

Abstract
We set out to define patterns of gene expression during kidney organogenesis by using high-density DNA array technology. Expression analysis of 8,740 rat genes revealed five discrete patterns or groups of gene expression during nephrogenesis. Group 1 consisted of genes with very high expression in the early embryonic kidney, many with roles in protein translation and DNA replication. Group 2 consisted of genes that peaked in midembryogenesis and contained many transcripts specifying proteins of the extracellular matrix. Many additional transcripts allied with groups 1 and 2 had known or proposed roles in kidney development and included LIM1, POD1, GFRA1, WT1, BCL2, Homeobox protein A11, timeless, pleiotrophin, HGF, HNF3, BMP4, TGF-α, TGF-β2, IGF-II, met, FGF7, BMP4, and ganglioside-GD3. Group 3 consisted of transcripts that peaked in the neonatal period and contained a number of retrotransposon RNAs. Group 4 contained genes that steadily increased in relative expression levels throughout development, including many genes involved in energy metabolism and transport. Group 5 consisted of genes with relatively low levels of expression throughout embryogenesis but with markedly higher levels in the adult kidney; this group included a heterogeneous mix of transporters, detoxification enzymes, and oxidative stress genes. The data suggest that the embryonic kidney is committed to cellular proliferation and morphogenesis early on, followed sequentially by extracellular matrix deposition and acquisition of markers of terminal differentiation. The neonatal burst of retrotransposon mRNA was unexpected and may play a role in a stress response associated with birth. Custom analytical tools were developed including “The Equalizer” and “eBlot,” which contain improved methods for data normalization, significance testing, and data mining.
Organogenesis is the result of a complex interplay of proliferation, cell-to-cell communication, inductive events, and cellular movements. It is widely held that stable changes in the state of the cell are accompanied by changes in gene expression and that cellular states during development progress from less to more differentiated, often in response to specific inductive signals from nearby cells. Although very useful, these constructs focus attention on the contribution of master regulatory genes and specific morphogens. A broad based and unbiased view of gene expression during mammalian organogenesis is lacking.
Advances in the technology for assaying RNA in a highly parallel fashion, coupled with the completion of several genome sequencing projects, make possible a complete description of gene regulatory systems during development. Here, we define the broad outlines of gene expression during kidney development, an example of organogenesis that involves mesenchymal-epithelial transformation, branching morphogenesis, and acquisition of organ-specific markers of terminal differentiation (1, 2). Of 8,740 genes, 873 were found to vary significantly (P < 0.0025) during kidney development. These genes clustered into five clear patterns or groups of gene expression, each of which was defined by a unique “personality” characterized by cluster-member gene function, tissue distribution, and embryonic expression. We have devised custom software algorithms that were critical to the analysis. Included are tools for data “equalization” by means of a continuously variable normalization vectors and for the creation of array-error models that permit assignment of statistical significance, as well as an electronic “eBlot” that uses information in the publicly available databases.
Methods
RNA Preparation and Analysis.
Pooled total RNA (5 μg) was isolated from embryonic rats at gestational day 13 (e13, n = 16), e15 (n = 8), e17 (n = 4), e19 (n = 2), newborn (n = 2), 1 week (n = 2), and nonpregnant adult (n = 2). Dissected tissues were immediately frozen in liquid nitrogen before RNA isolation. Total RNA was purified by using Strataprep Micro and Miniprep Total RNA isolation kits (Stratagene), according to the manufacturer's protocol. Reverse transcription, second-strand synthesis, and probe generation were all accomplished by the standard Affymetrix protocol (Affymetrix, Santa Clara, CA). In like fashion, Rat Genome U34A GeneChips (Affymetrix) were hybridized, washed, and scanned according to the standard Affymetrix protocol.
Data Analysis.
Array data were globally normalized by using “The Equalizer,” an application for global data normalization written in visual basic 6 (Microsoft). Additional analysis was performed with custom add-in applications for Microsoft excel, systat 9.0 (SPSS, Chicago), genespring (Silicon Genetics, San Carlos, CA), and a stand-alone implementation of the blast family of programs (NCBI, Washington, DC; ref. 3). Analytical methods are described in detail in the figure legends.
Data equalization.
