS. Expression, purification and characterisation of soluble GlfT and the identification of a novel galactofuranosyltransferase Rv3782 involved in priming GlfT-mediated galactan polymerisation in Mycobacterium tuberculosis. Protein Expr Purif. 2008;58:332–341. doi: 10.1016/j.pep.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y, Walker S. Remarkable structural similarities between diverse glycosyltransferases. Chem Biol. 2002;9:1287–1296. doi: 10.1016/s1074-5521(02)00295-8. [DOI] [PubMed] [Google Scholar]
- 43.Campbell JA, Davies GJ, Bulone V, Henrissat B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J. 1997;326:929–939. doi: 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osawa T, Sugiura N, Shimada H, Hirooka R, Tsuji A, Shirakawa T, Fukuyama K, Kimura M, Kimata K, Kakuta Y. Crystal structure of chondroitin polymerase from Escherichia coli K4. Biochem Biophys Res Commun. 2008;378:10–14. doi: 10.1016/j.bbrc.2008.08.121. [DOI] [PubMed] [Google Scholar]
- 45.Splain RA, Kiessling LL. Synthesis of galactofuranose-based acceptor substrates for the study of the carbohydrate polymerase GlfT2. Bioorg Med Chem. 2010;18:3753–3759. doi: 10.1016/j.bmc.2010.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rose NL, Zheng RB, Pearcey J, Zhou R, Completo GC, Lowary TL. Development of a coupled spectrophotometric assay for GlfT2, a bifunctional mycobacterial galactofuranosyltransferase. Carbohydr Res. 2008;343:2130–2139. doi: 10.1016/j.carres.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 47.Boeggeman E, Qasba PK. Studies on the metal binding sites in the catalytic domain of beta1,4-galactosyltransferase. Glycobiology. 2002;12:395–407. doi: 10.1093/glycob/cwf045. [DOI] [PubMed] [Google Scholar]
- 48.Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed Engl. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 49.Szczepina MG, Zheng RB, Completo GC, Lowary TL, Pinto BM. STD-NMR studies of two acceptor substrates of GlfT2, a galactofuranosyltransferase from Mycobacterium tuberculosis: epitope mapping studies. Bioorg Med Chem. 2010;18:5123–5128. doi: 10.1016/j.bmc.2010.05.069. [DOI] [PubMed] [Google Scholar]
- 50.Garinot-Schneider C, Lellouch AC, Geremia RA. Identification of essential amino acid residues in the Sinorhizobium meliloti glucosyltransferase ExoM. J Biol Chem. 2000;275:31407–31413. doi: 10.1074/jbc.M004524200. [DOI] [PubMed] [Google Scholar]
- 51.Keenleyside WJ, Clarke AJ, Whitfield C. Identification of residues involved in catalytic activity of the inverting glycosyl transferase WbbE from Salmonella enterica serovar Borreze. J Bacteriol. 2001;183:77–85. doi: 10.1128/JB.183.1.77-85.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCarter JD, Withers SG. Mechanisms of enzymatic glycoside hydrolysis. Curr Opin Struct Biol. 1994;4:885–892. doi: 10.1016/0959-440x(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida M, Itano N, Yamada Y, Kimata K. In vitro synthesis of hyaluronan by a single protein derived from mouse HAS1 gene and characterization of amino acid residues essential for the activity. J Biol Chem. 2000;275:497–506. doi: 10.1074/jbc.275.1.497. [DOI] [PubMed] [Google Scholar]
- 54.Forsee WT, Cartee RT, Yother J. A kinetic model for chain length modulation of Streptococcus pneumoniae cellubiuronan capsular polysaccharide by nucleotide sugar donor concentrations. J Biol Chem. 2009;284:11836–11844. doi: 10.1074/jbc.M900379200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saxena IM, Brown RM, Dandekar T. Structure–function characterization of cellulose synthase: relationship to other glycosyltransferases. Phytochemistry. 2001;57:1135–1148. doi: 10.1016/s0031-9422(01)00048-6. [DOI] [PubMed] [Google Scholar]
