

Abstract
Sickle cell disease (SCD) causes widely disseminated vaso-occlusive episodes. Building on evidence implicating invariant NKT (iNKT) cells in the pathogenesis of ischemia/reperfusion injury, recent studies demonstrate that blockade of iNKT cell activation in mice with SCD reduces pulmonary inflammation and injury. In patients with SCD, iNKT cells in blood are increased in absolute number and activated in comparison to healthy controls. iNKT cell activation is reduced by agonists of adenosine 2A receptors (A2ARs) such as the clinically approved coronary vasodilator, regadenoson. An ongoing multi-center, dose-finding and safety trial of infused regadenoson, has been initiated and is providing preliminary data about its safety and efficacy to treat SCD. Very high accumulation of adenosine may have deleterious effects in SCD through activation of adenosine 2B receptors that are insensitive to regadenoson. Future possible therapeutic approaches for treating SCD include selective A2BR antagonists and antibodies that deplete iNKT cells.
Keywords: iNKT cells, sickle cell disease, regadenoson
Introduction
Sickle cell disease (SCD) is the most common genetic disease among African Americans, affecting approximately 1 in 400 births in the United States. The basis for SCD is a non-synonomous mutation in the 6th position of the β globin gene (Glu→Val) that causes de-oxygenated sickle hemoglobin (ααSS) to polymerize, resulting in rigid sickle shaped erythrocytes. Historically, the pathogenesis of SCD has been attributed to deformed sickle erythrocytes that mechanically obstruct capillaries to cause microvascular occlusion (Hebbel et al., 1981). Emerging evidence, however, shows that vaso-occlusion is a complex, mutli-cellular process involving endothelial and platelet activation, a pro-coagulant state, leukocyte adhesion to endothelial cells and ischemia-reperfusion injury (IRI).
Vaso-occlusion secondary to IRI is the pathogenic basis for the two most common episodic morbidities in SCD, pain [1] and acute chest syndrome (ACS)[2]. End organ damage in people with SCD is due in part to recurrent episodes of disseminated microvascular IRI. Therapeutic options for pain and/or ACS episodes in the acute setting are currently limited to supportive care with fluids, antibiotics, opioids and, in the case of ACS, blood transfusion. Additional therapies for hospitalized patients with pain and ACS are needed. Hydroxyurea has been used successfully to increase the production of fetal hemoglobin and reduce red cell sickling [3]. Since data from several studies indicate that white cell and platelet activation accompanies red cell sickling and hemolysis, new strategies to dampen the frequency or severity of vaso-occlusive episodes include interrupting leukocyte adhesion to endothelial cells with intravenous immunoglobulins [4] or selectin inhibitors [5].
Murine models of sickle cell vaso-occlusion have provided preliminary evidence that IRI contributes to a profound pro-inflammatory environment causing leukocyte activation, migration and adhesion, thus sustaining and propagating vaso-occlusion initiated by sickled RBCs. Although the mechanism IRI in vaso-occlusion has not been fully elucidated, recent evidence implicates invariant NKT (iNKT) cells as key to propagating an inflammatory cascade associated with IRI. In this review we will examine evidence suggesting that iNKT cells contribute to IRI, vaso-occlusion and ultimately end organ damage in people with SCD. In a murine model of SCD, adenosine 2A receptor (A2AR) agonists have shown promise for interrupting iNKT cell activation and reducing pulmonary morbidity. We describe efforts that are currently underway to extend these findings to patients with SCD.