Briefly, before the normalization algorithm, here termed “equalization,” GeneChip expression data in many cases displayed marked systematic deviation from linearity in two-dimensional orthogonal projections of the original n-dimensional data matrix. The Equalizer was used to identify a group of points with similar rank order of signal intensity in any two gene expression lists; the group was then used to generate a continuously variable normalization vector that eliminated the systematic deviation (see Fig. 1 legend).
Figure 1.

Data equalization. (A) Before being subjected to the normalization algorithm (here termed equalization), GeneChip expression data in many cases displayed systematic deviation from linearity in two-dimensional orthogonal projections of the original n-dimensional data matrix. Genes with expression values near the readily apparent central tendency are invariant in the two comparison conditions, but may display raw signals that deviate significantly. The datum in the upper right corner is representative of a large number of points that, in this example, display an ≈60% systematic shift toward higher value along the y axis as compared with the x axis condition, despite their clear association with the central trend of the data. In the example, The Equalizer has identified a group of points with identical rank order of signal intensity (window of ±5) in the two gene expression lists (red points) and applied a locally weighted nonlinear regression “smoother” to generate a best-fit description of the central trend of the data. (B) The best-fit line was then used as a normalization vector, which, when applied to the data matrix, resulted in linearized data with a slope very near to 1. Note that before equalization, a description of the data by linear regression techniques yielded different answers depending on which variable was considered dependent or independent [unequal root mean square error (rmse)]. The Equalizer also provides for shifting of the data to positive values to allow for subsequent log transformations. All gene intensities were then shifted to the positive by an amount corresponding to the 1.5th percentile (a user-defined value) gene intensity value. The ≈1.5% of genes with shifted values less than noise were then set to the noise value.
Statistical modeling.
The scatter (log ratio) in the data was expressed as a function of baseline expression in identical replicate samples (Fig. 2). Each position in the scatter model was associated with a standard score Z which represented the deviation from identical expression in the context of baseline expression. Each pairwise comparison of experimental observations yielded a Z value. Multiple observations could be combined by averaging Zs, which could in turn be associated with a P value (see Fig. 2 legend). Array data and software are available at http://organogenesis.ucsd.edu/.
Figure 2.

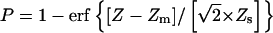 |
Results
Data Equalization.
DNA array experiments generate a data matrix in n-dimensional space (4). Such a data distribution is characterized by a central tendency or “line of identity” corresponding to genes with no change in expression in any condition. In the case that the central tendency is linear and characterized by a slope of 1, direct comparisons of raw numerical values yield valid results. Owing to variations in the hybridization and scanning process, the idealized case is essentially never realized. Various strategies, such as normalization and scaling, have been developed to compensate for this shortcoming. Such manipulations, based on average probe intensities and similar measures, involve simple linear transformations of the data matrix. Unfortunately, the central line of identity is also typically characterized by some degree of nonlinearity, particularly at higher levels of expression (Fig. 1A). The curved nature of the data distribution leads directly to an apparent increase in scatter at high levels of baseline expression, which, in previous work (though not specifically noted), has required the use of terms in statistical models that give great weight to the precision of any given pair of measurements (5, 6). To favor purely directional tendencies as opposed to precision, we have previously expressed the measure of differential expression as a vector quantity directed orthogonally away from the central tendency (4). The common, precision-based methods tend to overlook genes in which directional changes are identical but where absolute values of change are variable. However, our previous notion of differential expression as a vector in n-dimensional space rapidly becomes computationally expensive, particularly in the setting of curvilinear relationships. A data matrix in which the line of identity has been rendered linear permits a direct comparison of expression values and permits the use of statistical models that favor purely directional changes over the variations in absolute value (Fig. 1B).
Statistical Model.
Statistical algorithms employing t tests treat DNA array experiments as thousands of parallel experiments. Any such analysis performed thousands of times is subject to a significant multiple observation bias and fails to take into account the context of one measurement among many of thousands of similar measurements. We and others have modeled the variability in observations as a function of baseline expression in replicate samples (5). After appropriate equalization of the data, the increase in scatter observed at lower levels of baseline expression may be modeled by simple exponential equations (Fig. 2A). The error model created on replicate samples is then applied to experimental observations, and confidence values based on position in the error model are calculated from the error function. Multiple observations were readily combined into summary P values. A comparison matrix representing each possible pairwise comparison between different developmental days was analyzed. Data points that had no single summary P value more significant than P = 0.0025 represented those genes lying relatively close (in the context of baseline expression) to the central tendency in all dimensions and were excluded from further analysis (Fig. 2B).
