

Abstract
Synergistic catalysis is a synthetic strategy wherein both the nucleophile and the electrophile are simultaneously activated by two separate and distinct catalysts to afford a single chemical transformation. This powerful catalysis strategy leads to several benefits, specifically synergistic catalysis can (i) introduce new, previously unattainable chemical transformations, (ii) improve the efficiency of existing transformations, and (iii) create or improve catalytic enantioselectivity where stereocontrol was previously absent or challenging. This perspective aims to highlight these benefits using many of the successful examples of synergistic catalysis found in the literature.
1. Introduction
Catalysis is, without question, one of the most efficient and powerful mechanistic strategies for identifying or engineering new chemical reactions. Within the realm of chemical synthesis, traditional catalytic pathways generally rely on the interaction of a unique catalyst with a single substrate, thereby lowering the energetic barrier to bond formation with a second, unactivated substrate. While this mono-catalysis strategy has successfully delivered vast numbers of new reactions over many decades, multi-catalysis concepts have recently began to emerge that allow access to many difficult or unattainable transformations. In particular, synergistic catalysis, wherein two catalysts and two catalytic cycles work in concert to create a single new bond, has emerged as a powerful new mechanistic approach to reaction engineering. In its simplest form (Fig. 1), synergistic catalysis involves the concurrent activation of both a nucleophile and an electrophile using distinct catalysts. This simultaneously creates two reactive species, one with a higher HOMO (highest occupied molecular orbital) and the other with a lower LUMO (lowest unoccupied molecular orbital), in comparison to the respective ground state starting materials. If selected judiciously, these activated species can rapidly couple, in many cases enabling chemical reactions that are impossible or inefficient using traditional mono-catalysis methods.
Fig. 1.

The concept of synergistic catalysis.
Importantly, there are a number of multicatalysis mechanisms that do not fall under the classification of synergystic catalysis, and we feel it is important to define them herein. First, if both the nucleophile and electrophile are activated separately by discrete functional groups on the same catalyst, this has been classified as bifunctional catalysis (Fig 2A).1 When both catalysts work in concert to activate only one of the reacting partners, we term this double activation catalysis (Fig 2B).2 Similarly, if both catatysts activate the same reacting partner, but in a sequential fashion (i.e. an activated substrate produces an intermediate, which is further activated by the second catalyst), we have previously classified this strategy as cascade catalysis (Fig 2C).3 In contrast, and as defined above, when the nucleophile and electrophile are simultaneously activated by two separate catalysts to afford a single chemical transformation, we classify this strategy as synergistic catalysis (Fig 2D).
Fig. 2.

Classification of catalytic systems involving two catalysts.
While synergistic catalysis is now beginning to emerge as a recognized and valuable approach to bond forming processes, the concept itself is not a new one. Indeed, synergistic catalysis is prevalent in nature, as many enzymes function through the cooperation of two or more catalysts (or catalytic moieties within an active site) to afford a specific transformation.4 For example, tetrahydrofolate, an important coenzyme for the transfer of one-carbon units, is produced from the folic acid metabolite dihydrofolate via a hydride transfer to the imine moiety. The enzyme dihydrofolate reductase binds dihydrofolate and activates it towards hydride addition through protonation of the imine. A hydride is also catalytically activated by coenzyme NADP+, generating NADPH. This cofactor binds to the enzyme and delivers the hydride, producing tetrahydrofolate and regenerating NADP+ (Fig 3).5
Fig. 3.

Synergistic catalysis in dihydrofolate reductase.
There are some perceived challenges surrounding this strategy. While nature has the advantage of physical separation between catalytic sites in an enzyme, when applying the concept in synthesis the catalysts are free to interact with one another. This can potentially result in self-quenching, which renders both catalysts inactive. This can happen through a variety of processes, such as strong complexation of a Lewis acid and a Lewis base or a redox event. The key to overcoming this challenge is the judicious selection of appropriate catalyst combinations. For example, many Lewis acids and Lewis bases form labile complexes that exhibit reversible binding; these weak interactions can still allow respective substrate activation. Often combining hard Lewis acids with soft Lewis bases will accomplish this and avoid the formation of strongly coordinated complexes.6
Another perceived challenge of synergistic catalysis surrounds kinetics. At first glance, it may appear that any reaction between two catalytic intermediates that are present in low sub-stoichiometeric concentrations should be inherently difficult. This assumption neglects to consider an integral part of the rate equation, the rate constant k. When considering the reaction between two activated intermediates, the narrowing of the HOMO-LUMO gap that occurs in synergistic catalysis (ΔE, Fig 1) causes a large decrease in the activation energy, which in turn increases the rate constant to drive the desired pathway in preference to possible side reactions.
While we recognize the challenges in implementing synergistic catalysis, there have been many successful applications reported in the recent literature. Indeed, a survey of asymmetric catalysis based on this synergistic paradigm reveals a 10-fold increase in publication numbers over the last decade (Fig 4). We believe this emergence of synergistic catalysis in the commuity is founded upon the realization of the inherent utility and benefits provided by this multi-catalysis concept. In this perspective, we aim to illustrate these benefits through various literature examples that demonstrate the (i) development of new, previously unattainable chemical transformations; (ii) improvements of existing transformations; (iii) introduction or improvement of catalytic enantioselectivity in systems where stereocontrol was previously absent or difficult.
Fig. 4.

The total number of publications describing asymmetric synergistic catalysis from January 1993 to May 2011.
2. Carbonyl α-Functionalization
2.1 Synthesis of α-Allyl carbonyls via π-allyl intermediates
Palladium-catalyzed allylic alkylation (Tsuji-Trost allylation) is an attractive strategy to install an allyl subsitutent on a nucleophile via transient palladium(II) π-allyl intermediates.7 For aldehyde and ketone nucleophiles, traditional α-allylic alkylations often require stoichiometric preactivation to metal enolates, silyl enol ethers, or enamines prior to the allylation protocol. The concept of synergistic catalysis has enabled the development of several direct syntheses of α-allylic aldehydes and ketones through the merger of palladium cataysis with other catalytic cycles. Described below are various methods of catalytically activating carbonyls (or carbonyl precursors) to combine successfully with palladium(II) π-allyl complexes to forge α-allyl bonds.
An early example from the Ito group merges rhodium and palladium catalysis for enantioselective allylic alkylations of α-cyano carbonyls (Scheme 1).8 Chiral rhodium(I)-activated enolates combine with π-allylpalladium(II) complexes derived from allyl carbonates (2) to form optically active α-allyl-α-cyano esters (3), amides (4), and phosphonates (5) in high yield and selectivity. As shown in Scheme 2, chiral rhodium(I) complex 6 coordinates to the α-cyano group of 1 forming rhodium(I)-coordinated enolate 7. Simultaneously, the decarboxylative oxidative addition of allyl carbonate 2 to palladium(0) forms the π-allylpalladium(II) intermediate. The two catalytic cycles merge as rhodium-bound enolate 7 adds to the π-allyl electrophile, in a highly enantioselective fashion, to produce the stereogenic α-cyano-α-allyl carbonyl 3 and regenerate both catalysts.
Scheme 1.

Rhodium- and palladium-catalyzed enantioselective allylic alkylation of α-cyano carbonyls.
Scheme 2.

Mechanism of the rhodium- and palladium-catalyzed allylic alkylation of α-cyano carbonyls.
No reaction occurs in the absence of palladium, and while the reaction does proceed without the rhodium catalyst (with chiral ligand on palladium), the rate is considerably slower and racemic product is generated. These results indicate that a chiral environment around palladium imparts no stereocontrol on the transition state. This is not surprising as stabilized carbon nucleophiles attack the π-allyl electrophile from the face opposite the chiral palladium.7c The addition of the rhodium catalyst accelerates the rate of the reaction by activating the α-cyano carbonyl and also creates an opportunity to impart stereocontrol during the bond-forming step.
While the allylic alkylation of α-cyano carbonyls requires a synergistic strategy to create enantiocontrol in the transition state,α-allylic alkylations of unactivated aldehydes and ketones do not proceed without activation of both reacting partners, due to the weak nucleophilicity of the carbonyl compounds (a result of the low concentration of the moderately nucleophilic enol tautomer).
In 2006, Córdova and coworkers reported the direct α-allylic alkylation of unactivated aldehydes and cyclic ketones with allyl acetate 10 through the merger of enamine and palladium catalysis (Scheme 3).9 Using pyrrolidine to activate the carbonyl and tetrakis(triphenylphosphine)palladium(0) to activate allyl acetate 10, the reaction proceeds with high efficiency to yield the racemic α-allyl carbonyls (11).
Scheme 3.

Córdova’s direct α-allylation of unactivated aldehydes and ketones via enamine and palladium π-allyl intermediates.
The organocatalytic cycle begins as pyrrolidine condenses with 9 to form a transient pyrrolidine enamine 12. At the same time, allyl acetate 10 oxidatively adds to the palladium(0) complex, to form the π-allylpalladium(II) electrophile. The metal- and organocatalytic cycles intersect as the two intermediates combine to form α-allyl iminium ion 13. Hydrolysis releases α-allyl carbonyl 11 and regenerates both the amine and palladium catalysts.
Replacing pyrrolidine with a chiral amine catalyst was only moderately successful in the Córdova system for generating stereogenic α-allyl carbonyls. Enantiomeric excesses of up to 74% ee were obtained; however, reaction yields decreased dramatically. Zhang and coworkers, however, recently built upon Córdova’s synergistic strategy and were able to achieve an asymmetric varient of the α-allylic alkylation of unactivated aldehydes and ketones.10 Taking a different approach to enantioinduction than previously described (i.e. chiral carbonyl activation), palladium catalysts bearing C2-symmetric chiral metallocene-based ligands were used to activate secondary allyl acetates (14), generating chiral π-allyl elctrophiles (Scheme 5). The chiral environment created around palladium is able to induce high levels of enantioselectivity in the α-allyl products (15). Unfortunately, using catalytic quantities of pyrrolidine provides poor efficiency (≤ 50% yield) and a stoichiometric amount of the amine is needed.
Scheme 5.

An enantioselective α-allylation of aldehydes and ketones via enamine and palladium π-allyl intermediates.
 |
(1) |
In 2001, Takemoto and coworkers investigated a different approach to carbonyl activation and used a phase-transfer strategy to activate tert-butyl glycinate-benzophenone (16) to react with stryrenyl acetates 17 in the presence of palladium(0) (Scheme 6).11 As outlined in Scheme 7, the aqueous base deprotonates glycine imino ester 16 and the resulting enolate 19 associates with the chiral cinchonidium phase transfer catalyst in the organic phase of the biphasic mixture. The chiral phase transfer catalyst acts as the counterion to Schiff base enolate 19 and enables differentiation of the two faces of the prochiral nucleophile, allowing enantioselective addition to the π-allylpalladium(II) complex. The result is the highly enantioenriched α-allyl glycine imino ester 18 (up to 96% ee), which could not be generated using either catalyst individually.
Scheme 6.

Takemoto’s α-allylic alkylation of glycine imino esters.
Scheme 7.

α-Allylic alkylation of glycine imino esters via phase transfer and palladium catalysis.
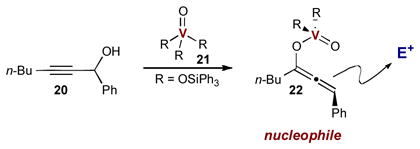
Until recently, synergistic catalysis strategies toward α-allyl carbonyls had focused exclusively on the direct α-allylation of saturated carbonyls. In 2011, however, Trost and Luan developed a unique synergistic approach to the synthesis of α-allyl-α,β-unsaturated ketones.12 In previous studies, it was demonstrated that oxyvanadium catalysts 21 induces a 1,3-transposition of propargylic alcohols via nucleophilic vanadium-allenoate complex 22 (Eq 1).13 The authors hypothesized that this catalytic nucleophile should also participate in a Tsuji-Trost allylation and couple with electrophilic π-allyl palladium complexes, forming α-allyl-α,β-unsaturated ketones. A challenging aspect of this transformation is that both vanadium-allenoate intermediate 21 and the π-allyl palladium complex can be trapped with a second propargyl alcohol (present in stoichiometric quantities), forming the Meyer–Schuster rearrangement product 24 (Scheme 8A) and propargylic ether 25 (Scheme 8B) respectively. The success of the desired synergistic transformation depends on the relative rates of the three potential reaction pathways. Despite the potential side reactions, the authors found that treating propargyl alcohol 20 and allyl carbonate 23 with a combination of vanadium and palladium catalysts provides the desired α-allyl enones 26 in excellent yield with no trace of the side products (Scheme 8C). Despite the low concentrations of both reactive intermediates, the desired α-allylation reaction outcompeted both possible side reactions.
Scheme 8.