Role of inflammation in Sickle Cell Disease
SCD causes sterile inflammation
SCD disease triggers hemolysis, ischemia and tissue necrosis that evoke sterile inflammation that is thought to be triggered by a combination of nitric oxide depletion [6] production by damaged cells of damage-associated molecular patterns (DAMPs) [7], ATP, ADP, cytokines [8] and lipid mediators [9]. A heightened state of inflammation in SCD is evidenced by increased levels of leukocytes, platelets, cytokines (TNF-α, IL-1, IL-6, IL-8) and leukotrienes (B4 and E4). Kaul et al. [10; 11] postulated that during vaso-occlusion, recurrent episodes of hypoxia followed by reoxygenation and exposure to oxidant species contributes to inflammation and promotes further vaso-occlusion. In SCD mice, hypoxia/reoxygenation increased leukocyte adhesion and emigration and increased oxidant production in vascular endothelial cells. Intravital microscopy analyses in SCD mice indicate that sickle RBCs interact primarily with adherent leukocytes (WBCs) in postcapillary and collecting venules leading to vascular obstruction [12]. A pan selectin inhibitor, GMI1070, inhibits RBCleukocyte interactions, and results in improved microcirculatory blood flow and improved survival [5]. Evidence of vascular injury includes increased circulating endothelial cells and soluble circulating adhesion molecules (ICAM-1, VCAM-1, E-selectin and P-selectin). ATP and ADP released from activated or damaged cells activates platelets via two G protein coupled ADP receptors (P2Y1 and P2Y12) and via ATP through the ligand-gated P2X1 receptor [13]. Platelet activation in SCD is associated with increased secretion of thrombospondin, β-thromboglobulin, platelet factor 4, and increased expression of P-selectin, glycoprotein IIbIIIa and CD40 ligand. Low nitric oxide levels in SCD probably contribute to platelet activation [14]. Platelet activation is associated with increased platelet adhesion to microvascular endothelium [15] and formation of platelet heteraggregates with erytorocytes [16] and leukocytes including neutrophils, monocytes and eosinophils [17]. In mouse and human blood, neutrophil activation is partly due to the formation of P selectin-mediated platelet-neutrophil heteroaggregates. Multi-spectral imaging identified a subpopulation of activated neutrophils with adhered platelets that exhibit greater oxidative activity than singlet neutrophils (Figure 1) [17]. Both the fraction of neutrophils with adhered platelets, and the number of platelets bound per neutrophil are increased as a result of human or murine SCD. Among individuals with SCD, increased WBC count is associated with a higher incidence of ACS, pain, silent and hemorrhagic strokes and premature death. Levels of soluble adhesion molecules ICAM-1 and VCAM-1 correlate with risk for pulmonary hypertension, and high leukotriene (B4 and E4) levels have been associated with an increased incidence rate of ACS and pain.
Figure 1. Effect of SCD on platelet-neutrophil aggregates in mouse blood.

Blood was collected from mice without and with SCD and analyzed to determine the % of neutrophils with 1 or more adhered platelets, and the average number of adhered platelets/neutrophil in heteroaggregates. Platelet-neutrophil aggregates were imaged by Multi-spectral Imaging Flow Cytometry using an ImageStream™ flow cytometer (Amnis Corporation, Seattle, WA). Images of platelet-neutrophil aggregates in blood derived from wild type (top) and SCD mice (bottom) indicates that SCD increases the number of platelets (red) bound/neutrophil (green). Reprinted from [17].
A limited number of clinical studies have examined therapies to decrease inflammation and potentially improve the course of vaso-occlusive pain and ACS. In separate randomized clinical trials, corticosteroids were shown to significantly decrease length of hospital stay; however, many of the participants treated with corticosteroids experienced rebound vaso-occlusion and up to 25% required re-hospitalization [18]. Thus, no anti-inflammatory therapy is currently standard treatment for vaso-occlusive episodes.
iNKT cells mediate IRI
A2A agonsits have also been found to reduce injury following ischemia or trauma in many tissues, including liver [19; 20; 21; 22; 23; 24], kidney [25; 26; 27; 28], skin [29], lung [30; 31; 32], heart [33; 34; 35; 36], intestine [37] and spinal cord [38; 39; 40]. The cellular targets of A2ARs initially were not clear. Neutrophils and macrophages express A2ARs that respectively inhibit oxidative burst and adhesion molecule expression [41] and cytokine production [42]. Reutershan et al. utilized mice with loxp sites flanking the first coding exon of the A2AR gene, adora2a, and crossed these mice to LysMCre mice. All lines were made congenic to C57BL/6J using marker-assisted selection. In LysM-Cre × A2ARf/f mice that selectively lack A2ARs in neutrophils and macrophages, A2AR activation was still highly effective at reducing liver or lung IRI [43]. Adoptive transfer of CD4+ (but not CD8+ T cells) to Rag1−/− mice reconstituted severe injury from IRI [44]. The A2A agonsit ATL146e inhibited this injury if the transferred cells had A2ARs, but not if they lacked A2ARs [35]. This result is striking because Rag1−/− mice reconstituted with A2AR−/− CD4+ T cells have a normal complement of A2ARs in all cells except the reconstituted T cells. The results indicate that despite the widespread distribution of A2ARs on platelets and leukocytes, A2A agonists reduce IRI primarily by their effects on T cells.
