

Abstract
Some small proteins, such as HP35, fold at submicrosecond timescale with low folding cooperativity. Although these proteins have been extensively investigated, still relatively little is known about their folding mechanism. Here, using single-molecule force spectroscopy and steered molecule dynamics simulation, we study the unfolding of HP35 under external force. Our results show that HP35 unfolds at extremely low forces without a well-defined unfolding transition state. Subsequently, we probe the structure of unfolded HP35 using the persistence length obtained in the force spectroscopy. We found that the persistence length of unfolded HP35 is around 0.72 nm, >40% longer than typical unstructured proteins, suggesting that there are a significant amount of residual secondary structures in the unfolded HP35. Molecular dynamics simulation further confirmed this finding and revealed that many native contacts are preserved in HP35, even its two ends have been extended up to 8 nm. Our results therefore suggest that retaining a significant amount of secondary structures in the unfolded state of HP35 may be an efficient way to reduce the entropic cost for the formation of tertiary structure and increase the folding speed, although the folding cooperativity is compromised. Moreover, we anticipate that the methods we used in this work can be extended to the study of other proteins with complex folding behaviors and even intrinsically disordered ones.
Introduction
Most proteins require correct and efficient folding from a mainly random coil conformation to a well-defined structure to achieve their biological functions. Such a folding process can be pictorially viewed as diffusion through a predefined funnel-like energy landscape (1). The folding cooperativity, describing how sharp and smooth the folding transition occurs, is essential for proteins to fold quickly, effectively, and to avoid misfolding and aggregation (2). Most intermediate or small proteins fold with high cooperativity and show a typical all-or-none (or two-state) feature. In a different way, a few very small proteins can fold at a submicrosecond timescale showing low folding cooperativity and low energy barriers (3). The C-terminal headpiece domain of Villin (HP35) (4) is a representative example of proteins showing such folding behavior. It contains three α-helices linked by two short loops (Fig. 1 a) (5). Simulations indicate that its folding does not follow a two-state fashion and the formation of the α-helices precedes the formation of the final tertiary structure (6–8). Experimentally, its folding has been studied using traditional chemical denaturation, temperature jump, and a few other newly developed methods (9–13). Although a low energy barrier was confirmed, the detailed folding processes such as when the secondary structures are formed are still under dispute.
Figure 1.
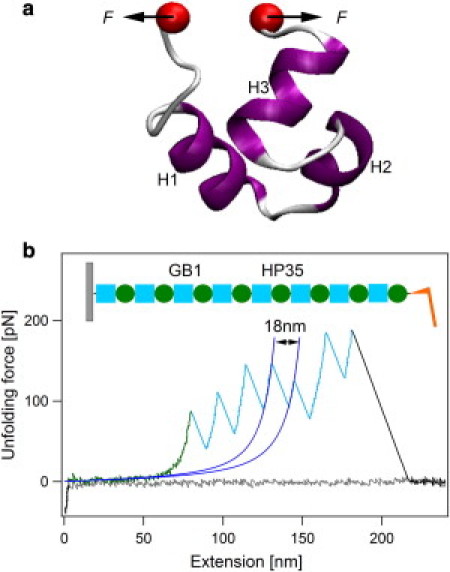
Single-molecule AFM reveals low unfolding forces and low unfolding cooperativity of HP35 (a) Three-dimensional structure of the C-terminal headpiece domain of human villin, HP35. H1, H2, and H3 refer to three helices of HP35 from N-terminus to C-terminus (PDB ID: 1UNC). The force is applied to both ends of the protein. (b) A typical set of approaching (gray line) and retraction traces for mechanical unfolding of HP35 using heteropolyprotein (GB1-HP35)8 at a pulling speed of 400 nm s−1. The schematic of the polyprotein is shown in the inset. Green cycles and cyan squares represent HP35 and GB1 domains, respectively. WLC fitting of consecutive unfolding events (blue line) identifies that all sawtooth-like peaks resulted from the unfolding of GB1 domains (highlighted in cyan). Thus, the featureless long spacer preceding the unfolding events of GB1 is attributed to the unfolding signature of HP35 (highlighted in green).
Here, we study the forced unfolding of HP35 using single-molecule atomic force microscopy (AFM) combined with steered molecular dynamics (SMD) simulation. Single-molecule AFM has become a powerful tool to study protein folding at the single-molecule level (14–23). Force can steer proteins from the native state to the unfolded state following a tilted free energy landscape, in which the folding is slowed down and unfolding is sped up. It allows directly measuring the mechanical features of both folded and unfolded proteins, from which the folding mechanism of proteins can be deciphered (16,17,24,25). Moreover, MD simulation provides the atomistic details about the folding and unfolding process probed by force (26,27), complementary to the single-molecule force spectroscopy techniques. Taking advantage of these two methods, many aspects of protein folding have been explored, such as intermediates in protein folding/unfolding (22,28–30), parallel folding/unfolding pathways (31,32), free energy landscape of unfolding along different pulling directions (33,34), folding under tensile force (21,35), temperature or solvent effect (20,36–40), and protein misfolding (31,41). These studies greatly enriched our current understanding on protein folding. Recently, Shank et al. (34) found that the folding cooperativity of a multiple domain protein T4-lysozyme depends on its topology using an optical tweezers-based single-molecule force spectroscopy assay. However, the folding cooperativity of small single-domain proteins has not been explored by single-molecule force spectroscopy techniques yet. Here, using single-molecule AFM and SMD simulation, we show that the forced unfolding of human Villin HP35 does not display pronounced free energy barriers, consistent with the previous findings. More strikingly, we directly detect the secondary structure elements in the unfolded HP35. This indicates that the individual secondary structure elements in HP35 are stable under residual force without the protection of the tertiary structure. Thus, we propose that retaining a significant amount of secondary structures in the unfolded state of HP35 may be an efficient way to reduce the entropic cost for the formation of tertiary structure and increase the folding speed, although the folding cooperativity is compromised.