- 56.Saxena IM, Brown RM, Fevre M, Geremia RA, Henrissat B. Multidomain architecture of beta-glycosyl transferases: implications for mechanism of action. J Bacteriol. 1995;177:1419–1424. doi: 10.1128/jb.177.6.1419-1424.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Charnock SJ, Henrissat B, Davies GJ. Three-dimensional structures of UDP-sugar glycosyltransferases illuminate the biosynthesis of plant polysaccharides. Plant Physiol. 2001;125:527–531. doi: 10.1104/pp.125.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pear JR, Kawagoe Y, Schreckengost WE, Delmer DP, Stalker DM. Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. Proc Natl Acad Sci USA. 1996;93:12637–12642. doi: 10.1073/pnas.93.22.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor NG, Laurie S, Turner SR. Multiple cellulose synthase catalytic subunits are required for cellulose synthesis in Arabidopsis. Plant Cell. 2000;12:2529–2540. doi: 10.1105/tpc.12.12.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiu CP, Watts AG, Lairson LL, Gilbert M, Lim D, Wakarchuk WW, Withers SG, Strynadka NC. Structural analysis of the sialyltransferase CstII from Campylobacter jejuni in complex with a substrate analog. Nat Struct Mol Biol. 2004;11:163–170. doi: 10.1038/nsmb720. [DOI] [PubMed] [Google Scholar]
- 61.Vonhippel PH, Fairfield FR, Dolejsi MK. On the processivity of polymerases. Ann N Y Acad Sci. 1994;726:118–131. doi: 10.1111/j.1749-6632.1994.tb52803.x. [DOI] [PubMed] [Google Scholar]
- 62.Härle J, Bechthold A. The power of glycosyltransferases to generate bioactive natural compounds. Methods Enzymol. 2009;458:309–333. doi: 10.1016/S0076-6879(09)04812-5. [DOI] [PubMed] [Google Scholar]
- 63.Langenhan JM, Griffith BR, Thorson JS. Neoglycorandomization and chemoenzymatic glycorandomization: two complementary tools for natural product diversification. J Nat Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]
- 64.Thibodeaux CJ, Melançon CE, Liu HW. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew Chem Int Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guan S, Clarke AJ, Whitfield C. Functional analysis of the galactosyltransferases required for biosynthesis of D-galactan I, a component of the lipopolysaccharide O1 antigen of Klebsiella pneumoniae. J Bacteriol. 2001;183:3318–3327. doi: 10.1128/JB.183.11.3318-3327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He XZ, Wang X, Dixon RA. Mutational analysis of the Medicago glycosyltransferase UGT71G1 reveals residues that control regioselectivity for (iso)flavonoid glycosylation. J Biol Chem. 2006;281:34441–34447. doi: 10.1074/jbc.M605767200. [DOI] [PubMed] [Google Scholar]
- 67.Gilbert M, Brisson JR, Karwaski MF, Michniewicz J, Cunningham AM, Wu Y, Young NM, Wakarchuk WW. Biosynthesis of ganglioside mimics in Campylobacter jejuni OH4384. Identification of the glycosyltransferase genes, enzymatic synthesis of model compounds, and characterization of nanomole amounts by 600-MHz 1H and 13C NMR analysis. J Biol Chem. 2000;275:3896–3906. doi: 10.1074/jbc.275.6.3896. [DOI] [PubMed] [Google Scholar]
- 68.Wakarchuk WW, Watson D, St Michael F, Li J, Wu Y, Brisson JR, Young NM, Gilbert M. Dependence of the bi-functional nature of a sialyltransferase from Neisseria meningitidis on a single amino acid substitution. J Biol Chem. 2001;276:12785–12790. doi: 10.1074/jbc.M011293200. [DOI] [PubMed] [Google Scholar]
- 69.Lairson LL, Watts AG, Wakarchuk WW, Withers SG. Using substrate engineering to harness enzymatic promiscuity and expand biological catalysis. Nat Chem Biol. 2006;2:724–728. doi: 10.1038/nchembio828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.