In 2005 Shimamura et al. [45] found that liver reperfusion injury was associated with an expansion and activation of NKT cells. Subsequently, Lappas et al. found that depletion of NKT and NK cells with PK136, an antibody that binds to NK1.1 found only on NKT and NK cells, or blockade of CD1d-restricted iNKT cell activation with an anti-CD1d antibody produces protection from liver IRI that is equivalent to and not additive with protection by ATL146e [46]. These studies indicate that the adenosine-sensitive T cells that mediate IRI are iNKT cells. The mechanisms by which iNKT cells are activated in IRI are not entirely clear, but recent studies suggest that tissue injury may result in the formation of a galactose-containing glycolipid that can activate the invariant TCR [9]. In addition, iNKT cell activation may be facilitated by the binding of phosphatidylserine on the surface of apoptotic cells to T cell Ig-like mucin-like-1 (TIM-1) receptors on NKT cells [47].
Role of iNKT cells in SCD
To determine whether iNKT cells play a role in SCD tissue damage, Wallace et al. compared the lungs of wild type and NY1DD mice. Pulmonary iNKT cells from NY1DD mice are increased in number and activated compared to C57BL/6 mice [48]. NY1DD lung iNKT cells displayed significantly increased levels CD69 and IFN-γ compared to C57BL/6 mice. The % of pulmonary iNKT cells positive for IFN-γ increased from 5% in wild type mice to 37% in NY1DD mice, a difference of 7.4-fold. Interrupting iNKT cell activation or migration into the lungs reduced pulmonary inflammation and improved pulmonary function in NY1DD SCD mice.
Wallace et al. discovered that there are high levels of IFN-γ in iNKT cells derived from NY1DD mouse lungs [48]. FACS analysis of pulmonary lymphocytes for cell surface CXCR3 revealed that the expression CXCR3 is significantly higher (% positive cells) on CD4 T-cells (6-fold), CD8 T-cells (7-fold), NK cells (4-fold), and iNKT cells (2-fold) from NY1DD mice than C57BL/6 controls. ELISAs of pulmonary tissue homogenate also revealed significantly increased levels of IFN-γ and the IFN-γ inducible chemokines CXCL9 and CXCL10 in lungs of NY1DD mice as compared to C57BL/6 mice [48]. Neutrilization of CXCR3 was found to significantly reduce numbers of PMNs, CD4+ cells, CD8+ cells, NK cells and NKT cells in the lungs of NY1DD mice. Furthermore, anti-CXCR3 treated NY1DD animals had significantly decreased vascular leak and increased arterial oxygen saturation as compared to NY1DD mice. Treatment of NY1DD mice with anti-CXCR3 antibodies significantly improved breathing parameters. These finding suggest that iNKT cells orchestrate an inflammatory cascade by involving IFN-γ and INF-γ-inducible chemokines. Hence, blocking CXCR3 signaling constitutes another potential therapeutic approach to treating SCD.
iNKT cells are the primary targets of A2AR activation in SCD
Wallace et al. reasoned that since A2AR activation inhibits the activation of iNKT cells and other leukocytes and platelets, that A2AR activation would reduce SCD lung injury. Administration of ATL146e for 3 days by subcutaneous Alzet minipumps produced a dose-dependent reduction in the number of lung iNKT cells, NK cells and neutrophils [49]. The optimal dose, 10 ng/kg/min, is similar to the optimum noted in liver, kidney and heart models of IRI, and is below the threshold dose that changes heart rate and blood pressure. When 10 ng/kg/min of ATL146e is continued for 3 days and then discontinued, lung function is improved on day 3 and somewhat on day 4, but reverts to baseline by day 7. This experiment demonstrates that the palliative effects of ATL146e on lung function in SCD does not desensitize over at least 3 days, nor is there a rebound effect after the compound is discontinued. However, the reduction in injury cause by A2AR activation is gradually reversed after the agonist is discontinued.
