

Abstract
Vasoactive intestinal peptide (VIP) is an anti-inflammatory immunomodulatory neuropeptide with therapeutic potential demonstrated for collagen-induced arthritis. The aim of this study was to characterise its potential anti-arthritic effect on human monocytes, macrophages, T cells, and rheumatoid arthritis synovial membrane cells. Monocytes, macrophages, and T cells derived from human peripheral blood were treated with VIP and compared with other cAMP-elevating drugs for a range of activating stimuli. Cytokine production was assessed for cell cultures and, in addition, the ability of VIPs to activate cAMP response element binding protein. VIP partially suppressed monocyte- and macrophage-derived tumour necrosis factor α (TNF-α) with no effect on IL-10, whereas VIP fails to regulate IL-10 and TNF-α production by T lymphocytes. No such modulation of cytokine profile was observed for rheumatoid arthritis synovial membrane cells. Elevation of intracellular cAMP, on the other hand, potently suppressed macrophage TNF-α production and modulated T-cell response by inhibiting TNF-α and IFN-γ. VIP's lack of effect on IL-10 and its slight effect on TNF-α results from cAMP being rapidly degraded as the phosphodiesterase IV inhibitor, rolipram, rescues cAMP-dependent activation of cAMP response element binding protein. Interestingly, macrophages stimulated with phorbol 12-myristate 13-acetate/ionomycin displayed an augmented IL-10 response upon addition of dibutyryl cAMP, with corresponding downregulation in TNF-α, suggesting a complex interaction between protein kinase C and protein kinase A in cytokine regulation. In conclusion, VIP may represent an efficaceous anti-arthritic treatment modulating macrophage and T-cell cytokine profiles when used alongside a phosphodiesterase inhibitor.
Keywords: IL-10, macrophage, T cells, TNF-α, VIP
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterised by the dysregulated expression of many proinflammatory cytokines including tumour necrosis factor α (TNF-α), with increased yet insufficient production of anti-inflammatory cytokines including IL-10 [1]. The validation of TNF-α as a therapeutic target in RA has encouraged the investigation of signalling pathways regulating its production by cells relevant to the pathophysiology of this disease. One pathway known to downregulate proinflammatory TNF-α production and, consequently, upregulate the anti-inflammatory cytokine IL-10 is that elicited by the second messenger cAMP [2,3]. This pathway may therefore represent a good therapeutic target due to its opposing effects on TNF-α and IL-10. Previously, we and others demonstrated that rolipram, a phosphodiesterase (PDE) IV inhibitor, reduced the clinical and histological severity of collagen-induced arthritis (CIA) [4,5]. These studies demonstrated the potential for the cAMP/protein kinase A (PKA) pathway in treatment of autoimmune diseases such as RA.
Another stimulator of the cAMP/PKA pathway whose principle immunomodulatory functions are anti-inflammatory is the vasoactive intestinal peptide (VIP). VIP is a 28-aminoacid neuropeptide belonging to the glucagon/secretin family, found in the nervous system and in the immune system, where it is detected in a variety of cell types including mast cells, neutrophils, and mononuclear cells. The effects of VIP are transduced via three known receptors, VPAC1, VPAC2, and PAC1, all of which are coupled to adenylate cyclase via heterotrimeric G proteins. In vivo, VIP has a therapeutic effect in the CIA mouse model [6,7] and protects from lipopolysaccharide (LPS) shock by suppression of TNF-α [8,9] and nuclear factor κB (NF-κB) activation [10]. Furthermore, in vitro studies showed that VIP inhibits the production of proinflammatory factors TNF-α, IL-6, IL-12 [11,12], chemokines [13,14], and nitric oxide (NO) [15] and stimulates the production of the anti-inflammatory cytokine IL-10 [16], most of these effects being apparently mediated through the VPAC1 receptor. In addition, neuropeptides such as VIP have been shown to inhibit activities both of stimulated T cells (VIP being described as a Th2 cytokine), effectively suppressing T helper cell type 1 (Th1) differentiation [17] and of macrophages [18] and to antagonise inflammatory mediators such as histamine, prostaglandin E2, leukotrienes, and neurokinins [19]. The mechanism by which VIP antagonises LPS-induced production of proinflammatory TNF-α and abrogates production of anti-inflammatory IL-10 is suggested to result from a fine balance between cAMP response element DNA binding factors where VIP increases the phosphorylation of PKA-dependent cAMP response element binding protein (CREB) and decreases the phosphorylation of c-Jun N-terminal kinase-dependent c-Jun phosphorylation, without affecting the amount of CRE binding: changes of CRE binding complexes from high c-Jun/low CREB (LPS treated) to low c-Jun/high CREB (VIP treated) leads to an inhibition of TNF-α mRNA expression, whereas the corresponding stimulation in IL-10 gene expression is due to an increase in CRE binding by VIP [[10]; reviewed in [20]].
