

Abstract
We describe monozygotic twins discordant for childhood leukemia and secondary thyroid carcinoma. We used bisulfite pyrosequencing to compare the constitutive promoter methylation of BRCA1 and several other tumor suppressor genes in primary fibroblasts. The affected twin displayed an increased BRCA1 methylation (12%), compared with her sister (3%). Subsequent bisulfite plasmid sequencing demonstrated that 13% (6 of 47) BRCA1 alleles were fully methylated in the affected twin, whereas her sister displayed only single CpG errors without functional implications. This between-twin methylation difference was also found in irradiated fibroblasts and untreated saliva cells. The BRCA1 epimutation may have originated by an early somatic event in the affected twin: approximately 25% of her body cells derived from different embryonic cell lineages carry one epigenetically inactivated BRCA1 allele. This epimutation was associated with reduced basal protein levels and a higher induction of BRCA1 after DNA damage. In addition, we performed a genome-wide microarray analysis of both sisters and found several copy number variations, i.e., heterozygous deletion and reduced expression of the RSPO3 gene in the affected twin. This monozygotic twin pair represents an impressive example of epigenetic somatic mosaicism, suggesting a role for constitutive epimutations, maybe along with de novo genetic alterations in recurrent tumor development.
Keywords: BRCA1, childhood cancer, DNA Methylation, epimutation, monozygotic twins, secondary cancer, somatic mosaicism
Introduction
Cancer is rare among children throughout the world. In developed countries, only about 0.5% of all cancers occur among children under 15 y. In contrast to adult malignancies, where carcinomas predominate, childhood cancers are histologically very diverse. There are 12 major groups including leukemias, lymphomas, brain and spinal tumors, sympathetic nervous system tumors, retinoblastoma, kidney tumors, liver tumors, bone tumors, soft tissue sarcomas, gonadal and germ-cell tumors, epithelial tumors, and other unspecified neoplasms. The International Classification of Childhood Cancer is based on morphology and topography.1 Diagnostic groups are defined in the second edition of the International Classification of Diseases (ICD) for Oncology.
Cancer formation is generally considered as a genetic disease involving alterations in DNA structure ranging from gene mutations to gross chromosomal rearrangements. However, dysregulation of gene expression can also be acquired by epigenetic abnormalities, which are a hallmark of cancer evolution at all stages.2,3 In contrast to the rest of the genome where most CpGs are methylated, CpG islands in 5′ cis-regulatory regions of genes are usually unmethylated. Methylation of these CpG islands during development or disease processes is associated with gene silencing.4,5 Inactivation of tumor suppressor genes by promoter hypermethylation provides an important mechanism for tumor initiation and progression.
The genomic caretaker BRCA1 is necessary for faithful rejoining of broken DNA ends and one mutated BRCA1 allele may already be sufficient to impair this process. The compromised genomic stability in BRCA1 germline mutation carriers may trigger the genetic changes necessary for neoplastic transformation in hereditary breast cancer patients. Methylation of the BRCA1 promoter occurs in approximately 20% of sporadic breast cancers.6,7 Sporadic breast tumors with BRCA1 promoter methylation are mainly estrogen- and progesterone-receptor negative and display similar pathological features as hereditary tumors with BRCA1 mutations.8
The concordance rates in monozygotic (MZ) and dizygotic pairs of twins allow one to estimate the heritability of complex phenotypic traits. MZ twins arise from the same zygote, which then divides into two genetically identical embryos. MZ twins discordant for monogenic disorders are generally thought to represent rare examples of somatic mosaicism due to genetic mutations after the twinning event, which are then propagated to subsets of cells from one twin. Here we report on a pair of MZ twins discordant for recurrent tumor development. Our results suggest that post-zygotic epimutations are another source of somatic mosaicism leading to different disease susceptibility in MZ twins.
Results
Case report. The twin sisters were born in 1977 by spontaneous delivery, seven weeks before term. No intensive care or icterus treatment were necessary. Until 1982 the development of both twins was normal. At the age of 4 y and 8 mo one sister was diagnosed with precursor B-cell lymphoblastic leukemia (ICD10:C910). Chemotherapy had to be discontinued because of intolerability. A first relapse of the leukemia occurred in 1984. The second chemotherapy could be completed, but another relapse made bone marrow transplantation from her healthy twin sister necessary. Following bone marrow transplantation at age 7 y she was monitored until 1988. From 1985 until 1990 she received human growth hormone. In 2002 at the age of 25 y she was diagnosed with thyroid carcinoma (ICD10:C730). She underwent thyroidectomy and since then receives steroid hormone replacement. She was not treated with radiation or radioactive iodine. In 2006 a type 2 diabetes became manifest, but otherwise she is healthy. In 2009 she gave birth to a healthy daughter. When re-examined at age 34 y, the affected twin and her sister did not show any clinical manifestation of cancer. Neither family history nor clinical examination gave any hints for a DNA repair syndrome or another hereditary disease. Apart from the affected twin there were no other cases of cancer in four generations. Considering her medical history, it is not unexpected that the affected twin differed from her sister in some features, including height (156 cm vs. 168 cm) and occipitofrontal circumference (52 cm vs. 51 cm). Monozygosity was confirmed by genotyping short tandem repeats. Chromosome banding analysis (at the 500 band level) of fibroblast cultures revealed normal female karyotypes in both sisters.
