

Abstract
Background
The AMP-activated protein kinase (AMPK) cascade is a sensor of cellular energy charge that acts as a 'metabolic master switch' and inhibits cell proliferation. Activation requires phosphorylation of Thr172 of AMPK within the activation loop by upstream kinases (AMPKKs) that have not been identified. Recently, we identified three related protein kinases acting upstream of the yeast homolog of AMPK. Although they do not have obvious mammalian homologs, they are related to LKB1, a tumor suppressor that is mutated in the human Peutz-Jeghers cancer syndrome. We recently showed that LKB1 exists as a complex with two accessory subunits, STRADα/β and MO25α/β.
Results
We report the following observations. First, two AMPKK activities purified from rat liver contain LKB1, STRADα and MO25α, and can be immunoprecipitated using anti-LKB1 antibodies. Second, both endogenous and recombinant complexes of LKB1, STRADα/β and MO25α/β activate AMPK via phosphorylation of Thr172. Third, catalytically active LKB1, STRADα or STRADβ and MO25α or MO25β are required for full activity. Fourth, the AMPK-activating drugs AICA riboside and phenformin do not activate AMPK in HeLa cells (which lack LKB1), but activation can be restored by stably expressing wild-type, but not catalytically inactive, LKB1. Fifth, AICA riboside and phenformin fail to activate AMPK in immortalized fibroblasts from LKB1-knockout mouse embryos.
Conclusions
These results provide the first description of a physiological substrate for the LKB1 tumor suppressor and suggest that it functions as an upstream regulator of AMPK. Our findings indicate that the tumors in Peutz-Jeghers syndrome could result from deficient activation of AMPK as a consequence of LKB1 inactivation.
Introduction
AMP-activated protein kinase kinase (AMPKK) and AMP-activated protein kinase (AMPK) are the upstream and downstream components, respectively, of a protein kinase cascade that acts as a sensor of cellular energy charge [1,2]. AMPK is activated by the elevation in cellular 5'-AMP that accompanies a fall in the ATP:ADP ratio due to the reaction catalyzed by adenylate kinase (2ADP ↔ ATP + AMP). This occurs during metabolic stresses such as hypoxia, ischaemia, glucose deprivation and, in skeletal and cardiac muscle, during contraction or exercise [1-3]. Once activated by stress, AMPK switches on the uptake of glucose and fatty acids and the oxidative metabolism of these fuels to generate ATP, while switching off biosynthetic pathways that consume ATP. It achieves this metabolic switching both by direct phosphorylation of metabolic enzymes and by longer-term effects on gene expression [1,2].
We have previously partially purified from rat liver an upstream kinase (AMPKK) that activates AMPK by phospho-rylation of AMPK residue Thr172 within the activation loop of the kinase domain [4], but we have been unable to identify the activity as a defined gene product. As an alternative approach, we searched for kinases upstream of the Saccharomyces cerevisiae homolog of AMPK (the SNF1 complex), taking advantage of genome-wide approaches available in that organism. This identified Elm1, Pak1 and Tos3 as alternative upstream kinases in yeast that can activate the SNF1 complex in vivo in a partially redundant manner [5]. The nearest relatives encoded by the human genome are calmodulin-dependent protein kinase kinase (CaMKK) and LKB1 (see Additional data file 1 with the online version of this article). We have previously shown that CaMKK purified from pig brain could activate AMPK in cell-free assays (albeit poorly in comparison to the extent to which it activates its known substrate, calmodulin-dependent protein kinase I); but the AMPKK previously purified from rat liver was not calmodulin-dependent [6]. LKB1 is a 50 kDa serine/threonine kinase that was originally discovered as the product of the gene mutated in the autosomal dominant human disorder, Peutz-Jeghers syndrome (PJS) [7,8]. People with PJS develop benign polyps in the gastrointestinal tract but also have a 15-fold increased risk of developing malignant tumors in other tissues [9,10]. Nearly 100 different PJS mutations have been reported, and most are expected to impair the kinase activity of LKB1 [11]. Several human tumor cell lines, including HeLa and G361 cells, lack expression of LKB1. Expression of wild-type LKB1, but not catalytically inactive LKB1 or PJS mutants, in G361 cells caused a G1-phase cell-cycle arrest [12] that was associated with the induction of the cyclin-dependent kinase inhibitor, p21, and was dependent on p53 [13]. Homozygous LKB1 knockout mice die of multiple defects at mid-gestation [14].
Heterozygotes are viable, but most develop polyps similar to those found in people with PJS by 45 weeks of age [15-18], although it is controversial as to whether these are caused by haploinsufficiency or loss of heterozygosity (reviewed in [11]). It has also been reported that a significant number of LKB1+/- mice over 50 weeks of age develop hepatocellular carcinomas that are associated with loss of LKB1 expression [19]. These results show that LKB1 acts as a tumor suppressor and that the catalytic activity of LKB1 is essential for this function, but the downstream substrate(s) that LKB1 phosphorylates to mediate the suppression of cell proliferation remained unknown.
Recently, we reported that LKB1 is associated with two accessory proteins called Ste20-related adaptor protein-α (STRADα) [20] and mouse protein 25-α (MO25α) [21], for each of which there is also a closely related isoform (STRADβ and MO25β) encoded in the human genome. Although STRADα and β are related to the Ste20 protein kinases, several of the residues expected in active protein kinases are not conserved, and they appear to be inactive 'pseudokinases' [20]. MO25α binds to the carboxyl-terminus of STRADα and stabilizes the association between STRADα and LKB1 [21]. The association of LKB1 with STRADα and MO25α increased the kinase activity of LKB1 against an artificial substrate (myelin basic protein) and also enhanced its cytoplasmic localization [20,21], which was previously implicated in the tumor suppressor function of LKB1 [13]. Here, on the basis of the sequence similarity between LKB1 and the upstream kinases identified for the yeast homolog of AMPK [5], we investigate whether LKB1:STRAD:MO25 complexes could play a role in activating AMPK in mammalian cells.
Results
Resolution of two AMPKKs from rat liver that both contain LKB1, STRADα and MO25α
While experimenting with different conditions to optimize recovery at the Q-Sepharose step of our previous purification protocol for AMPKK [4], we found that we were able to resolve two peaks of activity (Figure 1a). Because the second peak corresponds to the AMPKK originally purified [4], we refer to it as AMPKK1, with the first peak being termed AMPKK2. On size-exclusion chromatography on Sephacryl S-200, AMPKK1 and AMPKK2 eluted as proteins of large but distinct size, with estimated Stokes radii of 5.7 and 5.2 nm respectively. We probed blots of fractions across the Q-Sepharose column using antibodies against LKB1, STRADα and MO25α (Figure 1b). This revealed that the activity of AMPKK2 correlated with the presence of the LKB1 polypeptide (around 50 kDa), as well as those of STRADα (around 45/48 kDa) and MO25α (around 40 kDa). The monoclonal antibody against STRADα, which is specific for the α isoform, detected a doublet, as reported previously [20]; the explanation for this is not known. We did not obtain any signal of the correct molecular mass in these fractions using anti-MO25β antibody (not shown), consistent with previous observations that MO25β is not expressed in mouse liver [21]. We also obtained a faint signal for LKB1 and STRADα in fractions containing AMPKK1, but at this loading MO25α was below the limit of detection. However, the presence of LKB1, STRADα and MO25α in these fractions was confirmed by analyzing a higher loading (Figure 1b, bottom three panels). An interesting finding was that the LKB1 polypeptide migrated with a significantly faster mobility in AMPKK1 than in AMPKK2, while LKB1 in AMPKK2 appeared to run as a doublet (Figure 1b; see also Figures 1c, 2b and 2d). The results in Figure 1c suggest that this difference in mobility was not due to a difference in phosphorylation state of the LKB1 polypeptide. Incubation of the AMPKK1 and AMPKK2 fractions with MgATP, followed by treatment with or without the catalytic subunit of protein phosphatase 1γ (PP1γ) or the protein phosphatase 2A1 (PP2A1) holoenzyme, did not alter the mobility of any of the LKB1 polypeptides. The right-hand panel in Figure 1c shows that these protein phosphatases did dephosphorylate Thr172 on the α subunit of AMPK when incubated under identical conditions.
Figure 1.