It would appear from these studies that VIP has therapeutic potential based on its ability to ameliorate CIA in mice [6,7], this effect possibly mediated by cAMP. However, the effect of VIP on human cells and particularly on RA synovial cells is unknown. Thus the aim of this study was to examine the potential of VIP as a therapeutic agent in chronic inflammatory diseases such as RA by investigating its effects on human macrophages, T cells, and synovial cells – all of which play an important role in the pathology of RA – and compare the findings with murine VIP data already published.
Materials and methods
Reagents
Capture and detection antibodies for human TNF-α, IL-10, and IFN-γ ELISAs were purchased from Pharmingen International, Oxford, UK. Direct colorimetric immunoassay kit for detection of cAMP was purchased from Merck Biosciences, Nottingham, UK. Macrophage-colony stimulating factor (M-CSF) was obtained from Genetics Institute, Boston, MA, USA. Rolipram, a PDE IV inhibitor, was a gift from Dr Peter Scholz (Schering, Berlin, Germany). PDE-resistant dibutyryl cAMP and forskolin, an activator of adenylate cyclase, were purchased from Sigma, Poole, Dorset, UK. VIP was synthesised at the Advanced Biotechnology Centre, Imperial College School of Medicine at Charing Cross Hospital, London, UK. Antibodies to CREB and phospho-CREB were purchased from New England Biolabs, Beverly, MA, USA. All reagents used in these tissue-culture experiments were tested for the presence of LPS/endotoxin contamination and were found to be below the lower level of detection of the limulus amoebocyte assay (Cambrex BioScience, Wokingham, Berkshire, UK). In addition, rolipram, dibutyryl cAMP, forskolin, and VIP were tested for cytotoxicity and displayed no toxicity at the concentrations being used in this study as determined by (3-[4,5-dimethylhiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay and trypan blue exclusion.
Purification of T lymphocytes and monocytes
Human peripheral blood mononuclear cells (PBMCs) were obtained by density centrifugation of human venous blood buffy coats (purchased from the North London Blood Transfusion Service, Colindale, UK) through Ficoll/ Hypaque (specific density 1.077 g/ml; Nycomed Pharma AS, Oslo, Norway). The resulting PBMCs were centrifugally elutriated in 1% fetal calf serum (FCS) RPMI 1640 medium in a Beckman JE6 elutriator. Lymphocyte and monocyte purity was assessed by flow cytometric analysis of binding of fluorochrome-conjugated anti-CD3, anti-CD19, anti-CD14, and anti-CD45 antibodies (Becton Dickinson, Oxford, UK). T cells obtained were routinely of >90% purity and monocytes of >85% purity.
Differentiation of monocytes to macrophages
Peripheral blood monocytes obtained by centrifugal elutriation were seeded at 1 × 106 ml-1 in assay medium in T-75 medium tissue-culture flasks. M-CSF was added to a final concentration of 100 ng/ml. Cells were cultured for 7 days at 37°C in a 5% CO2 humidified atmosphere. Adherent cells were then washed twice in FCS-free RPMI 1640 and removed from the plastic with cell-dissociation medium (Sigma). The resulting cells were washed twice more and resuspended in RPMI 1640/10% FCS ready for use.
Isolation of RA synovial membrane mononuclear cells
RA synovial membrane cells (RA-SMCs) were obtained from synovial membrane tissue samples provided by the Rheumatology Clinic and the Department of Musculoskeletal Surgery, Charing Cross Hospital, London, UK. All patients gave their signed consent, and ethical approval was obtained from the Riverside Research Ethics Committee, London. Patients met the American College of Rheumatology (ACR) 1987 revised criteria for RA. Synovial membranes obtained were from patients who failed to respond to antirheumatic regimens currently available in the rheumatology clinic and will be discussed later in this manuscript. In brief, tissue was cut into small pieces and digested in medium containing 0.15 mg/ml DNAse type I (Sigma) and 5 mg/ml collagenase A (Roche, Lewes, Sussex, UK) for 1 to 1.5 hours at 37°C. Cell debris was excluded by passing cells through a nylon mesh, and cells were then washed and collected in RPMI-10% FCS at 1 × 106 cells/ml and used immediately for spontaneous cytokine production by RA-SMCs.