Methylation analysis. Because MZ twins are genetically identical, epigenetic differences are one plausible explanation for discordant phenotypes. To test this hypothesis, we analyzed the methylation patterns of several representative tumor suppressor genes (ATM, BRCA1, BRCA2, MLH1, RAD51C, and TP53). One of the studied genes, BRCA1, showed constitutive promoter hypermethylation in normal body cells of the affected twin, but not of the healthy twin. Bisulfite pyrosequencing is a rapid and highly accurate method for epimutation screening. It can exactly (± 2%) quantify the methylation of individual CpG sites located in the 30–50 bp 3′ from the sequencing primer.9 Our pyrosequencing assay measures the methylation levels of five adjacent CpG sites in the BRCA1 5′ promoter. Because the density of methylated CpGs in a cis-regulatory region rather than individual CpGs turn a gene on or off,5,10 the average methylation of all analyzed CpGs (in two independent DNA samples) was used as an epigenetic marker for BRCA1 promoter methylation.
By bisulfite pyrosequencing of exponentially growing fibroblasts, the affected twin showed an increased BRCA1 methylation level of 12% (Fig. 1), compared with 3% in her healthy sister. Primary skin fibroblasts were used for epimutation screening, because they constitute a homogenous cell population with intact cell cycle and DNA repair checkpoints. Ten additionally analyzed two-cancer patients, who survived a childhood malignancy and then, unrelated to the first event, developed a secondary cancer, as well as 10 carefully matched one-cancer patients without a second malignancy all showed normal methylation levels (range 0–3%) in fibroblasts. Induction of DNA damage by γ-irradiation (1 Gy) of primary fibroblast cultures did not affect the BRCA1 methylation levels. In the affected twin, we found 10% methylation at 0 h, 10% at 1 h, 9% at 4 h, 11% at 12 h, and 10% at 24 h after irradiation. The control twin displayed 4%, 4%, 3%, 2%, and 4% methylation, respectively.
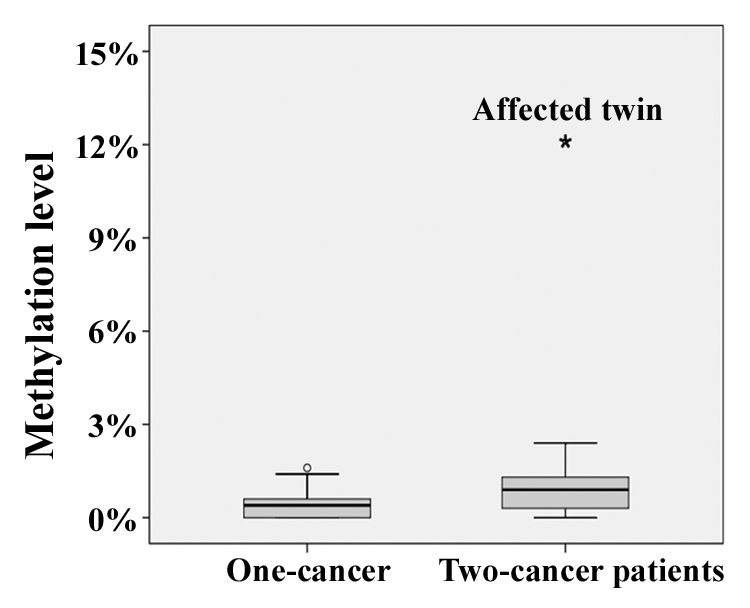
Figure 1. Box plots showing the distribution of BRCA1 methylation values in fibroblasts of 10 one-cancer and 10 two-cancer patients. The star symbol (extreme outlier) represents the affected twin. The median is represented by a horizontal line. The bottom of the box indicates the 25th percentile, the top the 75th percentile. The T bars extend from the boxes to 1.5 times the height of the box.
Lymphoblastoid cells of both the affected and the control twin exhibited equally low BRCA1 methylation levels (3% and 2%, respectively). However, because the affected twin was bone marrow-transplanted, her blood cells were also derived from the healthy twin’s stem cells. The normal range of BRCA1 methylation in blood cells of a large number (> 100) of controls was 0–5%. Similar to fibroblasts, saliva DNA of the affected twin showed a much higher BRCA1 methylation (9%) than that of her sister (2%). The normal methylation range in 10 saliva control samples was 2–4%. In addition to cells from buccal mucosa and salivary epithelium, saliva may also contain blood cells. This may explain the somewhat lower methylation level of saliva DNA compared with fibroblast DNA in the affected twin.