Materials and Methods
Protein engineering
The plasmid pUC19-GB1 and pQE80L-(GB1)8 were kindly provided by Prof. Hongbin Li of the University of British Columbia. The plasmid encoding of the whole human villin (including HP35 and HP67 proteins) was generously provided by Prof. Robert Robinson of the National University of Singapore. HP35 monomer, flanked with a 5′ BamHI restriction site and 3′ BglII and KpnI restriction sites, was amplified by polymerase chain reaction and digested with BamHI and KpnI. The copy vector pUC19-GB1 in which GB1 monomer was also flanked with a 5′ BamHI restriction site and 3′ BglII and KpnI restriction sites was digested with BglII and KpnI. On the basis of the identity of the sticky ends generated by BamHI and BglII restriction enzymes, after ligation the (GB1-HP35) was produced with a nonpalindromic site inside, which could not be digested by either BamHI or BglII, but still flanked with the 5′ BamHI restriction site and 3′ BglII and KpnI restriction sites. The (GB1-HP35)8 polyprotein gene was then constructed using an iterative approach of cloning (GB1-HP35) dimer into dimer and transferred into the expression vector pQE80L for the tetramer into tetramer and octamer into octamer cloning steps in Top10 (24,42). (GB1-HP35)8 was expressed in BL21 and purified by Ni2+-affinity chromatography. The purified polyprotein sample was kept at 4°C in Tris-HCl buffer at a concentration of ∼2 mg mL−1. Polyprotein (GB1-HP67)8 was engineered using the same method as for (GB1-HP35)8.
Single-molecule AFM
Single-molecule AFM experiments were carried out on a commercial AFM (ForceRobot 300, JPK, Berlin, Germany). All the force-extension experiments were carried out in Tris-HCl buffer (10 mM, pH 7.4, containing 10 mM NaCl). ∼2 μL of protein sample was directly deposited onto a freshly cleaved mica surface containing ∼50 μL buffer for ∼30 min; the sample chamber was then filled with ∼1 mL buffer before the measurement. Typically, experiments proceeded overnight to obtain enough force-extension traces. The spring constant of the AFM cantilevers (Biolever-RC-150VB-70 from Olympus) was calibrated using the equipartition theorem before each experiment with a typical value of 6 pN nm−1 and recalibrated every 4–6 h during each experiment. The pulling speed was 400 nm s−1 for all traces unless otherwise indicated. The detailed data analysis procedure can be found in the Supporting Material.
Statistical analysis
The comparisons of persistence lengths of different polyprotein constructs were carried out using a standard Student's t-test procedure based on an online t-test calculator, QuickCalcs, from GraphPad Software. (http://www.graphpad.com/quickcalcs/ttest1.cfm?Format=C).
MD simulation
The MD simulations were carried out using the NAMD package (version 2.7) with the CHARMM27 force field including CMAP correction to proteins (43). The TIP3P water model was used for the solvent. Solvation and ionization were performed using the Visual Molecular Dynamics program package. Stretching forces were applied to HP35, HP67, and GB1 using constant-velocity SMD protocols, with a pulling velocity v = 0.1 m s−1 and the harmonic constraint force constant k = 0.05 kcal mol−1 Å−2 = 34.7 pN nm−1. In all the simulations, the Cα atom of the C-terminal residue was kept fixed, and the Cα atom of the N-terminal residue was pulled along the direction that connects the initial positions of the N-terminus and the C-terminus. Constant volume control was used for all the SMD simulations. For the HP35 systems, the periodic cell was set as 119.0 Å × 37.0 Å × 29.0 Å throughout the entire simulations, and the pulling direction is along the longest dimension. The protein molecules were at least 0.8 Å from the nearest cell boundary during pulling to ensure that the unfolding process was not influenced by its images. Similar periodic boundary conditions were also applied for the HP67 and GB1 systems. Constant temperature control was disabled to avoid additional disturbing of the movement of the atoms. In total, 25 individual SMD simulations for HP35, five for HP67, and five for GB1 were performed. The simulation details can be found in the Supporting Material.