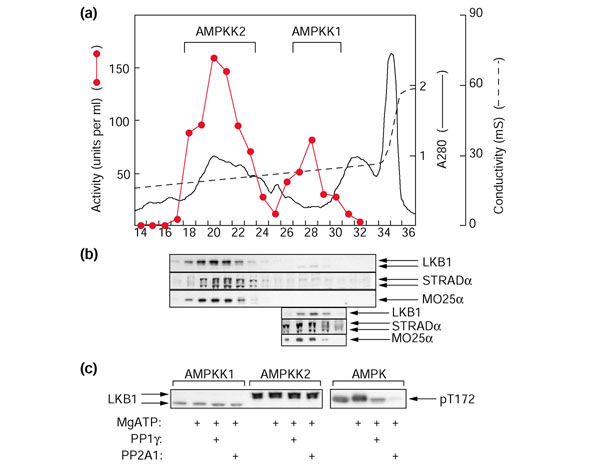
Two AMPKKs can be resolved from rat liver extracts and both contain LKB1, STRADα and MO25α. (a) Separation of two activities that activate the GST-AMPKα1 catalytic domain by Q-Sepharose chromatography. The graph shows AMPKK activity in 4.5 ml fractions (red circles and red line), absorbance at 280 nm (continuous black line) and conductivity in the eluate (dashed black line) plotted against fraction number. (b) Probing of blots of column fractions after SDS gel electrophoresis (1 μl per lane) using anti-LKB1, anti-STRADα or anti-MO25α antibodies. In the three bottom panels, fractions 26–30 were concentrated from 4.5 ml to 250 μl using Amicon Ultra-4 30,000 MWCO centrifugal concentrators, and reanalyzed by western blotting using 2 μl per lane. (c) The effect of protein phosphatase treatment on the mobility of LKB1. The peak fractions of AMPKK1 (0.2 units) or AMPKK2 (0.8 units) were incubated in a final volume of 20 μl with or without 5 mM MgCl2 and 200 μM ATP for 15 min at 30°C. Protein phosphatases (PP1γ, 8 mU; or PP2A1, 1 mU) or buffer were added and incubation continued for a further 15 min before stopping the reactions in SDS sample buffer and analyzing by SDS gel electrophoresis and western blotting using anti-LKB1 antibody.
Figure 2.
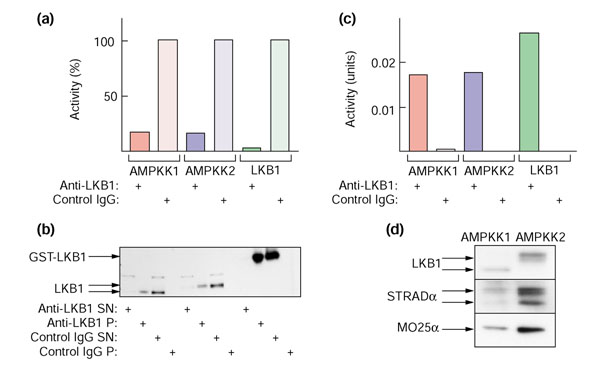
AMPKK activity (that is, the ability to activate AMPKα1 catalytic domain), and LKB1, STRADα and MO25α polypeptides, can be immunoprecipitated from rat liver AMPKK1 and AMPKK2 using anti-LKB1 antibody. (a) Depletion of AMPKK activity from supernatant. Sheep anti-human LKB1 or pre-immune control immunoglobulin (IgG) was prebound to Protein G-Sepharose beads and cross-linked with dimethylpimelimidate as described [47], except that a final wash of the beads with 100 mM glycine, pH 2.5, was performed. Bead-bound antibodies (40 μl) were incubated with the peak fraction of AMPKK1 (0.04 units), AMPKK2 (0.03 units) or recombinant GST-LKB1:STRADα:MO25α complex (0.06 units) for 120 minutes and the beads removed in a microcentrifuge (14,000 × g for 2 min). AMPKK activity was determined in the supernatants and is expressed as a percentage of the value obtained using the control IgG. (b) The pellets from the experiment in (a) were resuspended in the original volume and samples of the supernatants and pellets analysed by western blotting with anti-LKB1 antibody. The recombinant LKB1 migrates at a higher molecular mass because of the GST tag. (c) As in (a), except that the amounts of AMPKK1, AMPKK2 and recombinant GST-LKB1:STRADα:MO25α complex were increased to 0.44, 0.70 and 1.4 units, respectively, and the activities were determined in the resuspended pellets. In this experiment the amount of antibody was limiting, so only a fraction of the activity was precipitated. (d) The pellets from the experiment in (c) were resuspended and samples analyzed by western blotting with anti-LKB1, anti-STRADα and anti-MO25α antibodies.
AMPKK activity can be immunoprecipitated from AMPKK1 and AMPKK2
Using anti-LKB1 antibody but not a pre-immune control immunoglobulin, we were able to immunoprecipitate AMPKK activity from fractions containing both AMPKK1 and AMPKK2. Figure 2a shows results of an experiment where the amount of AMPKK1 or AMPKK2 was limiting and the antibody was in excess, and shows that we were able to remove more than 80% of the activity from the peak fractions containing AMPKK1 and AMPKK2 by immunoprecipitating with anti-LKB1 antibody, while no activity was removed using a pre-immune control immunoglobulin. We could remove more than 95% of the AMPKK activity of a recombinant tagged LKB1:STRADα:MO25α complex (see below) under the same conditions (Figure 2a). The small amount of activity remaining in the supernatants of the AMPKK1 and AMPKK2 immunoprecipitates could be accounted for by the fact that the immunoprecipitation was not quantitative, with a small amount of the LKB1 polypeptide remaining in the supernatant. No LKB1 polypeptide was precipitated using the pre-immune control immunoglobulin (Figure 2b).
Because of the small amount of AMPKK1 and AMPKK2 used in this experiment, it proved difficult to analyze the pellets for AMPKK activity and the presence of the other polypeptides. We therefore repeated the experiment with more AMPKK (the amount of antibody was now limiting) and analyzed the pellets only. This showed that we could recover a similar amount of AMPKK activity in the pellet from the peak fractions containing AMPKK1 and AMPKK2 as we could from the recombinant LKB1:STRADα:MO25α complex, with no activity being recovered in the pellet using the pre-immune control immunoglobulin (Figure 2c). Western blotting of the AMPKK1 and AMPKK2 pellets showed that they contained LKB1, STRADα and MO25α (Figure 2d).
Recombinant LKB1:STRADα:MO25α complexes activate AMPKα1 catalytic domain in cell-free assays
To examine whether LKB1 activated AMPK on its own or whether the accessory subunits STRADα/β and MO25α/β were required, we expressed LKB1 tagged with glutathione-S-transferase (GST), FLAG-tagged STRADα/β and Myc-tagged MO25α/β in various combinations in HEK-293T cells, and purified the complexes on glutathione-Sepharose. We also used a GST-tagged kinase-inactive mutant of LKB1 (D194A), and a plasmid expressing GST alone, as controls. The complexes were purified on glutathione-Sepharose and incubated with the AMPKα1 catalytic domain in the presence of MgATP, and activation of the catalytic domain as well as phosphorylation of Thr172 (using a phosphospecific anti-pT172 antibody) was measured. Figure 3a shows that LKB1 alone did not significantly increase the activity, or phosphorylation of Thr172, of the AMPKα1 catalytic domain above the basal activity observed in the presence of GST alone (compare lanes 1 and 14). The same result was obtained with LKB1 that had been co-expressed with MO25α or MO25β (lanes 4 and 5), which was expected as these proteins do not interact with LKB1 in the absence of STRADα/β [21]. An LKB1:STRADα complex did give a small but significant activation, and Thr172 phosphorylation, of the AMPKα1 catalytic domain above the basal value (compare lanes 1 and 2). To produce, however, a large activation and phosphorylation of the AMPKα1 catalytic domain, a heterotrimeric complex containing LKB1, STRADα or STRADβ, and MO25α or MO25β was required (lanes 6 to 9). With the heterotrimeric complexes the degree of activation was in the order LKB1:STRADα:MO25α > LKB1:STRADα:MO25β ≈ LKB1:STRADβ:MO25α > LKB1:STRADβ:MO25β. The ability of LKB1:STRAD:MO25 complexes to activate AMPKα1 was dependent on LKB1 catalytic activity, because complexes of a catalytically inactive mutant of LKB1 (D194A) with the various combinations of STRADα/β and MO25α/β (lanes 10–13) were unable to activate or phosphorylate AMPKα1. The degree of activation obtained with the various complexes of wild-type LKB1 correlated well with the phosphorylation of Thr172, as assessed by probing blots with a phosphospecific antibody (pT172). The bottom three panels in Figure 3a, probed with anti-GST, anti-FLAG or anti-Myc antibodies, confirm that the relevant STRAD and MO25 subunit co-precipitated with LKB1 when DNAs encoding these subunits had been co-transfected. When STRADα was co-expressed with LKB1 in the absence of a MO25 subunit, the amount of STRADα subunit co-precipitated with LKB1 was reduced (compare lanes 2 and 3 with lanes 6 and 7).
Figure 3.