Trost’s palladium- and vanadium-catalyzed synthesis of α-allyl-α,β-unsaturated ketones.
The mechanism of this remarkable transformation is outlined in Scheme 9. Vanadium catalyst 21 undergoes transesterification with proargyl alcohol 20 to form vanadium ester 27. A 1,3-transposition of the ester generates key vanadium-allenoate intermediate 22. At the same time, allyl carbonate 23 reacts with the palladium(0) catalyst to form the palladium π-allyl electrophile. Before either catalytic intermediate can react with unactivated propargyl alcohol 20, the two activated metal complexes combine to form the desired α-allyl enone 26.
Scheme 9.

Mechanism of the palladium- and vanadium-catalyzed α-allyl enone formation.
2.2 Enamine catalysis with metal-bound electrophiles
Traditional enamine catalysis relies on a relatively limited electrophile scope. An examination of these transformations reveals that electrophiles can generally be characterized as either unsaturated nucleophile acceptors (e.g. carbonyls, imines, enals) or electrophilic sources of heteroatoms.14 Using electrophiles outside these classes, enamine catalysis often suffers from side reactions, such as self-aldolization. To avoid this, the rate of enamine addition to the desired electrophile must be greater than addition to unreacted aldehyde. By combining enamine and transition metal catalysis, the desired chemoselectivity can increase through catalytic activation of the electrophile. This extends the traditional electrophile scope in order to develop new, previously unattainable chemical reactions.
The following transformations follow the same general mechanism (Scheme 10). The aldehyde (or ketone) condenses with an amine catalyst to form a transient enamine intermediate.
Scheme 10.

General mechanism of enamine catalysis with metal-bound electrophiles to generate α-substituted carbonyls.
A metal catalyst also activates an electrophile, generating a metal-bound electrophilic intermediate. This metal-bound electrophile interacts with the enamine, transferring the desired functional group, and creates an α-substituted iminium ion while regenerating the metal catalyst. Hydrolysis of the iminium ion releases the α-substituted product and returns the amine catalyst.
Shortly after Córdova reported the first example of merging transition metal catalysis and organocatalysis (Scheme 3), Wu and coworkers introduced a synergistic 3-component coupling reaction. 2-Alkynylbenzaldehydes, primary amines, and ketones combine under proline and Ag(I) catalysis to form 1,2-dihydroisoquinoline derivatives (31) via the first reported merger of enamine and π-acid catalysis (Scheme 11).15 They propose 2-alkynylbenzaldehyde 28 condenses with primary amine 29 to form 2-alkynylbenzimine 33 while ketone 30 condenses with proline to form enamine 32. Intermediate 33 coordinates AgOTf, which triggers a hydroamination/Mannich cascade to furnish the 1,2-dihydroisoquinoline framework. Hydrolysis of iminium 34 releases dihydroisoquinoline 31. Aryl substituted 2-alkynylbenzaldehydes and anilines react smoothly, however, when either of these components are alkyl substituted the yields obtained are much lower (≤ 35%). In addition, although proline forms chiral enamines, only racemic products are formed.
Scheme 11.

Wu’s synthesis of 1,2-dihydroisoquinoline derivatives.
Recently, our group has also become interested in the use of transition metal-activated electrophiles in enamine catalysis as a strategy to develop new asymmetric reactions. This approach enabled an enantioselective α-trifluoromethylation of aldehydes to be developed using Togni’s reagent (35), an electrophilic trifluoromethyl source (Scheme 12).16 Although Togni’s reagent has been shown to react efficiently with a variety of nucleophiles,17 when exposed to a transient chiral enamine, based on imidazolidinone catalyst 36, only low yields are observed (14% yield, 92% ee). As the most likely mechanism of trifluoromethylation is attack of the Togni reagent at iodine, followed by reductive elimination of the aryl iodide, it was reasoned that a Lewis acid should facilitate cleavage of the I–O bond and create activated electrophilic iodonium intermediate 39 (Scheme 12). Copper-bound 39 should render iodine more susceptable to nucleophilic attack by enamine 38 when compared to the unactivated Togni reagent. By adding catalytic CuCl as a Lewis acid, in conjuction with chiral imidazolidinone catalyst 36, excellent yield and enantioselectivity are obtained for the resultant α-trifluoromethyl aldehydes 37.
Scheme 12.

MacMillan’s organocatalytic α-trifluoromethylation of aldehydes using Togni’s reagent and CuCl.
By combining enamine and transition metal catalysis, the Nishibayashi group developed two complementary methods for enantioselective α-propargylation of aldehydes using propargyl alcohols 42 and benzoates 43 – classes of electrophiles previously outside the scope of enamine catalysis. Diruthenium and ligated copper catalysts activate 42 and 43, respectively, to form metal-allenylidene complex 46 that is electrophilic at the γ-position (Scheme 13).18 These metal-bound electrophiles are trapped by transient chiral enamine 47 to provide enantioenriched α-propargyl aldehyde 45, a product unattainable via enamine catalysis alone. Ruthenium appears to provide the product in higher yields,18a while copper delivers higher selectivities.18b
Scheme 13.

Nishibayashi’s enantioselective α-propargylation of aldehydes via enamine and transition metal catalysis.
The allenylidene intermediate 46 initially limited the scope of this transformation to terminal alkynes, as they cannot be formed with internal alkynes. Recently, however, Nishibayashi expanded the scope to include internal alkynes by using an alternative method of activating propargylic alcohols (Scheme 14).19 Using InBr3 as a Lewis acid, propargylic alcohol 49 is converted to the corresponding propargylic cation 51. These cations are trapped by transient enamines to provide enantioenriched α-propargyl aldehyde 50 bearing an internal alkyne.
Scheme 14.

Nishibayashi’s enantioselective α-propargylation of aldehydes using internal alkynes via enamine and indium catalysis.
In 2007, Sibi and Hasegawa reported an enantioselective α-oxyamination of aldehydes using FeCl3 and a chiral amine catalyst.20 Initially, this transformation was not described as an example of synergistic catalysis. It was initially proposed that FeCl3 oxidizes the transient enamine to produce a radical cation, which traps TEMPO at the α-position. Recently, through mechanistic studies performed in the MacMillan group this mechanism was shown to be incorrect; the transformation most likely proceeds via synergistic catalysis wherein the FeCl3 coordinates to TEMPO, creating a complex that is electrophilic at oxygen and therefore activated towards enamine addition.21 Based on this mechanistic insight, MacMillan and coworkers improved this transformation by switching to CuCl2 (known to form stable complexes with TEMPO) and using amine catalyst 53. This broadened the functional group tolerance of the reaction and increased both yield and enantioselectivity (Scheme 15). Under these conditions, TEMPO coordinates copper(II) to form electrophilic complex 56. Transient enamine 55 adds to the oxygen of the TEMPO-bound copper(II) complex 56, generating iminium ion 57 and a copper(I) salt. The copper(I) is reoxidized by ambient oxygen to regenerate the copper(II) catalyst.
Scheme 15.

MacMillan’s copper-catalyzed enantioselective α-oxyamination of aldehydes.
In all of the examples discussed, the transition metal catalyst is only directly involved with the electrophile, activating it towards enamine addition. An enamine, however, contains an electron-rich π-system and can also coordinate to a transition metal catalyst, helping to bring together the enamine and the electrophile to forge a bond at the α-position. Although less common in the literature, recently, the MacMillan group published an example using this synergistic strategy. Using copper catalysis in combination with a chiral amine catalyst, an enantioselective α-arylation of aldehydes was developed that uses diaryliodonium salts as the electrophilic arene source (Scheme 16).22 Although diaryliodonium salts are not electrophilic enough to react with a transient enamine under organocatalytic conditions alone, they can be activated by copper(I) salts to generate highly electrophilic arylcopper(III) intermediate 60.23 These highly reactive arylcopper(III) species react smoothly with enamines (derived from amine catalyst 58) to generate copper-bound iminium ion 61 that undergoes reductive elimination, followed by hydrolysis, to provide enantioenriched α-aryl aldehyde 59 in excellent yield and selectivity. A wide variety of arenes can be transferred by this protocol, ranging from electron rich to electron poor and even some heteroaromatics. Prior to this report, organocatalytic α-arylation reactions had only been developed for intramolecular substrates and intermolecular reactions with highly activated quinone substrates.24 Using synergistic catalysis a more general intermolecular reaction was developed through in situ activation of both reacting partners.
Scheme 16.

Organocatalytic α-arylation of aldehydes via the merger of enamine and copper catalysis.
2.3 Photoredox α-alkylation of aldehydes
In all of the synergistic catalysis examples outlined above, both catalysts have been directly involved in the key bond forming step. In an alternate mechanistic scenario, a catalyst may induce formation of a reactive intermediate, but not be involved in the actual coupling event; this strategy is key to the success of photoredox organocatalysis.25
The catalytic enantioselective α-alkylation of aldehydes has been an ongoing challenge in organic synthesis that has yet to be solved in a general sense.26 In enamine catalysis, which traditionally proceeds through a closed-shell, two-electron pathway, the use of alkyl halides has been generally unsuccessful, commonly leading to catalyst alkylation and/or self-aldolization.27 In an effort to circumvent these problems, MacMillan and Nicewicz reasoned that the electron-rich π-system of enamines should combine efficiently with electron-deficient radicals through a one-electron pathway.28 To implement this strategy, enamine catalysis could be merged with the catalytic formation of electron-deficient radicals through the reductive cleavage of alkyl halide C–X bonds (Eq 2). Recognizing that photoredox catalysis is an attractive method for reductive cleavage of C–X bonds via photo-induced electron transfer (PET) from Ru(bpy)3Cl2,29 the concepts
| (2) |
of enamine catalysis and photoredox catalysis were combined in an effort towards the enantioselective α-alkylation of aldehydes (Scheme 17).28
Scheme 17.

The reductive cleavage of electron-poor alkyl bromides for use in photoredox organocatalysis.
The general mechanism of organocatalytic photoredox α-alkylations is outlined in Scheme 18. When exposed to weak visible light, Ru(bpy)32+ accepts a photon and populates the metal-to-ligand charge transfer (MLCT) excited state, *Ru(bpy)2+. Initially, this excited state is quenched by a sacrificial amount of enamine 65 (0.5 mol%) to generate the highly reducing Ru(bpy)3+ (not shown). This reductant delivers an electron to alkyl halide 63, generating the required electron-deficient radical 64 and returning the photocatalyst to its original oxidation state. In the organocatalytic cycle, chiral transient enamine 65 is formed via condensation of the aldehyde with amine catalyst 62. The two catalytic cycles intersect and electron-deficient radical 63 adds to the accessible face of the electron-rich π-system of 65, achieving the key alkylation step. The coupling event results in α-amino radical 66, whose low barrier to oxidation allows for a second convergence of the two catalytic cycles. After accepting a photon, *Ru(bpy)32+ oxidizes 66 to provide an α-alkylated iminium ion and regenerate the strong reductant Ru(bpy)3+. Upon hydrolysis of iminium ion 67, the enantioenriched α-alkylated aldehyde is released and chiral amine catalyst 62 is returned. The first successful execution of this synergistic design was realized in 2008 for the α-alkylation of aldehydes using α-bromocarbonyls 68 as radical precursors.29 The combination of chiral amine organocatalyst 62 and Ru(bpy)32+ delivers the desired α-alkylated aldehydes 69 in excellent yields and enantioselectivities (Scheme 19A). This mild, yet powerful alkylation strategy supports high functional group tolerance and highly congested substrates as well as the formation of β-quaternary centers.
Scheme 18.

General mechanism for the photoredox enantioselective α-alkylation of aldehydes.
Scheme 19.

Examples of photoredox organocatalysis reported by MacMillan and coworkers.
Recognizing the generality of this synergistic strategy, MacMillan and coworkers turned to other α-alkyl substrate classes that have proven challenging under traditional catalysis. Shortly following the initial report, a photoredox strategy towards the α-trifluoromethylation and α-perfluoroalkylation of aldehydes was described using CF3I (or the corresponding perfluoroalkyl iodide) as the radical source (Scheme 19B).30 In this case, Ir(ppy)2(dtbbpy)PF6 is used as the photocatalyst, rather than Ru(bpy)3Cl2, since after single electron transfer, the reduced form, Ir(ppy)2(dtbbpy), is a stronger reductant than Ru(bpy)3+ (−1.51 V vs SCE in CH3CN for Ir(ppy)2(dtbbpy) compared to −1.33 V vs SCE in CH3CN for Ru(bpy)3+).29,30 The trifluoromethyl (or perfluoroalkyl) radical adds smoothly to the in situ generated enamine to furnish the α-CF3 or α-perfluoroalkylated aldehydes in excellent yields and enantioselectivities. To maintain the stereochemical integrity of these products, the reaction must be run under cryogenic conditions.
Another challenging alkylation that was addressed using photoredox organocatalysis is the enantioselective α-benzylation of aldehydes (Scheme 19C).31 Previous direct organocatalytic methods to form α-benzylated aldehydes require multiple and/or electron-rich aryl rings on the electrophilic coupling partner to stabilize intermediate benzylic carbocations.32 Since photoredox organocatalysis relies on the formation of electron-deficient radicals, this strategy complements existing benzylation protocols. The α-benzylation of aldehydes proceeds through a slightly different photoredox catalytic cycle than described above. This protocol uses the commercially available photocatalyst fac-Ir(ppy)3. In the excited state, fac-*Ir(ppy)3 is a strong enough reductant (−1.73 V vs SCE in CH3CN) to induce reductive cleavage of aryl and heteroaryl methylene halides 73 directly (Scheme 20). After single electron transfer and addition of benzyl radical 74 to enamine 75 (derived from chiral amine catalyst 71), fac-Ir(ppy)3 merges with the organocatalytic cycle to oxidize α-amino radical 76, which closes the photoredox cycle. The α-benzylation is successful for a range of electron-deficient aryl and heteroaryl methylene substrates and aldehyde functionality to form enantioenriched α-benzyl aldehydes in high yield and selectivity.
Scheme 20.

