

Abstract
Laser-coupled microphotoreactors were developed to bubble singlet oxygen [1O2 (1Δg)] into an aqueous solution containing an oxidizable compound. The reactors consisted of custom-modified SMA fiber-optic receptacles loaded with 150-μm silicon phthalocyanine glass sensitizer particles, where the particles were isolated from direct contact with water by a membrane adhesively bonded to the bottom of each device. A tube fed O2 gas to the reactor chambers. In the presence of O2, singlet oxygen was generated by illuminating the sensitizer particles with 669-nm light from an optical fiber coupled to the top of the reactor. The generated 1O2 was transported through the membrane by the O2 stream and formed bubbles in solution. In solution, singlet oxygen reacted with probe compounds (either 9,10-anthracene dipropionate dianion, trans-2-methyl-2-pentanoate anion, N-benzoyl-D,L-methionine, and N-acetyl-D,L-methionine) to give oxidized products in two stages. The early stage was rapid and showed that 1O2 transfer occurred via bubbles mainly in the bulk water solution. The later stage was slow, it arose only from 1O2-probe molecule contact at the gas/liquid interface. A mechanism is proposed that involves 1O2 mass transfer and solvation, where smaller bubbles provide better penetration of 1O2 into the flowing stream due to higher surface-to-volume contact between the probe molecules and 1O2.
Introduction
Our interest in developing a singlet oxygen [1O2 (1Δg)]-sparging reactor came from small-scale devices for disinfection of, for example, municipal and well water, but which used filtration, ozone and/or UV light.1,2 Low-cost water purification inventions that use visible light to generate 1O2 could be advantageous over ozone by using photocatalysts with high turnovers and over 4 decades' study of organic photooxidation product formation.3,4 Photophysical information has been generated using visible light for the photosensitized disinfection of water samples or stagnating wounds,5-8 but thus far, it is difficult to translate this information to handheld devices to deliver 1O2 as a biological toxin via bubbles at the gas-liquid interface.
We9-12 and others13 have reported the 1O2 production from hollow-tube configured devices. Our previous results established a singlet oxygen sensitization process with silica end-capped hollow-core fiber optic devices, utilizing the released 1O2 for Escherichia coli inactivation10 in a slow sparging system (9 ppm/h O2). Eisenberg et al.13 reported on a Pyrex tube bound-Rose Bengal photosensitizer, surrounded by lamps, rapidly flowing 3O2, 1O2 and N2 (30 L/min) in a gas-solid system. But unlike these previous systems, our desire was to produce singlet oxygen in a device that does not expose the photosensitizer to the water being purified. Since sensitizer molecules, themselves, may pose health risks, a means to isolate the sensitizer molecules from water was desired for water purification and/or applications where the device would come in contact with bodily fluids (e.g., surgery for cleansing and disinfecting wounds8).
One approach to increasing the rate of singlet oxygen production is using chemical oxygen-iodine lasers (COIL).14 These can produce gaseous 1O2 bubbles up to supersonic speeds. COIL is not catalytic, but the ratio of 1O2 to total oxygen concentrations is high, 30-50%, based on 2,5-dimethylfuran trapping studies.15 However, this approach is problematic as alkaline perhydroxyl ion (HO2-) and chlorine gas are required in high concentrations, several moles per liter of the former, and a few kPa pressure of the latter forming HCl as a by-product.
As part of an ongoing study of hand-held singlet oxygen 1O2-generating devices,9-12 we report here on a 1O2 sparging device which used photosensitized phthalocyanine particles isolated from bulk water by a hydrophobic micro-porous membrane. Figure 1 shows a cross-sectional schematic image of the device (3 versions of which were constructed). Singlet oxygen was generated in the photoreactor and flowed through the membrane into the surrounding aqueous solution where it was detected, trapped and analyzed. The sensitizer particles remain dry as the capillary pressure resulting from the submicron pores prevents water from diffusing through the membrane. Specifically, this paper describes: (1) the use of Si phthalocyanine, axially functionalized via a sol-gel process as a heterogeneous photosensitizer; (2) device construction including membrane selection and attachment to a flexible optical fiber; (3) performance of the device to photooxidize probe compounds in water and the effects of bubble sizes; and (4) a proposed gas-liquid photooxidation mechanism via O2 bubbles with mass transfer limitations.
Figure 1.
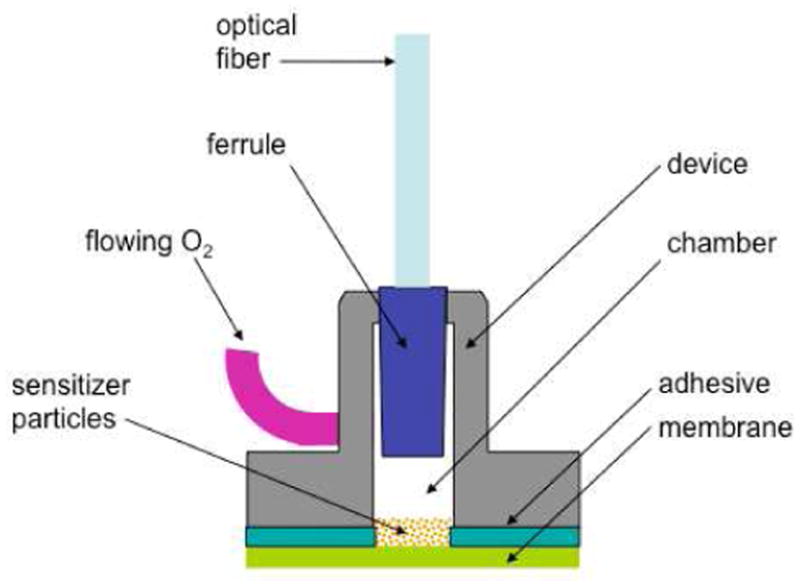
Geometry of the 1O2-sparging photogenerator. At the top is the optical fiber leading from the diode laser, and at the left is the O2 feed tube, connected to an oxygen gas tank. The lower part of the device, which contains a chamber for stockpiled silicon phthalocyanine sensitizer particles, was sealed with a microporous membrane.