Results
The unfolding kinetics of HP35
To study the unfolding of HP35 using single-molecule AFM, we engineered a heteropolyprotein (GB1-HP35)8 (Fig. 1 b, inset), in which the well-studied GB1 is used as the fingerprint and internal caliber to identify the mechanical unfolding signal of HP35 (42,44). The proper folding of both HP35 and GB1 in the polyprotein (GB1-HP35)8 is confirmed using circular dichroism and AFM (Fig. S1). Stretching (GB1-HP35)8 at a pulling speed of 400 nm s−1 results in force-extension curves that are characterized by a featureless long spacer (highlighted in green) followed by high unfolding force peaks of ∼150 pN and ΔLc of ∼18 nm that correspond to the unfolding of GB1 domains (Fig. 1 b) (42,44). Because HP35 alternates with GB1 in the heteropolyprotein, if the force-extension curve shows N unfolding events of GB1, it must contain signatures of the mechanical unfolding of N ± 1 unfolding events of HP35 (45,46). Hence, the long featureless spacer preceding GB1 unfolding events must result from the mechanical unfolding of HP35. This indicates that HP35 domains unfold at forces below the detection limit of our AFM (∼12 pN, estimated from the noise level of force-extension curves of ∼4 pN). This indicates the low mechanical stability of HP35. It is worthwhile noting that low mechanical stability does not necessarily imply low cooperativity of the mechanical unfolding process. Even for a two-state folder, either a low free energy barrier for mechanical unfolding or a long distance from the native state to the mechanical unfolding transition state can lead to a low mechanical unfolding force of a protein. Detailed MD simulations are required to fully elucidate the unfolding mechanism. However, a unique mechanical unfolding feature for cooperative ultrafast folding proteins is that it is possible to observe fast equilibrium between folded and unfolded states in the unfolding traces, which has been reported for many proteins, such as calmodulin (47) and solenoid proteins (48). Although it is not possible to resolve the forces of HP35 at different states, if fast equilibrium occurs, the apparent noise level for the unfolding traces would be greater, as proteins can adopt different states of distinct forces at the same extension (see the Supporting Material). In contrast, for the mechanical unfolding of HP35, the noise level corresponding to the HP35 unfolding region (green) remains the same as that of the rest part of the trace (Fig. 1 b). We also did not observe any fast unfolding-refolding equilibrium in the unfolding traces of HP35 even at a pulling speed as low as 10 nm s−1. These results imply that the folding of HP35 is not cooperative and the free energy barrier is not high enough to separate the unfolded and folded states. However, due to limited force resolution, it is not possible to quantitatively obtain the unfolding kinetics of HP35 from our single-molecule AFM data.
The refolding kinetics of HP35
We then probed the refolding kinetics of HP35 after the mechanical unfolding of HP35 by applying stretching-relaxation cycles to the same (GB1-HP35)8 molecule. Because HP35 is mechanically labile and always unfolds before the unfolding of GB1, we are able to selectively unfold HP35 in the polyprotein without unfolding GB1 by simply controlling the extension of the molecule. As shown in Fig. 2, the unfolding trace of HP35 is almost superimposable to the refolding trace, indicating that the unfolding of HP35 occurs close to equilibrium and the experimental condition is quasistatic. To further reduce the noise level of the force-extension curves, we average four stretching and relaxation traces (Fig. 2, inset). This reduces the noise level to <2 pN, yet, still no significant hysteresis can be found in the averaged traces. In our experiments, 21 individual (GB1-HP35)8 molecules were subjected to the stretching-relaxation cycles and none of them show pronounced hysteresis (two additional examples are shown in Fig. S2, a and b), suggesting that folding and unfolding of HP35 under force is reversible. As a control, if (GB1-HP35)8 is fully stretched, significant hysteresis between stretching and relaxation curves can be observed due to the nonequilibrium unfolding of GB1 domains under the same experimental condition (Fig. S2 c). Many proteins of fast folding rates show refolding force peaks in the relaxation traces (48–50) and the refolding forces are believed to be determined by the size and folding rate of a protein (50). Because HP35 folds at a submicrosecond timescale, at least 10 times faster than the proteins that have been reported to show detectable refolding force, it may be expected that folding of HP35 exhibits high refolding force if it follows typical two-state folding and unfolding (see the Supporting Material). However, the refolding force is not observed for HP35, which provides additional evidence that HP35 is not a typical two-state folder.
Figure 2.

Mechanical refolding of HP35. (GB1-HP35)8 is partially stretched to selectively unfold HP35 domains (black trace), and then the unfolded HP35 is relaxed to an initial position just above the substrate at the same pulling speed of 400 nm s−1 (dark gray trace in the printed version or red trace in the online version). The gray trace corresponds to the complete stretching of the molecule in the last cycle. The inset shows the averaged traces from four stretching and relaxation cycles to reduce the noise level of traces to <2 pN. The stretching and relaxation traces are almost superimposable, indicating unfolding of HP35 occurs close to equilibrium.