Mechanism of the photoredox organocatalytic α-benzylation of aldehydes.
Recently, Zeitler and coworkers also became interested in the concept of photoredox organocatalysis. Instead of turning to organometallic based photoredox catalysts, they spectulated that red and orange organic dyes, which absorb green wavelengths of visible light, may also perform successfully as photocatalysts. Through initial studies, eosin Y emerged as the optimal substitute for more typical organometallic photocatalysts.33 As a proof of concept with eosin Y, Zeitler was able to produce the same synergistic α-alkylation of aldehydes with α-bromocarbonyls as MacMillan’s seminal photoredox organocatalysis. The proposed mechanism mirrors that of MacMillan with photoexcited eosin Y (*EY) acting as the oxidant (requiring a sacrificial amount of enamine to initiate the photoredox cycle) and reduced eosin Y (EY·−) acting as the reductant (Scheme 21).
Scheme 21.

Zeitler’s photoredox protocol using organic photocatalysts.
2.4 Other Synergistic Strategies Involving Enamine Catalysis
A powerful advantage of using a synergistic approach to enamine catalysis is the ability to generate reactive electrophiles in situ from non-electrophilic precursors. Klussmann and coworkers recognized this advantage and developed a direct oxidative Mannich-type coupling of cyclic tertiary amines 78 with methyl ketones 79 using a combination of vanadium and proline as catalysts (Scheme 22).34a In situ oxidation of tertiary amine 78 with VO(acac)2 generates electrophilic iminium ion 81. After proline activates methyl ketone 79, transient enamine 82 adds to the cyclic iminium ion 81. Hydrolysis of 83 liberates proline and delivers the Mannich-type product 80 in moderate to good yields. To close the vanadium catalytic cycle, the reduced form of the catalyst is reoxidized by t-BuOOH. Unfortunately, despite using a chiral organocatalyst, little to no enantioinduction was observed with proline. Further attempts with chiral vanadium complexes were also unsuccessful at an asymmetric reaction. These observations are attributed to facile racemization of β-amino ketones.
Scheme 22.

Oxidative coupling reactions of cyclic tertiary amines and methyl ketones reported by Klussmann and Rueping.
Recently, the same oxidative coupling reaction was addressed by Rueping and coworkers. Instead of using catalytic VO(acac)2 to generate the intermediate iminium ion, photoredox catalysis with Ru(bpy)3(PF6)2 was used in conjuction with proline instead (Scheme 22).34b The oxidation is proposed to proceed through the same radical mechanism, where the photoexcited *Ru(bpy)3(PF6)2 oxidizes tertiary amine 78 to form iminium ion 81. Molecular oxygen oxidizes Ru(bpy)3+ to regenerate the photocatalyst. When compared to VO(acac)2 oxidation, photoredox catalysis provides a noticable increase in yield of the desired coupling product. Interestingly, use of 5W fluorescent light was found to be crucial to the high yields, as stronger light emission, whether from LED or UV light, resulted in diminished yields. This has been attributed to the interplay between the photoredox and organocatalytic cycles, since the strength of the light source can tune the rate of the photoredox cycle to match the rate of enamine formation. As with Klussmann’s protocol, Rueping’s photoredox oxidative coupling also provided nearly racemic products (8% ee).
Recently, Pihko and coworkers reported a three-component domino reaction to generate γ-nitro aldehydes 86 using a combination of chiral secondary amine catalyst 84 and a multiple hydrogen bond donor (MHBD) catalyst (Scheme 23).35 In this example, not only is the electrophile generated in situ through synergistic catalysis, it is subsequently consumed using the same combination of catalysts. Overall, the two step sequence involves the formation of nitroolefins from aldehydes and nitromethane, followed by the conjugate addition of the aldehyde to the nitroolefin via enamine catalysis. Attempting this domino process with a secondary amine catalyst alone results in aldol and aldol-type products, bypassing the nitromethane altogether. Furthermore, attempting just the second step of the domino sequence with pregenerated nitroolefins and an amine catalyst resulted in low yields of the γ-nitroaldehyde product 86 (< 20% yield), indicating both steps of this sequence benefit from synergistic catalysis.
Scheme 23.

An enantioselective three-component domino reaction via synergistic hydrogen-bond and enamine catalysis.
In the first step, adding a MHBD catalyst activates nitromethane to form hydrogen-bonded anion 87 for a Knoevenagel-type condensation with iminium ion 88 (formed via condensation of the aldehyde and amine catalyst 84). The in situ generation of nitroolefin 85 proceeds smoothly, even with challenging β-alkyl substitution. In the second step of the sequence, nitroolefin 85 is activated by the MHBD catalyst towards conjugate addition of chiral transient enamine 90 (derived from 84 and the second aldehyde). This synergistic domino process provides γ-nitroaldehydes 86 in good yields and excellent selectivities. The activation of reacting partners in both steps of this domino sequence allows for the desired reaction to proceed with high chemoselectivity over any aldol side products.
3. Reactions of α,β-unsaturated carbonyls
3.1 Organocatalytic activation of unsaturated carbonyls
Although synergistic catalysis involving enamine and transition metal catalysis has been known since Córdova’s allylic alkylation report in 1996,9 synergistic catalysis involving iminium catalysis is much more recent. Just as synergistic catalysis expands the electrophile scope in enamine catalysis, this strategy should also expand the nucleophile scope of iminium catalyzed reactions to include those that were previously unsuccessful. Córdova and coworkers recognized this and developed a catalytic enantioselective silyl conjugate addition to α,β-unsaturated aldehydes using Me2PhSi-B(pin) (92), an unlikely nucleophile in traditional iminium catalysis (Scheme 24).36 Treating 92 with CuCl induces transmetallation to form nucleophilic Cu(I)-silane 94. This complex intersects the iminium catalytic cycle and adds to the β-position of chiral transient iminium ion 95 (formed by condensation of the enal and amine catalyst 85). The resultant copper-bound β-silyl iminium ion 96 provides the desired enantioenriched β-silyl aldehyde 93 in good yields and selectivities after protonation and hydolysis of the amine catalyst. Although both aromatic and aliphatic enals react efficiently, selectivities are generally higher with aromatic enals.
Scheme 24.

Córdova’s catalytic enantioselective silyl conjugate addition to α,β-unsaturated aldehydes via copper and iminium catalysis.
Organocatalytic activation of enals is not limited to iminium catalysis. Condensation of an amine catalyst with an enal can also generate nucleophilic transient dienamines; this mode of activation has recently been combined synergistically with chiral Brønsted acid catalysis.37 While developing a γ-benzylation of α-branched enals, Melchiorre and coworkers found that trapping stabilized carbocations with chiral transient dienamines (derived from a cinchona alkaloid amine) provides only moderate enantioselectivities (up to 60% ee). Similarly, using a chiral phosphoric acid to induce carbocation formation, with achiral benzyl amine activating the enal, also leads to poor enantiocontrol (14% ee). When combined, however, the chiral cinchona alkaloid and chiral phosphoric acid together provide excellent yield and enantioselectivity for the desired γ-benzylated enal 99 (up to 95% ee) (Scheme 25). It is believed that the cinchona alkaloid catalyst activates enal 97 through dienamine formation, creating vinylogous nucleophilicity at the γ-position. At the same time, the chiral phosphoric acid catalyst protonates dibenzyl alcohol 98, inducing carbocation formation. The benzylic cation, with a chiral phosphate counterion, interacts with the dienamine through a network of non-covalent interactions, leading to a highly organized transition state (Scheme 25). The authors propose that the chiral phosphate counterion, associated with the benzylic cation, also participates in a hydrogen bond with the quinuclidine hydroxyl group at the 6′-position, presumably helping direct attack of the carbocation from the appropriate face of the dienamine. Evidence of the cooperation between the two catalysts can be seen with mismatched catalyst combinations, as this leads to dramatic loss in both reactivity and enantioselectivity (≤30% yield, ≤21% ee). Interestingly, in all cases the phosphoric acid catalyst determines the major enantiomer of the product obtained.
Scheme 25.

Direct γ-benzylic alkylation of α-branched enals via the merger of chiral dienamine and Brønsted acid catalysis.
The use of non-covalent ion-pairs in synergistic catalysis has also led to improvements in the organocatalytic enantioselective synthesis of tetrahydroxanthenones. In 2007, Córdova and coworkers reported the first catalytic enantioselective protocol for the synthesis of tetrahydroxanthenones 101 from salicylic aldehydes 100 and cyclohexenones via an iminium catalyzed oxa-Michael–Mannich domino reaction (Scheme 26A).38 The transformation provides the desired tricyclic product in modest yields (42–56%), but good selectivities (85–91% ee). Realizing the power of synergistic catalysis, Xu and coworkers sought to improve this transformation through organocatalytic activation of both salicylic aldehyde 100 and cyclohexenone with catalysts that could bring the reacting partners together via an ion-pair assembly (Scheme 26B).39 In Xu’s protocol, the cyclohexenone is activated by chiral pyrrolidine derivative 103, bearing a thiopyridine unit, to form iminium ion 104. At the same time, salicylic aldehyde 100 is activated by tert-leucine, forming imine 105. As a key design element, the two activated intermediates (104 and 105) form an ion-pair assembly through proton transfer from tert-leucine to the thiopyridine unit. This assembly brings together the two intermediates and ensures the domino oxa-Michael–Mannich reaction occurs with high efficiency and selectivity. Hydrolysis of catalyst 103 and subsequent elimination of tert-leucine regenerates both catalysts and delivers tetrahydroxanthenone 101 in excellent yields and enantioselectivites. Unsubstituted cyclohexenones provide significantly higher selectivites than that of the 4,4-disubstituted cyclohexenones (91–98% ee unsubstituted compared to 80% ee for 4,4-dimethylcyclohexenone).
Scheme 26.

Formation of tetrahydroxanthenones via organocatalytic enantioselective oxa-Michael–Mannich reaction.
3.2 Lewis acid activation of α,β-unsubstituted carbonyls
Chiral Lewis acid catalysis has been the cornerstone activation method for conjugate additions to α,β-unsaturated carbonyl systems. In 2003, Jacobsen and coworkers reported a conjugate addition of hydrogen cyanide to unsaturated imides 108 using a (salen)AlCl complex.40 Although excellent yields of highly enantioenriched cyanide adducts 109 were obtained, high catalyst loadings, long reaction times, and in some cases elevated temperatures were required. Through mechanistic studies, the authors found that the aluminum catalyst was activating both the imide and cyanide components. They hypothesized that the lower than desired reactivity was a result of inefficient activation of the cyanide component by the aluminum complex. To improve the conjugate cyanation protocol, Jacobsen and coworkers readdressed the same transformation, applying a synergistic catalysis strategy to effectively activate both reacting partners individually.41 Chiral (pybox)lanthanide complexes, such as (pybox)ErCl3, are known to activate cyanide towards nucleophilic additions, but fail to effectively promote the conjugate cyanation alone (< 3% yield); this indicates the complex can activate the cyanide, but not imide 108. Similarly, μ-oxo dimer (salen)Al2O is also ineffective as a single catalyst towards the conjugate cyanation (< 3% yield), however, this lack of reactivity is attributed to an unactivated cyanide component. Although alone, neither complex promotes the enantioselective conjugate cyanation, when used together to activate their respective component, the combination promotes a highly efficient and selective reaction (Scheme 27). By using synergistic catalysis, reaction times were reduced, catalyst loadings were lowered, and there was no longer a need to heat less reactive substrates.
Scheme 27.