Experimental
Reagents, Materials, and Instrumentation
Silicon phthalocyanine dichloride (SiPcCl2), 3-aminopropyltriethoxysilane (ATPS), 3-glycidyloxypropyl-trimethoxysilane (GPTMS), 9,10-anthracene dipropionic acid, trans-2-methyl-2-pentenoic acid, N-benzoyl-D,L-methionine, N-acetyl-D,L-methionine, sodium hydroxide, hydrochloric acid, ethanol, methanol, deuterium oxide-d2, chloroform-d1 were purchased from Sigma Aldrich (Allentown, PA). Deionized water was purified using a U.S. Filter Corporation deionization system (Vineland, NJ). All of the above materials and chemicals were used as received without further purification. The membranes were manufactured from ultra-high molecular weight polyethylene (UHMWPE) and are composed of fibrils linked together to form a membrane of interpenetrating pores with a nominal pore area of 85% for each membrane (Millipore SureVent UPE Membranes, Billerica, MA). For the D2O samples, proton NMR spectra were recorded at 400 MHz on Bruker DPX400 instrument. UV-VIS spectra were collected on a Hitachi UV-Vis U-2001 spectrophotometer.
Synthesis
The addition of SiPcCl2 (5.1 × 10-4 M) to APTS (0.178 M) was conducted with stirring for 50 h at 120 °C, yielding an SiPc-APTS complex. The addition of GPTMS to the SiPc-APTS complex was carried out in acidic aqueous ethanol at 60 °C for 1 h; the temperature was then adjusted to 25 °C for 72 h, followed by drying at 50 °C for 10 h. The concentration of Pc within the gel corresponded to ∼5.2 × 10-6 M based on UV-VIS spectroscopy.
Devices and Procedure for Photooxidations
Optical energy was delivered from a CW diode laser (669-nm output, 506 mW, model 7404, Intense Ltd., North Brunswick, NJ, USA) or a Minilase 10-Hz Nd:YAG Q-switched laser (355-nm, ∼4 ns fwhm, 1-3 mJ/pulse, New Wave Research, Fremont, CA) into a stainless steel multimode FT-400-EMT optical fiber with an SMA 905 connector (numerical aperture 0.39; 0.4 μm core diameter × 3 ft length, Thorlabs, Newton NJ). Ground Pc sensitizer particles were placed into the SMA receptacle chambers. The diode laser was used for the steady-state experiments with 2-5 (0.05 to 40 mM). The Nd:YAG laser was used for the lifetime measurements of singlet oxygen; it was connected to the optical fiber via a free-space PAF-SMA-5-A fiber port applicator (f=4.6 mm). All experiments were conducted with the devices placed into 3.0 mL solutions of H2O or D2O and oxygen flowed through the devices and into the solutions during the irradiation of the samples. An H10330A-45 photomultiplier tube (Hamamatsu Corp., Hamamatsu City, Japan) was used operating at -650 V. In front of the A10449 mechanical shutter of the detector was placed either a 25 mm diameter 1150-nm long pass filter (FEL1150, Thorlabs Inc.) or one of three 25-mm diameter NIR bandpass filters centered at 1220, 1270, and 1315 nm (OD4 blocking, FWHM = 15 nm, Omega, Brattleboro, VT). In D2O, the 1O2 luminescence intensity was measured to be 0.078 with the 1150-nm long pass filter, and 0.005, 0.08, and ∼0 mV with the 1220, 1270, and 1315 nm bandpass filters, respectively; subtractions of the signals was not performed. Singlet oxygen was monitored based on the spectra consisting of ∼1 million data points registered on a 600 MHz 62MXs-B oscilloscope (LeCroy, Chestnut Ridge, NY). The singlet oxygen decay lifetime was determined by nonlinear least squares curve-fitting with the equation: luminescence1270 (t) = A × [exp-(t/τ)], where 1/kobs = τ(1O2) lifetime. The data processing was performed with Microsoft Excel (version 12.3.1). The radiant power of the 355-nm and 669-nm light exiting the fiber or devices 1-3 was measured with a Newport power meter model 1918-C. Some of the laser light encountered the bubbles and was scattered. The bonded membranes were susceptible to aging after prolonged exposure times (e.g., >100 h with device 1 loaded with 35 mg sensitizer particles) led to increased membrane elasticity and increased laser power output measured outside of the membrane by ∼10% from 0.098 to 0.11. Careful inspection of the water samples after photolysis showed that no sensitizer particles had escaped the device so that the observed photooxidation could not be due to sensitizer particles within the water. Gas flowed from a compressed oxygen gas tank through a regulator, and subsequently a mass flow controller (GFC-17, Aalborg, Orangeburg, NY). The concentration of O2 in water was measured with a pO2 Sens-Ion6 oxygen electrode (Hach Co., Loveland, CO).
Results and Discussion
Photosensitizer Synthesis
It was desirable to use a heterogeneous sensitizer with a strong absorption in the 670-nm region to match the 669-nm output of our diode laser. Si phthalocyanine (Pc) was selected because it possessed a strong absorption in the red spectral region (extinction coefficients >105 M-1 cm-1), and the 1O2 quantum yield (ΦΔ) was reported to be ∼0.2.16,17
Composite (Pc 1) was prepared by a sol-gel process using a previously described procedure except with relatively low concentrations of Pc.18 Silicon Pc dichloride (SiPcCl2) reacted with 3-aminopropyltriethoxysilane [(NH2(CH2)3Si(OC2H5)3, APTS] in a 1:350 molar ratio at 120 °C producing SiPc[NH(CH2)3Si(OC2H5)3]2, which reacted with 3-glycidyloxypropyl-trimethoxysilane (GPTMS) to produce Pc 1, which contained an assortment of bonds cross-linked, such as Si-O-Si bonds from condensation, and polyether chains and dioxane rings via epoxide ring opening reactions.19 Drying of the composite was done at 50 °C for 10 h to avoid destruction of the confined phthalocyanine molecules, producing an aerogel that shrunk ∼10% where some, but not all adsorbed water was removed. Complete dehydration occurs between 100 and 180 °C.20 Low final Pc concentrations in the glass (∼5.2 × 10-6 M) were targeted because dye overloading or crowding can lower 1O2 yields.21,22 Pc 1 was ground and sieved to obtain 150±30 μm sized particles. The surface area of each 150-μm Pc 1 sensitizer particle was approximately 0.06971 mm2, based on the calculations of Skidmore and Powers,23 assuming a spherical non-porous surface. Spectroscopically, Pc 1 contained the desired 670-nm Q-band for overlap with the diode laser excitation wavelength and the lack of a redshifted absorption expected of monomeric Pc in the glassy matrix.