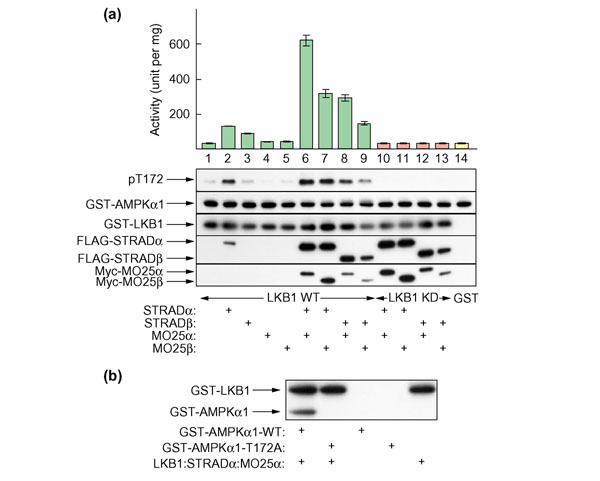
Recombinant LKB1:STRAD:MO25 complexes can efficiently activate the AMPKα1 catalytic domain via phosphorylation at Thr172. (a) The indicated combinations of GST-tagged wild-type LKB1 (WT, lanes 1–9), or kinase-dead (D194A; KD, lanes 10–13) LKB1 mutant, or GST-alone (lane 14), FLAG-tagged STRADα or STRADβ, and Myc-tagged MO25α or MO25β were coexpressed in HEK-293T cells, purified on glutathione-Sepharose and tested for their ability to activate GST-AMPKα1 catalytic domain (top panel). The results are expressed as the increase in the units of AMPK activity generated per mg full-length GST-AMPKα1 catalytic domain. Samples from each incubation were also analyzed by western blotting and probed using the indicated antibodies (from top to bottom): anti-pT172; anti-AMPKα1 catalytic domain (GST-AMPKα1); anti-GST to detect GST-LKB1; anti-FLAG to detect STRADα and STRADβ, and anti-Myc to detect MO25α and MO25β. All proteins migrated with the expected mobility, taking into account the epitope tags. The bottom three blots were conducted on blank reactions lacking GST-AMPKα1 catalytic domain, as the latter appeared to cause some interference with detection. (b) Recombinant GST-LKB1:STRADα:MO25α complex was used to phosphorylate wild-type GST-AMPKα1 catalytic domain (GST-α1-WT) or a T172A mutant (GST-α1-T172A) using [γ-32P]ATP as described in Materials and methods. The reaction was analyzed by SDS gel electrophoresis and autoradiography. Arrows show the migration of GST-LKB1 (which autophosphorylates) and GST-AMPKα1 catalytic domain.
Figure 3b provides evidence that Thr172 was the only site on the AMPKα1 catalytic domain phosphorylated by the LKB1:STRADα:MO25α complex. When the two proteins were incubated together in the presence of [γ-32P]ATP, the wild-type AMPKα1 catalytic domain became 32P-labeled, but a T172A mutant of the AMPKα1 catalytic domain did not.
AMPKK1, AMPKK2 and recombinant LKB1:STRAD:MO25 complexes also activate heterotrimeric AMPK complexes
Although most of the AMPKK assays in this study were conducted using the AMPKα1 catalytic domain as substrate, AMPKK1, AMPKK2 and the recombinant GST-LKB1:STRADα:MO25α complex also activated heterotrimeric AMPK complexes. We incubated rat liver AMPK (a mixture of α1 and α2 in complexes with β1 and γ1) with MgATP with or without each of the three AMPKK preparations. We then immunoprecipitated with anti-AMPKα1 or anti-AMPKα2 antibodies, and measured the activation of each isoform in the precipitate. The results (Figure 4a) show that the AMPKα1 and AMPKα2 complexes were activated by all three AMPKK preparations. Blotting of the three AMPKK preparations using anti-LKB1, anti-STRADα and anti-MO25α antibodies (Figure 4b) showed that activation of the heterotrimers was not simply proportional to the amount of these polypeptides in the preparation. Although the amounts of each AMPKK preparation used for Figure 4a had been chosen to yield a comparable degree of AMPK activation, there was much more LKB1, STRADα and MO25α in the recombinant LKB1:STRADα:MO25α complex than in either of the native complexes, and more of all three subunits in AMPKK2 than in AMPKK1. All three AMPKK preparations also activated recombinant α1β1γ1 and α2β1γ1 complexes prepared [22] by co-expression of recombinant DNA in CCL13 cells (not shown).
Figure 4.
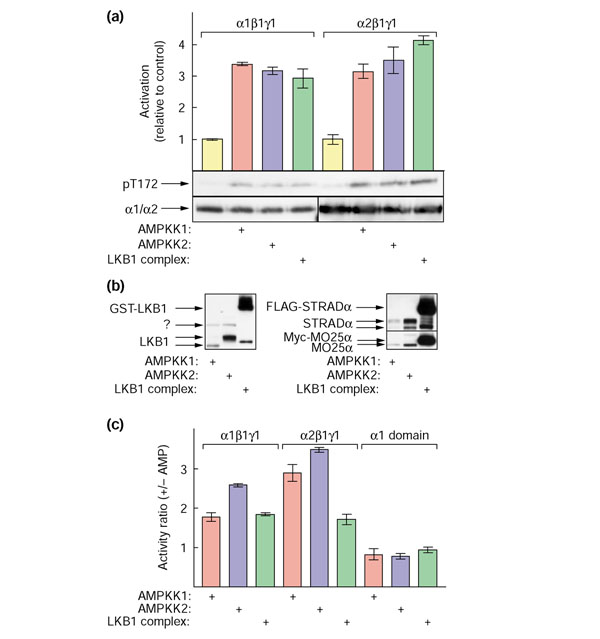
Activation and phosphorylation of heterotrimeric AMPK complexes by AMPKK1, AMPKK2 and recombinant GST-LKB1:STRADα:MO25α complexes, and the effect of AMP. (a) Activation of α1β1γ1 and α2β1γ1 complexes separated from purified rat liver AMPK. The AMPKα1- or AMPKα2-containing complexes were purified by immunoprecipitation and activation of the resuspended immunoprecipitates by the three AMPKK preparations examined. The results are expressed as activation relative to the control without added AMPKK. (b) Quantification by western blotting of the relative amounts of LKB1, STRADα and MO25α polypeptides in the three AMPKK preparations used in (a). A small amount of degradation is detectable due in part to the heavy loadings of the GST-LKB1 and FLAG-STRADα. The identity of the polypeptide labeled '?' in the anti-LKB1 blot is not known. (c) Effect of AMP on the activation of α1β1γ1 and α2β1γ1 heterotrimers of AMPK, and of GST-AMPKα1 catalytic domain, by AMPKK1, AMPKK2 and the recombinant GST-LKB1:STRADα:MO25α complex. AMPKK activity was measured as in Figure 3 with or without 200 μM AMP. The results are expressed as ratios of the activities obtained with and without AMP.
The assays in Figure 4a were conducted in the presence of 200 μM AMP. Figure 4c shows that when the AMPKα1β1γ1 or AMPKα1β1γ1 heterotrimers were used as substrate, the activation of all three AMPKK preparations was stimulated from 2- to 3.5-fold by AMP. When the AMPKα1 catalytic domain was used as substrate, however, the activation was not affected, or was even slightly inhibited, by AMP. The activity of the three AMPKK preparations was not significantly affected by the direct addition of phenformin to the assays up to 1 mM concentration, although concentrations above that started to inhibit AMPKK activity (not shown). These results indicate that neither AMP nor phenformin directly stimulates the LKB1:STRADα:MO25α complex.
Endogenous LKB1 that activates AMPK can be immunoprecipitated from 293 cells but not from HeLa cells
Figure 5a shows that AMPKK activity that activated the AMPKα1 catalytic domain above the basal activity, and phosphorylated Thr172, could be immunoprecipitated from untransfected HEK-293T cell extract using anti-LKB1 antibody (lane 1), but not pre-immune control immunoglobulin (lane 2). As reported previously [20,21], immunopre-cipitation of endogenous LKB1 resulted in the co-precipitation of STRADα and MO25α (lane 1). As a further control, we employed normal HeLa cells as an LKB1-null cell line, as it is known that LKB1 is not expressed in these cells [12]. Consistent with this, no LKB1, STRADα and MO25α subunits or AMPKK activity were immunoprecipitated from the same amount of HeLa cell-extract protein using anti-LKB1 antibody (lane 3). AMPKK activity and Thr172 phosphorylation, as well as detectable STRADα and MO25α subunits, were recovered following immunoprecipitation of LKB1 from a line of HeLa cells that stably express wild-type LKB1 [23] (lane 5). The LKB1, STRADα and MO25α polypeptides were still recovered in cells expressing a catalytically inactive mutant of LKB1, but AMPKK activity was not (lane 7). Although the LKB1 polypeptide was overexpressed to a large extent in the HeLa cells compared to the endogenous levels observed in 293 cells (compare lane 1 with lane 5 or 7), it is clear that the availability of STRADα and/or MO25α limits the activity in these cells. There was less AMPKK activity and Thr172 phosphorylation, as well as less co-precipitated STRADα and MO25α, in HeLa cells expressing LKB1 than in 293 cells, even though the LKB1 polypeptide was overexpressed. The AMPKK activity in the immunoprecipitates from 293 cells and HeLa cells expressing wild-type LKB1 correlated with the levels of STRADα and MO25α in the complex, rather than with the levels of LKB1.
Figure 5.