Jacobsen’s conjugate cyanation of unsaturated imides.
4. Carbonyl and imine 1,2-Addition
4.1 Asymmetric alkynylation of imines via metal acetylides
Among the most straightforward methods for producing enantioenriched propargylic amines, important chiral building blocks, is the addition of a metal acetylide to an imine or iminium ion. In 2005, Chan and coworkers reported the first asymmetric alkynylation of α-imino esters using chiral copper complexes.42a While high enantioselectivities can be obtained for aliphatic alkynes (up to 91% ee), aromatic alkynes resulted in much lower selectivities (up to 74% ee).42b Shortly after Chan’s report, two synergistic catalysis strategies were introduced for the asymmetric addition of metal acetylides, both aromatic and aliphatic, to imines under chiral Brønsted acid and transition metal catalysis.
The successful merger of transition metal and Brønsted acid catalysis for the asymmetric alkynylation of imines relies on the concurrent propagation and interaction of the two independent catalytic cycles without protonation of the metal acetylide (113) (Scheme 28). Imine 110 is protonated by a chiral Brønsted acid to form chiral ion-pair 111. Independently, terminal acetylene 112 interacts with the metal catalyst to form the activated metal acetylide nucleophile 113. Combination of the two intermediates delivers the chiral propargylic amine 114 and returns both catalysts to their respective catalytic cycles.
Scheme 28.

General mechanism for the addition of metal acetylides to Brønsted acid activated imines.
In 2007, Rueping and coworkers successfully implemented this strategy for the alkynylation of α-imino esters 115 using chiral binol-based phosphoric acid and AgOAc as catalysts (Scheme 29A).43 Complementing Chan’s method, the addition of electron-neutral and electron-rich aromatic acetylenes (116) to α-imino esters 115 provides the propargylic amines 117 in good yields and enantioselectivities (up to 92% ee). The use of AgOAc as the acetylene activator may be key to the success of this synergistic catalytic system. Alkynyl-silver derivatives are known to be stable in protic media, requiring strong acids (e.g. HCl, TfOH) for hydrolysis.44a In addition, acetylides activated by AgOAc do not add to non-activated imines, preventing an achiral background reaction.44b Although the authors believe this transformation proceeds through the general mechanism outlined above (Scheme 28), they also acknowledge a possible counterion exchange between the silver acetate and chiral phosphoric acid that could generate chiral silver complex 118 (Scheme 29A). To investigate this, experiments were performed with chiral silver-binol complexes and achiral diphenyl hydrogen phosphates; however, these resulted in racemic propargylic amines. This suggests that the Brønsted acid cycle is responsible for the stereoinduction, regardless of the nature of the silver acetylide involved.
Scheme 29.

Asymmetric synthesis of propargylic amines via the merger of transition metal catalysis with Brønsted acid catalysis.
Recently, Arndtsen and coworkers were the second group to report a similar synergistic catalysis protocol for the addition of acetylides to imines (Scheme 29B).45 Using N-Boc-proline as the chiral acid and ligated Cu(MeCN)2PF6 to activate acetylene 119, enantioenriched propargylic amines 121 are obtained in excellent yields and selectivities (up to 99% ee). Aryl-, vinyl- and alkyl-substituted acetylenes add to aryl- and tert-butylimines with little impact on yield or selectivity. While the authors also propose the general mechanism outlined above (Scheme 28), they recognize that an amino acid/copper secondary coordination might also be possible. Mechanistic studies, however, indicate that the reaction rate has a first-order dependence on N-Boc-proline and a zero-order dependence on the copper catalyst. This disparity in reaction order suggests separate roles for the copper and acid catalysts. As the authors point out, an advantage to this protocol is the flexibility and modularity of the catalyst system. Even though the chiral information is held in the N-Boc-proline catalyst, the steric bulk located on the copper acetylide (via the ligands) has a significant influence on the enantioselectivity of the reaction. As there are many commercially available α-amino acids and tertiary phosphines, this catalyst system is easily tuned for any given substrate.
4.2 Racemic addition of metal acetylides to carbonyls and imines
In their efforts toward developing Grignard-like reactions in water, Li and coworkers reported a synergistic bimetallic system for the preparation of racemic propargylic alcohols.46 Early in the investigation of metal acetylide addition to aldehydes, they observed that when a single transition metal catalyst was used, the acetylenes were consumed while the aldehydes remained unreacted. They speculated that water-tolerant Lewis acids may be needed to activate the carbonyl and this led to their successful RuCl3/In(OAc)3 bimetallic system (Scheme 30A). RuCl3, reduced in situ to Ru(II), activates the acetylene via oxidative addition of the acetylene C–H bond. The resultant Ru(IV) acetylide 125 adds to the In(OAc)3-activated carbonyl 124 to deliver the desired propargylic alcohol 122. Under this system, phenylacetylene adds to a broad range of non-enolizable aldehydes in moderate yields.
Scheme 30.

Li’s ruthenium- and copper-catalyzed synthesis of propargyl alcohols and amines.
Li and coworkers quicky extended this synergistic protocol to the preparation of propargylic N-aryl amines 123 through a three-component coupling reaction of non-enolizable aldehydes, anilines, and phenylacetylene (via the in situ formation of animine) (Scheme 30B).47 Although In(OAc)3 was the optimal Lewis acid for carbonyl activation, it was ineffective for in situ generated imines. Further investigation revealed CuBr as the best imine activator. Using the synergistic combination of RuCl3 and CuBr in water, propargylic amines 123 are obtained in excellent yields. This powerful synergistic catalyst system has even been extended to a five-component coupling reaction for the formation of dipropargyl amines (Eq 3).48
 |
(3) |
4.3 Asymmetric hydride transfer to imines
Asymmetric hydrogenation is a powerful technology for the synthesis of chiral compounds. The use of molecular hydrogen is particularly attractive due to its low cost and perfect atom economy. At this time, most asymmetric hydrogenations use precious late transition metal catalysts with specific chiral phosphines to impart stereocontrol.49 Recently, Beller and coworkers saw an opportunity for synergistic catalysis to provide a different approach to asymmetric hydrogenation of imines using molecular hydrogen. In their strategy, Knölker’s complex, a simple achiral iron hydrogenation catalyst, is used in combination with a chiral Brønsted acid to provide enantioenriched secondary amines 127 from corresponding imines 126 (Scheme 31).50 Knölker’s complex (129) is a molecularly defined iron complex that possess an iron hydride and an acidic hydrogen, both available for transfer to a polarized π-system.51 In this synergetic design, imine 126 is protonated by (S)-TRIP to form chiral ion-pair 128. This intermediate accepts the hydride from Knölker’s complex (129), producing enantioenriched secondary amine 127. The acidic hydrogen of Knölker’s complex is transferred to the (S)-TRIP anion, regenerating the Brønsted acid complex. The resultant 16-electron iron complex 130 coordinates a molecule of hydrogen and induces heterolytic cleavage to regenerate Knölker’s complex and complete the iron catalytic cycle. While this transformation is successful for a broad range of both aryl aromatic and aliphatic N-aryl ketimines, aromatic ketimines provided noticeably higher selectivities. This is the first example of a highly enantioselective catalytic hydrogenation of imines that uses an achiral metal complex to activate molecular hydrogen.
Scheme 31.

Beller’s catalytic asymmetric hydrogenation of imines using a simple achiral iron catalyst and a chiral Brønsted acid catalyst.
Along with the asymmetric hydrogenation of imines, synergistic catalysis has also provided a similar strategy for the direct asymmetric reductive amination of ketones. Previously, Xiao and coworkers found that the half-sandwich complex Cp*Ir(III)-diamine·(R)-TRIP (134) efficiently reduced N-aryl ketimines using hydrogen in high yields and selectivites.52 When they tried to apply the same system to the reductive amination of aromatic ketones, very little amine product was observed. They reasoned that formation of the ketimine was rate-limiting and adding (R)-TRIP as an additional Brønsted acid catalyst may promote imine formation. Pleasingly, the synergetic catalyst combination of (R)-TRIP and Cp*Ir(III)-diamine·(R)-TRIP (134) was effective in promoting the reductive amination of aromatic methyl ketones 131 and anilines with molecular hydrogen to yield stereogenic amines 132 in excellent yields and selectivities (Scheme 32).53 It should be noted that the synergistic strategy is only required in the case of aromatic ketones; aliphatic ketones undergo reductive amination in the presence of the iridium catalyst alone.
Scheme 32.

The reductive amination of methyl ketones catalyzed by a chiral iridium complex and a chiral Brønsted acid.
The mechanism of this transformation begins with the Brønsted acid-catalyzed condensation of ketone 131 and aniline to form the protonated ketimine 133 with a chiral TRIP phosphate counterion. At the same time, iridium complex 134 coordinates a molecule of hydrogen and induces heterolytic cleavage to protonate the TRIP phosphate counterion (returning the free Brønsted acid catalyst) and generate iridium hydride 135. Iridium hydride 135 transfers a hydride to chiral iminium phosphate 133, providing enantioenriched secondary amine 132 and regenerating iridium complex 134.
4.4 Mannich-type addition to imines
In 2005, Hu and coworkers introduced a Rh2(OAc)4-catalyzed three-component reaction to couple diazoacetates with imines and alcohols.54 The imine traps a rhodium oxonium ylide intermediate to build racemic β-amino-α-alkoxyesters. In order to suppress undesired side products, the scope was limited to highly electron-deficient imines and imines derived from 2-aminophenol (the phenol functionality activates the imine through an intramolecular hydrogen bond). Through the course of their study, the authors observed that the desired reaction is accelerated by Brønsted acids, presumably through activation of the imine. They reasoned that adding a Brønsted acid catalyst, in conjuction with rhodium, should expand the scope of this reaction to include less activated imines. Furthermore, incorporating a chiral Brønsted acid may lead to an asymmetric three-component reaction.
Implementing this synergistic catalysis strategy, Hu and coworkers found that Rh2(OAc)4 with chiral phosphoric acid catalysts results in a highly efficient and selective coupling of diazoacetate 136 with a variety of imines 137 and benzyl alcohols to yield syn-β-amino-α-alkoxyesters 138 in high yields and selectivities (Scheme 33).55a Electronically diverse imines can now be used with no apparent impact on yield or selectivity. This protocol was also extended to water as a nucleophile, producing syn-β-amino-α-hydroxyesters 139 in good yields and excellent selectivities (Scheme 33).55b
Scheme 33.

The three-component coupling of diazoacetates, alcohols, and imines catalyzed by chiral Brønsted acids and Rh2(OAc)4.
This three-component transformation proceeds through the following mechanism (Scheme 34). Rh2(OAc)4 catalyzes the decomposition of diazoacetate 136 to form a rhodium carbenoid 140. The alcohol nucleophile adds to carbenoid 140 and generates rhodium-bound oxonium ylide 141. At the same time, imine 137 is protonated by the chiral phosphoric acid catalyst, forming chiral ion-pair 142. Oxonium ylide 141 undergoes an enantioselective Mannich-type addition to 142, forming the enantioenriched β-amino-α-alkoxyester. Without sufficient activation of the imine, oxonium ylide 141 may undergo an undesired 1,2-hydride shift to form α-alkoxyester 143, dramatically lowering the yield.
Scheme 34.

Mechanism of the three-component coupling of diazoacetates, alcohols, and imines via rhodium- and Brønsted acid catalysis.
Hu and coworkers rationalized that along with activating the imine, the Brønsted acid catalyst might also induce its in situ formation from aldehyde 144 and amine 145, creating a four-component coupling reaction.56 This strategy would eliminate the need for prior imine preparation and isolation. The four component reaction proceeds smoothly under very similar conditions leading to high yields and selectivities (Scheme 35). Both electron-poor and electron-rich aromatic aldehydes perform well in the reaction (alkyl aldehydes suffer from low yield and selectivity); however, bulky 9-anthryl alcohol 146 is needed to increase both diastereoand enantioselectivity.
Scheme 35.

The four-component coupling of diazoacetates with alcohol, aldehydes, and amines to generate β-amino-α-hydroxyesters.
This robust synergistic system of a chiral phosphoric acid and Rh2(OAc)4 is also successful with carbamate nucleophile 147, generating α,β-diaminoester 148 in high yield and selectivity via the analogous ammonium ylide (Scheme 36).57 Initially, the more basic diamine products appeared to poison the phosphoric acid catalyst through bidentate coordination. Adding L-tartaric acid alleviates this effect by neutralizing the basic diamine product and regenerating the chiral phosphoric acid. Although L-tartaric acid is also chiral, the Brønsted acid is the only source of enantiocontrol. Replacing L-tartaric acid with the racemic variant delivers the same high level of enantioselectivity, while absence of the chiral phosphoric acid catalyst results in racemic product.
Scheme 36.