To distinguish between single CpG methylation errors, which are most likely stochastic errors without pathological consequences, and true epimutations (allele methylation errors), which can be expected to interfere with gene regulation, it is necessary to study the methylation patterns of individual DNA molecules. Classic bisulfite plasmid sequencing has the added advantage that it allows one to look at a larger number of CpG sites. Here we analyzed a BRCA1 amplicon with 13 CpG sites including the five sites targeted by the pyrosequencing assay (Fig. 2). Forty-seven clones (individual DNA molecules) were recovered from primary fibroblasts of each twin. Six (13%) clones of the affected twin exhibited epimutations, indicating that most (at least 70%) CpG sites on these DNA molecules were aberrantly methylated, typical for epigenetically silenced alleles. In contrast, all 47 alleles of the healthy twin displayed normal hypomethylated patterns. The BRCA1 epimutation rate was significantly (χ2 test; p = 0.01) higher in the affected twin, compared with her sister. Both sisters showed approximately 2% stochastic (single CpG) methylation errors: 10 of 531 CpG sites in the affected twin (excluding abnormal alleles) and 14 of 609 in the healthy twin were methylated. Collectively, our results suggest a constitutively increased methylation level at the BRCA1 promoter in normal body cells of the affected twin. Approximately 25% cells (skin fibroblasts, cells from buccal mucosa and salivary epithelium) derived from different embryonic lineages are endowed with one hypermethylated copy of the BRCA1 tumor suppressor gene, representing a first hit according to Knudson’s two-hit hypothesis of tumor development.

Figure 2. Methylation patterns of the BRCA1 promoter in fibroblasts of the affected twin and her healthy sister. Each line represents an individual allele (DNA molecule) analyzed by bisulfite plasmid sequencing. Filled circles indicate methylated CpG and open circles unmethylated CpG sites. Missing dots indicate CpG sites that could not be analyzed because of poor sequence quality. The five framed CpG sites correspond to those in the bisulfite pyrosequencing assay. Both twin sisters display alleles with single CpG (stochastic) methylation errors. Six of 47 analyzed alleles (indicated by arrows) in the affected twin represent epimutations with the majority of CpGs being aberrantly methylated, whereas all 47 alleles in the healthy twin are hypomethylated.
Because constitutional epimutations in different tumor suppressor genes have been linked to sequence variants in the 5′ cis-regulatory region,11-13 we sequenced a 3.4 kb upstream fragment including the BRCA1 promoter and exon 1. However, we did not find any genetic defect/sequence variant in the affected twin (data not shown).
In addition to BRCA1, we performed an epimutation screening by bisulfite pyrosequencing for several other tumor suppressor genes (ATM, BRCA2, MLH1, RAD51C, and TP53) in fibroblasts of the twin pair and the 20 one- or two-cancer patients. However, apart from the BRCA1 epimutation in the affected twin, all analyzed samples and promoters displayed normal (< 5%) methylation values (data not shown), indicating that constitutive epimutations in tumor suppressor genes are rare events in childhood-cancer patients.
Protein expression analysis. Customized antibody microarrays were used to compare the basal protein levels (without induction of DNA damage) of BRCA1 and several other genomic caretakers (ATM, BRCA2, MLH1, RAD51, and TP53) in exponentially growing primary fibroblasts of the affected twin vs. her healthy sister. Triplicate measurements (technical replicates) of the protein levels were performed on protein samples from three independent cell cultures each (biological replicates). The BRCA1 protein expression ratio between the affected and the healthy twin (z ratio normalized with log10 transformation and z scores) was 0.75 (Fig. 3A), consistent with constitutively reduced expression in the affected twin. ATM expression was slightly increased (1.25) in the affected twin, whereas ACTB (positive control), BRCA2, MLH1, RAD51, and TP53 all showed comparable protein levels in both twins. The BRCA1 protein levels were also quantified in fibroblast cells at 1 h and 4 h after 1 Gy γ-irradiation (Fig. 3B). In the healthy twin, the amount of BRCA1 protein increased 1.8-fold at 1 h after irradiation, compared with untreated cells, and reached basal levels at 4 h (the z ratio of treated vs. untreated cells was 0.9). In the affected twin, BRCA1 protein expression was induced 2.7-fold at 1 h and also back to basal levels (z ratio of 1.1) at 4 h.

Figure 3. (A) Constitutive protein expression of ACTB (control), ATM, BRCA1, BRCA2, MLH1, RAD51, and TP53 in untreated fibroblasts of the affected vs. the healthy twin. Relative protein expression (z ratio normalized by log10 transformation and z scores) was measured by antibody microarrays using three biological replicates. Note the decreased BRCA1 level (75% expression ratio) in the affected twin. (B) Differential induction of BRCA1 protein in fibroblasts of the healthy twin (gray bars) and the affected twin (black bars) at 1 h and 4 h after 1 Gy γ-irradiation. Relative protein expression in treated vs. untreated cells of each twin was measured by antibody microarrays using three biological replicates.
Molecular karyotype analysis. Microarray analysis revealed a 181 kb heterozygous deletion containing the RSPO3 gene on chromosome 6q22.33 and a 90 kb heterozygous deletion containing the open reading frame C5orf13 on chromosome 5q22.1 in primary fibroblasts of the affected twin. We did not see similar deletions in > 40 additionally studied childhood cancer patients and 100 normal healthy individuals (from the Gutenberg Heart Study14). We did not find evidence for a microdeletion or duplication affecting the BRCA1 cis-regulatory region in the affected twin. By quantitative real-time PCR (qPCR) we confirmed the presence of the heterozygous RSPO3 and C5orf13 deletions in a mosaic state (Fig. 4A). All qPCR experiments were performed on three independent DNA samples (biological replicates) of the affected twin and a mixture of control DNAs, respectively. Using RFC3 as reference for a normal diploid gene (with two copies), qPCR revealed a copy number of two for both RSPO3 and C5orf13 in the controls. In contrast, the affected twin displayed copy numbers around 1.5, indicating loss of one RSPO3 and one C5orf13 copy in approximately 50% of cells. This does not necessarily imply that both genes are deleted in the same cells. Expression analysis with GeneAtlas microarrays using four independent RNA samples (from different cell cultures of the affected and the healthy twin) demonstrated reduced (approximately 50%) RSPO3 mRNA levels in fibroblasts of the affected twin (Fig. 4B), whereas C5orf13 levels were comparable in both twins (data not shown).