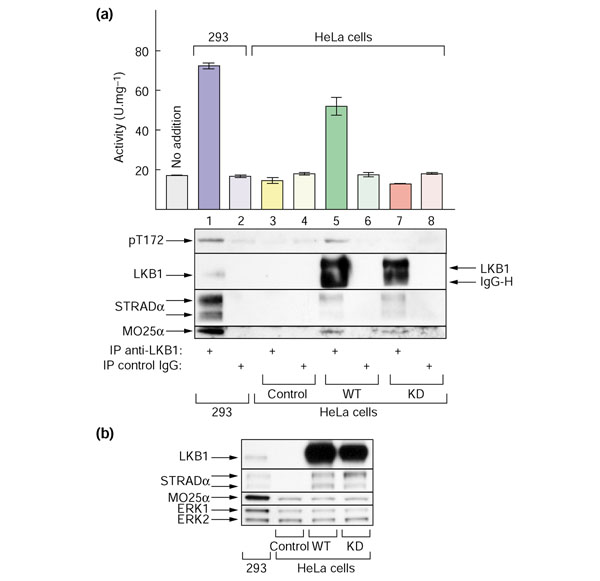
Endogenous AMPKK activity (that is, ability to activate AMPKα1 catalytic domain) can be immunoprecipitated from 293 cells using anti-LKB1 antibody, but activity can only be immunoprecipitated from HeLa cells if they stably express wild-type LKB1, but not a catalytically-inactive mutant. (a) LKB1 was immunoprecipitated from 0.5 mg cell extract derived from untransfected HEK-293T cells (lanes 1,2), untransfected HeLa cells (control; lanes 3,4), or HeLa cells stably expressing wild-type LKB1 (WT; lanes 5,6) or a kinase-dead LKB1 mutant (D194A; KD, lanes 7,8). Immunoprecipitation used anti-LKB1 (lanes 1, 3, 5, 7) or a pre-immune control immunoglobulin (IgG; lanes 2, 4, 6, 8). Samples of each immunoprecipitate were used to assay activation of GST-AMPKα1 catalytic domain, to analyze phosphorylation of GST-AMPKα1 catalytic domain on Thr172 (middle panel), and to determine by western blotting the recovery of LKB1 and its accessory subunits (bottom panels). In lanes 5 and 7 some immunoglobulin heavy chain (IgG-H) had eluted from the protein G-Sepharose despite the fact that it had been cross-linked: this explains why LKB1 may not appear to comigrate in lanes 1, 5 and 7. Also shown at left in the top panel is the basal activity obtained when the GST-AMPKα1-catalytic domain was incubated with MgATP on its own (no addition). (b) Whole cell lysates from the same cells were analyzed by SDS gel electrophoresis and blots probed using anti-LKB1, anti-STRADα, and anti-MO25α antibodies. They were also probed with anti-ERK1/2 antibodies as loading controls.
Figure 5b shows western blots of total lysates of the same cells. Although the expression of MO25α in HeLa cells was lower than in 293 cells, it was unaffected by overexpression of either wild-type or kinase-dead LKB1, as was expression of the protein kinases ERK1 and ERK2, used as loading controls. Interestingly, however, expression of the STRADα doublet was almost undetectable in the control HeLa cells but was readily detectable in cells stably expressing wild-type or kinase-dead LKB1.
Expression of LKB1 restores activation of AMPK in HeLa cells
The drug 5-aminoimidazole-4-carboxamide (AICA) riboside activates AMPK in intact cells by being taken up and immediately converted by adenosine kinase to AICA riboside monophosphate (ZMP), which mimics the effect of AMP on the AMPK system [24]. The anti-diabetic drug metformin activates AMPK in intact cells by a mechanism that is not known, although it does not involve changes in cellular adenine nucleotide content [25-27]. We have previously found (unpublished observations) that, although AMPK is expressed in HeLa cells, it is not activated either by AICA riboside or by metformin. A potential explanation for this is that HeLa cells do not express LKB1 ([12]; see also above). To examine whether expression of recombinant LKB1 might restore the ability of HeLa cells to respond to these drugs, we used the HeLa cell line that stably expresses wild-type LKB1 [23]. In these experiments we used phenformin, a close relative of metformin that we have found to activate AMPK more rapidly than metformin in other cell types. Figure 6a shows that neither AICA riboside nor phenformin activated AMPK above the basal level in control HeLa cells. In cells expressing wild-type LKB1 but not the kinase-inactive mutant, however, both AICA riboside and phenformin caused a robust activation, as well as a small increase in basal activity. The AMPK activity also correlated with the phosphorylation of Thr172 on AMPKα (shown by probing with the anti-pT172 antibody, Figure 6b) and with the phosphorylation of a downstream target of AMPK, acetyl-CoA carboxylase (ACC; shown by probing with a phosphospecific antibody against Ser-79, the primary AMPK site on that protein; Figure 6c). Interestingly, stable expression of wild-type LKB1 in the HeLa cells caused a small but reproducible degree of upregulation of expression of AMPKα1 and a marked down-regulation of expression of ACC (data not shown). These effects may be a consequence of the increase in basal AMPK activity shown in Figure 6a. Because the expression of these proteins was not uniform in these cells, in order to accurately quantify their phosphorylation status we simultaneously probed single blots of lysates using either anti-pT172 and anti-α1/α2 antibodies (to detect Thr172 phosphorylation and total AMPKα), or with anti-pACC and streptavidin (to detect Ser79 phosphorylation and total ACC). The two probing reagents used in each of these dual-labeling protocols were labeled with fluorescent dyes emitting in different regions of the infra-red spectrum, and the results were quantified in two separate channels using an infra-red laser scanner. In Figure 6b, these results are expressed as ratios of the signal obtained using the phosphospecific antibody to the signal obtained for the total protein, which corrects for different levels of expression of the proteins. This revealed that there was a good correlation between activation of AMPK and phosphorylation of Thr172. There was also a correlation with phosphorylation of ACC, although in this case AICA riboside appeared to have a larger effect than phenformin.
Figure 6.
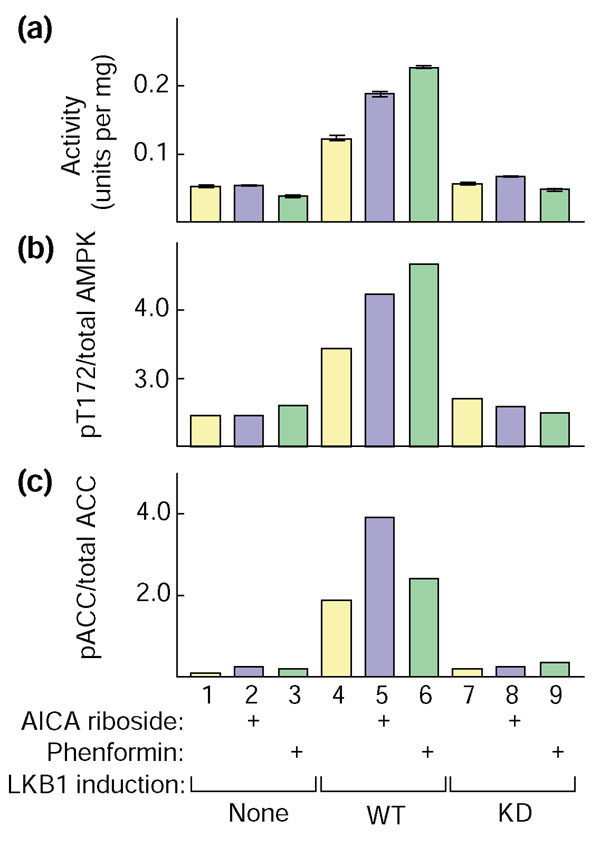
Restoration of the ability of AMPK to be activated, and AMPK and acetyl-CoA carboxylase to be phosphorylated, by AICA riboside and phenformin in HeLa cells following expression of LKB1. Control HeLa cells (lanes 1,2,3), HeLa cells expressing wild-type LKB1 (WT; lanes 4,5,6) or kinase-inactive mutant LKB1 (D194A; KD, lanes 7,8,9) were incubated for 60 min with no further addition, with 2 mM AICA riboside or 10 mM phenformin, and lysed. (a) Endogenous AMPK was immunoprecipitated from the cell extracts and assayed. (b) The cell lysates was immunoblotted with antibodies recognizing AMPKα1 phosphorylated at Thr172 or total AMPKα1; the results were analyzed using the LI-COR Odyssey™ IR imager as described in the Materials and methods section, and are expressed as a ratio of the two signals. (c) The cell lysates were analyzed by western blotting and the membranes probed with antibodies recognizing ACC phosphorylated at Ser79, or streptavidin to determine total AMPKα1. The results were analyzed using the LI-COR imager as for (b).
AMPK activation is defective in immortalized fibroblasts from LKB-/- mouse embryos
We performed further experiments with immortalized mouse embryo fibroblasts (MEF cells) from embryonic-day (E) 9.5 embryos of LKB1-/- knockout mice. In cells from control LKB1+/+ embryos, AICA riboside and phenformin caused twofold and three-fold activation of endogenous AMPK, but this was completely absent from the LKB1-/- cells (Figure 7). The basal activity of AMPK was also about 60% lower in the LKB1-/- cells. Figure 7 also confirms, by western blotting of cell lysates, that the expression of LKB1 was absent from LKB1-/- cells, that the expression of the AMPKα subunits was normal, and that the phosphorylation of Thr172 on the AMPKα subunits correlated with AMPK activity.