Device Construction
Devices were constructed to isolate the solid Pc 1 sensitizer particles from the surrounding water solution with an “internal” supply of light and flowing O2. A chamber within each device functioned as a reactor for the sensitizer particles, light, and O2, to generate 1O2.
Figure S4 (Supporting Information) shows the loading of device 2 with sensitizer particles, as well as the three devices without the optical fibers attached. Each device was fabricated from a chrome plated brass SMA receptacle with a SMA connector at one end of a cylindrical chamber (Amphenol). The dimensions of the chamber, and other device details, are listed in Table S1 (Supporting Information). Because device 3 was larger, the mg of Pc 1 particles that could be loaded into it was 740 mg, whereas devices 1 and 2 could only hold 75 mg. Table S2 shows the estimated total surface area of the particles and the number of particles that can be loaded into the devices. The term surface area refers only to the exterior surface area of the particle and does not consider internal pores. It is known that sol-gel glasses can be highly porous.24 The diode laser was connected by attaching the fiber SMA fitting to the device. The divergence angle of the red light exiting the fiber was not matched to the membrane area. The opposite, open end was sealed with the porous membrane. A hole was drilled into the cavity and a brass tube, 1/16″ o.d., was soldered in place to introduce the oxygen feed gas supply between the laser and the sensitizer.
Devices were fabricated with membranes of different pore sizes and thicknesses (Millipore). The membranes were manufactured from ultra-high molecular weight polyethylene (UHMWPE) and are composed of fibrils linked together to form a membrane of interpenetrating pores with a nominal pore area of 85% for each membrane. UHMWPE is biocompatible and used extensively for medical implants. The membranes were adhesively bonded to the bottom surface of the receptacle using a 3M pressure sensitive tape coated on both surfaces with a high bond strength adhesive. The pressure sensitive tape was die cut to form a ∼5 mm hole to allow the sensitizer to sit directly onto the membrane.
To insure that liquid does not penetrate the membrane and interact with the sensitizer, the membranes were selected such that the capillary pressure was sufficiently high to exclude water. The capillary pressure was calculated from the Young-Laplace equation:
| (1) |
where pc is the capillary pressure, γ is the liquid surface tension, θ is the contact angle between the liquid and the membrane material, and r is the pore radius. For water and UHMWPE, the values of γ and θ are 72 dynes/cm and 105° respectively. Thus the capillary pressure will be inversely proportional to the pore radius; the larger the radius the lower the pressure. Table S1 shows that decreasing the diameter of the pores in the membrane increased the capillary pressure and so keeps water from infiltrating the membrane at higher pressures. For example, the capillary pressure of a 0.44 μm pore is 12 PSI, whereas the capillary pressure of a 0.05 μm pore is 108 PSI. Thus, the device with a 0.44 μm pore membrane could be submerged to a depth of ∼8 meters of water before water ingress could occur, whereas the 0.05 μm pore membrane would prevent the sensitizer from leaching into solution at depths over 75 meters of water. In our experiments, no leaching of sensitizer was observed with any device, regardless of membrane pore size (see Experimental Section). For the membranes studied, the capillary pressure values range from 75 × 104 to 8.5 × 104 Pa (108-12 psi, as noted above). Thinner membranes with smaller pore diameters are also advantageous as the reduced thickness shortens the path over which 1O2 must diffuse before contacting water or being detected. However, the thinner membranes are somewhat fragile and may create a greater pressure drop for gas flow. Thicker membranes with larger pores are more robust.
Device Operation
The effect of membrane pore size, and of sensitizer particle loading, on the size of bubbles exiting the devices is shown in Table 1. The Pc particles tended to pool in the center of the membrane where it bulged from the O2 pressure. Individual bubbles ranged in diameter from 2 to 10 mm, where their sizes decreased with smaller membrane pores, in the order 0.05 μm < 0.22 μm < 0.44 μm. Higher loadings of sensitizer particles in the devices also led to smaller bubbles. Table 2 shows the volume and number of bubbles transmitted per experiment. Bubbles were mostly cylindrical and monodisperse, although some bubble clustering occurred, the bubble coalescence behavior at the membrane/water interface was not scrutinized. The 0.091, 0.14, and 0.46-mL bubbles that emerged from devices 1, 2, and 3, respectively provided agitation to the solution (Figure 2).
Table 1. Bubble Sizes Egressing into Aqueous Solution, and Power Measurements.
| Device number | Membrane pore size (μm) | Quantity of Pc 1 loaded into devices (mg) | Bubble diameter (mm)a,b | Power (mW) measured outside of the membranec |
|---|---|---|---|---|
| 0 | ∼ 5 | 3.5 | ||
| 1 | ∼4.8 | 2.9 | ||
| 3 | 4.2±0.8 | 2.5 | ||
| 1 | 0.05 | 10 | 3.6±0.5 | 0.15 |
| 35 | 2.8±0.4 | 0.098 | ||
| 50 | 2.8±0.4 | 0.0076 | ||
| 75 | ∼2 | 0.0085 | ||
| 0 | ∼5 | 2.5 | ||
| 1 | ∼5 | 2.0 | ||
| 3 | ∼4 | 1.5 | ||
| 2 | 0.22 | 10 | 3.4±0.5 | 0.76 |
| 35 | 3.2±0.4 | 0.16 | ||
| 50 | ∼3 | 0.0075 | ||
| 75 | ∼2 | 0.0060 | ||
| 0 | ∼10 | 5.0 | ||
| 1 | 7.6±1.5 | 3.0 | ||
| 3 | 0.44 | 3 | 7.4±1.8 | 2.7 |
| 10 | 5.8±1.1 | 1.5 | ||
| 35 | 4.8±1.3 | 0.14 | ||
| 50 | 4.6±0.9 | 0.0089 | ||
| 75 | 4.2±0.8 | 0.010 | ||
The bubble sizes effusing through the device membranes were measured from photographic images with ruler reference points and/or pixel size correlations. The values shown here are averages of 2 or more measurements.
The experiments were carried out flowing O2 at a rate of 60 mL/min with a regulator pressure of 35 PSI and a ∼2 mm height of water above the membrane.
The output of the diode laser (669 nm, 506 mW) was coupled to the fiber optic, where 383-mW laser light exited the fiber optic and entered the top portion of the devices at the fiber optic/SMA junction.