Detection of intracellular cAMP
A number of signals are known to stimulate the production of cAMP through the action of adenylate cyclase converting ATP to cAMP. Intracellular cAMP was measured using a colorimetric direct immunoassay in accordance with the manufacturer's instructions. Briefly, 5 × 105 monocytes/macrophages were incubated with test reagents, and then cells were lysed in 0.1 N HCl at room temperature for approximately 10 min. In the capture microtitre plate provided, 100 μl lysate and controls were added per well along with 50 μl conjugate and 50 μl antibody solution and incubated for 2 hours at room temperature on a plate shaker. The plate was then emptied and washed three times in wash buffer provided. Colour development was detected using 200 μl pNpp substrate solution and incubated for 1 hour at room temperature and stopped by the addition of 50 μl stop solution. These assays were read and quantified on a Labsystems Multiscan Bichromatic plate reader at 405 nm and analysed with a Deltasoft II programme (BioMetallics Inc, Princeton, NJ, USA). The minimal sensitivity of the assay was 0.078 pmol/ml cAMP. All results are expressed as the mean concentration of cAMP obtained per condition.
Cytokine determination by ELISA
Sandwich ELISAs were used to measure human IL-10, TNF-α, and IFN-γ. In the IL-10 assay, the anti-IL-10 monoclonal antibody 9D7 was used as the capture antibody, and biotinylated 12G8 was used as the detection antibody. The ELISA was performed as previously described, with a standard curve of recombinant human IL-10 from 10,000 to 13 pg/ml [21]. TNF-α ELISA was carried out as described using 61E71 as the coating antibody and a rabbit polyclonal anti-TNF-α antibody as the detection antibody. This polyclonal anti-TNF-α antibody was in turn detected by a horseradish-peroxidase-conjugated goat anti-rabbit IgG(H+L) (Jackson ImmunoResearch, West Grove, PA, USA). The standard curve of recombinant human TNF-α covered the range of 20,000 to 8 pg/ml [22]. In addition, human IFN-γ ELISA was carried out in accordance with the manufacturer's specifications (PharMingen International, Oxford, UK). These ELISAs were quantified by tetramethylbenzide dichloride activity in response to the horseradish peroxidase conjugate and read on a Labsystems Multiscan Bichromatic plate reader at 450 nm and analysed with the Deltasoft II programme (BioMetallics). The minimal sensitivity of the ELISAs were 8 pg/ml for human TNF-α and 13–40 pg/ml for the human IL-10 and IFN-γ ELISAs. All results are expressed as the mean concentration of cytokine ± SD obtained per condition.
Western blot analysis of phospho-CREB
Macrophages were seeded at 5 × 106 cells/ml in 24-well plates in RPMI 1640/10% FCS. To inhibit phosphodiesterase activity prior to VIP treatment, macrophages were pretreated for 1 hour with 10 μM rolipram and then stimulated for 30 min with 10 μM VIP before harvesting of cell lysates. The stimulation time was previously defined in our laboratory as optimal for activation of CREB. After stimulation, cells were lysed on ice for 15 min in lysis buffer (1% NP-40, 200 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol, 1 mM Na3VO4, 1 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 10 μg/ml aprotinin). Lysed samples (10 μg) were separated by electrophoresis on a 10% SDS–polyacrylamide gel and transferred to a nitrocellulose membrane. Phosphorylated proteins were detected using antibodies raised against phospho-CREB followed by anti-rabbit horseradish peroxidase conjugate and enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech UK Ltd, Little Chalfont, Buckinghamshire, UK). Protein bands were visualised by autoradiography using Hyperfilm (Amersham Pharmacia Biotech UK).
Statistical analysis
Comparison of data was assessed using GraphPad Prism version 3.0 (GraphPad Software Inc, San Diego, CA, USA). Statistical differences were determined with Student's t-test. Differences were regarded as significant when *P < 0.05, **P < 0.01, or ***P < 0.001.