Figure 4. (A) C5orf13 and RSPO3 copy numbers in fibroblast cells of the two-cancer twin, compared with healthy controls, as determined by qPCR (using the RFC3 gene copy number as reference). Standard deviations represent three independent DNA samples (biological replicates) of the affected twin and a mixture of control DNAs. (B) RSPO3 mRNA expression ratio between the affected twin and her healthy sister, as determined by GeneAtlas microarrays. Standard deviations represent RNA samples from four different cell cultures of the affected and the healthy twin, respectively.
Discussion
Traditionally, phenotypic discordances between MZ twin pairs have been attributed to environmental factors; however, accumulating experimental evidence also suggests a role for genetic and, more importantly, epigenetic differences between co-twins. Microarray-based karyotype analyses revealed somatic mosaicism for copy number variations (CNVs) in a number (estimated 10%) of both phenotypically discordant (for Parkinson disease, parkinsonism or Lewy body dementia) and concordant MZ twin pairs.15 Other molecular karyotype and genome sequence studies could not detect any CNV differences between MZ twins that were discordant for cleft lip palate16 or multiple sclerosis,17 suggesting that post-zygotic genomic alterations are at least not a common cause of phenotypic discordance. Several recent studies clearly demonstrated the existence of genome-wide epigenetic differences between MZ twins.18,19 Spontaneous epimutations may occur 10–100 times more frequently than somatic DNA mutations.20,21 This spontaneous epimutation rate can be modified by environmental factors, providing a link between lifestyle and phenotypic differences.22-24 In this light, stochastic and/or environmentally induced epimutations represent a major source of phenotypic variation and discordance for complex diseases between MZ twins. The accumulation of epigenetic differences may also explain the phenotypic divergence of MZ twins, as they age.18
Here we studied a MZ twin pair discordant for childhood cancer and a second primary cancer. Microarray analysis and qPCR revealed mosaic heterozygous deletions on chromosome 5q22.1 (C5orf13) and 6q22.33 (RSPO3) in the affected twin. C5orf13 encodes an 8-kDa intracytoplasmic protein initially identified in neurons and muscles.25 It contains a conserved PEST domain, which is important for targeting proteins for degradation by the ubiquitin/proteasome. R-Spondin family members are modulators of the Wnt/β-catenin signaling pathway which plays a crucial role in cell growth, development and disease pathogenesis.26 RSPO3 is frequently upregulated in human breast cancers and correlated with several tumor parameters, suggesting that it can function as a breast cancer oncogene.27 However, RSPO3 was downregulated in normal fibroblasts of the affected twin. Although C5orf13 and RSPO3 haploinsufficiency may contribute to the discordant cancer phenotype, they are unlikely to be the primary cause.
We propose that epigenetic changes, specifically hypermethylation of one BRCA1 allele constituted the first hit leading to inactivation of an important tumor suppressor gene and tumor initiation. Germline mutations in BRCA1 are associated with hereditary breast cancer, ovarian cancer and some other malignancies; however, so far they are generally not considered as a risk factor for childhood cancer.28 Our index patient developed precursor B-cell lymphoblastic leukemia at the age of 4 y and 8 mo. There are only few data linking BRCA1 to hematologic malignancies. In chronic myeloid leukemia, expression of the BCR-ABL fusion protein leads to posttranscriptional downregulation of BRCA1.29 One study reported partial hypermethylation and reduced expression of BRCA1 in primary and therapy-related acute myeloid leukemia,30 whereas others did not find abnormal BRCA1 methylation in leukemia samples.7,31
In the MZ twin pair studied here, only one sister exhibited a BRCA1 epimutation in a mosaic state in multiple tissues/cell types, whereas the unaffected twin did not. This epimutation most likely originated by a methylation error during early embryogenesis after the twinning event. It affects a substantial subset of cells derived from different embryonic cell lineages, resulting in somatic mosaicism. Genome-wide demethylation waves in the early embryo erase most germline methylation patterns, followed by de novo methylation and establishment of somatic methylation patterns.32 Most constitutive epimutations may occur during this vulnerable time window, where epigenetic genome reprogramming occurs, and represent stochastic and/or environmentally induced errors in the establishment or maintenance of an epigenetic state.33,34 Similar to our case, discordant methylation patterns at the KCNQ1OT1 locus (imprinting control region 2 on chromosome 11p15) have been described in a number of MZ twin pairs discordant for Beckwith-Wiedemann syndrome, which is characterized by tumor predisposition and multiple congenital abnormalities.35 Constitutive H19 epimutations (imprinting control region 1 on chromosome 11p12) were found in several patients with sporadic Wilms tumor without features of Beckwith-Wiedemann syndrome.36
Somatic gene mutations and mosaicism (with or without involvement of the germline) are an important etiological mechanism for monogenic disorders with high de novo mutations rates such as Duchenne muscular dystrophy, neurofibromatosis type 1 and retinoblastoma.37-39 In contrast, constitutive epimutations, characterized by soma-wide methylation abnormalities in functionally relevant cis-regulatory regions of disease genes, are an understudied phenomenon. Constitutive epimutations in the DNA mismatch repair gene MLH1 have been identified in a small number of mutation-negative cases of hereditary non-polyposis colorectal cancer (HNPCC).40 In rare familial cases with dominant transmission of mosaic MLH1 methylation, the constitutional epimutation was linked to a single nucleotide variant in the 5′ UTR of MLH1.13 Similarly, constitutional epimutations in the DNA mismatch repair gene MSH2, another rare cause of HNPCC, were linked to 3′ end deletions of EPCAM, a gene directly upstream of MSH2.12 Constitutive epimutations in the DAPK1 gene that predispose to B cell chronic lymphocytic leukemia are caused by a point mutation upstream of the DAPK1 promoter.11 Neither sequencing nor microarray analysis of the 5′ cis-regulatory region of BRCA1 revealed any detectable genetic differences between the discordant MZ twin pair studied here. Although we cannot exclude a causative genetic defect elsewhere, our findings are consistent with the view that the affected twin exhibits a true BRCA1 epimutation.