Table 2. Effect of Membrane Pore Size on Bubble Volume and Number Transmitteda.
| Device number | Sensitizer 1 loaded into device (mg) | Membrane pore size (μm) | Bubble volume (mL) | Number of bubbles transmittedb |
|---|---|---|---|---|
| 1 | 35 | 0.05 | 0.091 | 98,900 |
| 2 | 35 | 0.22 | 0.14 | 65,700 |
| 3 | 35 | 0.44 | 0.46 | 19,400 |
Devices were loaded with 0.35 mg Pc 1; O2 flow rate was 60 mL/min; solution was 3 mL D2O.
Over the course of a 2.5 h experiment, 9 L of O2 was consumed.
Figure 2.

(A) Device 1, with smaller pores than the other two devices, is seen here. It shows 1O2 bubbling from the distal end of the device. The membrane bulges due to oxygen pressure during the irradiation. The side and bottom were covered with the 0.05 μm membrane to improve adhesion. (B) Device 3 attached to the 0.44 μm membrane. At the bottom relatively larger O2 bubbles can be seen emerging from the membrane. The larger bubbles result in smaller surface-to-volume ratios and limited 1O2 contact, which may explain why this device was less efficient in oxidizing compounds in the surrounding water solution.
With the diode laser turned off, no apparent cooling of the aqueous solution occurred from the devices sparging O2 at a rate of 60 mL/min. In contrast, with the diode laser turned on, the solution temperature increased from 22 °C to 28 °C. We measured only a very small light output through the membrane (0.006-0.010 mW when loaded with sensitizer and 2.0-5.0 mW with no sensitizer) and so we estimated that most of the light (∼380 mW) was absorbed by the sensitizer particles and walls of the device, which subsequently transferred the heat to the solution in which it was immersed. There was some variability of light absorption in the devices, resulting from the different chamber sizes. Oxygen solubility is reported to decrease from 7.9 ppm O2 at 25 °C to 7.2 ppm O2 at 30 °C and its mass transfer coefficient increases.25
Effect of Device Geometry and Bubble Size on Product Yield
Chemical trapping of 1O2 was conducted in the surrounding aqueous solution with 9,10-anthracene dipropionate dianion (2), trans-2-methyl-2-pentenoic acid (3), N-benzoyl-D,L-methionine (4), and N-acetyl-D,L-methionine (5) (Figure 3).9,26-32
Figure 3.

Chemical agents used to trap 1O2 using (a) devices 1, 2, or 3; (b) in H2O or D2O; and (c) in D2O.
Devices 1-3 were connected to the 669-nm diode laser (fluence = 4128 J/cm2) via an optical fiber and an O2 gas tank (60 mL/min flow). Compounds 2-5 were photooxidized in 3.0 mL H2O or D2O downstream. Compounds 2 and 3 are specific quenchers of singlet oxygen, but 4 and 5 are not. The formation of 9,10-anthracene-9,10-endoperoxide dipropionate dianion (6) took place via a [4+2] cycloaddition of singlet oxygen with 2 (0.001 M, pH = 10), and the formation of 3-hydroperoxy-2-methylene pentanoate anion (7) occurred via an ‘ene’ reaction of singlet oxygen with 3 (0.04 M, pH = 10). Two moles of methionine sulfoxide formed per mole 1O2 in the reaction of the corresponding methionines (4 and 5) (ea. 0.04 M, pH = 10). For the methionines 4 and 5, the S-oxide products were detected, but neither the sulfones nor rearranged products, as seen in some mechanistic studies.33,34 Because the 1O2 lifetime is longer in D2O (τΔ = 65 μs) than in H2O (τΔ = 3.5 μs),35 the preferred use of D2O in the experiments in Table 3 was the result of shorter reaction times. Irradiation of 2-5 in the absence of sensitizer particles produced no products with all devices (cf. entry 1, 8, and 15).
Table 3. Photooxidation of Probe Compounds in D2Oa,b.
| device number | entry | sensitizer particles loaded into device (mg) | yield of endoperoxide 6, μmol (and %)c | yield of hydroperoxide 7, μmol (and %)d | yield of benzoyl methionine S-oxide 8, μmol (and %)d | yield of acetyl methionine S-oxide 9, μmol (and %)d |
|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | 0 | |
| 2 | 1 | 0 | 0 | 0 | 0 | |
| 3 | 3 | 0 | 0 | 0 | 0 | |
| 1 | 4 | 10 | 0 | 0 | 0.48 (0.8%) | 1.14 (1.9%) |
| 5 | 35 | 0.99 (33%) | 2.16 (1.8%) | 1.74 (2.9%) | 1.38 (2.3%) | |
| 6 | 50 | 1.23 (41%) | 4.68 (3.9%) | 2.82 (4.7%) | 2.16 (3.6%) | |
| 7 | 75 | 1.38 (46%) | 5.76 (4.8%) | 4.50 (7.5%) | 2.70 (4.5%) | |
| 8 | 0 | 0 | 0 | 0 | 0 | |
| 9 | 1 | 0 | 0 | 0 | 0 | |
| 10 | 3 | 0 | 0 | 0 | 0 | |
| 2 | 11 | 10 | 0.12 (4.0%) | 0 | 0 | 0.6 (1.0%) |
| 12 | 35 | 0.42 (14%) | 1.80 (1.5%) | 0.66 (1.1%) | 0.96 (1.6%) | |
| 13 | 50 | 0.69 (23%) | 3.84 (3.2%) | 1.02 (1.7%) | 1.35 (2.7%) | |
| 14 | 75 | 0.78 (26%) | 4.20 (3.5%) | 1.55 (3.1%) | 1.60 (3.2%) | |
| 15 | 0 | 0 | 0 | 0 | 0 | |
| 16 | 1 | 0 | 0 | 0 | 0 | |
| 17 | 3 | 0 | 0 | 0 | 0 | |
| 3 | 18 | 10 | 0 | 0 | 0 | 0 |
| 19 | 35 | 0 | 1.20 (1.0%) | 0.42 (0.7%) | 0 | |
| 20 | 50 | 0.72 (24%) | 1.80 (1.5%) | 0.72 (1.2%) | 0.60 (1.0%) | |
| 21 | 75 | 0.96 (32%) | 4.50 (2.5%) | 1.44 (2.4%) | 1.20 (2.0%) | |
All samples were illuminated at 669 nm (fluence = 4128 J/cm2) under an O2 flow rate of 60 mL/min for 2.5 h at 28 °C. Over the course of each experiment 9 L of O2 was consumed.