Since A2ARs are found not only on iNKT cells, but also on most leukocytes and platelets and on various other tissues, Wallace et al. sought to determine the relative importance of A2A receptors on various cells in mediating lung protection in SCD. In order to evaluate the role of A2ARs on iNKT cells we crossed NY1DD mice with SCD to Rag1−/− mice that lack mature T cells including NKT cells. NY1DD × Rag1−/− mice displayed reduced pulmonary injury that was restored by adoptive transfer of 106 purified iNKT cells. Reconstituted lung injury was reversed by the A2AR agonsit, ATL146e, unless the adoptively transferred iNKT cells were pretreated with the A2AR alkylating antagonist, 5-amino-7-[2-(4-fluorosulfonyl)phenylethyl]-2-(2-furyl)-pryazolo[4,3- ]-1,2,4-triazolo[1,5-c]pyrimidine (FSPTP) which completely prevented protection [49]. These data indicate that despite the widespread expression of A2ARs on most cells of the immune system, activation of A2ARs on iNKT cells is required for effective inhibition of pulmonary injury by A2AR agonsits in the NY1DD mouse model of SCD. The pivitol role of A2ARs on iNKT cells may be related to the fact that A2AR mRNA is induced on these cells when they are activated as a consequence of SCD [49].
Activation of iNKT cells in patients with sickle cell disease
There is an expansion and activation of iNKT cells in patients with SCD compared to healthy African American controls (Figure 2) [49]. Given the general leukocytosis of patients with SCD, it is notable that there is selective expansion of iNKT cells among lymphocytes, from < 1% in control blood, to an average of 6% in the blood of SCD patients. We also examined the activation state of iNKT cells in human blood. SCD causes a significant increase in the % iNKT cells positive for CXCR3 and CD69, and an increase in intracellular IFN-γ. On the basis of these data we concluded that SCD produces a similar expansion and activation of iNKT cells in mice and patients.
Figure 2. Effects of SCD on human iNKT cells.
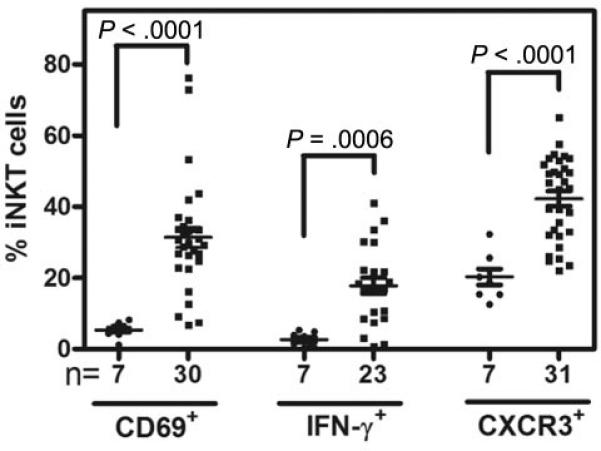
Blood from patients with HbSS SCD or African American controls was collected into EDTA and analyzed by flow cytometry. Each point show iNKT cell activation markers in controls (●) or individuals with HbSS at baseline (■). Reproduced from [49].
Deleterious effects of adenosine
Although adenosine accumulation in SCD can produce beneficial effects by activating anti-inflammatory signaling in iNKT cells and other leukocytes and platelets, very high accumulation of adenosine that may occur during severe vaso-occlusive episodes can activate other adenosine receptor subtypes and may produce deleterious effects. For instance, activation of renal A1 receptors can reduce glomerular filtration and produce a potentially damaging anti-diruesis [50]. In addition, in the severe Berkeley mouse model of SCD, activation of A2B receptors has been implicated as contributing priapism [51], penile fibrosis [52] and red blood cell sickling [53]. It remains to be determined how frequently adenosine accumulation that is sufficient to activate A1 and A2B receptors occurs in patients with SCD. Fortunately, regadenoson, which is being tested as an A2AR agonist to treat SCD, is selective for the A2AR over the A1 or A2B subtypes. It should be possible to find a therapeutic dose of regadenoson that activates anti-inflammatory A2A receptors without producing detrimental effects that might be produced by activating other adenosine receptor subtypes. It will be interesting in the future to consider combinations of A2A agonists and A2B antagonists as a strategy to optimally prevent and treat vaso-occlusive events.