Our study revealed the existence of a constitutive BRCA1 epimutation only in the affected sibling of a MZ twin pair. Compared with her healthy sister, the two-cancer twin showed reduced basal BRCA1 protein levels (in untreated fibroblasts) and a higher induction of protein expression by DNA damage. Upregulation of BRCA1 in irradiated cells of both the affected and the healthy twin was not mediated by demethylation of the BRCA1 promoter. In the affected twin the percentage of aberrantly methylated alleles remained constant (at about 10%) after DNA damage and in the healthy twin the BRCA1 promoter was already completely demethylated in untreated cells. It is well known that the methylation of CpGs in 5′ promoters that are usually protected from methylation in somatic tissues can suppress gene expression during development, differentiation and disease processes.4,5 However, other mechanisms, i.e., gene body methylation, may be more important for regulating the cell-context specific efficiency of transcription.41 The basal ATM protein levels were also increased in the affected twin, although both twins showed equally low promoter methylation levels (around 1%) of the ATM gene. We speculate that ATM is constitutively upregulated by a compensation mechanism to counterbalance the reduced BRCA1 levels. ATM mediates the cellular DNA damage response. BRCA1 is regulated by an ATM-dependent mechanism and, on the other hand, essential for the recruitment of previously activated ATM to the sites of DNA damage.42 Although we cannot exclude the formal possibility of a chance coincidence or a late (i.e., therapy-related) somatic event, it is plausible to assume that epigenetic silencing of one copy of the BRCA1 tumor suppressor gene in 25% body cells and differences in the DNA damage response contributed to the discordant cancer phenotype in the affected twin. Preliminary evidence suggests that BRCA1 promoter hypermethylation can also be found in blood cells of a subset of breast cancer patients.43,44 Collectively, these results suggest that constitutive BRCA1 epimutations occur in normal tissues and may be associated with an increased cancer risk. Our pyrosequencing assay may prove useful for high-throughput screening of larger patient populations.
The DNA methylation analysis performed in this study was limited to a number of key tumor suppressor genes. Considering the enormous epigenetic variability due to stochastic methylation errors or extrinsic variations during post-zygotic genome reprogramming,20,21,34,45 we cannot rule out the possibility that epimutations in the affected twin in genes other than those studied are responsible for the discordant phenotype. Nevertheless, 10% soma-wide promoter methylation in an essential tumor suppressor gene such as BRCA1 can be expected to contribute to a tumor susceptibility phenotype.
Materials and Methods
Patients and cell substrates. The study was approved by the Ethics Committee of the Medical Association of Rhineland-Palatinate [(Nr. 837.440.03(4102)]. With the help of the German Childhood Cancer Registry, we recruited a discordant MZ twin pair. One twin suffered from childhood leukemia and later on thyroid carcinoma, whereas her twin sister was completely healthy (without malignancy). In addition, we recruited 10 two-cancer patients as well as 10 carefully matched one-cancer patients (same sex, same primary cancer, equal age at first diagnosis) who did not develop a second malignancy.
Primary fibroblasts from skin biopsies were cultured in minimal essential medium with Earle’s salts (Invitrogen, Karlsruhe, Germany), supplemented with 10% fetal bovine serum, vitamins and antibiotics. The cell cultures of the twin sisters were always passaged in parallel and harvested in a subconfluent stage. To induce DNA damage, subconfluent cultures were γ-irradiated with a dose of 1 Gy using a GammaCell 2000 (Cs137) irradiator. Cells were harvested at 1 h, 4 h, 12 h, and 24 h after irradiation. Lymphoblastoid cell lines were established by Epstein-Barr virus transformation of peripheral blood lymphocytes.