Mechanical features of unfolded HP35
We then probed the secondary structural features of unfolded HP35 using its characteristic mechanical properties by single-molecule AFM. In the worm-like chain (WLC) model of polymer elasticity, persistence length (p) is a parameter that related to the stiffness of a polymer (51). Therefore, formation of secondary structures could increase the stiffness of a peptide and thus the persistence length (52). Before the unfolding of the first GB1 domain, the contour length of the polyprotein chain is mainly contributed by the unfolded HP35 domains. We measured the persistence length of this region as the persistence length of unfolded HP35 (p1, Fig. 3 a, blue line). The distribution of p1 from different (GB1-HP35)8 is shown in Fig. 3 d. The average persistence length of unfolded HP35 is 0.72 ± 0.15 nm, which is clearly larger than that for random coil sequences. For comparison, we constructed a polyprotein, (GB1-HP67)8. HP67 is the villin headpiece containing both the N- and C-terminal domains. Previous experiments indicated that the N-terminal domain of HP67 is unstructured in the absence of C-terminal domain, HP35 (53). The representative force-extension curve of (GB1-HP67)8 is shown in Fig. 3 b. Because the N-terminal part of HP67 adopts random coil conformation, we therefore expected a shorter persistence length of unfolded HP67 measured from the force-extension curve of (GB1-HP67)8 (p1, Fig. 3 b, blue line). Indeed, the persistence length of HP67 is 0.62 ± 0.15 nm, less than that of HP35 (Fig. 3 e). To rule out the possibility that the increased persistence length of HP35 measured using (GB1-HP35)8 construct is due to the contribution from folded GB1 domains in the polyprotein, we also measured the persistence length of (GB1)8 before the unfolding of the first GB1 domain (Fig. 3 c). The p1 for (GB1)8 is 0.56 ± 0.14 nm (Fig. 3 f), lower than that of both (GB1-HP35)8 and (GB1-HP67)8. A direct comparison of p1 for (GB1-HP35)8, (GB1-HP67)8, and (GB1)8 is shown in Fig. 3 g. Clearly, such difference is not due to experimental error but truly because of the presence of secondary structures in unfolded HP35. Moreover, we also measured the persistence length of these three constructs after the unfolding of all GB1 domains (p2, red lines in Fig. 3, a, b, and c). The data are shown in Fig. 3, h, i, and j, respectively. Because GB1 adopts mainly random-coiled structure in the unfolded conformation, the unfolding of GB1 increases the amount of unstructured sequence in the polyprotein and thereby leads the p2 for all three constructs lower than p1. However, due to the presence of a large amount of unfolded GB1 in the polyprotein, the difference among p2 for these three constructs is less significant compared with p1 (Fig. 3 k), although it seems that there are still a certain amount of secondary structures in (GB1-HP35)8. This is probably because the secondary structure of HP35 may partially refold in the traces up to the position where p2 is measured, given that the folding rate of HP35 is extremely fast (1.38 × 106 s−1) (4) and the formation of secondary structures of HP35 is at the timescale of ∼100 ns (6). Our MD simulation on HP35 also confirms that its secondary structures can indeed survive at stretching forces of 120–150 pN (Fig. S3). Although the pulling speed in our single-molecule AFM experiments is much lower than that in the simulation, the unfolding forces for the secondary structures of HP35 is still ∼70–100 pN, as estimated using the Bell-Evans model (54), by assuming the potential width for the unfolding of flexible α-helical secondary structures of HP35 is ∼1 nm (based on our simulation results). However, this can only be considered as a rough estimation, because the potential width is experimentally unknown and the unfolding mechanism of individual helices by force may not follow the simple Bell-Evans model. More precise experimental characterization is required to fully verify this estimation. It is also worth mentioning that the reformation of the secondary structures of HP35 in the single-molecule AFM traces may not be complete after the force on the polyprotein drops upon GB1 unfolding, because the residual force on the polyprotein is still very high (∼50 pN, as estimated based on the WLC model; see Fig. S4 and discussions in the Supporting Material for details) significantly slowing down the folding. Moreover, the limited force ranges available for WLC fitting to obtain p2 could also lead to higher uncertainly in p2 data. The detailed p1 and p2 values for all constructs are summarized in Table S1.
Figure 3.

Long persistence length of unfolded HP35 corroborates the presence of secondary structures in HP35 after mechanical unfolding. (a–c) Representative force-extension traces of (GB1-HP35)8, (GB1-HP67)8, and (GB1)8. Gray lines, (blue and red lines in the online version) correspond to the WLC fitting to the force-extension traces before and after the unfolding of GB1 domains and the corresponding persistence lengths are denoted as p1 and p2. (d–f) Distribution of p1 for (GB1-HP35)8, (GB1-HP67)8, and (GB1)8, respectively. (g) Comparison of p1 for these three constructs. (h–j) Distribution of p2 for (GB1-HP35)8, (GB1-HP67)8, and (GB1)8, respectively. (k) Comparison of p2 for these three constructs. p-value corresponds to the statistical significance between two pairs of data calculated using Student's t-test.
SMD simulation on the unfolding of HP35
To obtain more molecular insight into the unfolding process of HP35, we performed all-atom SMD simulation on HP35 (55). As shown in Fig. 4, pulling HP35 at a speed of 0.1 m s−1 results in low force plateaus and low force peaks barely above the noise level, suggesting low free energy barriers in the mechanical unfolding pathway of HP35. Moreover, the unfolding trajectories reveal a rather continuous structural change of HP35 upon stretching. The third α-helix rotates along the force direction leading to a partial opening of the tertiary structure at ∼0.2–0.5 nm extension. This process results in large fluctuations in the force-extension traces at forces around 10 pN. The break of the interaction between the C-terminal leucine residue and the hydrophobic cluster formed by three phenylalanine and other hydrophobic residues leads to the first major peak at ∼50 pN and ∼2 nm. The complete rupture of the tertiary structure then occurs at ∼50 pN and ∼3.5 nm, corresponding to the separation of the leucine residue of the third α-helix and the three phenylalanine residues from the first and second α-helices. After that, the local secondary structures of HP35 gradually open up at elevated forces. The complete melting of the secondary structures requires forces as high as 120 pN. These results indicate that the unfolding of HP35 is not a representative two-state process with a well-developed transition state, consistent with the single-molecule AFM data. It is worthwhile noting that the pulling speed used in the simulation is 250,000 times faster than that in single-molecule AFM experiments. Even at such a high pulling speed, the unfolding force for HP35 is still <60 pN, which is significantly lower than that of well-studied two-state folders and some proteins that were considered as mechanically weak (55–59).
Figure 4.