Results
VIP, rolipram, and dibutyryl cAMP suppress LPS-induced monocyte TNF-α production without upregulating IL-10
First, we compared the effects of VIP, rolipram, and dibutyryl cAMP on the production of TNF-α and IL-10 by monocytes. Spontaneous production of IL-10 and TNF-α by monocytes could not be detected; cytokine production was induced, however, by addition of LPS (control samples). VIP dose-dependently inhibited LPS-induced TNF-α production from LPS-stimulated control levels of 508 ± 65 pg/ml to 226 ± 11 (P = 0.021) at 10-6 M with a median inhibitory concentration (IC50) value of 1.45 nM (range 0.83 to 2 nM for n = 4 experiments) (Fig. 1b). In contrast, the anti-inflammatory cytokine IL-10 is not significantly regulated by VIP: the LPS-stimulated control value was 420 ± 41 pg/ml, versus 371 ± 82 pg/ml at 10-5 M (Fig. 1a). The effects of VIP are reported to be mediated by the cAMP/PKA pathway – a pathway that potently regulates TNF-α and IL-10 production. Thus, the contribution of cAMP to cytokine production was investigated using the PDE IV inhibitor rolipram and the PDE-resistant dibutyryl cAMP. Inhibition of PDE by rolipram had little effect on LPS-induced IL-10 production (Fig. 1c), whereas rolipram potently inhibited LPS-stimulated TNF-α production (IC50 = 350 nM) (Fig. 1d). In addition, LPS-stimulated TNF-α production was potently inhibited by dibutyryl cAMP (IC50 = 4 μM) (Fig. 1f). IL-10, on the other hand, was only partially suppressed: control 747 ± 13 pg/ml, versus 428 ± 8 pg/ml at 100 μM (Fig. 1e). This effect on IL-10 production was thought to be a consequence of TNF-α suppression, as endogenous TNF-α has been demonstrated to regulate LPS-induced IL-10 production in monocytes [23]. In fact, the addition of a neutralising anti-TNF-α antibody only served to inhibit IL-10 by approximately 30%; the simultaneous addition of VIP had no effect on IL-10 production. Control experiments were carried out to determine the effectiveness of VIP on the basis of its ability to potently inhibit LPS-induced monocyte production of IL-8; in our hands, VIP suppressed IL-8 production, resulting in a mean IC50 = 11 nM for three separate experiments (range 2 to 25 nM; data not shown).
Figure 1.
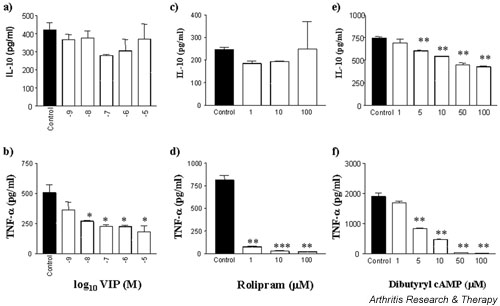
VIP suppresses LPS induction of monocyte TNF-α but has no effect on IL-10 production. Fresh, elutriated human monocytes were plated out at 2 × 105 cells per well in a U-bottomed 96-well plate and pretreated with VIP (a,b), rolipram (c,d), or dibutyryl cAMP (e,f) for 1 hour prior to stimulation with 1 ng/ml LPS and incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for TNF-α and IL-10 by ELISA. Data are mean cytokine levels in pg/ml of triplicate culture supernatants ± SD, showing a representative of n = 4 replicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001. LPS, lipopolysaccharide; TNF-α, tumour necrosis factor α; VIP, vasoactive intestinal peptide.
VIP, rolipram, and dibutyryl cAMP suppress LPS-induced macrophage TNF-α production without upregulating IL-10
VIP has been shown to differentially modulate proinflammatory and anti-inflammatory cytokine production by murine macrophages [8,9,12,13,16]. Thus it was desirable to compare human monocytes with monocyte-derived macrophages obtained by M-CSF treatment of peripheral blood monocytes, this cell type being more representative of tissue macrophages present in the rheumatoid joint. Again, spontaneous cytokine production could not be detected in the absence of an activating stimulus. The effects of VIP on macrophage IL-10 and TNF-α were comparable and not significant. VIP inhibited LPS-induced TNF-α (Fig. 2b), with IC50 values ranging between 7 and 50 nM for n = 7 experiments, and partially suppressed IL-10 production (Fig. 2a). This trend was repeated by treatment with rolipram and dibutyryl cAMP, where LPS-induced TNF-α production was suppressed, resulting in values of IC50 = 50 nM (Fig. 2d) and IC50 = 2.5 μM (Fig. 2f), respectively. LPS-induced IL-10 production was partially suppressed by rolipram (Fig. 2c) and dibutyryl cAMP (Fig. 2e). This partial suppression of IL-10 was independent of endogenous TNF-α expression, as blockade by anti-TNF-α antibodies failed to abrogate this partial suppression by VIP, as did the addition of exogenous TNF-α (Table 1). LPS-induced macrophage IL-10 production was suppressed by 12.7% by 10-6M VIP, which, upon neutralisation of TNF-α, apart from the expected decrease in IL-10 production (48%), resulted in 17.8% suppression at the same concentration of VIP. Exogenous TNF-α had little effect on IL-10 production and the lack of modulation by VIP. LPS-induced IL-10 was partially modulated by 10-6M VIP (12.7% suppression), which upon addition of exogenous TNF-α resulted in 8.4% suppression by VIP. Conversely, VIP suppressed LPS-induced macrophage TNF-α by 55%, which upon neutralisation of IL-10, apart from the expected increase in TNF-α production (2.17-fold), resulted in 28% suppression at the same concentration of VIP (Table 2). Exogenous IL-10 suppressed total TNF-α production by 91% but had no effect on VIP modulation, which resulted in a 26% suppression (Table 2). The effect of IL-10 neutralisation or addition of exogenous IL-10 significantly modulated LPS-induced TNF-α production by macrophages. However, modulation of TNF-α production by VIP was not significantly different in these groups; a separate set of data showed suppressions of 26%, 36%, and 58%, versus 55%, 28%, and 26% for control, endogenous, and exogenous IL-10, respectively. Control experiments were carried out to determine the effectiveness of VIP based on its ability to potently inhibit LPS-induced monocyte production of IL-8; in our hands, VIP suppressed macrophage IL-8 production, resulting in a mean IC50 = 38 nM for three separate experiments (range 9 to 79 nM).