Figure 7.
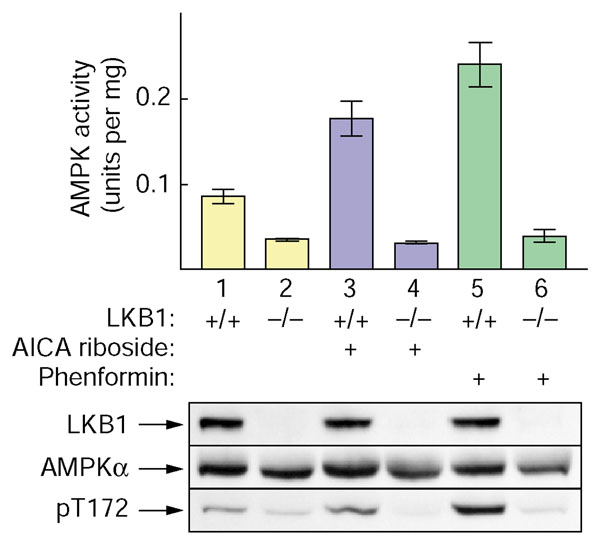
AMPK is activated and phosphorylated in response to AICA riboside and phenformin in LKB1+/+ but not in LKB1-/- MEF cells. Cells immortalized from control embryos LKB1+/+ and LKB1-/- knockout embryos [14], were incubated with AICA riboside (2 mM) or phenformin (10 mM) for 1 hour. Lysates were prepared and AMPK activity (expressed as units per mg total lysate protein) determined in immunoprecipitates made using a mixture of anti-AMPKα1 and anti-AMPKα2 antibodies. Lysates were also analyzed by SDS gel electrophoresis and blots probed using anti-LKB1, anti-AMPKα1/β2, and anti-pT172 antibodies.
Discussion
Our results provide strong evidence that LKB1:STRAD:MO25 complexes represent the major upstream kinases acting on AMPK, although they do not rule out the possibility that the complex might contain additional components. The key evidence may be summarized as follows. First, during previous extensive efforts to purify from rat liver extracts activities that activate dephosphorylated AMPK, ([4] and subsequent unpublished work), we have not detected any activities other than AMPKK1 and AMPKK2, at least under the assay conditions used. Second, both AMPKK1 and AMPKK2 purified from rat liver contained LKB1, STRADα and MO25α that were detectable by western blotting and whose presence correlated with AMPKK activity across the column fractions (Figure 1). Third, the ability of the AMPKK1 and AMPKK2 fractions to activate AMPK was almost completely eliminated by immunoprecipitation with anti-LKB1 antibody, but not a control immunoglobulin. Activity was also detected, along with the LKB1, STRADα and MO25α polypeptides, in the anti-LKB1 immunoprecipitates but not in the control immunoprecipitates (Figure 2). Fourth, the AMPKK activity in AMPKK1 and AMPKK2 was not a contaminant that co-precipitated with anti-LKB1 antibody, because recombinant complexes of GST-LKB1, STRAD and MO25 expressed in 293 cells and purified on glutathione-Sepharose also activated the AMPKα1 catalytic domain efficiently, and phosphorylated Thr172 (Figure 3). Complexes formed from a catalytically inactive mutant LKB1 failed to activate or phosphorylate AMPK. Phosphorylation of the AMPKα1 catalytic domain by this recombinant complex occurred exclusively at Thr172, because the wild-type AMPKα1 catalytic domain, but not a T172A mutant, could be phosphorylated using [γ-32P]ATP and the GST-LKB1:STRADα:MO25α complex. Fifth, although most of the experiments in this study were conducted using the bacterially expressed AMPKα1 catalytic domain as substrate, AMPKK1, AMPKK2 and recombinant LKB1-STRADα-MO25α complexes also efficiently activated heterotrimeric AMPK complexes, both the α1β1γ1 and α2β1γ1 isoforms (Figure 4). Sixth, HeLa cells, unlike HEK 293T cells, do not express LKB1 (Figure 5) and therefore represent a natural 'knockout' cell line. The drugs AICA riboside and phen-formin, which activate AMPK in other cell types via distinct mechanisms [24,27], did not activate AMPK in HeLa cells. In cells stably transfected with DNA that expressed wild-type LKB1 (but not a catalytically inactive mutant), however, the ability of AICA riboside and phenformin to activate AMPK, to phosphorylate Thr172 on the AMPKα subunit, and to cause phosphorylation of a downstream target (ACC) was restored (Figure 6). This experiment proves that (in the presence of STRADα and MO25α) LKB1 is sufficient for AMPK activation, but does not prove that it is necessary, because expression of upstream kinases other than LKB1 might also be defective in HeLa cells. Figure 5 also confirms that STRADα and MO25α are necessary to generate an active complex because, although the LKB1 polypeptide was greatly overexpressed in the stably transfected HeLa cells compared to the endogenous level in 293 cells, the AMPKK activity, and the amounts of STRADα and MO25α, in the anti-LKB1 immunoprecipitate, were actually less. This suggests that the amount of active LKB1 was limited by the availability of STRADα and MO25α. Seventh, in LKB1+/+ MEF cells, AMPK became activated in response to both AICA riboside and phenformin. In LKB1-/- MEF cells, however, the basal activity of AMPK was lower and AICA riboside and phenformin failed to activate AMPK. These results show that LKB1 is both necessary and sufficient for AMPK activation, at least in MEF cells.
All of the assays of the activity of LKB1 and its complexes described in this article, whether using the AMPKα1 catalytic domain or AMPK heterotrimers as substrate, utilized MgATP as the co-substrate. Previous studies of the kinase activity of LKB1, whether utilizing autophosphorylation [12], or p53 [28] or myelin basic protein [20] as substrates, had used MnATP as co-substrate and had reported that there was no activity with the more physiological MgATP complex. Thus, unlike previously used substrates, AMPK is a good substrate for LKB1 complexes even using the physiologically relevant divalent metal ion.
We have previously reported that the activation of AMPK by AMPKK1 (called at that time AMPKK) was stimulated by AMP, and presented evidence favoring the hypothesis that AMP acted not only by binding to the downstream kinase and making it a better substrate, but also by activating the upstream kinase [6]. The results in Figure 4c support the first hypothesis but do not support the second. Using α1β1γ1or α2β1γ1 AMPK complexes as substrate, activation by AMPKK1, AMPKK2 or LKB1 was stimulated from 2-to 3.5-fold by AMP. When using the AMPKα1 catalytic domain as substrate, however, AMP had no effect, or even slightly inhibited activation. The AMPKα1 catalytic domain is not allosterically activated by AMP [29], and AMP binding appears to be a function of the γ subunit ([30], and unpublished observations). Taken together with previous results [6,24,31], these data support the idea that the effects of AMP on the kinase cascade are all mediated through binding to the downstream kinase, AMPK. The previous report that AMP stimulated the upstream kinase was obtained using a less pure AMPKK preparation [6], and we have been unable to reproduce this with the more purified preparations utilized here.
Both AMPKK1 and AMPKK2 appear to contain LKB1, STRADα and MO25α, and thus it is not clear at present why they resolve on Q-Sepharose chromatography. One interesting difference is that the LKB1 polypeptide in AMPKK1 migrated significantly faster on SDS gels than that in AMPKK2 (Figures 1 and 2). LKB1 is known to be phosphorylated at up to eight sites, and is also farnesylated at Cys433, near the carboxyl-terminus [23,28,32,33], suggesting that the difference in mobility might be due to a difference in covalent modification. It did not appear to be due to differential phosphorylation, however, because neither incubation with MgATP, nor protein phosphatase treatment, produced a shift in mobility of the LKB1 polypeptides in either AMPKK1 or AMPKK2 (Figure 1c). Another difference between AMPKK1 and AMPKK2 was their Stokes radii estimated by size exclusion chromatography (5.7 versus 5.2 nm respectively). By combining estimates of Stokes radius and sedimentation coefficient, we previously estimated the molecular mass of AMPKK1 to be 195 kDa [4], and assuming a similar shape our estimate of the Stokes radius of AMPKK2 would suggest a mass of about 175 kDa. These values are close to, although slightly larger than, the calculated mass of 140 kDa for a 1:1:1 LKB1:STRADα:MO25α complex. Although we cannot rule out the possibility that AMPKK1 and/or AMPKK2 contain additional associated protein(s) other than LKB1, STRADα/β and MO25α/β, it is also possible that differences in covalent modification might affect the shape of the complex and hence the Stokes radius. Whatever the reason for the difference in electrophoretic and chromatographic behavior of AMPKK1 and AMPKK2, a clear conclusion from Figure 4 is that, for the same amount of LKB1, STRADα and MO25α polypeptides, the former was more active than the latter. Although further work is required to explain these differences, they might be caused by the same covalent modifications that alter the mobility on SDS gels. Figure 4 also shows that, for the same amount of LKB1, STRADα and MO25α polypeptides, both AMPKK1 and AMPKK2 were much more active than the recombinant complex. The low activity of the latter might be explained by the presence of the purification tag on each subunit, by imperfect folding or assembly, or by an altered level of covalent modification, when the complex is overexpressed. As mentioned above, our data do not rule out the possibility that the recombinant LKB1 complex may be lacking additional subunit(s) present in the endogenous AMPKK1 and AMPKK2 complexes.