DNA sequence analysis. For Sanger sequencing of the 5′ UTR and exon 1 of the BRCA1 gene, a 3.4 kb fragment on chromosome 17 (41.277.112–41.280.530 bps, Ensembl release 64) was divided into 11 amplicons (Table S1). For amplicons 1–5 and 8, the PCR reaction mixture (25 μl) consisted of 2.5 μl 10x PCR buffer, 20 mM MgCl2, 0.5 μl 10 mM dNTP mix, 1 μl (10 pmol) of each forward and reverse primer, 0.2 μl (1 U) FastStart Taq DNA Polymerase (Roche Diagnostics, Mannheim, Germany), 18.8 μl PCR-grade water, and 1 μl (~100 ng) template DNA. For amplicons 6, 7, 8, 10, and 11, the PCR mixture (25 μl) contained 25 mM ammonium sulfate, 750 mM TRIS-HCl, 0.1% Tween 20, 240 µM dNTPs, 2.4 mM magnesium sulfate, 2.4x PCRX Enhancer Solution (Invitrogen), 0.4 µM of each primer, 1 Unit of Platinum Taq (Invitrogen), and 1 μl (~100 ng) template DNA. All PCR amplifications were performed with an initial denaturation step at 95°C for 5 min, 40 cycles of 95°C for 30 sec, primer-specific annealing temperature (Table S1) for 30 sec, 72°C for 45 sec, and a final extension step at 72°C for 7 min. Sequencing of the resulting PCR products was done on an ABI 3130 x l automated sequencer.
Methylation analysis. Genomic DNA from fibroblasts and EBV-transformed lymphoblasts was extracted using the Qiagen Mini DNA kit (Qiagen, Hilden, Germany). Saliva DNA was extracted using the ORAGENE DNA extraction kit (DNA Genotek, Kanata, Ontario, Canada). Bisulfite conversion of genomic DNA was performed with the EpiTect Bisulfite kit (Qiagen).
Bisulfite pyrosequencing was performed on a PyroMarkTMQ96 MD Pyrosequencing System with the PyroMark Gold Q96 CDT Reagent kit (Qiagen). An assay quantifying the methylation levels of five representative CpG sites in the BRCA1 promoter was designed using the PyroMark Assay Design 2.0 software (Qiagen). A 232 bp amplicon (HSA17: 41,277,292–41,277,523 bps; Ensembl release 60) was PCR amplified from bisulfite-treated DNA using forward primer 5′-ATTTAGAGTAGAGGGTGAAGG-3′ and biotinylated reverse primer 5′-TCTATCCCTCCCATCCTCTAATT-3′. The reaction mixture consisted of 2.5 μl 10x PCR buffer, 20 mM MgCl2, 0.5 μl 10 mM dNTP mix, 1 μl (10 pmol) of each primer, 0.2 μl (1 U) FastStart Taq DNA Polymerase (Roche Diagnostics, Mannheim, Germany), 18.8 μl PCR-grade water, and 1 μl (~100 ng) template DNA. PCR amplifications were performed with an initial denaturation step at 95°C for 5 min, 35 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 45 sec, and a final extension step at 72°C for 5 min. Sequencing was performed with primer 5′-TTGAGAAATTTTATAGTTTGTTTT-3′. The Pyro Q-CpG software (Qiagen) was used for data analysis.
For classical bisulfite plasmid sequencing BRCA1 PCR products were cloned into pCR2.1-TOPO vector using T4-DNA ligase, the TA cloning kit and One Shot TOP10 chemically competent Escherichia coli (Invitrogen). Plasmid DNA of individual clones was isolated with the ZR Plasmid Miniprep Classic Kit (Zymo Research, Freiburg, Germany). Insert-containing clones were sequenced using dye terminator cycle sequencing with M13 primers on an ABI 3130xl automated sequencer (Life Technologies, Frankfurt, Germany). Sequences were analyzed with BiQ Analyzer software tool.46
Protein expression analyses. Cells were resuspended two times in 500 μl of 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, pH 7.9 and incubated on ice for 10 min. After 10 sec centrifugation at maximum speed the supernatant was discarded. The pellet was resuspended in 100 µl of 20 mM HEPES, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, pH 7.9 and homogenized. After incubation on ice for 30 min and centrifugation for 30 min at maximum speed at 4°C, the supernatant containing the nuclear protein extract was separated from the cytoplasmic pellet and stored at -80°C. The protein concentration was measured according to Bradford, using Roti Quant (Roth, Karlsruhe, Germany).
Customized antibody microarrays for quantification of BRCA1 (antibody #sc-1553, Santa Cruz Biotechnology, Heidelberg, Germany) and several other DNA repair-associated proteins (ATM, BRCA2, MLH1, RAD51, and TP53) were prepared by spotting triplicate drops, each containing approximately 0.5 pg antibody onto nitrocellulose-coated slides (Oncyte, nitrocellulose 16 multi-pad slides, Grace Bio-Labs, Bend, OR, USA), using a sciFLEXARRAYER 3 non-contact spotter (Scienion, Berlin, Germany). Antibodies against β-actin (ACTB) (#A5441, Sigma-Aldrich, Munich, Germany) served as positive, spotting buffer as negative control. Slides were stored at 4°C in dry condition. Nuclear proteins were labeled with an amine reactive fluorine dye, which forms a covalent amide bond between the primary amines of proteins. Two micrograms of protein and 0.12 μl fluorescent dye (Dylight 649 NHS Ester, Pierce, Rockford, USA) were incubated for 1 h at room temperature in the dark. Then, excess fluorescent dye was inactivated by adding 100 mM glycine to the reaction. Prior to use, antibody microarrays were covered with 16-pad FAST frame hybridization chambers (Whatman, Maidstone, UK). Unspecific binding sites were blocked for 1 h at 4°C with 120 μl PBS containing 4% non-fat dry milk per subarray, followed by three washes with 120 μl PBS each for 10 min. Labeled protein samples were incubated on subarrays overnight at 4°C. Afterwards, the slides were washed two times for 15 min with PBS, 5% Tween 20 and two times for 15 min with HPLC-grade water. Finally, the slides were dried in a SpeedVac and scanned with a high-resolution confocal Affymetrix array scanner 428 TM (Affymetrix, High Wycombe, UK). Slide images were analyzed using the TM4 Spotfinder (version 3.1.1) software (Dana Faber Cancer Institute, Boston, USA). Background subtraction was performed according to the formula: spot intensity = mean intensity SP - (sum bkg – sum top25bkg)/(number of pixelSP - number of pixel top25bkg), where SP represents any spot, bkg the corresponding background and top25bkg the top 25% of background pixel. Microarray data were analyzed using log10 transformation, z score and z ratio calculations.47 Three biological replicates (using protein samples from different cell cultures of the affected and the healthy twin) were performed for each experiment.