Clustering.
The stringent filtering of the data for significant and consistent changes greatly facilitated identification of biologically relevant gene clusters. Those genes in which at least a single pairwise comparison yielded a significant difference could, in principal, constitute a heterogeneous mix of genes in which a single spurious result interrupted an otherwise housekeeping pattern. Likewise, it was formally possible that many of the genes would display “zigzag” patterns that resulted from multiple spurious measurements and that were nonsensical in a biological sense. Therefore, we applied hierarchical clustering to the filtered data to visualize patterns of gene expression globally (Fig. 3). The clustering algorithm grouped the embryonic gene expression vectors separately from the neonatal group, both of which were grouped separately from the adult.
Figure 3.

Hierarchical clustering. There were 873 genes identified as changing significantly at some point in kidney development. These 873 genes were clustered [by using the hierarchical clustering algorithm, genespring (Silicon Genetics)] in two dimensions according to their gene expression and experimental vectors in Euclidian space after compressing the equalized data to a target maximum value of 3. Numbers at the bottom indicate group numbers derived from k-means clustering. Group 1 genes are up-regulated (red) in the early embryonic period and decrease thereafter. Group 2 genes rise to a mid-late embryonic peak. Group 3 genes peak in the neonatal period. Group 4 genes rise somewhat linearly throughout development. Group 5 genes display a distinct peak in the adult vs. all earlier times. 13, 15, 17, 19, embryonic days; N, newborn; W, 1 week old; A, adult.
Clustering on the gene axis produced five major groups that corresponded to groups also identified through k-means clustering. Each cluster was characterized by an idealized gene expression vector (Fig. 4). The groups were named 1 through 5 based on the timing of their peak expression during development. Notably lacking were patterns inconsistent with a role in development or that likely reflected spurious data. Thus, group 1 consisted of genes that had very high relative levels of expression in early development. Group 2 consisted of genes that were relatively low initially but that peaked and declined during prenatal life. Group 3 was composed of genes that peaked in the neonatal period. Group 4 consisted of genes that had a relatively steady increase in expression throughout development. Finally, group 5 consisted of genes that had low levels of expression throughout development but that were significantly up-regulated in the adult. The canonical clusters identified here make biological sense, both in terms of their respective shapes and in terms of function of their member genes and previously observed tissue distribution (see below).
Figure 4.

Temporal gene expression profiles during kidney development. Data are expressed as the mean at each time for clusters of genes as defined by k-means clustering (1–5). The distribution of individual profiles is also shown for the most heterogeneous group (2, all). Identities of representative genes are shown in Table 1. 13, 15, 17, 19, embryonic days; N, newborn; W, 1 week old; A, adult.
eBlot Database of Gene Functions and Distribution.
A complete annotation of gene function, cellular location, and other information is lacking for genes present in the arrays. Moreover, there is not yet a common gene terminology in place to allow comparisons of curated gene information across multiple databases. Despite the lack of common terminology, one field—the sequence—is present, or referenced, in most databases and is insensitive to minor “spelling” changes between database or species. Therefore, we created a database (Fig. 5), termed eBlot, that links array target sequences with gene sequences/accessions present in public databases (7–10). Briefly, the “target sequences” associated with each probe, which may have changed in identity or completeness, were updated and, in most cases, completed through automated blast searches against Unigene and against the nonredundant GenBank nucleotide database. This database consolidation procedure yielded curated mRNA sequences for most probes, updated accession numbers, and identifications of what previously were unknown expressed sequences tag (EST) sequences. The updated sequence fields were then linked to Gene Ontology (GO) records and ontologies through the sequence field via blast comparison (3, 8).
Figure 5.

eBlot database schema. The Affymetrix target sequences were updated by comparison to Unigene sequences and associated by sequence similarity with entries in publicly available databases. Individually curated gene function information was derived for genes without high scoring matches in Gene Ontology Consortium-linked databases. SGD, Saccharomyces Genome Database; FB, Flybase; MGI, Mouse Genome Informatics; dbEST, EST database (National Center for Biotechnology Information); GO, Gene Ontology Consortium, DEV, developmental stage; ID, identifier; SEQ, sequence; LIB, library; CA, cancer.