Our present results confirm, using a probable physiological substrate, previous findings using an artificial substrate (myelin basic protein) that a STRAD subunit stimulates the kinase activity of LKB1 [20], and that the MO25 subunit stimulates the activity further, probably by stabilizing the LKB1:STRAD complex [21]. No AMPKK activity was obtained with recombinant LKB1 unless a STRAD subunit was also expressed, and the activity was increased substantially by the additional presence of a MO25 subunit (Figure 3). It was also noticeable that the amount of STRADα and STRADβ that co-precipitated with LKB1 was greatly enhanced by the co-expression of MO25α or MO25β (Figure 3), consistent with previous findings [21]. Another new result in this article is that STRADα protein (unlike MO25α) was not detectable in HeLa cells unless either wild-type or kinase-dead LKB1 was stably expressed (Figure 6b). These results suggest that STRADα is normally complexed with LKB1 in the cell, and that STRAD is unstable in the absence of LKB1. The exact mechanism by which the STRAD and MO25 subunits activate LKB1 remains unclear, but these accessory subunits introduce scope for additional regulation of the kinase. It is already known that LKB1 phosphorylates STRADα at two distinct sites [20], and that STRADα and MO25α form a complex that retains LKB1 in the cytoplasm [21].
People with PJS are heterozygous for mutations in LKB1, and further work is required to establish whether loss of one allele of LKB1 could affect AMPK activation in these patients. An interesting unanswered question is whether activation of AMPK can explain the ability of LKB1 to act as a tumor suppressor and to arrest cell growth and proliferation. This certainly seems plausible, because apart from the fact that AMPK is a general inhibitor of biosynthesis [1,2], there is accumulating evidence that it can regulate cell proliferation and apoptosis. For example, activation of AMPK inhibits proliferation of HepG2 cells by stabilizing p53 [34]. Interestingly, expression of LKB1 in G361 cells that normally lack expression of the kinase causes an arrest in G1 phase of the cell cycle that is associated with an induction of p21 and is dependent on p53 [12,13].
Another exciting possibility is that LKB1:STRAD:MO25 complexes might also act as upstream kinases for other protein kinases, in the same manner that PDK1 phosphorylates threonine residues in the activation loop of a number of kinases of the 'AGC' subfamily [35]. A dendrogram showing the relationships between catalytic domain sequences of 518 human protein kinases encoded in the human genome [36] shows that the AMPKα1 and AMPKα2 subunits lie on a small sub-branch also containing eight other protein kinases (NuaK1, NuaK2, BrsK1, BrsK2, SIK, QIK, QSK and MELK), most of which either have not been studied previously or have very little known about them. An alignment of the activation loop sequences of these kinases is shown in Additional data file 2 with the online version of this article and show that, as well as conservation of the threonine residue phosphorylated by LKB1 in AMPK, they have other conserved motifs that are not present in other protein kinases known to be activated by other upstream kinases. It remains to be determined whether the other kinases in the AMPK subfamily are activated by phosphorylation of the conserved threonine residue by LKB1:STRAD:MO25 complexes, but if this is the case these other kinases might mediate some of the tumor suppressor functions of LKB1.
A significant number of inherited forms of PJS found in certain families do not exhibit mutations in the LKB1 gene [37,38], indicating that there could be other causative loci for PJS. On the basis of the results presented here it would be very interesting to examine whether mutations in the genes encoding STRADα or β, MO25α or β, or any of the subunits of AMPK or of the AMPK-like subfamily of kinases, are found in PJS patients who do not have mutations in the LKB1 gene.
While this article was under review, two papers were published that are relevant to our results. Hong et al. [39] reported that FLAG-tagged LKB1 expressed in, and purified from, COS7 cells would activate a recombinant AMPK heterotrimer, and phosphorylate the α subunit at Thr172, in cell-free assays. This is consistent with our results, although these authors provided no evidence that LKB1 acts on AMPK in vivo, or that LKB1 required the presence of the STRAD and MO25 subunits to phosphorylate AMPK. Spicer et al. [40] reported evidence, based on expression of recombinant LKB1 in cultured cells, suggesting that it might act upstream of the PAR1A protein kinase. PAR1A (also known as MARK-3 [41]) lies with three closely related protein kinases (MARK-1, MARK-2, MARK-4) on a branch of the human kinase tree [36] immediately adjacent to AMPK-α1 and -α2 and the eight AMPK-like kinases discussed above. Although Spicer et al. [40] did not provide evidence that LKB1 directly phosphorylated PAR1A, the results of our study suggest that this might be the case. The sequence of the activation segment of PARIA is given in Additional data file 2 (available with the online version of this article).
Conclusions
Our results provide strong evidence, both in cell-free assays and in intact cells, that complexes between LKB1, STRADα/β and MO25α/β constitute the long sought-after upstream kinases that activate AMPK via phosphorylation at Thr172 in the activation loop. Because it is already known that pharmacological activation of AMPK causes a general inhibition of biosynthesis, as well as a p53-dependent arrest in G1 phase of the cell cycle, activation of AMPK by LKB1 might explain, at least in part, the ability of LKB1 to act as a tumor suppressor. LKB1 may also act as an upstream kinase for other members of the AMPK-like subfamily of protein kinases.
Materials and methods
Materials, proteins and antibodies
Protein G-Sepharose, glutathione-Sepharose and prepacked Q-Sepharose columns were from Amersham Pharmacia Biotech, Little Chalfont, UK. The GST-AMPKα1 catalytic domain, and a T172A mutant, were expressed in Escherichia coli and purified as described previously [29]. Sheep antibodies against the α1 and α2 subunits of AMPK [22], human LKB1, MO25α and MO25β [21], and phosphospecific antibody against the Thr172 site on the AMPKα subunit (anti-pT172) [42] were described previously. Sheep antibody against AMPKα1 catalytic domain was raised against the peptide CDPMKRATpIKDIRE (cysteine + residues 252–264 of rat AMPKα1 given in the single-letter code for amino acids; Tp, phosphothreonine) using methods described for anti-pT172 [42]. Although designed as a phosphospecific antibody, it recognizes the GST-AMPKα1 catalytic domain expressed in bacteria and recognition is not affected by protein phosphatase treatment. The monoclonal antibody against STRADα was described previously [20]. Monoclonal anti-GST and anti-FLAG epitope antibodies were from Sigma (Poole, UK). Anti-Myc antibodies were prepared by ammonium sulfate precipitation of medium from Myc1-9E10 hybridoma cells grown in RPMI 1640 medium supplemented with 2 mM glutamine and 15% (v/v) fetal bovine serum. Anti-Erk1/2 antibodies were from Cell Signaling Technology (New England Biolabs, Hitchin, UK). The DNA constructs encoding GST-LKB1 (wild-type and kinase-dead) in the pEBG-2T vector [28], and FLAG-STRADα, FLAG-STRADβ, Myc-MO25α and Myc-MO25β in the pCMV5 vector [21] have been described previously. PP1γ was expressed in E. coli [43], and PP2A1 was purified from rabbit skeletal muscle [43]. Sources of other materials and proteins were as described previously [4].
Enzyme assays
AMPK [4], PP1γ and PP2A1 [43] were assayed, and units defined, as described previously. AMPKK was assayed as follows (based on [27]). A fusion protein between the kinase domain of the α1 subunit of AMPK and glutathione-S-transferase (GST-AMPKα1) was expressed in E. coli [29]. Although some preparations of GST-AMPKα1 show evidence of proteolytic degradation, only the full length GST-AMPKα1 is phosphorylated (J.L.R., unpublished observations). The amount of full-length GST-AMPKα1 was quantified by den-sitometry of Coomassie-Blue-stained gels, using bovine serum albumin as standard. The E. coli lysate expressing GST-AMPKα1 was adsorbed onto glutathione-Sepharose beads (Amersham-Pharmacia) such that the final concentration of kinase after maximal activation using MgATP and AMPKK in the assay below was 1 unit in the standard kinase assay per 5 μl of beads. The slurry was washed with 4 × 1 ml of IP buffer (50 mM Tris-HCl, pH 7.4 at 4°C, 50 mM NaF, 5 mM Na pyrophosphate, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM dithiothreitol (DTT), 1 mM benzamidine, 0.1 mM phenylmethane sulfonyl fluoride, 1 M NaCl) to remove unbound proteins. It was then washed in 3 × 1 ml of assay buffer (50 mM Na Hepes, pH 7.4, 1 mM DTT, 0.02% Brij-35).