Figure 2.
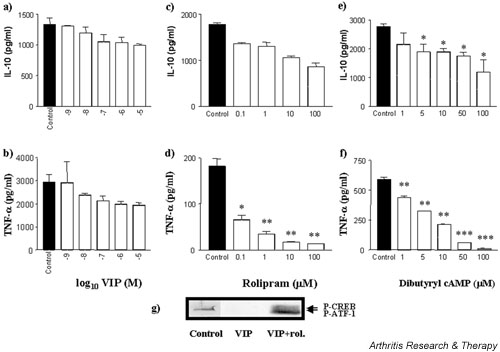
VIP suppresses LPS induction of macrophage TNF-α with little effect on IL-10 production. Human-monocyte-derived macrophages were plated out at 1 × 105 cells per well in a flat-bottomed 96-well plate and pretreated with VIP (a,b), rolipram (c,d) or dibutyryl cAMP (e,f) for 1 hour prior to stimulation with 1 ng/ml LPS and were incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for TNF-α and IL-10 by ELISA. Data are mean cytokine levels in pg/ml of triplicate culture supernatants ± SD, showing a representative of n = 7 replicate experiments. Western blot analysis of activated phospho-CREB (g) shows VIP modulation in the presence or absence of PDE inhibition. Lane 1, LPS-stimulated macrophage control; 2, LPS-stimulated macrophage + 10-6 M VIP; 3, LPS-stimulated macrophage + 10-6 M VIP + 10 μM rolipram. Data are representative of n = 3 replicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001. CREB = cAMP response element binding protein; LPS, lipopolysaccharide; P-ATF, activating transcription factor-1; P-CREB, phospho-CREB; rol., rolipram; TNF-α, tumour necrosis factor α; VIP, vasoactive intestinal peptide.
Table 1.
TNF-α fails to modulate VIP regulation of LPS-induced macrophage IL-10
| IL-10 | ||
| Treatment | Control | + VIP |
| Control | 2182 ± 317.7 | 1904 ± 255.7 (12.7%) |
| Anti-TNF-α | 1123 ± 23.92 | 922.7 ± 49.93 (17.8%) |
| Exogenous TNF-α | 1481 ± 494.6 | 1606 ± 906.3 (-8.4%) |
Macrophage-colony stimulating factor (M-CSF)-primed monocyte-derived macrophages, plated at 1 × 105 cells/well, were stimulated with 1 ng/ml LPS in the presence or absence of 10-6M VIP. TNF-α modulation of VIP regulation of IL-10 production was assessed by addition of 10 μg/ml neutralising anti-TNF-α (A2) or 10 ng/ml TNF-α. Results with an isotype-matched control antibody did not differ significantly from the control sample presented in this table. The resulting cultures were incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for IL-10 by ELISA. Data are mean IL-10 levels in pg/ml and percentage suppression by VIP in parentheses of triplicate culture supernatants ± SD, showing a representative of n = 3 experiments. LPS, lipopolysaccharide; TNF-α, tumour necrosis factor α; VIP, vasoactive intestinal peptide.
Table 2.
IL-10 fails to modulate VIP regulation of LPS-induced macrophage TNF-α
| TNF-α | ||
| Treatment | Control | +VIP |
| Control | 1400 ± 62.02 | 630.3 ± 61.55 (55%) |
| Anti-IL-10 | 3041 ± 624.9 | 2195 ± 224.9 (28%) |
| Exogenous IL-10 | 119.5 ± 6.944 | 88.26 ± 14.48 (26%) |
Macrophage-colony stimulating factor (M-CSF)-primed monocyte-derived macrophages, plated at a density of 1×105 cells/well, were stimulated by 1 ng/ml LPS in the presence or absence of 10-6M VIP. IL-10 modulation of VIP regulation of TNF-α production was assessed by addition of 10 μg/ml neutralising anti-IL-10 (9D7) or 10 ng/ml IL-10. Results with an isotype-matched control antibody did not differ significantly from the control sample presented in this table. The resulting cultures were incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for TNF-α by ELISA. Data are mean TNF-α levels in pg/ml and percentage suppression by VIP in parentheses of triplicate culture supernatants ± SD, showing a representative of n = 3 experiments. LPS, lipopolysaccharide; TNF-α, tumour necrosis factorα; VIP, vasoactive intestinal peptide.