The concentrations of 3-hydroperoxy-2-methylene pentanoate anion 7, N-benzoy-D,L-methionine S-oxide 8, N-acetyl-D,L-methionine S-oxide 9 were determined by 1H NMR by the appearance of singlets at 2.71 ppm (s, 3H), at 5.56 ppm (s, 1H), and at 5.94 ppm (s, 1H), respectively. The concentration of endoperoxide 6 was estimated by UV-VIS by the disappearance of 9,10-anthracene dipropionate dianion 2 absorption at 378 nm.
The starting concentration of 2 was 1 mM (0.003 mmol).
The starting concentrations of 3-5 was 0.04 M (0.12 mmol).
As shown in Table 3, higher photooxidation yields were observed from smaller bubbles. We attributed this to the enhanced contact between trap molecule and 1O2 due to higher surface-to-volume ratios resulting from smaller bubbles. Irradiation of 2 (1.0 mM) for 2.5 h led to 0.99, 0.42, and 0 μmol of endoperoxide 6 with devices 1, 2, and 3, respectively, each loaded with 35 mg of Pc 1 particles where bubble diameters were 2.8, 3.2, and 4.8 mm, respectively, as shown in Table 3.
Higher loadings of sensitizer particles also led to increased product formation 6-9 likely due to an increase in exposed sensitizer surface area within the reactor chamber. In the case of 75 mg particle loading, the reaction yielded 1.38, 0.78, and 0.96 μmol of endoperoxide 6. For all devices, a minimum quantity of sensitizer was required before photooxidized products could be detected; for devices 1 and 2, greater than 10 mg of sensitizer particles was required, for device 3, 35 mg was required.
Regarding the photoreactor design, it is important to note that melting of the sensitizer occurred when the laser-head was in close proximity to the particles. Since the melting point of Pc 1 is 65 °C, the temperature of the sensitizer was >25 °C. Thus, it was advantageous to use reduced loadings (e.g., 35 mg loadings) to increase the distance between the laser-head and the sensitizer particles to prevent excess heating of the sensitizer. On the other hand, the reduced product yields, compared to 75 mg loadings, was a disadvantage (Table 3).
Mechanism of Singlet Oxygen Mass Transfer
The formation of 1O2 was further examined with device 1 loaded with 35 mg sensitizer particles in H2O and D2O. The data are consistent with the mechanism in Figure 4, where Ia is the rate of light absorption by the Pc 1 particles, km is the device membrane deactivation rate constant, kr is the trapping rate constant, kq is physical quenching rate constant by the trapping agent, kd is the decay rate constant by H2O or D2O. In H2O, the O2 concentration measured at t=0 min was 1.5×10-4 M (4.7 ppm), according to a Clarke type oxygen electrode. Upon sparging O2 via device 1, successive readings of O2 concentrations were constant after 40 min when oxygen saturation was reached 8.3×10-4 M (26.6 ppm) (Figure 5). We initially thought that oxygen would saturate this volume more quickly than 40 min, but the effect is likely due to the small bubbles generated from device 1.
Figure 4.
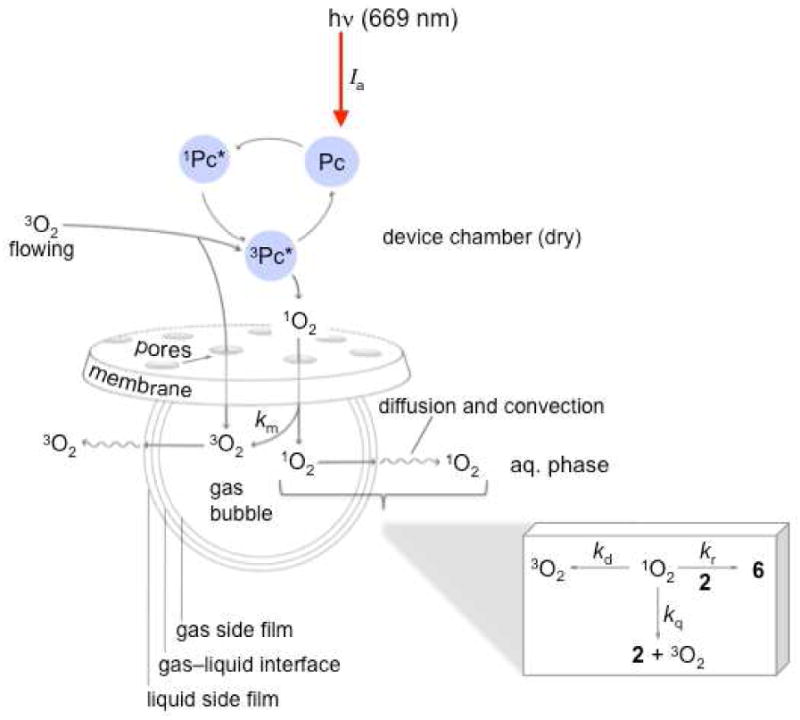
Proposed photooxidation mechanism.
Figure 5.
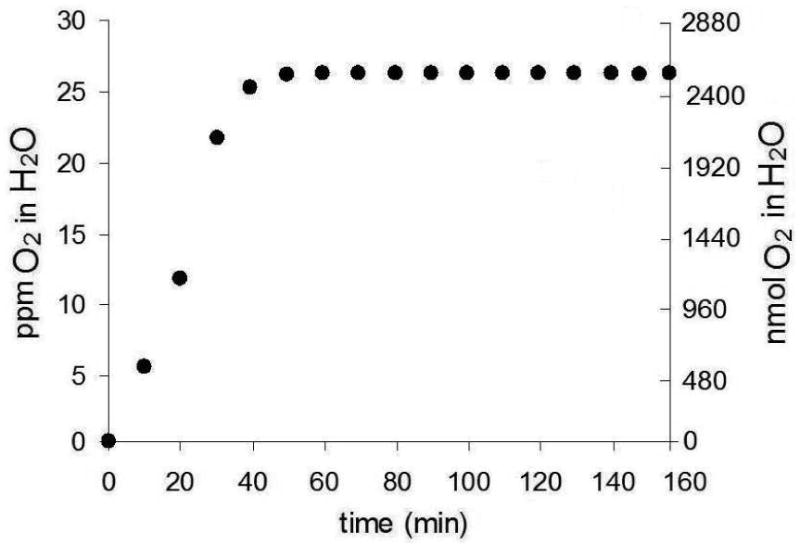
Solubility of O2 in 3-mL H2O as a function of time using device 1 loaded with 0.35 mg sensitizer particles. The flow rate was 60 mL/min.