For the kinase kinase assay, the AMPKK preparation was incubated with 10 μl of a 50% slurry of the glutathione-Sepharose beads with bound GST-AMPKα1, plus 200 μM AMP, 200 μM ATP, 5 mM MgCl2 in assay buffer in a final volume of 25 μl. After incubation for 20 min at 30°C on a rotary shaker, the beads were washed with 4 × 1 ml of IP buffer and 3 × 1 ml of assay buffer prior to a standard AMPK assay. The units of AMPKK are the units of AMPK generated in the assay, expressed per mg of full length GST-AMPKα1 protein used. Rapid lysis of cells for AMPK assays was as described previously [44]. AMPK and AMPKK assays were carried out in triplicate and results are expressed as mean ± standard deviation.
Purification of AMPKK1 and AMPKK2
AMPKK was purified to the Blue-Sepharose stage as described previously [4]. The flow-through from this column was adjusted to 160 mM NaCl by dilution in buffer A (50 mM Hepes, 10% (w/v) glycerol, 0.02% (w/v) Brij-35, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM benzamidine, 0.1 mM PMSF, 1 μg/ml soybean trypsin inhibitor), and applied to a high performance Q-Sepharose HiLoad 16/10 column in buffer A plus 160 mM NaCl at 3 ml/min. The column was washed in buffer A plus 160 mM NaCl until the A280 was < 0.05, and AMPKK activity was then eluted with a linear gradient (120 ml) from 160–400 mM NaCl in buffer A.
Expression of recombinant LKB1 complexes in HEK-293T cells
Various combinations of GST-tagged LKB1, FLAG-tagged STRADα or STRADβ, and Myc-tagged MO25α or MO25β were expressed in HEK293 cells and the complexes purified on glutathione-Sepharose as described previously [21].
Phosphorylation of GST-α1 catalytic domain using [γ-32P]ATP
GST-AMPKα1 catalytic domain (50 μg/ml), either wild-type or a T172A mutant [29] was incubated for 30 min at 30°C with 5 mM MgCl2 and [γ-32P]ATP (200 μM; approximately 750 cpm/pmole) in the presence or absence of the GST-LKB1:STRADα:MO25α complex (20 units/ml). The reaction was terminated by the addition of SDS sample buffer (Invitrogen, Paisley, UK), the polypeptides resolved by SDS gel electrophoresis and the dried gel subjected to autoradiography.
Preparation of and activation of AMPK heterotrimers
AMPK was purified from rat liver as described previously [4]. Dephosphorylation, with PP2A, addition of okadaic acid to inhibit the phosphatase, and incubation with AMPKK was as described previously [4]. The reaction was stopped by adding 5 μl of 0.5 M EDTA to 20 μl of the dephosphorylated AMPK, and 20 μl of the mixture was then incubated for 2 h at 4°C with 75 μl of a 15% suspension of anti-AMPKα1 or anti-AMPKα2 antibodies bound to protein G-Sepharose [44] plus 200 μl of IP buffer. The beads were recovered by centrifugation (14,000 × g for 2 min) and washed twice with IP buffer and twice with 50 mM Na Hepes buffer, pH 7.4. AMPK assays were then carried out on the resuspended immunoprecipitates [44]. To obtain recombinant AMPK, plasmids encoding Myc-AMPKα1 or Myc-AMPKα2, AMPKβ1 and AMPKγ1 were co-expressed in CCL13 cells [22], and cells harvested by the rapid lysis method [44]. Lysates were immunoprecipitated with anti-Myc antibody and resuspended in 50 mM Na HEPES, 1 mM dithiothreitol, 0.02% (w/v) Brij-35, pH 7.5, and assayed as above.
Immunoprecipitation of endogenous LKB1
Immunoprecipitation of endogenous LKB1 using anti-human antibody and protein G-Sepharose has been described previously [21]. The kinase kinase assays were conducted using GST-AMPKα1 catalytic domain as substrate in shaking incubators as described previously for immuno-precipitate assays of AMPK [44].
HeLa cells expressing LKB1
The generation and culture conditions of HeLa cells stably expressing inducible (Tet-ON) wild-type or kinase-inactive mutant LKB1, and conditions for their culture, has been described previously [23].
Production of immortalized mouse embryo fibroblasts
Wild-type and LKB1-/-E9.5 embryos [14] were minced into small fragments and placed in culture in Dulbecco's modified Eagle's Medium supplemented with penicillin, streptomycin, glutamine, 10% fetal bovine serum (AutogenBioclear, Santa Cruz, USA), and 10% conditioned medium collected from day-3 cultures of wild-type fibroblasts. The cultures were subsequently allowed to expand for 5 days, after which they were passaged according to a modified 3T3 protocol [45]. High-passage cultures that expanded were considered immortalized.
Protein analysis and electrophoresis
Protein concentration was determined using the dye-binding method of Bradford [46]. SDS gel electrophoresis used precast Bis-Tris 4–12% gradient polyacrylamide gels, in the MOPS buffer system (Invitrogen), except for analysis of acetyl-CoA carboxylase, where pre-cast 3–8% Tris-acetate gels (Invitrogen) were used. Proteins were transferred to nitrocellulose membranes (BioRad, Hemel Hempstead, UK) using the Xcell II Blot Module (Invitrogen).
Detection of western blots by infra-red imaging
To analyze phosphorylation of ACC, membranes were incubated in LI-COR Odyssey™ Blocking buffer for 1 h. Anti-pACC antibody (1.46 μg/ml in blocking buffer containing Tween-20 0.2% v/v) was then added and left shaking for 1 h. The membranes were washed 6 × 5 min with TBS (10 mM Tris-HCl, pH 7.4, 0.5 M NaCl) plus Tween-20 (0.2% v/v). The membranes were immersed in blocking buffer containing Tween-20 (0.2% v/v) and 1 μg/ml anti-sheep IgG conjugated to IR dye 680 (Molecular Probes, Leiden, The Netherlands) and 1 μg/ml streptavidin conjugated to IR Dye 800 (Rockland Inc., from Lorne Laboratories, Reading, UK) and left shaking for 1 h, protected from light. The membranes were then washed 6 × 5 min using TBS-Tween (0.2%) and 1 × 5 min in PBS. The membranes were scanned in two different channels using the Odyssey IR imager, the results quantified using Odyssey software and expressed as a ratio of the signal obtained with the pACC antibody to that obtained with streptavidin. Analysis of phosphorylation of GST-α1 catalytic domain was similar except that the 4–12% Bis-Tris gels were used, and the membranes were simultaneously probed for 1 h with the sheep anti-pT172 and anti-AMPKα1 catalytic domain antibodies, directly labeled with the IR dye 680 and IR dye 800 respectively, according to manufacturers' instructions.
Additional data files
The following additional materials are available with the online version of this article: Additional data file 1, showing an alignment of the kinase domains of Tos3, Pak1, CaMKKβ, LKB1 and Elm1, and Additional data file 2, showing an alignment of the activation loop of the kinase subgroup phylogenetically close to LKB1.
Supplementary Material
Alignment of the amino acid sequences of Tos3, Pak1 and Elm1 from Saccharomyces cerevisiae, and LKB1 and CaMKKβ (β3 variant) from humans. The alignment was created using PILEUP from the GCG suite [48] and the consensus sequence determined, and color added, using BOXSHADE 3.2.1 [49]. Residues that are identical in at least half of the sequences are colored red, with conservative changes in blue. The consensus sequence is represented by an upper case letter if the residue at a particular position is identical in all sequences, and by a lower case letter if at least half are conserved. Only the conserved central regions containing the kinase domains are shown (the kinase domain of LKB1 is approximately residues 44-309).
Sequence alignment of the activation loop sequences of AGC subfamily kinases (PKAα and PKCα, which are not thought to be activated by LKB1) with the activation loop residues of the AMPK/SNF1 subfamily of protein kinases. The methods used and color-coding are as in Additional data file 1. The threonine residues corresponding to Thr172 of AMPK ('P') and other residues mentioned in the text are indicated with downward arrows. All sequences are from humans, except AtSnRK1-α1, AtSnRK1-α1 and ScSnf1, which are the Arabidopsis thaliana (AtSnRK1, SNF1-related kinase-1) and Saccharomyces cerevisiae (ScSnf1) homologs of AMPK. Although the human, A. thaliana and S. cerevisiae homologs, as well as the PKAα and PKCα kinases, are known to be activated by phosphorylation of the threonine residue equivalent to Thr172, it has yet to be established whether the other AMPK subfamily members (NuaK1, NuaK2, BrsK1, BrsK2, SIK, QIK, QSK, MELK and PAR1A) are activated by phosphorylation at this residue, and whether LKB1:STRAD:MO25 complexes can mediate this phosphorylation.
Acknowledgments
Acknowledgements
This study was supported by the Wellcome Trust (D.G.H.) the UK Medical Research Council (D.G.H. and D.R.A.), the Association for International Cancer Research (D.R.A), Diabetes UK (D.R.A.), and by the Finnish Cancer Organization, Sigrid Juselius Foundation, and the Academy of Finland (T.P.M.). J.L.R. was supported by a studentship from the MRC and L.U. is a student of the Helsinki Biomedical Graduate School. We thank Philip Cohen for helpful discussions, Moustapha Aoubala for preparation of antibodies, Agnieszka Kieloch for assistance with tissue culture, Debbie Mander for preparation of LKB1:STRAD:MO25 complexes, Annette Baas and Hans Clevers for providing us with the STRADα cDNA and STRADα antibodies, Greg Stewart for the preparation of the T172A mutant of the AMPKα1 catalytic domain, and Antti Ylikorkala and Derrick Rossi for helping to generate the MEF cultures.