Clinical trial of the A2A agonist, regadenoson
Our group is currently conducting a phase I safety study of regadenoson in adults and children with SCD. Regadenoson is a selective A2AR agonist that is FDA approved for myocardial perfusion imaging in individuals unable to undergo adequate exercise stress. The strategy of testing an A2AR agonist to treat SCD is based on the reduction of pulmonary inflammation and injury we observed after inhibiting iNKT cell function in murine models of SCD with CD1d-blocking antibodies or A2AR agonists. Among the challenges of administering regadenoson to patients with SCD for the purpose of reducing inflammation is to determine the optimal dose and duration of treatment. For cardiac stress tests, regadenoson is administered as a 400 μg bolus over 10 seconds and produces coronary vasodilation and, not infrequently, transient hypotension and tachycardia. The rationale for using regadenoson as an A2AR agonist is that the anti-inflammatory effects of A2ARs are more potent than the cardiovascular toxicities. Since the ultimate goal is to administer regadenoson during a pain or ACS episode, regadenoson's short half-life necessitates that we administer the drug as a continuous infusion. In the absence of dosing guidelines for a biological meaningful effect on iNKT cells, we estimated dosing and cardiovascular toxicities from animal studies. Based on binding to recombinant human A2A adenosine receptors we determined that regadenoson is about 15 times less potent than ATL146e (the A2AR agonist used in animal studies). However, the terminal half life of regadenoson in man is about 12 times longer than ATL146e (2 hours vs. 10 min), and during continuous infusion, regadenoson is expected to reach steady state blood levels about 12 time higher than ATL146e. Hence we estimated that regadenoson and ATL146e will have similar potencies during infusions and will both achieve maximally effective anti-inflammatory effects in the range of 10 ng/kg/min. We also estimated that the threshold for cardiovascular side effects will be about 100 ng/kg/min.
An ongoing study of regadenoson in SCD is comprised of 4 stages. In stage 1, we will determine the maximally tolerated and biologically effective dose of regadenoson during a 12-hour infusion. Stage 2 will determine the safety of a 24 hour infusion. Stages 3 and 4 will examine the safety of regadenoson in adults and children with SCD, respectively, during a vaso-occlusive episode. iNKT cell activation markers will be measured before, during and after the infusion. If we determine a safe and biologically effective dose, we will pursue studies to determine whether regadenoson is efficacious for the treat of pain episodes or ACS. In the future we anticipate that in additional to A2AR activation, other means of depleting or inhibiting the activation of iNKT cells will be utilized to prevent or treat acute exacerbations of SCD. In addition, inhibition of platelet and neutrophil activation may also prove to be effective.
Targeting iNKT cells in future clinical trials of SCD
A2A agonists such as regadenoson represent one approach to transiently inhibiting the activation of iNKT cells. There are a number of additional approaches that could be tried in the future to produce a more persistent blockade. It may be possible to disrupt activation of human iNKT cells with antibodies that target CD1d. Although such antibodies have recently been shown to activate antigen presenting cells to produce pro-inflammatory responses [58], this approach may still be effective in SCD patients. Another strategy is to deplete iNKT cells using antibodies that target the human invariant TCR [54]. For use in SCD it will be important to demonstrate that such antibodies do not produce transient iNKT cell activation. Finally, it may be possible to selectively inhibit iNKT cell activation through the use of glycolipid antagonists that bind to CD1d but do not activate iNKT cells [55]. These approaches will require careful investigation because of growing evidence that iNKT cells play an important role in immune resistence to certain pathogents [56; 57].
Acknowledgments
Grant Support: Supported in part by the National Heart, Lung, and Blood Institute award R01HL095704 (JL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, Kinney TR. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11–6. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- 2.Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, Gillette P, Vera JC, Levy PS. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84:643–9. [PubMed] [Google Scholar]