Evidence suggested that 1O2 transfer occurred via bubbles into bulk water prior to oxygen saturation of the solution. Firstly, by monitoring the emission at 1270 nm, the lifetime of 1O2 sparged into D2O was found to be 60±3 μs, which matched the value expected of 1O2 in bulk D2O, but increased to ∼1 ms in air (Figure S7, Supporting Information). Secondly, rapid photooxidation of anthracene 2 was observed prior to O2 saturation (0-40 min, Figure 6). The starting concentration of 2 was 0.05 mM (150 nmol 2). Lines were fitted to the fast stage of the plot, where the rate of formation of 6 was 1.1 nmol/min in D2O and 0.2 nmol/min in H2O from 0 to 40 min.
Figure 6.
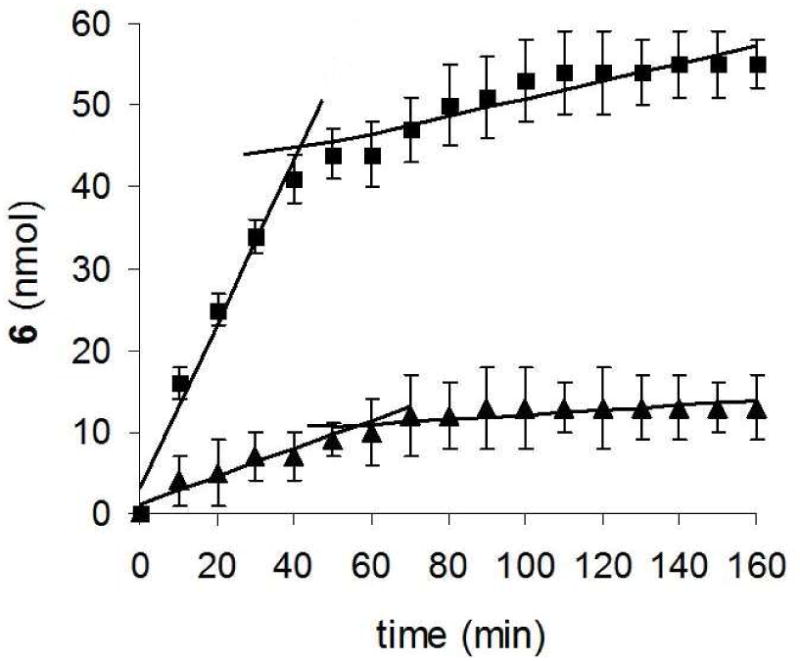
Nanomoles of photoproduct 6 as a function of time from device 1 loaded with 0.35 mg sensitizer particles with an O2 flow rate was 60 mL/min into 3-mL D2O (▪) and H2O (▴). The starting concentration of 2 was 0.05 mM (150 nmol 2).
After the solution was saturated with O2, the sensitivity of the slope reduced by 10 fold. This slower stage for the photooxidation of 2 might arise from contact of singlet oxygen and 2 at the gas-liquid interface with reduced 1O2 transfer into bulk solution due to the O2 equilibrium reached between the gas and liquid phases. The rate of formation of 6 was 0.12 nmol/min in D2O and 0.02 nmol/min in H2O from the period of ∼40 to 160 min. After 160 min, 34% conversion of 2 was reached in D2O. Control experiments showed that D2O was saturated with O2 about 2 times more rapidly than H2O, and the solubility was slightly greater (cf. 32.5 ppm in D2O with 26.6 ppm in H2O). These two facets can help explain why the inflection point in Figure 6 occurs at about 40 min in D2O and 60 min in H2O.
We believe that singlet oxygen continues to transfer into the bulk water even after saturation. The water may saturate with O2, but it is not static. O2 would be vaporizing from the surface (at both the bulk liquid-air interface as well as liquid bubble interface) and new 1O2 and O2 would dissolve to replace, but the rate will be lower than before saturation. There is ample precedent that when an O2 equilibrium exists in the gas and liquid phases O2 exchange still occurs, concentrations of O2 are linearly related in both phases (Henry's Law), but there is no net change in O2 concentration, which is driven by a concentration gradient (Fick's Law).36 There is a large literature on how gas in bubbles interacts with aqueous solutions.37 However, the two stages of the reaction indicate that the movement of the probe molecules in solution (convection) were caused by the bubbles to overcome threshold quantity of product yields imposed by equilibrium.
The sensitivity of the slope of product formation in D2O compared to H2O prior to or after O2 saturation of the solution was consistent with the longer lifetime of 1O2 in the former.35 Table 4 shows the ratio of endoperoxide 6 molecules formed to 3O2 molecules transmitted, which translates (roughly) to the number of oxidized molecules that arose from each sensitizer particle. A lower-limit of the number of 1O2 molecules within the bubbles was ∼3 ppm for device 1, ∼2 ppm for device 2, and ≪1 ppm for device 3. Averaged over 2.5 h, the rate of 6 formation was ∼8 nmol/min for device 1, ∼4 nmol/min for device 2, and for device 3, no product was detected. The nanomole per minute rates we observe are about 100 fold less efficient than photooxidation batch reactors,38-40 but for the batch reactors the photosensitizer must be soluble in solution and then separated (e.g., via permeation chromatography). In contrast, our devices use a membrane, which effectively keeps the sensitizer dry and separated from the solution and so there is no concern with sensitizer removal after the reaction. Interest has surrounded the quenching of photosensitizers by O2 at solution/solid and gas/solid interfaces41-46 for clean external production of 1O2,47 and improved 1O2 transmission would be conceivable in device membranes containing C-D or C-F bonds, since C-H bonds are more effective in the vibrational deactivation of 1O2 in small organic molecules.48,49 Isolating the photosensitizer from the solution avoids the possibility of ground-state hydrogen abstraction or electron transfer Type I photooxidation processes4 for clean transmission of 1O2 across the membrane.
Table 4. Membrane Pore Sizes, Ratio of Oxygen Transmitted to Endoperoxide 6 Formeda,b.
| Device Numberc | Membrane pore size (μm) | Ratio of endoperoxide 6 molecules to 3O2 transmitted (ppm) | Nanomoles 6 formed per sensitizer particled |
|---|---|---|---|
| 1 | 0.05 | 2.6 | 67 |
| 2 | 0.22 | 1.1 | 41 |
| 3 | 0.44 | <0.1 | <5 |
Devices were loaded with 0.35 mg Pc 1; O2 flow rate was 60 mL/min; solution was 3 mL D2O. The starting concentration of 2 was 1.0 mM (3 μmol 2).