The five canonical gene expression patterns varied considerably with respect to the general gene functions of cluster members (Fig. 6; Table 1). Group 1 (n = 323), which is significantly up-regulated in early embryogenesis, contained a preponderance of genes that function in protein synthesis (n = 46), DNA replication or structure (n = 36), and RNA synthesis or processing (n = 48). This group also contained a number of genes with previously recognized roles in organogenesis. Among the genes of group 2 (n = 70), which peaked in mid-late embryogenesis, were genes for structural proteins of the extracellular matrix (ECM) (n = 19) or cytoskeleton (n = 6), as well as genes with previously identified morphogenetic roles. Group 3 genes (n = 48) also contained genes for ECM proteins (6) but was most notably populated by RNA species specifying retrotransposons (n = 15). Group 4 genes (n = 262) increased in a relatively linear fashion (relative to many other housekeeping genes) and included most prominently genes specifying proteins involved in energy production (n = 65) and transport (n = 29). Group 5 genes (n = 172), which also included genes involved in transport (n = 15), shared a similar temporal pattern of developmental expression with those of group 4 but exhibited a significant bias toward relatively high levels of expression only in the adult. The group 5 cluster of genes also included genes involved in detoxification (n = 19), immune recognition (n = 20), amino acid catabolism (n = 12), and defense against oxidative stress (n = 15).
Figure 6.

Functional associations of gene clusters. Gene clusters varied remarkably in terms of major functional classifications of component genes. Key (Lower right) indicates major gene functional classifications: B, biosynthetic; CA, cell adhesion; C, catabolism-small molecules; CC, cell cycle; CS, cytoskeletal; DE, defense; D, DNA structure or replication; E, extracellular matrix; ED, endocytosis; EN, energy metabolism; H, homeostasis of the organism; M, morphogenetic; P, protein synthesis or processing; R, RNA synthesis or processing; HS, heat-shock proteins; DT, detoxification of exogenous substances; RD, protection against oxidative stress; T, transport; RT, retrotransposon; U, unknown function. The icons preceding the group names were derived from Fig. 4 and display the associated temporal expression profile. Group 1 expressed earlier in nephrogenesis was most notable for genes involved in DNA replication (D), RNA production (R), protein synthesis (P), and morphogenesis (M), consistent with an actively proliferating tissue. Group 2 (which peaked in midnephrogenesis) was most notable for genes of the extracellular matrix (E) as well as morphogenetic genes (M). Group 3 (with a peak in neonatal life) was dominated by retrotransposon transcripts (RT). Group 4 was most notable for transport (T) and energy metabolism (EN) related genes. Group 5 genes (significantly up-regulated in the adult vs. all previous times) was more heterogeneous and included genes specifying catabolic enzymes (C), defense and immune recognition (DE), homeostasis of the organism as a whole (H), detoxification (DT), oxidative stress (RD), and transport (T).
Table 1.