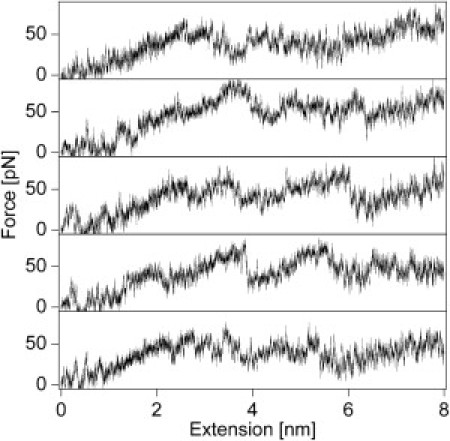
Five representative force-extension profiles of HP35 from SMD simulations at a constant stretching velocity of 0.1m s−1. The simulations were stopped after 80 ns, when the extension reached 80 Å. Instead of showing pronounced unfolding force peaks, these trajectories show continuous low force plateaus and a few low force peaks (<60 pN) with variable positions. These are clear signatures for noncooperative unfolding.
To further acquire the molecular detail of the secondary structures in the unfolded HP35, we analyzed the simulated unfolding trajectories of HP35. Fig. 5 a shows the contact maps of HP35 at different extensions. Most of the tertiary contacts disappear at an extension of ∼4 nm (highlighted in black cycle). However, a majority of the secondary structures are still presented at this extension. The contacts correspond to first, second, and third α-helices marked in orange, yellow, and cyan, respectively. With further extension, the C-terminal part of the third helix becomes unstructured at an extension of 6 nm. However, the C-terminal part of the first α-helix and the N-terminal part of the third α-helix can retain their secondary structure even at an extension of 8 nm. The evolution of the structure of HP35 is pictorially illustrated in Fig. 5 b. Such a continuous change in secondary structure indicates low unfolding cooperativity of HP35. For comparison, we analyzed the unfolding trajectories of HP67 and GB1 (Fig. S5). The N-terminal domain of HP67 loses its secondary structure simultaneously with the tertiary structure, consistent with the single-molecule AFM data. Moreover, as a representative two-state folder, the secondary structures of GB1 are completely disrupted at an extension of 6 nm, although its size is much longer than HP35. Therefore, the presence of a large amount of secondary structures in the unfolded form may be a unique feature of HP35. Such a feature accounts for its fast folding rate, because it can efficiently reduce the entropic cost for the folding process and allows the folding starts locally.
Figure 5.

Contact maps and representative snapshots of HP35 at different extensions during the SMD simulations. (a) Contact maps of HP35 with extension x = 0 nm (top), 2 nm (middle, upper half), 4 nm (middle, lower half), 6 nm (bottom, upper half), and 8 nm (bottom, lower half). The value of each dot represents the percentage of the formed contact between a pair of residues, defined as p = contact number/native contact number, where the contact number between two residues is the number of atom pairs that are within 4.5 Å of each other and from different residues. Local contacts of <3 residues are removed for clarity. Tertiary contacts are highlighted in a black cycle for extensions of 0, 2, and 4 nm. Contacts of the first, second, and third α-helices are marked by orange, yellow, and cyan circles, respectively. Clearly, secondary contacts of HP35 are well retained after the disappearance of tertiary contacts. (b) Representative snapshots of HP35 evolved under different extensions. The Cα atom of the last C-terminal residue was kept fixed, and the Cα atom of the first N-terminal residue was pulled in the simulation.
Discussion
As a paradigm, the folding of HP35 has been characterized using many different experimental techniques (9–13) as well as simulations (6–8). All-atom simulations of protein folding by Duan et al. and Lei et al. (6,7) indicate that formation of the final tertiary structure goes through various intermediate structures with a low free energy barrier. Our results on forced unfolding of HP35 are consistent with this picture and provide additional evidences for the formation of large local secondary structure elements before the formation of final tertiary structure. Recently, using backbone amide-to-ester mutation and Φ value analysis, Gai and co-workers were able to experimentally probe the formation of α-helix during the folding process for the first time (10). They concluded that hydrogen bond-mediated α-helix formation occurs after the surmounting of a folding transition state, because these mutations do not notably change the folding rate but lower the overall stability. Their conclusions are made based on the two-state folding scheme for HP35 (10). Taking account of our forced unfolding data on HP35, an alternative explanation may be that the formation of α-helices takes place before crossing the major folding transition. The helices have already been formed in both the unfolded state (or named as the intermediate state) and the transition state. Therefore, amide-to-ester mutation destabilizes both the unfolded state and the transition state by a similar amount, and does not affect the net free energy barrier. Our interpretation is also in line with recent NMR studies on a truncated HP35, which only contains the first two helices (60,61). They found that there are a significant amount of native-like structures in this peptide, consistent with our finding that the helices in HP35 can be maintained after the tertiary interactions between the H1 and H3 of HP35 are disrupted. Moreover, significant structures in unfolded proteins have also been reported for many other ultrafast folding proteins (62–64). Hence, the formation of native-like structures in the unfolded proteins may be a general strategy for proteins to fold at an ultrafast timescale.
As proposed by Daggett and Fersht (65) the interplay between the formation of secondary and tertiary interactions underlines the structure of the folding transition state and the folding mechanism of a protein. For most proteins, the secondary structure is only marginally stable in the absence of tertiary structure. Thus, the formation of a native-like transition state is the key step for the folding, making the folding process cooperative. However, for a protein with stable secondary structure, like HP35, the folding process becomes less cooperative and more hierarchical, following a framework mechanism. Our results on the forced unfolding of HP35 directly support this view. Moreover, the preformed secondary structure is actually essential for the fast folding of HP35, as it can restrict the initial conformational search. However, such a mechanism may not be applicable to larger proteins. Because the secondary structure propensities of a peptide sequence are not unique, preformed secondary structures could lead to unwelcome nonnative conformations, misfolding, and even aggregation.