Over the course of experiment 9 L of O2 was consumed.
Number of bubbles transmitted over the course of the experiment: 98,900 (device 1); 65,700 (device 2); 19,400 (device 3).
150±30 μm sensitizer particles; 2.5 h reaction time.
We believe that the 1O2 yield is significantly greater than the measured yield of oxidized acceptors. For the 1O2 that was transported across the membrane, some 1O2 is lost to other quenching processes. But the measured yield was severely limited by mass transport50 across the membrane as well as into the solution. This results in much of the 1O2 being released to the air when bubbles reach the bulk liquid-air interface, as shown in Figure S8, which has been recognized previously;51 thus, the device appears to operate within gas-water mass transfer limitations.
Conclusion
We report on the fabrication and properties of a singlet oxygen-generating device, in which a solid Pc photosensitizer was isolated from an aqueous solution by using a porous membrane in a laser-coupled device. A sol-gel technique was used to synthesize the Pc photocatalyst within a glass matrix. Due to the high capillary pressure of the membrane, the sensitizer remains dry within the device as it is irradiated with laser light in the presence of an oxygen flow. Within the device, O2 was sensitized by excited Pc sites in the particles. Singlet oxygen molecules were then transported across the membrane, forming bubbles at the membrane-water interface.
Not only do the smaller diameter pores in the membrane prevent water ingress at higher pressures, but the smaller pores also generate smaller bubbles and thus increase the device efficiency. Reaction rates between singlet oxygen and four probe compounds were measured and the rates were proportional to sensitizer particle loading and inversely proportional to the membrane pore diameter. Bubble diameter was correlated to pore diameter, and rates increased when smaller bubbles were observed. A mechanism is proposed whereby the oxidation of probe compounds is limited by transport of 1O2 across the bubble-liquid interface. Given the flow is held constant in all experiments, smaller bubble diameters result in larger oxygen-water interfacial areas. In addition, the reaction rate slows by a factor of ∼10 after the solution becomes saturated with O2. Oxygen saturation reduces the rate of 1O2 transport from the bubble into the solution.
Water purification and wound disinfection are our long-term goals, and the first step in this paper was to demonstrate 1O2 delivery from a photosensitizer isolated from water. Future experiments are planned, including evaluation of the effectiveness of the technique for inactivation of bacteria and oxidation of groundwater contaminants.
Supplementary Material
Acknowledgments
DB, DA, and AG acknowledge support from the National Institute of General Medical Sciences (NIH SC1GM093830). AML acknowledges support from the NYS Empire State Development's Division of Science, Technology & Innovation (NYSTAR). BG acknowledges support from the National Science Foundation STEM Talent Expansion via Applied Mathematics (STEAM) (Grant #0653056). We also thank Alison Domzalski for the photography work and Leda Lee for the graphic arts work. This paper is dedicated to the memory of Prof. Ronald Bentley of the University of Pittsburgh.
Footnotes
Supporting Information Available: Fluorescence and UV-VIS spectra of Pc 1, images of the SMA receptacle, photomicrographs of the membranes, luminescence signal of singlet oxygen in D2O and in air, and an image of the loss of singlet oxygen in bubbles that reach the air interface. This material is available free of charge via the Internet at http://pubs.acs.org/.
Contributor Information
Alan M. Lyons, Email: alan.lyons@csi.cuny.edu.
Alexander Greer, Email: agreer@brooklyn.cuny.edu.
References
- 1.Gavasci R, Chiavola A, Spizzirri M. Water Sci Technol. 2010;62:1371–1378. doi: 10.2166/wst.2010.337. [DOI] [PubMed] [Google Scholar]
- 2.Loeb BL. Ozone: Sci Eng. 2011;33:329–342. [Google Scholar]
- 3.Foote CS. Acc Chem Res. 1968;1:104–110. [Google Scholar]
- 4.Greer A. Acc Chem Res. 2006;39:797–804. doi: 10.1021/ar050191g. [DOI] [PubMed] [Google Scholar]
- 5.Manjón F, Villén L, García-Fresnadillo D, Orellana G. Environ Sci Technol. 2008;42:301–307. doi: 10.1021/es071762y. [DOI] [PubMed] [Google Scholar]
- 6.Benabbou AK, Guillard C, Pigeot-Rémy S, Cantau C, Pigot T, Lejeune P, Derriche Z, Lacombe S. J Photochem Photobiol A: Chem. 2011;219:101–108. [Google Scholar]
- 7.Remucal CK, McNeill K. Environ Sci Technol. 2011;45:5230–5237. doi: 10.1021/es200411a. [DOI] [PubMed] [Google Scholar]
- 8.Kammerlander G, Assadian O, Eberlein T, Zweitmuller P, Luchsinger S, Andriessen A. J Wound Care. 2011;20:149–158. doi: 10.12968/jowc.2011.20.4.149. [DOI] [PubMed] [Google Scholar]