The lack of any great effect of VIP on monocyte-derived macrophages and monocytes themselves led us to postulate that there was an endogenous phosphodiesterase activity intrinsic to these human cells. This was confirmed by the inability of VIP to activate/phosphorylate CREB, a downstream effector molecule to the cAMP-dependent PKA pathway, in M-CSF-treated macrophages (Fig. 2g, lane 2). Of particular interest is the fact that simultaneous addition of VIP and the PDE IV inhibitor rolipram restored activation of CREB (Fig. 2g, lane 3), suggesting that in the absence of a PDE inhibitor these cells quickly and efficiently break down cAMP produced in response to VIP. Rolipram on its own inhibits PDE IV activity (cAMP breakdown) but does not stimulate cAMP production and as such was not included in this phospho-western result. This control, however, failed to activate CREB by phosphorylation of residue Ser 133 on all blots tested. Of particular interest, however, is the observation that activating transcription factor-1 (ATF-1) is activated by LPS (ATF-1 is also recognised by the CREB antibody used) (lane 1), an effect that is abrogated in the presence of VIP (lane 2), and that the combination of VIP and rolipram activates both CREB and ATF-1 upon LPS stimulation (lane 3).
VIP fails to modulate T-cell production of IL-10, TNF-α, and IFN-γ
T cells are thought to play a role in perpetuating the chronic inflammatory response in the rheumatoid joint. T cells in the rheumatoid joint are in close proximity to macrophages and can regulate the activation of such cells. However, macrophages can themselves regulate T-cell functions. Thus it was desirable to investigate the regulatory role of VIP on activated T lymphocytes. Phorbol 12-myristate 13-acetate (PMA)/ionomycin-stimulated T cells produced IL-10 and TNF-α over a 24-hour culture period. The addition of VIP, however, failed to modulate the production of either IL-10 (Fig. 3a) or TNF-α (Fig. 3b). In comparison, treatment of PMA/ionomycin-stimulated T cells with dibutyryl cAMP failed to modulate IL-10 production (Fig. 3d) but modestly suppressed TNF-α production (IC50 = 8.8 μM) (Fig. 3e).
Figure 3.
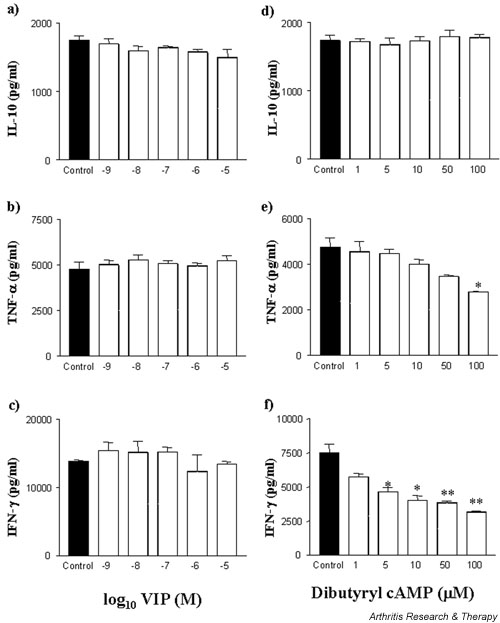
VIP fails to suppress PMA/ionomycin-stimulated T-cell induction of TNF-α, IL-10, and IFN-γ production. Fresh, elutriated human T lymphocytes were plated out at 1 × 105 cells per well in a U-bottomed 96-well plate and pretreated with VIP (a,b,c), or dibutyryl cAMP (d,e,f) for 1 hour prior to stimulation with 50 ng/ml PMA and 0.5 μg/ml ionomycin and incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for IL-10 (a,d), TNF-α (b,e), and IFN-γ (c,f) by ELISA. Data are mean cytokine levels in pg/ml of triplicate culture supernatants ± SD, showing a representative of n = 4 replicate experiments. *P < 0.05; **P < 0.01. PMA, phorbol 12-myristate 13-acetate; TNF-α, tumour necrosis factor α; VIP, vasoactive intestinal peptide.