- 3.Goldberg MA, Brugnara C, Dover GJ, Schapira L, Charache S, Bunn HF. Treatment of sickle cell anemia with hydroxyurea and erythropoietin. N Engl J Med. 1990;323:366–72. doi: 10.1056/NEJM199008093230602. [DOI] [PubMed] [Google Scholar]
- 4.Chang J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse acute vasoocclusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood. 2008;111:915–23. doi: 10.1182/blood-2007-04-084061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood. 2010;116:1779–86. doi: 10.1182/blood-2009-12-260513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–9. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 7.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 9.Darmoise A, Teneberg S, Bouzonville L, Brady RO, Beck M, Kaufmann SH, Winau F. Lysosomal alpha-galactosidase controls the generation of self lipid antigens for natural killer T cells. Immunity. 2010;33:216–28. doi: 10.1016/j.immuni.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–20. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pritchard KA, Jr., Ou J, Ou Z, Shi Y, Franciosi JP, Signorino P, Kaul S, Ackland-Berglund C, Witte K, Holzhauer S, Mohandas N, Guice KS, Oldham KT, Hillery CA. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L705–14. doi: 10.1152/ajplung.00288.2002. [DOI] [PubMed] [Google Scholar]
- 12.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047–51. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oury C, Toth-Zsamboki E, Vermylen J, Hoylaerts MF. The platelet ATP and ADP receptors. Curr Pharm Des. 2006;12:859–75. doi: 10.2174/138161206776056029. [DOI] [PubMed] [Google Scholar]
- 14.Marcondes S, Cardoso MH, Morganti RP, Thomazzi SM, Lilla S, Murad F, De Nucci G, Antunes E. Cyclic GMP-independent mechanisms contribute to the inhibition of platelet adhesion by nitric oxide donor: a role for alpha-actinin nitration. Proc Natl Acad Sci U S A. 2006;103:3434–9. doi: 10.1073/pnas.0509397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brittain HA, Eckman JR, Swerlick RA, Howard RJ, Wick TM. Thrombospondin from activated platelets promotes sickle erythrocyte adherence to human microvascular endothelium under physiologic flow: a potential role for platelet activation in sickle cell vaso-occlusion. Blood. 1993;81:2137–43. [PubMed] [Google Scholar]
- 16.Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111:474–81. doi: 10.1046/j.1365-2141.2000.02353.x. [DOI] [PubMed] [Google Scholar]
- 17.Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear AR, Linden J. P-Selectin-Mediated Platelet-Neutrophil Aggregate Formation Activates Neutrophils in Mouse and Human Sickle Cell Disease. Arterioscler Thromb Vasc Biol. 2010;30:2392–2399. doi: 10.1161/ATVBAHA.110.211615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernini JC, Rogers ZR, Sandler ES, Reisch JS, Quinn CT, Buchanan GR. Beneficial effect of intravenous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood. 1998;92:3082–9. [PubMed] [Google Scholar]
- 19.Alchera E, Tacchini L, Imarisio C, Dal Ponte C, De Ponti C, Gammella E, Cairo G, Albano E, Carini R. Adenosine-dependent activation of hypoxia-inducible factor-1 induces late preconditioning in liver cells. Hepatology. 2008;48:230–9. doi: 10.1002/hep.22249. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Ari Z, Pappo O, Sulkes J, Cheporko Y, Vidne BA, Hochhauser E. Effect of adenosine A2A receptor agonist (CGS) on ischemia/reperfusion injury in isolated rat liver. Apoptosis. 2005;10:955–62. doi: 10.1007/s10495-005-0440-3. [DOI] [PubMed] [Google Scholar]
- 21.Cao Z, Yuan Y, Jeyabalan G, Du Q, Tsung A, Geller DA, Billiar TR. Preactivation of NKT cells with alpha-GalCer protects against hepatic ischemia-reperfusion injury in mouse by a mechanism involving IL-13 and adenosine A2A receptor. Am J Physiol Gastrointest Liver Physiol. 2009;297:G249–58. doi: 10.1152/ajpgi.00041.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day YJ, Li Y, Rieger JM, Ramos SI, Okusa MD, Linden J. A2A adenosine receptors on bone marrow-derived cells protect liver from ischemia-reperfusion injury. J Immunol. 2005;174:5040–6. doi: 10.4049/jimmunol.174.8.5040. [DOI] [PubMed] [Google Scholar]
- 23.Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol. 2004;286:G285–93. doi: 10.1152/ajpgi.00348.2003. [DOI] [PubMed] [Google Scholar]