Representative group members
| Group 1: Translation and ribosomal proteins |
| (eif-2, J02646); (eif-2, L10652); (eiF3.9, AA875205); (eiF3s7, NM_018749); (factor 2C2, H31692); (ER ribosomal binding protein p34, D13623); (rp L10a, X93352); (rp L12, X53504); (rp L13, X78327); (rp L15, X78167); (rp L17, X58389); (rp L18a, X14181); (rp L21, X15216); (rp L22, X60212); (rp L27, X07424); (rp L27a, X52733); (rp L29, X68283); (rp L3, X62166); (rp L35a, X05705); (rp L41, X82550); (rp L6, X87107); (rp L8, X62145); (rp S10, X13549); (rp S11, AB028894); (rp S15, NM_017151); (rp S19, X51707); (rp S24, M89646); (rp S3, X51536); (rp S6, M29358); (rp S7, X53377); (rp S9, X66370) |
| Group 2: ECM related proteins |
| (Agrin, M64780); (COL1A1, Z78279, M27207, U75405); (COL3A1, M21354, X70369); (COL5A2, AJ224880); (Decorin, X59859); (FN, L00191, U82612, X05834); (Follistatin, U06864); (Gel-A, U65656); (osteonectin, Y13714, U75929); (gelatinase-A, U65656) |
| Group 3: Transposons |
| (LINE3 cds 1, M13100#1); (LINE3 cds 2, M13100#2); (LINE3 cds 3, M13100#3); (LINE3 cds 4, M13100#4); (LINE3 cds 5, M13100#5); (LINE3 cds 6, M13100#6); (LINE4, M13101); (L1 transposon, U83119, X61295); (2.4kb transposon cds 1, X05472#1); (2.4kb transposon cds 2, X05472#2); (2.4kb transposon cds 3, X05472#3); (L1 Rn, X07686); (LINE, X53581) |
| Group 4: Energy related |
| (aldolase, AA892395, X02284, X02291); (fructose-1,6-bisphosphatase, M86240); (hexokinase, AA858607); (lactate DH, U07181); (phosphoenolpyruvate carboxykinase, K03243); (argininosuccinate synthetase, X12459); (dihydrolipoamide succinyltransferase, D90401); (glutamate DH, AI179613, AI233216); (malate DH, AF093773); (methylmalonate semialdehyde DH, M93401); (NADP Isocitrate DH, AA892314); (pyruvate DH-1, AA799598, Z12158); (pyruvate DH-2, U10357); (ADP/ATP translocase, D12771); (COX Va, X15030); (CYT B, J01436); (CYT B5, AF007107, AA945054); (CYT B558, U18729); (CYT C OX, L48209); (CYT C OX-II, AI010292); (DICARB, AJ223355); (H+ATPase, D10874, D13127); (NADH-Q oxidoreductase B22, AI171542); (NADH-Q reductase, AI009390, S46798); (NADH-Q oxidoreductase CI, AI176491); (SUCCINATE-Q oxidoreductase, AA800250) |
| Group 5: Transporters |
| (Na channel b1, M91808); (cation transporter, X78855); (ROMK2b, S78154); (K channel, D86039); (Kir5 channel, AF249676); (CHIP28, X67948); (Aquaporin, D13906); (Na/HCO3 exchanger, AF004017); (integral membrane transport protein 1, AF028739); (SDCT sodium-dicarboxylate cotransporter 1, AF058714); (UST1r integral membrane transport protein, Y09945); (RATSTRAP Stimulates Transport of Amino Acids Protein, M80804); (ROSIT renal osmotic stress induced transporter, U12973); (OCT1a organic cation transporter, U76379); (LSTP liver-specific transport protein, L27651) |
In each citation in parentheses, the first item is the common name and the second item is the accession number. rp, ribosomal protein; DH, dehydrogenase; COX, cytochrome oxidase; CYT, cytochrome.
A number of genes with known and suspected morphogenetic roles in kidney development and elsewhere were observed to be differentially regulated. Most fell into group 1 and included IGF-II, X17012; pleiotrophin, NM_017066.1; activin type IIB R, M87067; ganglioside GD3, NM_012811.1; transforming growth factor (TGF)-β2, M96643; frizzled, L02530; and platelet derived growth factor (PDGF)-R, NM_011058. Group 2 included jagged, L38483; LIM1, S71523; POD1, AF061752,NM_011545; and IGFBP2, M91595. Group 4 contained hepatocyte growth factor (HGF)-activator-inhibitor, AF099020, and group 5 contained epidermal growth factor (EGF), U04842, and EGFR, X12748. In addition, a number of known morphogenetic effectors displayed trends that allied them with a particular group but that were not included by the present stringent threshold. Among these effectors were, in group 1, Timeless, retinoic acid receptor (RAR)-α, RAR-β, bcl2, Homeobox protein A11, GFRA1, and Wilm's tumor (WT)1; in group 2, HGF, HNF-3, BMP4, TGF-α, met, fibroblast growth factor (FGF)7, and BMP4; and in group 4, BMP7. Thus, the array data revealed important changes in expression of several transcription factors, secreted factors, and receptors with putative roles in kidney development; these genes generally fell into groups 1 and 2. A few important morphoregulatory molecules such as Pax2 and Emx2 were not present on the arrays.