Molecular karyotype analysis. High-resolution screening for microdeletions and duplications was performed with the Affymetrix GeneChip Genome Wide Human SNP array 6.0 and the GeneChip Genome Wide SNP Sty Assay Kit 5.0/6.0, following the protocol developed by the manufacturer. Copy numbers were determined using the Affymetrix Genotyping Console 4.0 and Chromosome Analysis Suite 1.0.1.
Quantitative real-time PCR with the Universal Probe Library Set 04683633001 (Roche Diagnostics) was used to validate copy number changes of C5orf13 (UPL probe #24) and RSPO3 (#23). PCR was performed on an ABI 7500 Fast Real-Time PCR system (Life Technologies) with one cycle of 95°C for 10 min and 45 cycles of 95°C for 30 sec and 60°C for 60 sec. Copy number calculation was performed with the ∆∆-CT method, using the RFC3 (UPL probe #32) gene copy number as reference. All experiments were performed on three biological replicates.
Genome-wide expression analyses were performed with RNA samples from four fibroblast cell cultures of each twin using the GeneAtlas Personal Microarray System (Affymetrix, Santa Clara, CA, USA). Data were analyzed with the Affymetrix Expression Console program. Data were corrected for background noise and normalized with the RMA and Quantil algorithms.
Supplementary Material
Supplementary PDF file supplied by authors.
Acknowledgments
This work was supported by the Stiftung Rheinland-Pfalz für Innovation (project no. 698). First of all, we thank the patients whose participation made this study possible. We also thank Annalisa Tiozzo (Affymetrix), Tanja Zeller (Clinical Chemistry and Laboratory Medicine, University Medical Center Mainz), Niels Boehm and Franz Grus (Experimental Ophthalmology, Ocular Proteomics and Immunology Center, University Medical Center Mainz) for help with the DNA and antibody arrays, respectively.
Glossary
Abbreviations:
- CNV
copy number variation
- HNPCC
hereditary non-polyposis colorectal cancer
- ICD
international classification of diseases
- MZ
monozygotic
- qPCR
quantitative real-time PCR
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/18814
References
- 1.Kramárová E, Stiller CA. The international classification of childhood cancer. Int J Cancer. 1996;68:759–65. doi: 10.1002/(SICI)1097-0215(19961211)68:6<759::AID-IJC12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 2.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502–17. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 5.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–66. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 6.Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18:1957–65. doi: 10.1038/sj.onc.1202509. [DOI] [PubMed] [Google Scholar]
- 7.Bianco T, Chenevix-Trench G, Walsh DC, Cooper JE, Dobrovic A. Tumour-specific distribution of BRCA1 promoter region methylation supports a pathogenetic role in breast and ovarian cancer. Carcinogenesis. 2000;21:147–51. doi: 10.1093/carcin/21.2.147. [DOI] [PubMed] [Google Scholar]
- 8.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–9. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 9.Tost J, Dunker J, Gut IG. Analysis and quantification of multiple methylation variable positions in CpG islands by pyrosequencing. Biotechniques. 2003;35:152–6. doi: 10.2144/03351md02. [DOI] [PubMed] [Google Scholar]
- 10.Sontag LB, Lorincz MC, Georg Luebeck E. Dynamics, stability and inheritance of somatic DNA methylation imprints. J Theor Biol. 2006;242:890–9. doi: 10.1016/j.jtbi.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 11.Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129:879–90. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41:112–7. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 13.Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, et al. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5′UTR. Cancer Cell. 2011;20:200–13. doi: 10.1016/j.ccr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Wild PS, Sinning CR, Roth A, Wilde S, Schnabel RB, Lubos E, et al. Distribution and categorization of left ventricular measurements in the general population: results from the population-based Gutenberg Heart Study. Circ Cardiovasc Imaging. 2010;3:604–13. doi: 10.1161/CIRCIMAGING.109.911933. [DOI] [PubMed] [Google Scholar]