- 24.Harada N, Okajima K, Murakami K, Usune S, Sato C, Ohshima K, Katsuragi T. Adenosine and selective A(2A) receptor agonists reduce ischemia/reperfusion injury of rat liver mainly by inhibiting leukocyte activation. J Pharmacol Exp Ther. 2000;294:1034–42. [PubMed] [Google Scholar]
- 25.Day YJ, Huang L, McDuffie MJ, Rosin DL, Ye H, Chen JF, Schwarzschild MA, Fink JS, Linden J, Okusa MD. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–91. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288:F722–31. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 27.Okusa MD, Linden J, Huang L, Rosin DL, Smith DF, Sullivan G. Enhanced protection from renal ischemia-reperfusion [correction of ischemia:reperfusion] injury with A(2A)-adenosine receptor activation and PDE 4 inhibition. Kidney Int. 2001;59:2114–25. doi: 10.1046/j.1523-1755.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 28.Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am J Physiol. 1999;277:F404–12. doi: 10.1152/ajprenal.1999.277.3.F404. [DOI] [PubMed] [Google Scholar]
- 29.Peirce SM, Skalak TC, Rieger JM, Macdonald TL, Linden J. Selective A(2A) adenosine receptor activation reduces skin pressure ulcer formation and inflammation. Am J Physiol Heart Circ Physiol. 2001;281:H67–74. doi: 10.1152/ajpheart.2001.281.1.H67. [DOI] [PubMed] [Google Scholar]
- 30.Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, Kron IL, Yang Z. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemiareperfusion injury. J Thorac Cardiovasc Surg. 2010;139:474–82. doi: 10.1016/j.jtcvs.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gazoni LM, Laubach VE, Mulloy DP, Bellizzi A, Unger EB, Linden J, Ellman PI, Lisle TC, Kron IL. Additive protection against lung ischemia-reperfusion injury by adenosine A2A receptor activation before procurement and during reperfusion. J Thorac Cardiovasc Surg. 2008;135:156–65. doi: 10.1016/j.jtcvs.2007.08.041. [DOI] [PubMed] [Google Scholar]
- 32.Rivo J, Zeira E, Galun E, Einav S, Linden J, Matot I. Attenuation of reperfusion lung injury and apoptosis by A2A adenosine receptor activation is associated with modulation of Bcl-2 and Bax expression and activation of extracellular signal-regulated kinases. Shock. 2007;27:266–73. doi: 10.1097/01.shk.0000235137.13152.44. [DOI] [PubMed] [Google Scholar]
- 33.Xi J, McIntosh R, Shen X, Lee S, Chanoit G, Criswell H, Zvara DA, Xu Z. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J Mol Cell Cardiol. 2009;47:684–90. doi: 10.1016/j.yjmcc.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rork TH, Wallace KL, Kennedy DP, Marshall MA, Lankford AR, Linden J. Adenosine A2A receptor activation reduces infarct size in the isolated, perfused mouse heart by inhibiting resident cardiac mast cell degranulation. Am J Physiol Heart Circ Physiol. 2008;295:H1825–33. doi: 10.1152/ajpheart.495.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–64. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 36.Patel RA, Glover DK, Broisat A, Kabul HK, Ruiz M, Goodman NC, Kramer CM, Meerdink DJ, Linden J, Beller GA. Reduction in myocardial infarct size at 48 hours after brief intravenous infusion of ATL-146e, a highly selective adenosine A2A receptor agonist. Am J Physiol Heart Circ Physiol. 2009;297:H637–42. doi: 10.1152/ajpheart.00705.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Paola R, Melani A, Esposito E, Mazzon E, Paterniti I, Bramanti P, Pedata F, Cuzzocrea S. Adenosine A2A receptor-selective stimulation reduces signaling pathways involved in the development of intestine ischemia and reperfusion injury. Shock. 2010;33:541–51. doi: 10.1097/SHK.0b013e3181c997dd. [DOI] [PubMed] [Google Scholar]
- 38.Reece TB, Tribble CG, Okonkwo DO, Davis JD, Maxey TS, Gazoni LM, Linden J, Kron IL, Kern JA. Early adenosine receptor activation ameliorates spinal cord reperfusion injury. J Cardiovasc Med (Hagerstown) 2008;9:363–7. doi: 10.2459/JCM.0b013e3282eee836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Oskouian RJ, Day YJ, Rieger JM, Liu L, Kern JA, Linden J. Mouse spinal cord compression injury is reduced by either activation of the adenosine A2A receptor on bone marrow-derived cells or deletion of the A2A receptor on non-bone marrow-derived cells. Neuroscience. 2006;141:2029–39. doi: 10.1016/j.neuroscience.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Cassada DC, Tribble CG, Young JS, Gangemi JJ, Gohari AR, Butler PD, Rieger JM, Kron IL, Linden J, Kern JA. Adenosine A2A analogue improves neurologic outcome after spinal cord trauma in the rabbit. J Trauma. 2002;53:225–9. doi: 10.1097/00005373-200208000-00005. discussion 229-31. [DOI] [PubMed] [Google Scholar]