In addition, VIP has been shown to modulate T-cell function in the murine system of CIA, shifting a Th1 cytokine response to a Th2-like response [6]. We investigated this modulation in the context of the human T cells stimulated by PMA/ionomycin. Unlike its effect in the murine system, VIP failed to modulate human T-cell activity. PMA/ionomycin-induced T-cell IFN-γ production was not significantly affected by VIP (Fig. 3c). Unlike VIP however, elevation of intracellular cAMP did modulate IFN-γ production. The addition of cell-permeable dibutyryl cAMP suppressed IFN-γ production from PMA/ionomycin-stimulated T cells (IC50 = 9 μM) (Fig. 3f). In a separate set of experiments, IFN-γ was also suppressed by the adenylate cyclase activator forskolin (data not represented graphically) from PMA/ionomycin-stimulated control levels of 6963 ± 230 pg/ml to 5691 ± 265 pg/ml (P = 0.027) and 4968 ± 372 pg/ml (P = 0.025) at concentrations of 10 μM and 20 μM respectively (IC50 = 6 μM), and by rolipram, the PDE IV inhibitor, from 6963 ± 230 pg/ml to 2685 ± 204 (P = 0.002) and 2262 ± 94 (P = 0.002) at 1 μM and 10 μM respectively (IC50 = 2 μM) (data not represented graphically). In addition, these data were reproducible for concanavalin-A-stimulated T cells from human peripheral blood.
cAMP modulates PMA/ionomycin stimulated macrophage cytokine profile
Unlike its effect in the murine system, elevation of cAMP in LPS-activated macrophages failed to augment the human anti-inflammatory IL-10 response but potently inhibited the TNF-α response. In this study, macrophages were stimulated by PMA/ionomycin activating protein kinase C (PKC). Elevation of intracellular cAMP, by the addition of the phosphodiesterase-resistant dibutyryl cAMP, augmented IL-10 production with a corresponding decrease in TNF-α production (Fig. 4). Dibutyryl cAMP augmented IL-10 production (ED50 = 6.4 μM; Fig. 4a) whereas TNF-α production was inhibited (IC50 = 6 μM; Fig. 4b). These data were confirmed by use of the PDE inhibitor rolipram and the adenylate cyclase activator forskolin. Rolipram and forskolin augmented IL-10 to 779% and 767% whereas TNF-α was inhibited by 50% and 55% at 100 μM and 10 μM, respectively (data not shown). VIP failed to modulate IL-10 production on its own but could in the presence of rolipram (see paragraph below and Fig. 6a below). In addition, the phosphodiesterase-resistant dibutyryl cAMP costimulated the downstream effector molecule to the cAMP-dependent PKA pathway, CREB, which was phosphorylated upon PKC activation by PMA/ionomycin (Fig. 4c, lane 3). Neither stimulus, on its own, activated CREB.
Figure 4.
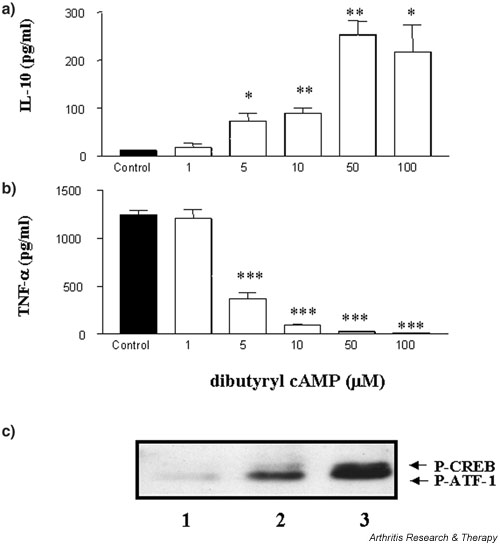
Elevation of intracellular cAMP augments PMA/ionomycin-stimulated macrophage IL-10 production and suppresses TNF-α. Human-monocyte-derived macrophages were plated out at 1 × 105 cells per well in a flat-bottomed 96-well plate and pretreated with dibutyryl cAMP for 1 hour prior to stimulation with 50 ng/ml PMA and 0.5 μg/ml ionomycin and incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for IL-10 (a) and TNF-α (b) by ELISA. Data are mean cytokine levels in pg/ml of triplicate culture supernatants ± SD, showing a representative of n = 7 replicate experiments. Western blot analysis of activated phospho-CREB (c) shows cAMP modulation of CREB upon macrophage stimulation by PMA/ionomycin. Lane 1, macrophage control; 2, macrophage + PMA/ionomycin; 3, macrophage + PMA/ ionomycin + cAMP. Data are representative of n = 3 replicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001. CREB, cAMP response element binding protein; P-ATF, activating transcription factor-1; P-CREB, phospho-CREB; PMA, phorbol 12-myristate 13-acetate; TNF-α, tumour necrosis factor α.