- 9.Zamadar M, Aebisher D, Greer A. J Phys Chem B. 2009;113:15803–15806. doi: 10.1021/jp907945c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aebisher D, Zamadar M, Mahendran A, Ghosh G, McEntee C, Greer A. Photochem Photobiol. 2010;86:890–894. doi: 10.1111/j.1751-1097.2010.00748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahendran A, Kopkalli Y, Ghosh G, Ghogare A, Minnis M, Kruft BI, Zamadar M, Aebisher D, Davenport L, Greer A. Photochem Photobiol. 2011;87:1330–1337. doi: 10.1111/j.1751-1097.2011.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zamadar M, Ghosh G, Mahendran A, Minnis M, Kruft BI, Ghogare A, Aebisher D, Greer A. J Am Chem Soc. 2011;133:7882–7891. doi: 10.1021/ja200840p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisenberg WC, Taylor K, Murray RW. J Phys Chem. 1986;90:1945–1948. [Google Scholar]
- 14.Nikolaev VD, Svistun MI, Zagidullin MV, Hager GD. Appl Phys Lett. 2003;86:231102. [Google Scholar]
- 15.Spalek O, Kodymová J, Hirsl A. J Appl Phys. 1987;62:2208–2211. [Google Scholar]
- 16.Aoudia M, Cheng G, Kennedy VO, Kenney ME, Rodgers MAJ. J Am Chem Soc. 1997;119:6029–6039. [Google Scholar]
- 17.Rodríguez-Córdoba W, Noria R, Guarín CA, Peon J. J Am Chem Soc. 2011;133:4698–4701. doi: 10.1021/ja1113547. [DOI] [PubMed] [Google Scholar]
- 18.Xia H, Nogami M, Hayakawa T, Imazumi D. J Mat Sci Lett. 1999;18:1837–1839. [Google Scholar]
- 19.Innocenzi P, Brusatin G, Babonneau F. Chem Mater. 2000;12:3726–3732. [Google Scholar]
- 20.Hench LL, West JK. Chem Rev. 1990;90:33–72. [Google Scholar]
- 21.Campo MA, Babriel D, Kucera P, Gurny R, Lange N. Photochem Photobiol. 2007;83:958–965. doi: 10.1111/j.1751-1097.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 22.Lovell JF, Chen J, Liu TWB, Zheng G. Chem Rev. 2010;110:2839–2857. doi: 10.1021/cr900236h. [DOI] [PubMed] [Google Scholar]
- 23.Skidmore EL, Powers DH. Soil Sci Am J. 1982;46:1274–1279. [Google Scholar]
- 24.Bonhomme C, Coelho C, Baccile N, Gervais C, Azaïs, Babonneau F. Acc Chem Res. 2007;40:738–746. doi: 10.1021/ar600030j. [DOI] [PubMed] [Google Scholar]
- 25.Peterson J. Minerals Eng. 2010;23:504–510. [Google Scholar]
- 26.Sysak PK, Ching TY, Foote CS. Photochem Photobiol. 1977;26:19–27. [Google Scholar]
- 27.Lindig BA, Rodgers MAJ, Schaap AP. J Am Chem Soc. 1980;102:5590–5593. [Google Scholar]
- 28.Di Mascio P, Sies H. J Am Chem Soc. 1989;111:2909–2914. [Google Scholar]
- 29.Martinez GR, Ravanat JL, Medeiros MHG, Cadet J, Di Mascio P. J Am Chem Soc. 2000;122:10212–10213. [Google Scholar]
- 30.Aubry JM, Pierlot C, Rigaudy J, Schmidt R. Acc Chem Res. 2003;36:668–675. doi: 10.1021/ar010086g. [DOI] [PubMed] [Google Scholar]
- 31.Fudickar W, Linker T. Langmuir. 2010;26:4421–4428. doi: 10.1021/la904299n. [DOI] [PubMed] [Google Scholar]
- 32.Turro NJ, Ramamurthy V, Scaiano JC. Modern Molecular Photochemistry of Organic Molecules. University Science Books; Sausalito, CA: 2010. pp. 1001–1040. [Google Scholar]
- 33.Toutchkine A, Aebisher DA, Clennan EL. J Am Chem Soc. 2001;123:4966–4973. doi: 10.1021/ja004188y. [DOI] [PubMed] [Google Scholar]
- 34.Liu F, Fang Y, Chen Y, Liu J. J Phys Chem B. 2011;115:9898–9909. doi: 10.1021/jp205235d. [DOI] [PubMed] [Google Scholar]
- 35.Jensen RL, Arnbjerg J, Ogilby PR. J Am Chem Soc. 2010;132:8098–8105. doi: 10.1021/ja101753n. [DOI] [PubMed] [Google Scholar]
- 36.Tromans D. Ind Eng Chem Res. 2000;39:805–812. [Google Scholar]
- 37.Poling B, Prausnitz J, O'Connell J. The Properties of Gases and Liquids. McGraw-Hill; New York: 2001. [Google Scholar]
- 38.Levesque F, Seeberger PH. Org Lett. 2011;13:5008–5011. doi: 10.1021/ol2017643. [DOI] [PubMed] [Google Scholar]
- 39.Yavorskyy A, Shvydkiv O, Nolan K, Hoffmann N, Oelgemoller M. Tetrahedron Lett. 2011;52:278–280. [Google Scholar]
- 40.Maurya RA, Park CP, Kim DP. Beilstein J Org Chem. 2011;7:1158–1163. doi: 10.3762/bjoc.7.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harper J, Sailor MJ. Langmuir. 1997;13:4652–4658. [Google Scholar]
- 42.Fuchter MJ, Haffman BM, Barrett AGM. J Org Chem. 2006;71:724–729. doi: 10.1021/jo052156t. [DOI] [PubMed] [Google Scholar]
- 43.Griesbeck AG, Bartoschek A, Neudoerfl J, Miara C. Photochem Photobiol. 2006;82:1233–1240. doi: 10.1562/2006-03-03-RA-832. [DOI] [PubMed] [Google Scholar]
- 44.Naito K, Tachikawa T, Cui SC, Sugimoto A, Fujisuka M, Majima T. J Am Chem Soc. 2006;128:16430–16431. doi: 10.1021/ja066739b. [DOI] [PubMed] [Google Scholar]
- 45.Rossi LM, Silva PR, Vono LLR, Fernandes AU, Tada DB, Baptista MS. Langmuir. 2008;24:12534–12538. doi: 10.1021/la800840k. [DOI] [PubMed] [Google Scholar]
- 46.Llansola Portolés MJ, Gara PMD, Kotler ML, Bertolotti S, San Román E, Rodríguez HB, Gonzalez MC. Langmuir. 2010;26:10953–10960. doi: 10.1021/la100980x. [DOI] [PubMed] [Google Scholar]
- 47.Midden WR, Wang SY. J Am Chem Soc. 1983;105:4129–4135. [Google Scholar]
- 48.Sivaguru J, Solomon MR, Poon T, Jockusch S, Bosio SG, Adam W, Turro NJ. Acc Chem Res. 2008;41:387–400. doi: 10.1021/ar7001254. [DOI] [PubMed] [Google Scholar]
- 49.Wilkinson F, Helman WP, Ross AB. J Phys Chem Ref Data. 1995;24:663–1021. [Google Scholar]
- 50.Bourne RA, Han X, Poliakoff M, George MW. Angew Chem Int Ed. 2009;48:5322–5325. doi: 10.1002/anie.200901731. [DOI] [PubMed] [Google Scholar]
- 51.Evans DF, Upton MW. J Chem Soc Dalton Trans. 1985;6:1141–1145. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.