Correspondence regarding LKB1 should be addressed to Dario Alessi and regarding AMPK to Grahame Hardie.
Contributor Information
Dario R Alessi, Email: d.r.alessi@dundee.ac.uk.
D Grahame Hardie, Email: d.g.hardie@dundee.ac.uk.
References
- Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/S0014-5793(03)00560-X. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. BioEssays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- Coven DL, Hu X, Cong L, Bergeron R, Shulman GI, Hardie DG, Young LH. Physiologic role of AMP-activated protein kinase (AMPK) in the heart: graded activation during exercise. Am J Physiol Endocrinol Metab. 2003;285:E629–E636. doi: 10.1152/ajpendo.00171.2003. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver, and identification of threonine-172 as the major site at which it phosphorylates and activates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJR, Scmidt MC, Hardie DG. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol. 2003;13:1299–1305. doi: 10.1016/S0960-9822(03)00459-7. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 53-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270:27186–27191. doi: 10.1074/jbc.270.45.27186. [DOI] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- Hemminki A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell Mol Life Sci. 1999;55:735–750. doi: 10.1007/s000180050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, Offerhaus JA. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- Boudeau J, Sapkota G, Alessi DR. LKB1, a protein kinase regulating cell proliferation and polarity. FEBS Lett. 2003;546:159–165. doi: 10.1016/S0014-5793(03)00642-2. [DOI] [PubMed] [Google Scholar]
- Tiainen M, Ylikorkala A, Makela TP. Growth suppression by LKB1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum Mol Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- Ylikorkala A, Rossi DJ, Korsisaari N, Luukko K, Alitalo K, Henke-meyer M, Makela TP. Vascular abnormalities and deregulation of VEGF in LKB1-deficient mice. Science. 2001;293:1323–1326. doi: 10.1126/science.1062074. [DOI] [PubMed] [Google Scholar]
- Jishage K, Nezu J, Kawase Y, Iwata T, Watanabe M, Miyoshi A, Ose A, Habu K, Kake T, Kamada N, et al. Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci USA. 2002;99:8903–8908. doi: 10.1073/pnas.122254599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharp-less NE, Loda M, Carrasco DR, DePinho RA. Loss of the LKB1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Nakau M, Ishikawa TO, Seldin MF, Oshima M, Taketo MM. Gastrointestinal hamartomatous polyposis in LKB1 heterozygous knockout mice. Cancer Res. 2002;62:2261–2266. [PubMed] [Google Scholar]
- Rossi DJ, Ylikorkala A, Korsisaari N, Salovaara R, Luukko K, Launonen V, Henkemeyer M, Ristimaki A, Aaltonen LA, Makela TP. Induction of cyclooxygenase-2 in a mouse model of Peutz-Jeghers polyposis. Proc Natl Acad Sci USA. 2002;99:12327–12332. doi: 10.1073/pnas.192301399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Hepatocellular carcinoma caused by loss of heterozygosity in LKB1 gene knockout mice. Cancer Res. 2002;62:4549–4553. [PubMed] [Google Scholar]
- Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutowski M, Prescott AR, Clevers HC, Alessi DR. MO25 isoforms interact with the STE20-related pseudokinase STRADα/β and enhance their ability to bind, activate and localise the LKB1 tumour suppressor in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Salt I, Scott J, Hardie DG, Carling D. The α1 and α2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996;397:347–351. doi: 10.1016/S0014-5793(96)01209-4. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Deak M, Kieloch A, Morrice N, Goodarzi AA, Smythe C, Shiloh Y, Lees-Miller SP, Alessi DR. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J. 2002;368:507–516. doi: 10.1042/BJ20021284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside: a specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI200113505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The anti-diabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell growth. J Biol Chem. 2001;276:19469–19482. doi: 10.1074/jbc.M009953200. [DOI] [PubMed] [Google Scholar]
- Scott JW, Norman DG, Hawley SA, Kontogiannis L, Hardie DG. Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J Mol Biol. 2002;317:309–323. doi: 10.1006/jmbi.2001.5316. [DOI] [PubMed] [Google Scholar]
- Cheung PCF, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase γ subunit iso-forms and their role in AMP binding. Biochem J. 2000;346:659–669. doi: 10.1042/0264-6021:3460659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Helps NR, Cohen PTW, Hardie DG. 5'-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2Cα and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- Collins SP, Reoma JL, Gamm DM, Uhler MD. LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem J. 2000;345:673–680. doi: 10.1042/0264-6021:3450673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J. 2002;362:481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5'-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- Alessi DR. Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem Soc Trans. 2001;29:1–14. doi: 10.1042/0300-5127:0290001. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Buchet-Poyau K, Mehenni H, Radhakrishna U, Antonarakis SE. Search for the second Peutz-Jeghers syndrome locus: exclusion of the STK13, PRKCG, KLK10, and PSCD2 genes on chromosome 19 and the STK11IP gene on chromosome 2. Cytogenet Genome Res. 2002;97:171–178. doi: 10.1159/000066620. [DOI] [PubMed] [Google Scholar]
- Resta N, Stella A, Susca FC, Di Giacomo M, Forleo G, Miccolis I, Rossini FP, Genuardi M, Piepoli A, Grammatico P, Guanti G, et al. Two novel mutations and a new STK11/LKB1 gene isoform in Peutz-Jeghers patients. Hum Mutat. 2002;20:78–79. doi: 10.1002/humu.9046. [DOI] [PubMed] [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer J, Rayter S, Young N, Elliott R, Ashworth A, Smith D. Regulation of the Wnt signalling component PAR1A by the Peutz-Jeghers syndrome kinase LKB1. Oncogene. 2003;22:4752–4756. doi: 10.1038/sj.onc.1206669. [DOI] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Sugden C, Crawford RM, Halford NG, Hardie DG. Regulation of spinach SNF1-related (SnRK1) kinases by protein kinases and phosphatases is associated with phosphorylation of the T loop and is regulated by 5'-AMP. Plant J. 1999;19:1–7. doi: 10.1046/j.1365-313X.1999.00532.x. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Street AJ, Cohen P, Cohen PTW. Inhibitor-2 functions like a chaperone to fold 3 expressed isoforms of mammalian protein phosphatase-1 into a conformation with the specificity and regulatory properties of the native enzyme. Eur J Biochem. 1993;213:1055–1066. doi: 10.1111/j.1432-1033.1993.tb17853.x. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Salt IP, Davies SP. Analysis of the role of the AMP-activated protein kinase in the response to cellular stress. Methods Mol Biol. 2000;99:63–75. doi: 10.1385/1-59259-054-3:63. [DOI] [PubMed] [Google Scholar]
- Denhardt DT, Edwards DR, McLeod M, Norton G, Parfett CL, Zimmer M. Spontaneous immortalization of mouse embryo cells: strain differences and changes in gene expression with particular reference to retroviral gag-pol genes. Exp Cell Res. 1991;192:128–136. doi: 10.1016/0014-4827(91)90167-s. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOXSHADE 3.21 http://www.ch.embnet.org/software/BOX_form.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignment of the amino acid sequences of Tos3, Pak1 and Elm1 from Saccharomyces cerevisiae, and LKB1 and CaMKKβ (β3 variant) from humans. The alignment was created using PILEUP from the GCG suite [48] and the consensus sequence determined, and color added, using BOXSHADE 3.2.1 [49]. Residues that are identical in at least half of the sequences are colored red, with conservative changes in blue. The consensus sequence is represented by an upper case letter if the residue at a particular position is identical in all sequences, and by a lower case letter if at least half are conserved. Only the conserved central regions containing the kinase domains are shown (the kinase domain of LKB1 is approximately residues 44-309).
Sequence alignment of the activation loop sequences of AGC subfamily kinases (PKAα and PKCα, which are not thought to be activated by LKB1) with the activation loop residues of the AMPK/SNF1 subfamily of protein kinases. The methods used and color-coding are as in Additional data file 1. The threonine residues corresponding to Thr172 of AMPK ('P') and other residues mentioned in the text are indicated with downward arrows. All sequences are from humans, except AtSnRK1-α1, AtSnRK1-α1 and ScSnf1, which are the Arabidopsis thaliana (AtSnRK1, SNF1-related kinase-1) and Saccharomyces cerevisiae (ScSnf1) homologs of AMPK. Although the human, A. thaliana and S. cerevisiae homologs, as well as the PKAα and PKCα kinases, are known to be activated by phosphorylation of the threonine residue equivalent to Thr172, it has yet to be established whether the other AMPK subfamily members (NuaK1, NuaK2, BrsK1, BrsK2, SIK, QIK, QSK, MELK and PAR1A) are activated by phosphorylation at this residue, and whether LKB1:STRAD:MO25 complexes can mediate this phosphorylation.