The three-component coupling of diazoacetates, carbamates, and imines using a chiral Brønsted acid and Rh2(OAc)4.
An interesting element of stereocontrol is observed when the substitution of the binol-based phosphoric acid is altered. 3,5-Bis(trifluoromethyl)phenyl substituted binol provides the syn product, while changing to the larger triphenylsilyl substituted binol provides the anti product. The authors propose that with the 3,5-bis(trifluoromethyl)phenyl substitution, the phosphoric acid may form a bridge between the ammonium ylide and the imine, which leads to the syn product (Scheme 36, syn-TS). On the other hand, the much larger triphenylsilyl group cannot accommodate both the imine and the ylide and forms an open-chain transition state, leading to the anti product (Scheme 36, anti-TS).
The activation of imines in synergistic catalysis is not restricted to Brønsted acids. Chiral Lewis acids are traditionally designed to shield either the Re- or the Si-face of an electrophile, thereby restricting the nucleophile to attack the accessible face. For chiral tertiary anion nucleophiles there is the added challenge of differentiating between the easily interchangable enantiomers of the pyramidal anion. This can often lead to high enantioselectivity, but little to no diastereoselectivity. However, synergistic activation of both the electrophile and the nucleophile by chiral catalysts can create a diastereomeric pair of activated chiral intermediates, which reduces the task of controlling selectivity to finding a matched set of catalysts.
Using this design strategy for the aza-Henry reaction with tertiary α-nitroester 149, Jørgensen and coworkers activated α-iminoester 150 with chiral bisoxazoline-copper(II) complexes.58 In order to create a diastereomeric pair of intermediates, the tertiary nitroester was activated by the chiral organic base quinine, creating chiral ammonium ion pair 152 (Scheme 37). Under these conditions, the anionic nucleophile added efficiently and selectively to the activated α-iminoester providing the aza-Henry adduct 151 in 90% yield, 14:1 dr, and 98% ee. Through optimization and control reactions, Jørgensen and coworkers showed that both catalysts are, in fact, needed for efficiency and they must both be chiral for high enantioselectivity and diastereoselectivity. Absence of either catalyst leads to very little or no reaction (< 5% yield). Substituting the chiral BOX ligand with achiral 1,10-phenanthroline or 2,2′-bipyridine leads to the aza-Henry adduct with good diastereoselectivity (8:1 and 7:1 dr respectively), but no enantioselectivity. On the other hand, achiral Hünig’s base, in conjunction with chiral Ph-BOX-Cu(OTf)2, leads to poor diastereoselectivity (2:1 dr) but good enantioselectivity (80 and 82% ee). This illustrates the synergistic relationship between the two chiral catalysts to control both the diastereoselectivity and the enantioselectivity.
Scheme 37.

Enantioselective aza-Henry reaction via chiral organic and Lewis acid catalysis.
4.5 Lewis acid and Lewis base catalyzed enantioselective cyanation of carbonyls
In 1993, Corey and Wang reported one of the earliest examples of asymmetric synergistic catalysis.59 In an effort towards a practical and efficient asymmetric cyanohydrin formation, they developed a dual catalytic system wherein a chiral Lewis acid activates an aldehyde while a chiral Lewis base creates a chiral cyanide nucleophile (Scheme 38). Initial reactions with cyclohexane carboxaldehyde and TMSCN show that 20 mol% of a chiral magnesium cyanobisoxazoline (Mg-BOX) complex delivers the cyanohydrin adduct in 85% yield, but with only moderate selectivity (65% ee). Selectivity dramatically increases (94% ee) when 12 mol% of a chiral Lewis basic bisoxazoline (BOX) is added. It became clear that both catalysts are required for this highly selective reaction as the BOX catalyst used alone results in racemic cyanohydrin adducts. Furthermore, there are clear matched and mismatched catalyst interactions; changing the enantiomer of just one catalyst results in considerably lower enantioselectivity (38% ee with the opposite enantiomer of the BOX catalyst). Mechanistically, the authors reason that advantitious traces of water hydrolyze TMSCN to form small amounts of HCN. The BOX catalyst activates HCN to create the chiral cyanide 154. The chiral nucleophile forms a diastereomeric pair with the activated aldehyde 155 to react in a highly selective manner. The resulting cyanohydrin reacts with TMSCN to form cyanohydrin TMS ether 153 and generate HCN for another cycle. This transformation is highly selective for aliphatic, non-conjugated aldehydes (90–95% ee), but lower selectivities are observed for aryl- and α,β-unsaturated aldehydes (52–87% ee).
Scheme 38.

Corey’s enantioselective cyanation of aldehydes using TMSCN with Lewis acid and Lewis base catalysts.
A decade after Corey’s report, Feng and coworkers also developed asymmetric Lewis acid/Lewis base catalyzed cyanation of ketones using TMSCN (Scheme 39).60 In their first published report, a salen-Ti(IV) Lewis acid is combined with dimethylphenyl amine N-oxide (156) to provide the cyanohydrin adduct in moderate to good yields and selectivity (up to 83% yield, up to 84% ee).60a,b Subsequent improvements are observed in both yield and selectivity when the Lewis acid is changed to a salen-Al(III) complex (the Lewis base remains the same); excellent yields and selectivities are now seen for both aliphatic and aromatic ketones (up to 99% yield, up to 94% ee).60c
Scheme 39.

The enantioselective cyanation of ketones catalyzed by metal-salen complexes and dimethylphenylamine N-oxide.
In this system, the Lewis acid activates the ketone via carbonyl coordination to the metal (Scheme 40A). The amine N-oxide is believed to activate TMSCN through coordination of the N-oxide oxygen to silicon (Scheme 40B) As the intermediates approach each other, the silicon of TMSCN also coordinates the ketone carbonyl, forming an organized transition state (Scheme 40C). The N-oxide complex transfers the cyanide to the ketone with concurrent TMS trapping to form the cyanohydrin TMS ether.
Scheme 40.

Proposed transition states for the Lewis acid and Lewis base catalyzed cyanation of ketones.
5 Lewis Base-Catalyzed Cycloadditions and Cyclizations
5.1 Hetero [2 + 2] cycloadditions
Inspired by the importance of β-lactams in pharmacetical science and biochemistry,61 Lectka and coworkers reported several studies towards the catalytic, asymmetric synthesis of these important synthons.62 In their protocol, chiral amine cataysts (generally cinchona alkaloid derivatives) activate acyl chlorides to form ketene-derived zwitterionic enolates that subsequently add to electrophilic imines, a process that is mechanistically distinct from the Staudinger reaction. Although enantioselectivities were excellent (generally >95% ee), the yields were consistently low, ranging from 40–65%. The authors believed the low yields are a result of the zwitterionic enolate being consumed through side reactions before addition to the imine can occur. To address this, they envisaged adding a catalytic Lewis acid to activate the imine and increase the rate of the desired reaction and suppress any side products. They found that adding hard Lewis acidic metals with low azophilicity (e.g. Sc3+, Al3+, Zn2+, In3+) leads to a significant increase in yield while maintaining high levels of selectivity. After optimization, the combination of In(OTf)3 and benzoylquinine emerged as the optimal Lewis acid/Lewis base pair for their synergistic strategy towards the synthesis of β-lactams from acyl chlorides and α-iminoesters (Scheme 41).63
Scheme 41.

The asymmetric synthesis of β-lactams catalyzed by benzoylquinine and In(OTf)3.
Mechanistic studies64 suggest that the transformation begins with addition of the benzoylquinine to acyl chloride 157, creating ketene-derived zwitterionic enolate 160. At the same time, In(OTf)3 activates α-iminoester 158, through bidentate coordination, increasing its electrophilicity. Zwitterionic enolate 160 adds to metal-bound imine 161, furnishing the new carbon-carbon bond. Subsequent transacylation of 162 regenerates both catalysts and releases β-lactam 159 in excellent yield (> 90% yield) and selectivities (>9:1 dr, >95% ee).
Following Lectka’s report, similar synergistic systems were developed for the preparation of β-lactones65 and β-sultones66 (Scheme 42). Calter and coworkers reported the synthesis of β-lactones 163 from acyl chlorides and benzaldehydes using quinidine trimethylsilyl ether and a Lewis acidic metal triflate (Scheme 42A). The reaction proceeds through a similar mechanism as the β-lactam synthesis, wherein ketene-derived zwitterionic enolates add to Lewis acid-activated aldehydes. For alkyl substituted acyl chlorides, Sc(OTf)3 provides the best yield and selectivity, while Er(OTf)3 is optimal with α-oxy acyl chlorides. It should be noted that due to the coordination ability of oxygen, the α-oxy acyl chlorides provide predominately the cis product (163-syn) while the alkyl substituted acyl chlorides provide predominately trans (163-trans).
Scheme 42.

The asymmetric synthesis of (A) β-lactones and (B) β-sultones using cinchona alkaloid and metal Lewis acid catalysts.
Recently, Peters and Koch also applied this synergistic Lewis acid/Lewis base strategy to the preparation of β-sultones from sulfonyl chlorides and trichloroacetaldehyde (Scheme 42B).66 Sulfonyl chloride 164 is deprotonated to generate sulfene 167, which reacts with the quinuclidine catalyst, (DHQ)2PYR, to form the zwiterionic nucleophile 168. Analogous to the previously described mechanism, zwitterion 168 adds to the Lewis acid-activated trichloroacetaldehyde 165 to provide β-sultone 166.
5.2 N-Heterocyclic carbene-catalyzed cyclizations
Just as cinchona alkaloid catalysts are able to create nucleophiles from acyl chlorides, nucleophilic Lewis base catalysts can also render α,β-unsaturated aldehydes or ketones nucleophilic rather than electrophilic. N-Heterocyclic carbenes (NHC), for example, can combine transiently with an α,β-unsaturated aldehyde (or ketone) to generate homoenolate equivalents, which are nucleophilic at the β-position. Scheidt and coworkers used this strategy in a synergistic route to stereogenic γ-lactams 171 (Scheme 43).67 Treating an α,β-unsaturated aldehyde and an N-acyl hydrazone with a chiral NHC catayst alone provides the γ-lactam in only moderate yields. Further examination revealed incomplete consumption of the hydrazone, indicating electrophilicity may be the problem. To increase the electrophilicity of the hydrazone, the authors persued a synergistic strategy and screened a variety of Lewis acidic metal salts. Previously, NHCs have not been used synergistically with Lewis acids, presumably because these species are excellent ligands for late transition metals.68 Combining the NHC with Mg(Ot-Bu)2, however, increased both the yield and the stereoselectivity of the [3 + 2] annulation, leading to a highly efficient reaction. In this catalytic system, chiral azolium precatalyst 175 is deprotonated by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) to form the active NHC catalyst 176. NHC 176 adds to α,β-unsaturated aldehyde 170, forming homoenolate equivalent 173 that is nucleophilic at the β-position. At the same time, Mg(Ot-Bu)2 activates hydrazone 170 through bidentate coordination. Transient homoenolate 173 adds to activated hydrazone 172 and subsequent cyclization of 174 furnishes γ-lactam 171 in good yields and stereoselectivities.
Scheme 43.

Scheidt’s γ-lactam formation via a [3 + 2] annulation catalyzed by an N-heterocyclic carbene and a Lewis acid.
6 Synergistic Cross-Coupling Reactions
6.1 Synergistic catalysis in traditional Pd-catalyzed cross coupling reactions
For several decades, cross-coupling has been considered a cornerstone technology to form carbon–carbon bonds in organic chemistry. It should not be surprising that the earliest examples of synergistic catalysis are found within this realm. The most well known example of synergism in cross coupling is the Sonogashira reaction. This transformation, which couples terminal acetylenes with aryl or vinyl halides, was discovered by Sonogashira, Tohda, and Hagihara in 1975 and is arguably one of the first examples of synergistic catalysis in organic synthesis.69 Just prior to Sonogashira’s report, Cassar along with Dieck and Heck published simultaneous accounts of the same transformation using palladium alone, however, these single catalyst methods generally required high temperatures.70 Sonogashira and coworkers realized the power of a synergistic strategy and added catalytic copper(I) iodide to initiate a copper-mediated transmetalation of the acetylide to the palladium. The result of this synergistic system is a much milder and robust method for the alkynylation of arenes and alkenes. This transformation is believed to proceed through the two synergistic catalytic cycles described in Scheme 44. The palladium is assumed to participate in a traditional C–C cross coupling mechanism wherein the active Pd(0) complex oxidatively inserts into the Ar–X bond of 177, generating Pd(II) intermediate 180. While this is occuring, acetylene 178 is activated by the Cu(I) catalyst to presumably generate transient copper acetylide 181 (possibly through coordination of the Cu(I) and subsequent deprotonation by the amine base). The two catalytic cycles intersect as the intermediates transmetalate to form alkynylpalladium(II) complex 182, which upon reductive elimmation, delivers alkynylated product 179.
Scheme 44.