The tissue distribution of genes provides important functional clues. A wealth of information exists in the printed record regarding tissue distribution but remains inaccessible in any systematic way by electronic means. Nevertheless, the EST database (http://www.ncbi.nlm.nih.gov/dbEST/), consisting of more than 6 million RNA sequences from ≈7,000 libraries, by definition, contains accessible information for tissue distribution and, in many cases, for developmental stage. The database also distinguishes between cancer and normal tissue (11). Therefore, we parsed the EST database for human, mouse, and rat sequences and generated a custom database associating the tissue source, developmental stage, and derivation from tumor/normal tissue (information available for each library of origin) with each EST sequence (Fig. 5). This database was associated with the GeneChip sequences, as was done for functional annotations.
The results summarize the association of the gene clusters with tissues of origin in the EST database (Fig. 7). Thus, member genes of group 5 (adult group) were strikingly more common in source libraries from tissues characterized by the presence of branching ductal epithelial structures such as kidney, lung, liver, and pancreas. This finding is in marked contrast to the other groups (1–3) in which the member genes had more heterogeneous tissue associations. Group 4, like group 5, was characterized by transport proteins, but the tissue associations as a whole were less biased toward epithelial tissues. In addition, many EST source library descriptions include information as to the developmental stage of the source tissue and its derivation from tumor/normal tissue. A striking tendency toward embryonic tissue association was found for the genes of group 1; group 2, characterized by ECM and morphogenetic genes, was most closely associated with tumor tissues (not shown).
Figure 7.

Tissue distribution/association of cluster member genes in the EST database. Group member genes were associated with EST database entries by sequence similarity and the tissue associations were summarized as in Fig. 6. When kidney-, colon-, lung-, pancreas-, and liver-derived genes were found homologous (blast bit score ≥100) to any of the 873 significantly changed genes, they tended to come from group 4 and, particularly, group 5. The results provide independent evidence for the validity of the canonical clusters; genes that appeared late in kidney development (groups 4 and 5) were associated with EST source libraries consisting of branching ductal epithelial tissue. Interestingly, group 3 (dominated by retrotransposon RNA species) was more frequently associated with adrenal and islet tissues. Abbreviations: K, kidney; CO, colon; LU, lung; PN, pancreas; LI, liver; SN, skin; B, brain, ER, ear; EY, eye; T, testis; OV, ovary; BM, bone marrow; SP, spleen; AD, adrenal; IS, islets; H, heart; SK, skeletal muscle; EN, endothelium.
The database (Fig. 5) identifies the intersection of the previous analytical efforts for the efficient prioritization of genes for additional studies. It is possible, for instance, to select a subset of genes belonging to groups 1 or 2 (early in development, Figs. 3 and 4), with unknown function (Fig. 6), which have a high frequency of association with kidney and/or embryonic tissue sources and/or tumor tissue. Had their developmental roles as yet been unsuspected (functional classification = unknown), this strategy would have identified known developmental genes such as LIM1 and POD1. In fact, a small number of such candidate genes were identified.
Discussion
We have described the broad outlines of gene expression during organogenesis of the kidney. The data frame the main themes in kidney development, as evidenced by five characteristic clusters of significantly and consistently differentially regulated genes: cellular proliferation and morphogenesis (group 1), followed by continued morphogenesis and ECM production (group 2), and terminal differentiation and ability to respond to oxidative and osmotic stress (groups 4 and 5). We also have uncovered a non-germ-line retrotransposon transcriptional burst that may be a response to neonatal cellular stress (group 3). The data are the result of our system of normalization, error modeling, and statistical analysis aimed at a precise characterization of DNA array performance. The gene clusters identified were readily interpretable in biological and temporal terms and, to the degree that such genes were present in the arrays, included a number of recognized master regulatory or morphogenetic genes such as LIM1, POD1, WT1, homeobox gene A11, and GFRA1. In addition, a number of genes with unknown function were identified in all groups.