- 15.Bruder CE, Piotrowski A, Gijsbers AA, Andersson R, Erickson S, Diaz de Ståhl T, et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet. 2008;82:763–71. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimani JW, Yoshiura K, Shi M, Jugessur A, Moretti-Ferreira D, Christensen K, et al. Search for genomic alterations in monozygotic twins discordant for cleft lip and/or palate. Twin Res Hum Genet. 2009;12:462–8. doi: 10.1375/twin.12.5.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–6. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–5. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 20.Bennett-Baker PE, Wilkowski J, Burke DT. Age-associated activation of epigenetically repressed genes in the mouse. Genetics. 2003;165:2055–62. doi: 10.1093/genetics/165.4.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goyal R, Reinhardt R, Jeltsch A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res. 2006;34:1182–8. doi: 10.1093/nar/gkl002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutherland JE, Costa M. Epigenetics and the environment. Ann N Y Acad Sci. 2003;983:151–60. doi: 10.1111/j.1749-6632.2003.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 23.Whitelaw NC, Whitelaw E. How lifetimes shape epigenotype within and across generations. Hum Mol Genet. 2006;15:R131–7. doi: 10.1093/hmg/ddl200. [DOI] [PubMed] [Google Scholar]
- 24.Dolinoy DC, Jirtle RL. Environmental epigenomics in human health and disease. Environ Mol Mutagen. 2008;49:4–8. doi: 10.1002/em.20366. [DOI] [PubMed] [Google Scholar]
- 25.Studler JM, Glowinski J, Levi-Strauss M. An abundant mRNA of the embryonic brain persists at a high level in cerebellum, hippocampus and olfactory bulb during adulthood. Eur J Neurosci. 1993;5:614–23. doi: 10.1111/j.1460-9568.1993.tb00527.x. [DOI] [PubMed] [Google Scholar]
- 26.Kim KA, Zhao J, Andarmani S, Kakitani M, Oshima T, Binnerts ME, et al. R-Spondin proteins: a novel link to beta-catenin activation. Cell Cycle. 2006;5:23–6. doi: 10.4161/cc.5.1.2305. [DOI] [PubMed] [Google Scholar]
- 27.Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J, et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 2007;39:759–69. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- 28.Brooks GA, Stopfer JE, Erlichman J, Davidson R, Nathanson KL, Domchek SM. Childhood cancer in families with and without BRCA1 or BRCA2 mutations ascertained at a high-risk breast cancer clinic. Cancer Biol Ther. 2006;5:1098–102. doi: 10.4161/cbt.5.9.3167. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch E, Jarrousse S, Buet D, Dugray A, Bonnet ML, Vozenin-Brotons MC, et al. Downregulation of BRCA1 in BCR-ABL-expressing hematopoietic cells. Blood. 2003;101:4583–8. doi: 10.1182/blood-2002-10-3011. [DOI] [PubMed] [Google Scholar]
- 30.Scardocci A, Guidi F, D'Alo' F, Gumiero D, Fabiani E, Diruscio A, et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br J Cancer. 2006;95:1108–13. doi: 10.1038/sj.bjc.6603392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–9. [PubMed] [Google Scholar]
- 32.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 33.Haaf T. Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol. 2006;310:13–22. doi: 10.1007/3-540-31181-5_2. [DOI] [PubMed] [Google Scholar]
- 34.Schneider E, Pliushch G, El Hajj N, Galetzka D, Puhl A, Schorsch M, et al. Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic Acids Res. 2010;38:3880–90. doi: 10.1093/nar/gkq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, et al. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum Mol Genet. 2002;11:1317–25. doi: 10.1093/hmg/11.11.1317. [DOI] [PubMed] [Google Scholar]
- 36.Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet. 2008;40:1329–34. doi: 10.1038/ng.243. [DOI] [PubMed] [Google Scholar]
- 37.Lohmann DR, Gerick M, Brandt B, Oelschlager U, Lorenz B, Passarge E, et al. Constitutional RB1-gene mutations in patients with isolated unilateral retinoblastoma. Am J Hum Genet. 1997;61:282–94. doi: 10.1086/514845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–58. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 39.Erickson RP. Somatic gene mutation and human disease other than cancer. Mutat Res. 2003;543:125–36. doi: 10.1016/S1383-5742(03)00010-3. [DOI] [PubMed] [Google Scholar]
- 40.Hitchins MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet. 2009;46:793–802. doi: 10.1136/jmg.2009.068122. [DOI] [PubMed] [Google Scholar]
- 41.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol. 2005;70:99–109. doi: 10.1101/sqb.2005.70.002. [DOI] [PubMed] [Google Scholar]
- 43.Snell C, Krypuy M, Wong EM. kConFab investigators, Loughrey MB, Dobrovic A. BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res. 2008;10:R12. doi: 10.1186/bcr1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Moghrabi N, Al-Qasem AJ, Aboussekhra A. Methylation-related mutations in the BRCA1 promoter in peripheral blood cells from cancer-free women. Int J Oncol. 2011;39:129–35. doi: 10.3892/ijo.2011.1021. [DOI] [PubMed] [Google Scholar]
- 45.El Hajj N, Trapphoff T, Linke M, May A, Hansmann T, Kuhtz J, et al. Limiting dilution bisulfite (pyro)sequencing reveals parent-specific methylation patterns in single oocytes and early embryos. Epigenetics. 2011;6:1176–88. doi: 10.4161/epi.6.10.17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067–8. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- 47.Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary PDF file supplied by authors.