The Sonogashira coupling reaction first disclosed in 1975 by Sonogashira, Tohda, and Hagihara.
Since Sonogashira’s first report in 1975, there have been a vast number of catalyst systems developed for many different substrate types. The scope has moved well beyond the original reported scope of aryl iodides, pyridyl bromides, and vinyl bromides. It now includes, but is not limited to, aryl chlorides and diazonium salts, numerous aromatic and non-aromatic heterocycles, alkynyl halides, vinyl tosylates, and even primary and secondary alkyl bromides (Fig 4).71 In addition to expanding the substrate scope, considerable work has been devoted towards the development of mild single catalyst systems; however, most Sonogashira reactions still use the synergistic combination of palladium and copper.
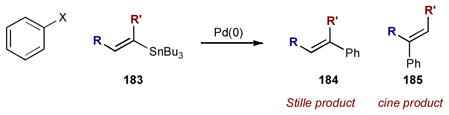
Although Stille coupling is generally considered to be a reaction involving one metal catalyst and one stoichiometric metallic reagent, this transformation has also benfitted from synergistic catalysis. Traditional Stille coupling involves the union of aryl or vinyl halides (or triflates) and organostannanes under palladium catalysis. For certain substrates, Stille couplings can prove to be troublesome with low yields and undesired side products. In particular, sterically hindered 1-substituted vinylstannanes 183 had remained a challenging substrate class, often leading to low yields of desired product 184 and/or as a mixture with cine type coupling product 185 (Eq 4).72 To overcome these challenges, copper(I) salts can be added to help promote Stille reactions that will not proceed effectively via palladium catalysis alone.73 This palladium/copper system was first formally studied by Liebeskind and coworkers; interestingly they found that the role of the copper(I) salt is solvent dependent.73b In ethereal solvents (such as THF or dioxane), the copper(I) is proposed to scavenge free ligand that otherwise decreases the rate-determining transmetalation step. This role of copper is not within the realm of synergistic catalysis. In polar solvents such as DMF or NMP, however, the copper is believed to transmetalate with the organostannane 186 to produce the more reactive organocopper intermediate 188; this species enters the palladium catalytic cycle for a second transmetalation with arylpalladum(II) intermediate 187 (Scheme 45). As transmetalation is responsible for the group transfer selectivity of unsymmetrical stannanes, an intermediate transmetalation to copper could be expected to affect the transfer ratio. In their studies of copper catalysis in Stille coupling,
Scheme 45.

Palladium- and copper-catalyzed Stille coupling reaction.
 |
(4) |
Liebeskind and coworkers observed a large selectivity increase by the addition of copper(I), lending support to the transmetalation mechanism (Eq 5).73b
Both Corey and Baldwin are among the many that have exploited this synergistic relationship to significantly enhance challenging Stille reactions.74 Corey and coworkers developed a Pd(PPh3)4/CuCl/LiCl system in DMSO for coupling 1-substituted vinylstannanes 190 with aryl triflate or nonaflates 189 (Scheme 46A), a reaction that generally results in low yields and mixtures of position-isomeric coupling products.74a By adding CuCl and LiCl, the resulting styrenyl products 191 are obtained in excellent yields under mild conditions. The authors propose the Sn/Cu transmetalation to generate the more reactive vinylcopper species. It is also believed that LiCl suppresses homocoupling of the vinylstannanes, which otherwise leads to diminished yields.
Scheme 46.

Palladium- and copper-catalyzed Stille coupling reactions.
 |
(5) |
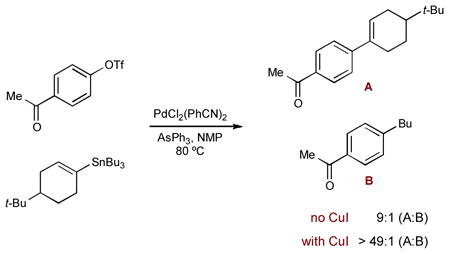
Baldwin and coworkers also developed a similar system to enhance difficult Stille couplings, whether the difficulty stems from sterics or electronics. They found that a Pd(PPh3)4/CuI/CsF system could effectively promote the coupling between aryl iodides or triflates and a wide variety of aryl- and vinylstannanes (including 1-substituted vinylstannanes) (Scheme 46B).74b A similar system of PdCl2/Pt-Bu3/CuI/CsF was also developed for aryl bromides and electron-deficient aryl chlorides. The desired coupling products were obtained in excellent yields under very mild conditions and generally short reaction times. Through optimizing the coupling of 4-iodotoluene (192) and 4-(tri-n-butylstannyl)nitrobenzene (193), the large positive effect of CuI became clear. Using palladium as a single catalyst, the reaction only provides 2% of coupling product 194. Adding CuI increases the yield of 194 to 46%, while combining CuI and CsF results in nearly quantitative conversion (98% yield). It should also be noted that both Pd(PPh3)4 and CsF alone fail to promote the reaction efficiently, as does CuI/CsF without palladium. CsF is believed to remove the Bu3SnCl byproduct through formation of the insoluble Bu3SnF. When used with the copper catalyst, this should help drive the formation of the more reactive organocopper species.
Palladium-catalyzed cyanation of aryl halides offers a practical alternative to the Rosenmund-von Braun reaction, which cyanates aryl halides via stoichiometric CuCN and high reaction temperatures (150–250 °C).75 Although the palladium-catalyzed variant offers much milder conditions than its predecessor, it experiences the inherent problem that cyanide interferes with the oxidative addition of the aryl halide to palladium(0); this requires minimizing the exposure of palladium(0) to free cyanide. One successful approach is to use stoichiometric metal cyanide complexes of a more covalent nature, such as Zn(CN)2 and CuCN; unfortunately, this leads to significant amounts of metallic waste. Andersen and coworkers at Eli Lilly and Company found that catalytic amounts of CuI (or ZnI2) work synergistically with palladium(0) to promote the same transformation using KCN or NaCN (Scheme 47A).76 Catalytic copper(I) salts act as a cyanide shuttle between the sparingly soluble alkali-metal cyanide 195 and the arylpalladium(II) intermediate 196. This shuttle mechanism functions through sequential transmetalations involving the copper catalyst. First, the copper(I) interacts with alkali-metal cyanide 195 to form a substoichiometric amount of copper cyanide 197. This intermediate transmetalates with arylpalladium(II) 196 (produced via aryl halide oxidative addition to palladium(0)). The resulting cyano(aryl)palldium(II) intermediate 198 reductively eliminates to deliver the benzonitrile product with excellent yields.
Scheme 47.

Synergistic palladium-catalyzed cyanation of aryl halides.
Following this report, Yang and Williams at Merck Research Laboratories disclosed a similar synergistic palladium-catalyzed cyanation of aryl halides and triflates, this time using catalytic Bu3SnCl as the cyanide shuttle.77 Prior attempts at using stoichiometric Me3SnCN as the cyanide source were ineffective, as rapid reaction between Me3SnCN and Pd(0) quickly terminated the cataytic cycle.78 Through further exploration, the authors found that with very low catalytic loadings, Bu3SnCl acts as a highly effective cyanide shuttle between KCN and arylpalldium(II), through a similar mechanism as described above (Scheme 47B). Based on NMR studies, the authors believe that the active cyano species is tin ate complex 199, formed through the addition of KCN to Bu3SnCl without the loss of chloride (Eq 6). This may be the key to the success of this reaction, as ate complex 199 may favor transmetalation with arylpalladium(II), while suppressing deactivation of palladium(0). With this protocol, a wide variety of benzonitriles are produced with just 0.5 mol% Pd2(dba)3 and 0.14 mol% Bu3SnCl.
 |
(6) |
6.2 Synergistic catalysis in non-traditional cross-coupling reactions
Synergistic catalysis has enabled many cross-coupling reactions that do not fall under the traditional named reactions. For example, palladium-catalyzed coupling reactions involving transient copper acetylides extend beyond the traditional Sonogashira reaction. Recently, Kang and coworkers at Johnson & Johnson Pharmaceutical Research and Development reported a direct dehydrative coupling of tautomerizable heterocycles with alkynes using synergistic palladium and copper catalysts (Scheme 48).79 This coupling proceeds via chemoselective activation of C–H and C–OH bonds to achieve carbon-carbon bond formation. This transformation is termed a dehydrative cross-coupling since the substrates together formally lose an equivalent of water to form a carbon–carbon bond. By directly activating C–H and C–OH bonds via synergistic catalysis, the need to prepare carbon–halogen or carbon–metal substrates is eliminated, along with the harmful waste they generate.
Scheme 48.

Direct dehydrative cross-coupling of tautomerizable heterocycles and alkynes via palladium and copper catalysis.
This efficient transformation proceeds via a Sonogashira-type coupling mechanism. Before coupling takes place, the C–OH bond of the phenol tautomer of heterocycle 200 is activated by phosphonium 202 to generate pseduo-aryl halide 203. Phosphonium-activated heterocycle 203 enters the palladium catalytic cycle and oxidatively adds to palladium(0). Resulting palladium(II) phosphonium 204 transmetalates with copper acetylide 205 (generated in the copper catalytic cyle) to produce aryl(alkynyl)palladium(II) intermediate 206. This species reductively eliminates to deliver alkynylated heterocycle 201.
While copper acetylides are common intermediates in cross-coupling reactions, Cheng and coworkers show that other organocopper species are also useful in non-traditional cross-couplings. Using benzynes, allylic epoxides, and terminal alkynes, they developed a palladium- and copper-catalyzed three-component coupling reaction to form 1,2-disubstituted arenes 207 (Scheme 49).80 Using only palladium to catalyze this coupling, the product is isolated as a mixture of regioisomers (Eq 7). During efforts to improve selectivity, the authors found that adding catalytic CuI, in conjunction with the palladium, allows coupling product 207 to be obtained as a single regioisomer.
Scheme 49.

Three-component coupling of benzyne, allylic epoxides, and terminal acetylenes.
This coupling is proposed to take place via the synergistic mechanism outlined in Scheme 49. Catalytic CuI activates the acetylene to form copper acetylide 208. This acetylide undergoes an alkynylcupration with in situ-generated benzyne 209 to generate key arylcopper species 210. Simultaneously, the allylic epoxide oxidatively adds to palladium(0), forming π-allyl intermediate 211. At this point, the copper and palladium cycles intersect as arylcopper 210 adds to 211 at the least hindered position, regenerating the copper(I) catalyst and forming palladium-bound 1,2-disubstituted arene 212. Dissociation of the palladium catalyst delivers desired product 207.
 |
(7) |
The enhancement of regioselectivity by CuI is most likely the result of a change in mechanism. When catalyzed by palladium alone, the authors propose an alternate mechanism wherein palladium π-allyl intermediate 211 adds across benzyne to produce a mixture of linear and branched arylpalladium species (213 and 214), which subsequently react with the acetylene (Eq 8).81 Attack of an unsubstituted benzyne is much less sterically hindered than the 1,2-disubstituted arylcopper intermediate proposed for the dual catalyzed system, which leads to poor regioselectivity.
Copper is not the only metal to be used synergistically with palladium-catalyzed cross-coupling. As an alternative to
 |
(8) |
carbonylative coupling, Chang and coworkers developed a palladium- and ruthenium-catalyzed coupling of aryl halides with 2-pyridylmethyl formate (215) to form corresponding benzoates 216 (Scheme 50).82 In previous studies, the authors discovered that Ru3(CO)12 catalyzed the decarbonylation of formate 215, generating the corresponding carbinol and CO, a process driven by coordination of the pyridine nitrogen to ruthenium.83 The authors hypothesized that the same ruthenium intermediate (221), bearing the chelated carbinol and CO, could also be used in a palladium-catalyzed coupling mechanism to generate aryl esters without the need for a CO atmosphere.
Scheme 50.

Palladium- and ruthenium-catalyzed coupling of 2-pyridylmethyl formate with aryl and vinyl halides.
The interaction of chelated ruthenium intermediate 221 with palladium-catalyzed coupling proceeds through the intersecting cycles outlined in Scheme 50. The aryl halide oxidatively adds to palladium(0) and the resulting arylpalladium(II) intermediate 220 transmetalates with ruthenium carbinol 221 as CO inserts into the Pd-carbinol bond. The resulting palladium(II) complex 222 reductively eliminates to deliver ester benzoate 216. The authors were pleased to find that the synergistic combination of Ru3(CO)12 and PdCl2 successfully coupled 2-pyridylmethyl formate (215) with a variety of aryl iodides, as well as vinyl iodides and imidoyl triflates, providing the ester products in excellent yields (217–219).
Shortly after their report with 2-pyridylmethyl formate, Chang and workers expanded this concept to similar couplings with N-(2-pyridyl)formamide (223) and 8-quinolinecarboxaldehyde (224) (Scheme 51).84 Using the same catalytic system, the corresponding benzamides 225 and aryl ketones 226 were also obtained in excellent yields. At this time, it is unclear whether these couplings proceed through ruthenium-catalyzed decarbonylation prior to reacting with the arylpalladium(II) intermediate, or if the key ruthenium intermediate is the five-membered chelate (via insertion into the C-H bond of the formyl group, see 227 and 228) formed prior to decarboxylation.
Scheme 51.