A picture emerges that is somewhat different in emphasis than much of the work focusing on knockout experiments. For example, early in kidney development, the work of the kidney seems to be largely to grow. A sizeable fraction of genes up-regulated in this period (group 1) are devoted to chromosome, nucleolus, and ribosome production (Table 1). Also, as mentioned above, early kidney development is the period when a number of likely morphogenetic effector genes are expressed. Expression of morphoregulatory genes is a theme that continues temporally into the period of expression of group 2 genes. As the period of cellular proliferation wanes, a burst of ECM and ECM-modifying genes are expressed, suggesting that the newly generated cells secondarily invest a great deal of their resources modifying the local environment. ECM-modifying proteins such as MT-MMP were present in group 1, and group 2 genes are also present in the early embryo (and only peak later); these facts, taken together, are consistent with the continuing importance of ECM deposition and modification, which begins very early and peaks in midnephrogenesis (12–15).
The gene clusters were distinct with respect to member gene functions, none more so than those up-regulated solely in the neonatal period (group 3). It was somewhat surprising to find that group 3 was dominated by members of the LINE family of retrotransposons. In one case, probe sets are present targeting six separate ORFs from the long interspersed repetitive DNA sequence LINE3 (16). Each separate RNA species was not only differentially regulated but clustered together in group 3, indicating that the relevant RNA might be coordinately regulated; indeed, these RNA were originally found in rat liver as a single transcript (16). The precise function of LINE sequences in mammalian cells remains elusive, although their germ-line contributions to the evolution of telomerase and antibody VDJ recombination are well known (17). It has been suggested that high levels of otherwise inactive transposon RNA serve to saturate and inactivate dsRNA-induced protein kinase (PKR; refs. 18–20) and thus maintain protein translation. PKR would otherwise interrupt translation during cellular stress. Retrotransposon transcription has also been shown to increase in insect cells under stress (21). The role of retrotransposon transcription in the neonatal period is unknown, but it provides an example of a set of new hypotheses generated through broad surveys of gene expression. Group 3 also contained genes with known roles in cytoprotection, particularly those involved in dealing with oxidative stress. This group includes glutathione peroxidase, NM_008161; glutathione S-transferase Yc2, S82820; and thioredoxin interacting factor, U30789. The development of glutathione- and metallothionein-related genes is a theme that continues through group 4 and reaches maximal levels in adulthood (group 5).
After cellular proliferation and somewhat concurrent with ECM deposition, the kidney up-regulates a group of transport proteins (group 4). That many genes with roles in energy production are coordinately up-regulated suggests that additional energy is required to subserve transport processes. Another group of transport proteins present in group 5 were up-regulated only after birth and, indeed, after the neonatal period. The transporters present in groups 4 and 5 were similar in character with one notable difference: group 4 (linearly increasing throughout development) alone contained subunits of the Na,K-ATPase, in keeping with its general role in cell volume regulation and many secondary transport processes. Both groups 4 and 5 contained examples of water channels (Aquaporins 1, 3, 5, and 7), inorganic (NaK2Cl, ROMK2/2b, CLCK2, NHE2, and NaPi-2-γ), and organic substance transporters (NKT/OAT1, GLUT5, SGLT2, and CD98). Notably, group 5 (adult) contained ROSIT (renal osmotic stress-induced NaCl organic solute cotransporter), suggesting that the adult kidney is subject to more osmotic stimulus to transporter repertoire than are embryonic or preweaning, neonatal kidneys (22). In concert with the increase in transporter expression in groups 4 and 5, increases in a number of enzymes involved in the catabolism of peptides and amino acids were observed; included were meprin, dipeptidyl peptidases 1 and 4, cathepsins D, H, and L, proline oxidase, branched chain keto acid dehydrogenase E1, and several kynurenine-related enzymes in the tryptophan degradative pathway leading to NAD/NADP cofactor synthesis.
Efficient prioritization of these genes, particularly those in group 1, will likely be facilitated by broad surveys of gene expression in in vitro models of organogenesis in which specific cell types may be analyzed in isolation, and in an inductive context where specific aspects of organogenesis (branching, epithelialization, and differentiation) may be observed (23, 24). Future array analyses will provide ever more discriminating power and allow finer dissection of subtle changes in gene expression. The normalization strategy described here will facilitate the ongoing development and interpretation of a continuously updated developmental database (http://organogenesis.ucsd.edu/).
Acknowledgments
We thank William Wachsman, Linda Feng, the Veterans Affairs San Diego Health Care Systems GeneChip Core, and the University of California San Diego Cancer Center Microarray Shared Resource for performing the GeneChip assays. R.O.S. is supported by the Medicine Education and Research Foundation and by National Institutes of Health Grant K08-DK02392. K.T.B. is supported by American Heart Association Grant 9730096N. S.K.N. is supported by National Institutes of Health Grants PO1 DK54711 and RO1 DK49517.