Traditional chemical or thermal denaturing experiments are extremely powerful to probe the unfolding and refolding of the tertiary structure of proteins. However, presence of chemical denaturant or high temperature inevitably affects the study of the residual secondary structures in an unfolded protein, because they also destabilize the residual secondary structures after the tertiary structure of a protein is unfolded. Thus, developing methods that can independently probe the secondary structure and tertiary structure could be useful for the study of the folding mechanism of proteins. In this work, we show that combining single-molecule AFM and SMD simulation, the folding mechanism of proteins can be understood in more detail. In particular, the mechanical features of the unfolded peptide chain can be used as signals for residual secondary structures. Moreover, this technique can be easily extended to study the residual structures in intrinsic disordered proteins and their structural change upon binding (66). We anticipate it can eventually become a general tool to study the conformational change of proteins and other biomacromolecules.
Conclusion
In summary, in this work, we study the unfolding of HP35 using single-molecule force spectroscopy and SMD simulation. Our results show that HP35 unfolds at forces <12 pN without a well-defined unfolding transition state, providing direct evidence of its low folding cooperativity. Moreover, we probe the structure of unfolded HP35 using the persistence length obtained in the force spectroscopy. We found that the persistence length of unfolded HP35 is around 0.72 nm, >40% longer than typical unstructured proteins, suggesting that there are a significant amount of residual secondary structures in the unfolded HP35. MD simulation further confirmed this finding and revealed that many native contacts are maintained in HP35; even its two ends have been extended up to 8 nm. Thus, we proposed that retaining a significant amount of secondary structures in the unfolded state of HP35 may be an important reason to account for its ultrafast folding, because it can efficiently reduce the entropic cost for the formation of tertiary interactions and speed up the folding process, although the folding cooperativity is compromised. This is distinct from the folding of larger proteins with high folding cooperativity. To pinpoint the detailed folding process and stability of each individual α-helix in HP35 will be our next endeavor.
Acknowledgments
This work is supported by the National Natural Science Foundation of China under grant Nos. 11074115, 81121062, 10834002, 10904064, and 31170813, the Natural Science Foundation of Jiangsu Province under grant No. BK2009008, the program for New Century Excellent Talents in University, and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Contributor Information
Yi Cao, Email: caoyi@nju.edu.cn.
Wei Wang, Email: wangwei@nju.edu.cn.
Supporting Material
References
- 1.Dill K.A., Chan H.S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Muñoz V. Conformational dynamics and ensembles in protein folding. Annu. Rev. Biophys. Biomol. Struct. 2007;36:395–412. doi: 10.1146/annurev.biophys.36.040306.132608. [DOI] [PubMed] [Google Scholar]
- 4.Kubelka J., Eaton W.A., Hofrichter J. Experimental tests of villin subdomain folding simulations. J. Mol. Biol. 2003;329:625–630. doi: 10.1016/s0022-2836(03)00519-9. [DOI] [PubMed] [Google Scholar]
- 5.Vermeulen W., Vanhaesebrouck P., Borremans F.A. Solution structures of the C-terminal headpiece subdomains of human villin and advillin, evaluation of headpiece F-actin-binding requirements. Protein Sci. 2004;13:1276–1287. doi: 10.1110/ps.03518104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duan Y., Kollman P.A. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 7.Lei H., Wu C., Duan Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2007;104:4925–4930. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piana S., Lindorff-Larsen K., Shaw D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zagrovic B., Pande V.S. Simulated unfolded-state ensemble and the experimental NMR structures of villin headpiece yield similar wide-angle solution x-ray scattering profiles. J. Am. Chem. Soc. 2006;128:11742–11743. doi: 10.1021/ja0640694. [DOI] [PubMed] [Google Scholar]
- 10.Bunagan M.R., Gao J., Gai F. Probing the folding transition state structure of the villin headpiece subdomain via side chain and backbone mutagenesis. J. Am. Chem. Soc. 2009;131:7470–7476. doi: 10.1021/ja901860f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu K.N., Yau W.M., Tycko R. Detection of a transient intermediate in a rapid protein folding process by solid-state nuclear magnetic resonance. J. Am. Chem. Soc. 2010;132:24–25. doi: 10.1021/ja908471n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung J.K., Thielges M.C., Fayer M.D. Dynamics of the folded and unfolded villin headpiece (HP35) measured with ultrafast 2D IR vibrational echo spectroscopy. Proc. Natl. Acad. Sci. USA. 2011;108:3578–3583. doi: 10.1073/pnas.1100587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cellmer T., Buscaglia M., Eaton W.A. Making connections between ultrafast protein folding kinetics and molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2011;108:6103–6108. doi: 10.1073/pnas.1019552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rief M., Gautel M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez J.M., Li H. Force-clamp spectroscopy monitors the folding trajectory of a single protein. Science. 2004;303:1674–1678. doi: 10.1126/science.1092497. [DOI] [PubMed] [Google Scholar]