Palladium- and ruthenium-catalyzed coupling of aryl formates with aryl halides.
7 Conclusions and Future Outlook
Although examples of synergistic catalysis have been known for more than 35 years, when thought of as an area of catalysis the concept is still in its infancy. We believe that synergistic cataysis will continue to grow and be recognized as a powerful catalysis strategy. As the examples in this perspective illustrate, the synergistic interaction of two catalytic cycles brings about several benefits, specifically (i) introducing new, novel reactivity not attainable with a single catalyst; (ii) improving existing reactions, often by suppressing side reactions; (iii) creating or improving stereocontrol through highly organized transition states. Although this concept may be perceived to have inherent problems, such as catalyst compatibility or kinetics involving small concentration of key intermediates, we hope this perspective has illustrated these challenges are easily overcome with the right catalyst combination. For example, Lewis acids and Lewis bases are compatible in synergistic systems when together they form labile complexes, which avoids catalyst quenching.
Currently, the vast majority of synergistic catalysis strategies use systems where at least one catalytic intermediate is formed reversibly and/or is relatively stable. While these strategies are powerful, we believe this is just the beginning and more is possible with respect to this concept. Synergistic catalysis also has the potential to couple many types of irreversibly-formed, fleeting intermediates, as exemplified with Trost and Luan’s palladium-and vanadium-catalyzed synthesis of α-allylated-α,β-unsaturated ketones (Section 2.1, Scheme 8).12 Both reactive intermediates are formed irreversibly, but still find each other and react to form the desired product before either engage in side reactions. As the concept of synergistic catalysis expands, the number of new and novel transformations this strategy provides will continue to grow. This will lead to new bond disconnections and recognized synthons in organic synthesis, expanding our synthetic toolbox. Traditional catalysis changed the way we approach the synthesis of molecules in a laboratory and the concept of synergistic catalysis will continue to increase the power of catalysis.
Fig. 5.

Fig 4 Examples of Sonogashira substrates that undergo alkynylation using synergistic palladium- and copper-catalysis.71
Scheme 4.