Abbreviations
- En
embryonic days n from gestation
- EST
expressed sequences tag
- ECM
extracellular matrix
References
- 1.Saxen L. Organogenesis of the Kidney. Cambridge, U.K.: Cambridge Univ. Press; 1987. [Google Scholar]
- 2.Pohl M, Stuart R O, Sakurai H, Nigam S K. Annu Rev Physiol. 2000;62:595–620. doi: 10.1146/annurev.physiol.62.1.595. [DOI] [PubMed] [Google Scholar]
- 3.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavlova A, Stuart R O, Pohl M, Nigam S K. Am J Physiol. 1999;277:F650–F663. doi: 10.1152/ajprenal.1999.277.4.F650. [DOI] [PubMed] [Google Scholar]
- 5.Roberts C J, Nelson B, Marton M J, Stoughton R, Meyer M R, Bennett H A, He Y D, Dai H, Walker W L, Hughes T R, et al. Science. 2000;287:873–880. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- 6.Golub T R, Slonim D K, Tamayo P, Huard C, Gaasenbeek M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 7.The FlyBase Consortium. Nucleic Acids Res. 1999;27:85–88. [Google Scholar]
- 8.Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry J M, Davis A P, Dolinski K, Dwight S S, Eppig J T, et al. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ball C A, Dolinski K, Dwight S S, Harris M A, Issel-Tarver L, Kasarskis A, Scafe C R, Sherlock G, Binkley G, Jin H, et al. Nucleic Acids Res. 2000;28:77–80. doi: 10.1093/nar/28.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blake J A, Eppig J T, Richardson J E, Bult C J, Kadin J A. Nucleic Acids Res. 2001;29:91–94. doi: 10.1093/nar/29.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boguski M S, Lowe T M, Tolstoshev C M. Nat Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 12.Ekblom P, Alitalo K, Vaheri A, Timpl R, Saxen L. Proc Natl Acad Sci USA. 1980;77:485–489. doi: 10.1073/pnas.77.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekblom P. J Cell Biol. 1981;91:1–10. doi: 10.1083/jcb.91.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pohl M, Sakurai H, Stuart R O, Nigam S K. Dev Biol. 2000;224:312–325. doi: 10.1006/dbio.2000.9783. [DOI] [PubMed] [Google Scholar]
- 15.Kanwar Y S, Carone F A, Kumar A, Wada J, Ota K, Wallner E I. Kidney Int. 1997;52:589–606. doi: 10.1038/ki.1997.372. [DOI] [PubMed] [Google Scholar]
- 16.D'Ambrosio E, Waitzkin S D, Witney F R, Salemme A, Furano A V. Mol Cell Biol. 1986;6:411–424. doi: 10.1128/mcb.6.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smit A F. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 18.Li T, Spearow J, Rubin C M, Schmid C W. Gene. 1999;239:367–372. doi: 10.1016/s0378-1119(99)00384-4. [DOI] [PubMed] [Google Scholar]
- 19.Schmid C W. Nucleic Acids Res. 1998;26:4541–4550. doi: 10.1093/nar/26.20.4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W M, Chu W M, Choudary P V, Schmid C W. Nucleic Acids Res. 1995;23:1758–1765. doi: 10.1093/nar/23.10.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura R H, Choudary P V, Schmid C W. Nucleic Acids Res. 1999;27:3380–3387. doi: 10.1093/nar/27.16.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wasserman J C, Delpire E, Tonidandel W, Kojima R, Gullans S R. Am J Physiol. 1994;267:F688–F694. doi: 10.1152/ajprenal.1994.267.4.F688. [DOI] [PubMed] [Google Scholar]
- 23.Qiao J, Sakurai H, Nigam S K. Proc Natl Acad Sci USA. 1999;96:7330–7335. doi: 10.1073/pnas.96.13.7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakurai H, Barros E J, Tsukamoto T, Barasch J, Nigam S K. Proc Natl Acad Sci USA. 1997;94:6279–6284. doi: 10.1073/pnas.94.12.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]