- 16.Harris N.C., Song Y., Kiang C.H. Experimental free energy surface reconstruction from single-molecule force spectroscopy using Jarzynski's equality. Phys. Rev. Lett. 2007;99:068101. doi: 10.1103/PhysRevLett.99.068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borgia A., Williams P.M., Clarke J. Single-molecule studies of protein folding. Annu. Rev. Biochem. 2008;77:101–125. doi: 10.1146/annurev.biochem.77.060706.093102. [DOI] [PubMed] [Google Scholar]
- 18.Schwaiger I., Schleicher M., Rief M. The folding pathway of a fast-folding immunoglobulin domain revealed by single-molecule mechanical experiments. EMBO Rep. 2005;6:46–51. doi: 10.1038/sj.embor.7400317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlierf M., Rief M. Surprising simplicity in the single-molecule folding mechanics of proteins. Angew. Chem. Int. Ed. Engl. 2009;48:820–822. doi: 10.1002/anie.200804723. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Manyes S., Dougan L., Fernández J.M. Osmolyte-induced separation of the mechanical folding phases of ubiquitin. Proc. Natl. Acad. Sci. USA. 2009;106:10540–10545. doi: 10.1073/pnas.0902090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Manyes S., Dougan L., Fernández J.M. Direct observation of an ensemble of stable collapsed states in the mechanical folding of ubiquitin. Proc. Natl. Acad. Sci. USA. 2009;106:10534–10539. doi: 10.1073/pnas.0901213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nunes J.M., Hensen U., Muller D.J. A “force buffer” protecting immunoglobulin titin. Angew. Chem. Int. Ed. Engl. 2010;49:3528–3531. doi: 10.1002/anie.200906388. [DOI] [PubMed] [Google Scholar]
- 23.Puchner E.M., Gaub H.E. Force and function: probing proteins with AFM-based force spectroscopy. Curr. Opin. Struct. Biol. 2009;19:605–614. doi: 10.1016/j.sbi.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Carrion-Vazquez M., Oberhauser A.F., Fernandez J.M. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H.B. Mechanical engineering of elastomeric proteins: toward designing new protein building blocks for biomaterials. Adv. Funct. Mater. 2008;18:2643–2657. [Google Scholar]
- 26.Paci E., Karplus M. Unfolding proteins by external forces and temperature: the importance of topology and energetics. Proc. Natl. Acad. Sci. USA. 2000;97:6521–6526. doi: 10.1073/pnas.100124597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sotomayor M., Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316:1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 28.Bertz M., Rief M. Mechanical unfoldons as building blocks of maltose-binding protein. J. Mol. Biol. 2008;378:447–458. doi: 10.1016/j.jmb.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 29.Li L., Huang H.H., Fernandez J.M. Mechanical unfolding intermediates observed by single-molecule force spectroscopy in a fibronectin type III module. J. Mol. Biol. 2005;345:817–826. doi: 10.1016/j.jmb.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Damaghi M., Köster S., Müller D.J. One β hairpin follows the other: exploring refolding pathways and kinetics of the transmembrane β-barrel protein OmpG. Angew. Chem. Int. Ed. Engl. 2011;50:7422–7424. doi: 10.1002/anie.201101450. [DOI] [PubMed] [Google Scholar]
- 31.Peng Q., Fang J., Li H. Kinetic partitioning mechanism governs the folding of the third FnIII domain of tenascin-C: evidence at the single-molecule level. J. Mol. Biol. 2011;412:698–709. doi: 10.1016/j.jmb.2011.07.049. [DOI] [PubMed] [Google Scholar]
- 32.Mickler M., Dima R.I., Rief M. Revealing the bifurcation in the unfolding pathways of GFP by using single-molecule experiments and simulations. Proc. Natl. Acad. Sci. USA. 2007;104:20268–20273. doi: 10.1073/pnas.0705458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dietz H., Berkemeier F., Rief M. Anisotropic deformation response of single protein molecules. Proc. Natl. Acad. Sci. USA. 2006;103:12724–12728. doi: 10.1073/pnas.0602995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shank E.A., Cecconi C., Bustamante C. The folding cooperativity of a protein is controlled by its chain topology. Nature. 2010;465:637–640. doi: 10.1038/nature09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hyeon C., Morrison G., Thirumalai D. Refolding dynamics of stretched biopolymers upon force quench. Proc. Natl. Acad. Sci. USA. 2009;106:20288–20293. doi: 10.1073/pnas.0905764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang G., Cecconi C., Bustamante C. Solid-state synthesis and mechanical unfolding of polymers of T4 lysozyme. Proc. Natl. Acad. Sci. USA. 2000;97:139–144. doi: 10.1073/pnas.97.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlierf M., Rief M. Temperature softening of a protein in single-molecule experiments. J. Mol. Biol. 2005;354:497–503. doi: 10.1016/j.jmb.2005.09.070. [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi Y., Brockwell D.J., Kawakami M. The effect of temperature on mechanical resistance of the native and intermediate states of I27. Biophys. J. 2008;95:5296–5305. doi: 10.1529/biophysj.108.141275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma L., Xu M., Oberhauser A.F. Naturally occurring osmolytes modulate the nanomechanical properties of polycystic kidney disease domains. J. Biol. Chem. 2010;285:38438–38443. doi: 10.1074/jbc.M110.183913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao Y., Li H. How do chemical denaturants affect the mechanical folding and unfolding of proteins? J. Mol. Biol. 2008;375:316–324. doi: 10.1016/j.jmb.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 41.Oberhauser A.F., Marszalek P.E., Fernandez J.M. Single protein misfolding events captured by atomic force microscopy. Nat. Struct. Biol. 1999;6:1025–1028. doi: 10.1038/14907. [DOI] [PubMed] [Google Scholar]