Mechanism of the enantioselective α-allylation of aldehydes and ketones via enamine and palladium π-allyl intermediates.
Acknowledgments
Financial support was provided by NIHGMS (R01 GM078201-05) and kind gifts from Merck, Amgen, Abbott Research Laboratories and Bristol-Myers Squibb.
Notes and references
- 1.For reviews on bifunctional catalysis, see: Shibasaki M, Kanai M, Matsunaga S, Kumagai N. Acc Chem Res. 2009;42:1117. doi: 10.1021/ar9000108.Breslow R. J Molec Catal. 1994;91:161.Shibasaki M, Yoshikawa N. Chem Rev. 2002;102:2187. doi: 10.1021/cr010297z.Wang Y, Deng L. In: Catalytic Asymmetric Synthesis. 3. Ojima I, editor. John Wiley & Sons; New Jersey: 2010. p. 59.Shibasaki M, Kanai M. In: New Frontiers in Asymmetric Catalysis. Mikami K, Lautens M, editors. John Wiley & Sons; New Jersey: 2007. p. 383.
- 2.For examples of double activation catalysis, see: Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN. Science. 2010;327:986. doi: 10.1126/science.1182826.Rubina M, Conley M, Gevorgyan V. J Am Chem Soc. 2006;128:5818. doi: 10.1021/ja060085p.Mukherjee S, List B. J Am Chem Soc. 2007;129:11336. doi: 10.1021/ja074678r.Shi Y, Peterson SM, Haberaecker WW, III, Blum SA. J Am Chem Soc. 2008;130:2168. doi: 10.1021/ja710648b.Park YJ, Park JW, Jun CH. Acc Chem Res. 2008;41:222. doi: 10.1021/ar700133y.
- 3.For examples of cascade catalysis, see: Belot S, Vogt KA, Besnard C, Krause N, Alexakis A. Angew Chem Int Ed. 2009;48:8923. doi: 10.1002/anie.200903905.Han ZY, Xiao H, Chen XH, Gong LZ. J Am Chem Soc. 2009;131:9182. doi: 10.1021/ja903547q.Sorimachi K, Terada M. J Am Chem Soc. 2008;130:14452. doi: 10.1021/ja807591m.Cai Q, Zhao ZA, You SL. Angew Chem Int Ed. 2009;48:7428. doi: 10.1002/anie.200903462.Simmons B, Walji AM, MacMillan DWC. J Am Chem Soc. 2009;48:4349. doi: 10.1002/anie.200900220.Lathrop SP, Rovis T. J Am Chem Soc. 2009;131:13628. doi: 10.1021/ja905342e.
- 4.Strater N, Lipscomb WN, Klabunde T, Krebs B. Angew Chem Int Ed. 1996;35:2024. [Google Scholar]
- 5.(a) Brown KA, Kraut J. Faraday Discuss. 1992;93:217. doi: 10.1039/fd9929300217. [DOI] [PubMed] [Google Scholar]; (b) Polshakov VI. Russ Chem Bull, Int Ed. 2001;50:1733. [Google Scholar]
- 6.(a) Pearson RG. J Am Chem Soc. 1963;85:3533. [Google Scholar]; (b) Woodward S. Tetrahedron. 2002;58:1017. [Google Scholar]
- 7.Tsuji J, Takahashi H, Morikawa M. Tetrahedron Lett. 1965;6:4387.Trost BM, Fullerton TJ. J Am Chem Soc. 1973;95:292.For a review on transition metal-catalyzed asymmetric allylic alkylations, see: Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.
- 8.Sawamura M, Sudoh M, Ito Y. J Am Chem Soc. 1996;118:3309. [Google Scholar]
- 9.Ibrahem I, Córdova A. Angew Chem Int Ed. 2006;45:1952. doi: 10.1002/anie.200504021. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X, Liu D, Xie F, Liu Y, Zhang W. Org Biomol Chem. 2011;9:1871. doi: 10.1039/c0ob00915f. [DOI] [PubMed] [Google Scholar]
- 11.Nakoji M, Kanayama T, Okino T, Takemoto Y. Org Lett. 2001;3:3329. doi: 10.1021/ol016567h.Nakoji M, Kanayama T, Okino T, Takemoto Y. J Org Chem. 2002;67:7418. doi: 10.1021/jo0260645.Another report of a phase-transfer- and palladium-catalyzed allylic alkylation of glycine iminoesters was reported just prior to the work of Takemoto and coworkers, however, significantly lower selectivities were observed, see: Chen G, Deng Y, Gong L, Mi A, Cui X, Jiang Y, Choi MCK, Chan ASC. Tetrahedron: Asymmetry. 2001;12:1567.
- 12.(a) Trost BM, Luan X. J Am Chem Soc. 2011;133:1706. doi: 10.1021/ja110501v. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Luan X, Miller Y. J Am Chem Soc. 2011;133:12824. doi: 10.1021/ja204817y. [DOI] [PubMed] [Google Scholar]
- 13.Trost BM, Oi S. J Am Chem Soc. 2001;123:1230. doi: 10.1021/ja003629a. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 15.Ding Q, Wu J. Org Lett. 2007;9:4959. doi: 10.1021/ol7020669. [DOI] [PubMed] [Google Scholar]
- 16.Allen AE, MacMillan DWC. J Am Chem Soc. 2010;132:4986. doi: 10.1021/ja100748y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Eisenberger P, Gischig S, Togni A. Chem–Eur J. 2006;12:2579. doi: 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]; (b) Kieltsch I, Eisenberger P, Togni A. Angew Chem Int Ed. 2007;46:754. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; (c) Eisenberger P, Kieltsch I, Armanino N, Togni A. Chem Commun. 2008:1575. doi: 10.1039/b801424h. [DOI] [PubMed] [Google Scholar]; (d) Kieltsch I, Eisenberger P, Stanek K, Togni A. Chemia. 2008;62:260. [Google Scholar]; (e) Koller R, Stanek K, Stolz D, Aardoom R, Niedermann K, Togni A. Angew Chem Int Ed. 2009;48:4332. doi: 10.1002/anie.200900974. [DOI] [PubMed] [Google Scholar]
- 18.(a) Ikeda M, Miyake Y, Nishibayashi Y. Angew Chem Int Ed. 2010;49:7289. doi: 10.1002/anie.201002591. [DOI] [PubMed] [Google Scholar]; (b) Yoshida A, Ikeda M, Hattori G, Miyake Y, Nishibayashi Y. Org Lett. 2011;13:592. doi: 10.1021/ol1027865. [DOI] [PubMed] [Google Scholar]
- 19.Motoyama K, Ikeda M, Miyake Y, Nishibayashi Y. Eur J Org Chem. 2011:2239.Shortly after Nishibayashi’s report, Cozzi and coworkers published a very similar account with slightly higher diastereomeric ratios. They also previously demonstrated this amine/InBr3 strategy is successful for the α-allylic alkylation of aldehydes using allyl alcohols, see: Sinisi R, Vita MV, Gualandi A, Emer E, Cozzi PG. Chem Eur J. 17:7404. doi: 10.1002/chem.201100729.Capdevila MG, Befatti F, Zoli L, Stenta M, Cozzi PG. Chem Eur J. 2010;16:11237. doi: 10.1002/chem.201001693.
- 20.Sibi MP, Hasegawa M. J Am Chem Soc. 2007;129:4124. doi: 10.1021/ja069245n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Van Humbeck JF, Simonovich SP, Knowles RR, MacMillan DWC. J Am Chem Soc. 2010;132:10012. doi: 10.1021/ja1043006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Simonovich SP, Van Humbeck JF, MacMillan DWC. Chem Sci. 2011 doi: 10.1039/C1SC00556A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen AE, MacMillan DWC. J Am Chem Soc. 2011;133:4260. doi: 10.1021/ja2008906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lockhart TP. J Am Chem Soc. 1983;105:1940.(b) The MacMillan group has performed mechanistic studies that provide evidence to support this mechanism. Results of this study are forthcoming.
- 24.(a) Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J Am Chem Soc. 2009;131:11640. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alemán J, Cabrera S, Maerten E, Overgaard J, Jørgensen KA. Angew Chem Int Ed. 2007;46:5520. doi: 10.1002/anie.200701207. [DOI] [PubMed] [Google Scholar]
- 25.For a review on photoredox catalysis, see: Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n.
- 26.For selected carbonyl α-alkylation examples, see: Ooi T, Maruoka K. Angew Chem Int Ed. 2007;46:4222. doi: 10.1002/anie.200601737.Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. J Am Chem Soc. 1994;116:8829.Doyle AG, Jacobsen EN. J Am Chem Soc. 2005;127:62. doi: 10.1021/ja043601p.Dai X, Strotman NA, Fu GC. J Am Chem Soc. 2008;130:3302. doi: 10.1021/ja8009428.
- 27.Intramolecular organocatalytic asymmetric α-alkylation of aldehydes using alkyl iodides has been accomplished for specific substrate scopes, see: Vignola N, List B. J Am Chem Soc. 2004;126:450. doi: 10.1021/ja0392566.Enders D, Wang C, Bats JW. Angew Chem Int Ed. 2008;47:7539. doi: 10.1002/anie.200802532.
- 28.Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.For references on the reductive cleavage of alkyl halides using photoredox catalysis, see: Fukuzumi S, Mochizuki S, Tanaka T. J Phys Chem. 1990;94:722.
- 30.Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih H-W, Vander Wal MN, Grange RL, MacMillan DWC. J Am Chem Soc. 2010;132:13600. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Shaikh RR, Mazzanti A, Petrini M, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2008;47:8707. doi: 10.1002/anie.200803947. [DOI] [PubMed] [Google Scholar]; (b) Cozzi PG, Benfatti F, Zoli L. Angew Chem Int Ed. 2009;48:1313. doi: 10.1002/anie.200805423. [DOI] [PubMed] [Google Scholar]; (c) Brown AR, Kuo WH, Jacobsen EN. J Am Chem Soc. 2010;132:9286. doi: 10.1021/ja103618r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann M, Füldner S, König B, Zeitler K. Angew Chem Int Ed. 2011;50:951. doi: 10.1002/anie.201002992. [DOI] [PubMed] [Google Scholar]
- 34.(a) Sud A, Sureshkumar D, Klussmann M. Chem Commun. 2009:3169. doi: 10.1039/b901282f. [DOI] [PubMed] [Google Scholar]; (b) Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem Commun. 2011;47:2360. doi: 10.1039/c0cc04539j. [DOI] [PubMed] [Google Scholar]
- 35.Rahaman H, Madarász Á, Pápai I, Pihko PM. Angew Chem Int Ed. 2011;50:6123. doi: 10.1002/anie.201101835. [DOI] [PubMed] [Google Scholar]
- 36.Ibrahem I, Santoro S, Himo F, Córdova A. Adv Synth Catal. 2011;353:245. [Google Scholar]
- 37.Bergonzini G, Vera S, Melchiorre P. Angew Chem Int Ed. 2010;49:9685. doi: 10.1002/anie.201004761. [DOI] [PubMed] [Google Scholar]
- 38.Rios R, Sundén H, Ibrahem I, Córdova A. Tetrahedron Lett. 2007;48:2181. [Google Scholar]
- 39.Xia A-B, Xu D-Q, Luo S-P, Jiang J-R, Tang J, Wang Y-F, Xu Z-Y. Chem Eur J. 2010;16:801. doi: 10.1002/chem.200902540. [DOI] [PubMed] [Google Scholar]
- 40.Sammis GM, Jacobsen EN. J Am Chem Soc. 2003;125:4442. doi: 10.1021/ja034635k. [DOI] [PubMed] [Google Scholar]
- 41.Sammis GM, Danjo H, Jacobsen EN. J Am Chem Soc. 2004;126:9928. doi: 10.1021/ja046653n. [DOI] [PubMed] [Google Scholar]
- 42.(a) Ji JX, Wu J, Chan ASC. Proc Natl Acad Sci U S A. 2005;102:11196. doi: 10.1073/pnas.0502105102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shao ZH, Wang J, Ding K, Chan ASC. Adv Synth Catal. 2007;349:2375. [Google Scholar]
- 43.Rueping M, Antonchick AP, Brinkmann C. Angew Chem Int Ed. 2007;47:6903. doi: 10.1002/anie.200702439. [DOI] [PubMed] [Google Scholar]
- 44.(a) Halbes-Letinois U, Weibel JM, Pale P. Chem Soc Rev. 2007;36:759. doi: 10.1039/b602151b. [DOI] [PubMed] [Google Scholar]; (b) Ji JX, Au-Yeung TTL, Wu J, Yip CW, Chan ASC. Adv Synth Catal. 2004;346:42. [Google Scholar]
- 45.Lu Y, Johnstone TC, Arndtsen BA. J Am Chem Soc. 2009;131:11284. doi: 10.1021/ja904185b. [DOI] [PubMed] [Google Scholar]
- 46.Wei C, Li C-J. Green Chemistry. 2002;4:39. [Google Scholar]
- 47.Li C-J, Wei C. Chem Commun. 2002:268. doi: 10.1039/b108851n. [DOI] [PubMed] [Google Scholar]
- 48.Bonfield ER, Li C-J. Org Biomol Chem. 2007;5:435. doi: 10.1039/b613596j. [DOI] [PubMed] [Google Scholar]
- 49.For reviews of asymmetric hydrogenations, see: Xie JH, Zhu SF, Zhou QL. Chem Rev. 2011;111:1713. doi: 10.1021/cr100218m.Tang W, Zhang X. Chem Rev. 2003;103:3029. doi: 10.1021/cr020049i.
- 50.Zhou S, Fleischer S, Junge K, Beller M. Angew Chem Int Ed. 2011;50:5120. doi: 10.1002/anie.201100878. [DOI] [PubMed] [Google Scholar]
- 51.For the isolation of Knölker’s complex, see: Knölker H-J, Baum E, Goesmann H, Klauss R. Angew Chem Int Ed. 1999;38:2064. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2064::AID-ANIE2064>3.0.CO;2-W.For its development as a hydrogenation catalyst, see: Casey CP, Guan H. J Am Chem Soc. 2007;129:5816. doi: 10.1021/ja071159f.
- 52.Li C, Wang C, Villa-Marcos B, Xiao J. J Am Chem Soc. 2008;130:14450. doi: 10.1021/ja807188s. [DOI] [PubMed] [Google Scholar]
- 53.Li C, Villa-Marcos B, Xiao J. J Am Chem Soc. 2009;131:6967. doi: 10.1021/ja9021683. [DOI] [PubMed] [Google Scholar]
- 54.Lu CD, Liu H, Chen ZY, Hu WH, Mi AQ. Org Lett. 2005;7:83. doi: 10.1021/ol0478483.Huang H, Guo X, Hu W. Angew Chem Int Ed. 2007;46:1337. doi: 10.1002/anie.200604389.For the three-component reaction of diazoacetates, imines, and anilines, see: Wang Y, Zhu Y, Chen Z, Mi A, Hu W, Doyle MP. Org Lett. 2003;5:3923. doi: 10.1021/ol035490p.
- 55.(a) Hu W, Xu X, Zhou J, Liu WJ, Huang H, Hu J, Yang L, Gong LZ. J Am Chem Soc. 2008;130:7782. doi: 10.1021/ja801755z. [DOI] [PubMed] [Google Scholar]; (b) Qian Y, Jing C, Shi T, Ji J, Tang M, Zhou J, Zhai C, Hu W. ChemCatChem. 2011;3:653. [Google Scholar]
- 56.Xu X, Zhou J, Yang L, Hu W. Chem Commun. 2008:6564. doi: 10.1039/b816104f. [DOI] [PubMed] [Google Scholar]
- 57.Jiang J, Xu H-D, Xi J-B, Ren B-Y, Lv F-P, Guo X, Jiang L-Q, Zhang Z-Y, Hu W-H. J Am Chem Soc. 2011;133:8428. doi: 10.1021/ja201589k. [DOI] [PubMed] [Google Scholar]
- 58.Knudsen KR, Jørgensen KA. Org Biomol Chem. 2005;3:1362. doi: 10.1039/b500618j. [DOI] [PubMed] [Google Scholar]
- 59.Corey EJ, Wang Z. Tetrahedron Lett. 1993;34:4001. [Google Scholar]
- 60.(a) Chen FX, Feng X, Qin B, Zhang G, Jiang Y. Org Lett. 2003;5:949. doi: 10.1021/ol034158a. [DOI] [PubMed] [Google Scholar]; (b) Chen FX, Qin B, Feng X, Zhang G, Jiang Y. Tetrahedron. 2004;60:10449. [Google Scholar]; (c) Chen FX, Zhou H, Liu X, Qin B, Feng X, Zhang G, Jiang Y. Chem Eur J. 2004;10:4790. doi: 10.1002/chem.200400506. [DOI] [PubMed] [Google Scholar]
- 61.Alcaide B, Almendros P, Aragoncillo C. Chem Rev. 2007;107:4437. doi: 10.1021/cr0307300. [DOI] [PubMed] [Google Scholar]
- 62.(a) Taggi AE, Wack H, Hafez AM, France S, Lectka T. Org Lett. 2002;4:627. doi: 10.1021/ol0172525. [DOI] [PubMed] [Google Scholar]; (b) Dudding T, Hafez AM, Taggi AE, Wagerle TR, Lectka T. Org Lett. 2002;4:387. doi: 10.1021/ol017087t. [DOI] [PubMed] [Google Scholar]; (c) Hafez AM, Taggi AE, Dudding T, Lectka T. J Am Chem Soc. 2001;123:10853. doi: 10.1021/ja016556j. [DOI] [PubMed] [Google Scholar]; (d) Hafez AM, Taggi AE, Wack H, Drury WJ, III, Lectka T. Org, Lett. 2000;2:3963. doi: 10.1021/ol006659r. [DOI] [PubMed] [Google Scholar]; (e) Taggi AE, Hafez AM, Wack H, Young B, Drury WJ, III, Lectka T. J Am Chem Soc. 2000;122:7831. doi: 10.1021/ja005791j. [DOI] [PubMed] [Google Scholar]
- 63.France S, Wack H, Hafez AM, Taggi AE, Watsil DR, Lectka T. Org Lett. 2002;4:1603. doi: 10.1021/ol025805l. [DOI] [PubMed] [Google Scholar]
- 64.France S, Shah MH, Weatherwax A, Wack H, Roth JP, Lectka T. J Am Chem Soc. 2005;127:1206. doi: 10.1021/ja044179f. [DOI] [PubMed] [Google Scholar]
- 65.Calter MA, Tretyak OA, Flaschenriem C. Org Lett. 2005;7:1809. doi: 10.1021/ol050411q. [DOI] [PubMed] [Google Scholar]
- 66.Koch FM, Peters R. Angew Chem Int Ed. 2007;46:2685. doi: 10.1002/anie.200604796. [DOI] [PubMed] [Google Scholar]
- 67.Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nature Chem. 2010;2:766. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.(a) Burgess K, Perry MC. Tetrahedron: Asymmetry. 2003;14:951. [Google Scholar]; (b) Lin JCY, Huang RTW, Lee CS, Bhattacharyya A, Hwang WS, Lin IJ. Chem Rev. 2009;109:3561. doi: 10.1021/cr8005153. [DOI] [PubMed] [Google Scholar]; (c) Glorius FE. N-Heterocyclic Carbenes in Transition Metal Catalysis. Springer-Verlag; Berlin Heidelberg: 2010. [Google Scholar]
- 69.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467. [Google Scholar]
- 70.(a) Cassar L. J Organomet Chem. 1975;93:253. [Google Scholar]; (b) Dieck H, Heck F. J Organomet Chem. 1975;93:259. [Google Scholar]
- 71.For reviews on Sonogashira coupling, see: Sonogashira K. J Organomet Chem. 2002;653:46.Chinchilla R, Nájera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x.Doucet H, Hierso JC. Angew Chem Int Ed. 2007;46:834. doi: 10.1002/anie.200602761.Heravi MM, Sadjadi S. Tetrahedron. 2009;65:7761.Chinchilla R, Nájera C. Chem Soc Rev. 2011 doi: 10.1039/c1cs15071e.
- 72.For a review on Stille coupling, see: Farina V, Krishnamurthy V, Scott WJ. Org React. 1997;50:1.
- 73.Liebeskind LS, Fengl RW. J Org Chem. 1990;55:5359.Farina V, Kapadia S, Krishnan B, Wang C, Liebeskind LS. J Org Chem. 1994;59:5905.Marino and Long had previously documented using PdCl2(PPh3)2 and CuI to couple a vinylstannane and a vinyl tosylate in a footnote, but never elaborated on this finding, see: Marino JP, Long JK. J Am Chem Soc. 1988;110:7916.
- 74.(a) Han X, Stoltz BM, Corey EJ. J Am Chem Soc. 1999;121:7600. [Google Scholar]; (b) Mee SPH, Lee V, Baldwin JE. Angew Chem Int Ed. 2004;43:1132. doi: 10.1002/anie.200352979. [DOI] [PubMed] [Google Scholar]
- 75.For a recent review on paladium-catalyzed cyanation of aryl halides, see: Anbarasan P, Schareina T, Beller M. Chem Soc Rev. 2011 doi: 10.1039/c1cs15004a.
- 76.Anderson BA, Bell EC, Ginah FO, Harn NK, Pagh LM, Wepsiec JP. J Org Chem. 1998;63:8224. [Google Scholar]
- 77.Yang C, Williams JM. Org Lett. 2004;6:2837. doi: 10.1021/ol049621d. [DOI] [PubMed] [Google Scholar]
- 78.(a) Kingsbury WD, Boehm JC, Jakas DR, Holden KG, Hecht SM, Gallagher G, Caranga MJ, McCabe FL, Faucette LF, Johnson RK, Hertzberg RP. J Med Chem. 1991;34:98. doi: 10.1021/jm00105a017. [DOI] [PubMed] [Google Scholar]; (b) Nagamura S, Kobayashi E, Gomi K, Saitoa H. Bioorg, Med Chem. 1996;4:1379. doi: 10.1016/0968-0896(96)00132-0. [DOI] [PubMed] [Google Scholar]
- 79.Kang FA, Lanter JC, Cai C, Sui Z, Murray WV. Chem Commun. 2010;46:1347. doi: 10.1039/b917777a. [DOI] [PubMed] [Google Scholar]
- 80.Jeganmohan M, Bhuvaneswari S, Cheng C-H. Angew Chem Int Ed. 2009;48:391. doi: 10.1002/anie.200804873. [DOI] [PubMed] [Google Scholar]
- 81.This mechanism was proposed for a very similar palladium-catalyzed three-component coupling reaction of benzyne, allyl chlorides,, alkynylstannanes see: Jeganmohan M, Cheng C-H. Org Lett. 6:2821. doi: 10.1021/ol048891t.
- 82.Ko S, Lee C, Choi M-G, Na Y, Chang S. J Org Chem. 2003;68:1607. doi: 10.1021/jo026591o. [DOI] [PubMed] [Google Scholar]
- 83.Ko S, Na Y, Chang S. J Am Chem Soc. 2002;124:750. doi: 10.1021/ja017076v. [DOI] [PubMed] [Google Scholar]
- 84.(a) Ko S, Han H, Chang S. Org Lett. 2003;5:2687. doi: 10.1021/ol034862r. [DOI] [PubMed] [Google Scholar]; (b) Ko S, Kang B, Chang S. Angew Chem Int Ed. 2005;44:455. doi: 10.1002/anie.200462006. [DOI] [PubMed] [Google Scholar]