- 41.Sullivan GW, Fang G, Linden J, Scheld WM. A2A adenosine receptor activation improves survival in mouse models of endotoxemia and sepsis. J Infect Dis. 2004;189:1897–904. doi: 10.1086/386311. [DOI] [PubMed] [Google Scholar]
- 42.Murphree LJ, Sullivan GW, Marshall MA, Linden J. Lipopolysaccharide rapidly modifies adenosine receptor transcripts in murine and human macrophages: role of NF-kappaB in A(2A) adenosine receptor induction. Biochem J. 2005;391:575–80. doi: 10.1042/BJ20050888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179:1254–63. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 44.Zhai Y, Shen XD, Hancock WW, Gao F, Qiao B, Lassman C, Belperio JA, Strieter RM, Busuttil RW, Kupiec-Weglinski JW. CXCR3+CD4+ T cells mediate innate immune function in the pathophysiology of liver ischemia/reperfusion injury. J Immunol. 2006;176:6313–22. doi: 10.4049/jimmunol.176.10.6313. [DOI] [PubMed] [Google Scholar]
- 45.Shimamura K, Kawamura H, Nagura T, Kato T, Naito T, Kameyama H, Hatakeyama K, Abo T. Association of NKT cells and granulocytes with liver injury after reperfusion of the portal vein. Cell Immunol. 2005;234:31–8. doi: 10.1016/j.cellimm.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 46.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203:2639–48. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HH, Meyer EH, Goya S, Pichavant M, Kim HY, Bu X, Umetsu SE, Jones JC, Savage PB, Iwakura Y, Casasnovas JM, Kaplan G, Freeman GJ, DeKruyff RH, Umetsu DT. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. J Immunol. 2010;185:5225–35. doi: 10.4049/jimmunol.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallace KL, Marshall MA, Ramos SI, Lannigan JA, Field JJ, Strieter RM, Linden J. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114:667–76. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood. 2010 doi: 10.1182/blood-2010-06-290643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hocher B. Adenosine A1 receptor antagonists in clinical research and development. Kidney Int. 2010;78:438–45. doi: 10.1038/ki.2010.204. [DOI] [PubMed] [Google Scholar]
- 51.Mi T, Abbasi S, Zhang H, Uray K, Chunn JL, Xia LW, Molina JG, Weisbrodt NW, Kellems RE, Blackburn MR, Xia Y. Excess adenosine in murine penile erectile tissues contributes to priapism via A2B adenosine receptor signaling. J Clin Invest. 2008;118:1491–501. doi: 10.1172/JCI33467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen J, Jiang X, Dai Y, Zhang Y, Tang Y, Sun H, Mi T, Phatarpekar PV, Kellems RE, Blackburn MR, Xia Y. Increased adenosine contributes to penile fibrosis, a dangerous feature of priapism, via A2B adenosine receptor signaling. FASEB J. 2010;24:740–9. doi: 10.1096/fj.09-144147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H, Tao L, Lu G, Alexander DC, Milburn MV, Carter-Dawson L, Lewis DE, Eltzschig HK, Kellems RE, Blackburn MR, Juneja HS, Xia Y. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17:79–86. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Exley MA, Hou R, Shaulov A, Tonti E, Dellabona P, Casorati G, Akbari O, Akman HO, Greenfield EA, Gumperz JE, Boyson JE, Balk SP, Wilson SB. Selective activation, expansion, and monitoring of human iNKT cells with a monoclonal antibody specific for the TCR alpha-chain CDR3 loop. Eur J Immunol. 2008;38:1756–66. doi: 10.1002/eji.200737389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lombardi V, Stock P, Singh AK, Kerzerho J, Yang W, Sullivan BA, Li X, Shiratsuchi T, Hnatiuk NE, Howell AR, Yu KO, Porcelli SA, Tsuji M, Kronenberg M, Wilson SB, Akbari O. A CD1d-dependent antagonist inhibits the activation of invariant NKT cells and prevents development of allergen-induced airway hyperreactivity. J Immunol. 2010;184:2107–15. doi: 10.4049/jimmunol.0901208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Felio K, Nguyen H, Dascher CC, Choi HJ, Li S, Zimmer MI, Colmone A, Moody DB, Brenner MB, Wang CR. CD1-restricted adaptive immune responses to Mycobacteria in human group 1 CD1 transgenic mice. J Exp Med. 2009;206:2497–509. doi: 10.1084/jem.20090898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arrenberg P, Halder R, Kumar V. Cross-regulation between distinct natural killer T cell subsets influences immune response to self and foreign antigens. J Cell Physiol. 2009;218:246–50. doi: 10.1002/jcp.21597. [DOI] [PMC free article] [PubMed] [Google Scholar]