- 42.Cao Y., Li H. Polyprotein of GB1 is an ideal artificial elastomeric protein. Nat. Mater. 2007;6:109–114. doi: 10.1038/nmat1825. [DOI] [PubMed] [Google Scholar]
- 43.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao Y., Lam C., Li H. Nonmechanical protein can have significant mechanical stability. Angew. Chem. Int. Ed. Engl. 2006;45:642–645. doi: 10.1002/anie.200502623. [DOI] [PubMed] [Google Scholar]
- 45.Li H., Oberhauser A.F., Fernandez J.M. Atomic force microscopy reveals the mechanical design of a modular protein. Proc. Natl. Acad. Sci. USA. 2000;97:6527–6531. doi: 10.1073/pnas.120048697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao Y., Li H. Engineered elastomeric proteins with dual elasticity can be controlled by a molecular regulator. Nat. Nanotechnol. 2008;3:512–516. doi: 10.1038/nnano.2008.168. [DOI] [PubMed] [Google Scholar]
- 47.Junker J.P., Ziegler F., Rief M. Ligand-dependent equilibrium fluctuations of single calmodulin molecules. Science. 2009;323:633–637. doi: 10.1126/science.1166191. [DOI] [PubMed] [Google Scholar]
- 48.Kim M., Abdi K., Marszalek P.E. Fast and forceful refolding of stretched alpha-helical solenoid proteins. Biophys. J. 2010;98:3086–3092. doi: 10.1016/j.bpj.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee G., Abdi K., Marszalek P.E. Nanospring behaviour of ankyrin repeats. Nature. 2006;440:246–249. doi: 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- 50.Schlierf M., Berkemeier F., Rief M. Direct observation of active protein folding using lock-in force spectroscopy. Biophys. J. 2007;93:3989–3998. doi: 10.1529/biophysj.107.114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marko J.F., Siggia E.D. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 52.Li H., Oberhauser A.F., Fernandez J.M. Multiple conformations of PEVK proteins detected by single-molecule techniques. Proc. Natl. Acad. Sci. USA. 2001;98:10682–10686. doi: 10.1073/pnas.191189098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho J.H., O'Connell N., Palmer A.G., 3rd Phi-value analysis for ultrafast folding proteins by NMR relaxation dispersion. J. Am. Chem. Soc. 2010;132:450–451. doi: 10.1021/ja909052h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans E., Ritchie K. Strength of a weak bond connecting flexible polymer chains. Biophys. J. 1999;76:2439–2447. doi: 10.1016/S0006-3495(99)77399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu H., Schulten K. The key event in force-induced unfolding of Titin's immunoglobulin domains. Biophys. J. 2000;79:51–65. doi: 10.1016/S0006-3495(00)76273-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brockwell D.J., Beddard G.S., Radford S.E. Mechanically unfolding the small, topologically simple protein L. Biophys. J. 2005;89:506–519. doi: 10.1529/biophysj.105.061465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Best R.B., Li B., Clarke J. Can non-mechanical proteins withstand force? Stretching barnase by atomic force microscopy and molecular dynamics simulation. Biophys. J. 2001;81:2344–2356. doi: 10.1016/S0006-3495(01)75881-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ng S.P., Rounsevell R.W., Clarke J. Mechanical unfolding of TNfn3: the unfolding pathway of a fnIII domain probed by protein engineering, AFM and MD simulation. J. Mol. Biol. 2005;350:776–789. doi: 10.1016/j.jmb.2005.04.070. [DOI] [PubMed] [Google Scholar]
- 59.Sharma D., Feng G., Li H. Stabilization provided by neighboring strands is critical for the mechanical stability of proteins. Biophys. J. 2008;95:3935–3942. doi: 10.1529/biophysj.108.134072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang Y., Goger M.J., Raleigh D.P. NMR characterization of a peptide model provides evidence for significant structure in the unfolded state of the villin headpiece helical subdomain. Biochemistry. 2006;45:6940–6946. doi: 10.1021/bi052484n. [DOI] [PubMed] [Google Scholar]
- 61.Meng W., Shan B., Raleigh D.P. Native like structure in the unfolded state of the villin headpiece helical subdomain, an ultrafast folding protein. Protein Sci. 2009;18:1692–1701. doi: 10.1002/pro.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Myers J.K., Oas T.G. Preorganized secondary structure as an important determinant of fast protein folding. Nat. Struct. Biol. 2001;8:552–558. doi: 10.1038/88626. [DOI] [PubMed] [Google Scholar]
- 63.Ferguson N., Sharpe T.D., Fersht A.R. Ultra-fast barrier-limited folding in the peripheral subunit-binding domain family. J. Mol. Biol. 2005;353:427–446. doi: 10.1016/j.jmb.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 64.Chowdhury S., Lei H., Duan Y. Denatured-state ensemble and the early-stage folding of the G29A mutant of the B-domain of protein A. J. Phys. Chem. B. 2005;109:9073–9081. doi: 10.1021/jp0449814. [DOI] [PubMed] [Google Scholar]
- 65.Daggett V., Fersht A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 66.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 67.Crampton N., Alzahrani K., Brockwell D.J. Mechanically unfolding protein L using a laser-feedback-controlled cantilever. Biophys. J. 2011;100:1800–1809. doi: 10.1016/j.bpj.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dietz H., Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc. Natl. Acad. Sci. USA. 2004;101:16192–16197. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arnold K., Bordoli L., Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 70.Schwede T., Kopp J., Peitsch M.C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 72.Bustamante C., Marko J.F., Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 73.Best R.B., Hummer G. Protein folding kinetics under force from molecular simulation. J. Am. Chem. Soc. 2008;130:3706–3707. doi: 10.1021/ja0762691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.