Figure 6.
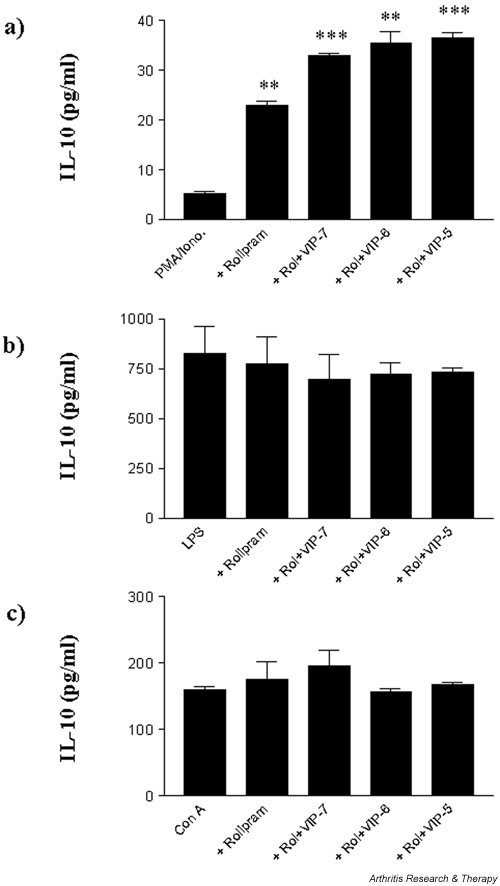
Rolipram and VIP augment IL-10 production in a stimulus- and cell-specific manner. Human-monocyte-derived macrophages and T cells were plated out at a density of 1 × 105 cells per well in a flat-bottomed 96-well plate and pretreated with 10 μM rolipram and indicated concentrations of VIP for 1 hour prior to stimulation. Macrophages were stimulated with (a) 50 ng/ml PMA and 0.5 μg/ml ionomycin or (b) 1 ng/ml LPS, and T cells were stimulated with (c) 10 μg/ml concanavalin A and incubated for 24 hours at 37°C in a 5% CO2humidified atmosphere, after which time supernatants were harvested and assayed for IL-10 by ELISA. Data are mean cytokine levels in pg/ml of triplicate culture supernatants ± SD, showing a representative of n = 3 replicate experiments. **P < 0.01; ***P < 0.001. Iono, ionomycin; LPS, lipopolysaccharide; PMA, phorbol 12-myristate 13-acetate; Rol, rolipram; VIP, vasoactive intestinal peptide.
Macrophages have a high endogenous PDE IV activity: VIP induction of cAMP is augmented by rolipram
VIP induces the release of cAMP. We have suggested earlier in this article that cAMP levels are not likely to persist, because of a high endogenous activity of PDE IV in macrophages. This has been avoided by the utilisation of the PDE-resistant form of cAMP, dibutyryl cAMP, which previously potently suppressed macrophage TNF-α production and specifically augmented PMA/ionomycin-stimulated macrophage IL-10 production. We wished to investigate VIP regulation of cAMP levels in monocytes and macrophages upon stimulation by LPS and PMA/ionomycin. VIP regulation of cAMP was augmented by the PDE IV inhibitor rolipram, where LPS-stimulated monocytes resulted in 0.833 pmol/ml and PMA/ionomycin stimulation resulted in 0.367 pmol/ml (see Fig. 5a). LPS- and PMA/ionomycin-stimulated macrophages, on the other hand, produced much higher (10- to 20-fold) levels of cAMP upon treatment with VIP and rolipram (11.67 pmol/ml and 16.60 pmol/ml respectively; see Fig. 5b) compared with monocytes, a finding that would confirm the higher level of endogenous PDE activity observed in macrophages. Thus, in the presence of high endogenous PDE activity, VIP is incapable of maintaining a prolonged elevation of cAMP, which would suggest the relatively modest effect of VIP on TNF-α production when compared with PDE-resistant dibutyryl cAMP and the distinct lack of modulation of IL-10 production. Rolipram treatment alone failed to exhibit any induction of intracellular cAMP over that observed for stimulation controls, which confirms the finding that VIP induces a cAMP-dependent response. Positive controls were measured for addition of dibutyryl cAMP, where PMA/ionomycin-stimulated macrophages resulted in intracellular levels of 28.18 pmol/ml and LPS-stimulated macrophages, 21.38 pmol/ml, versus monocyte levels of 39.81 pmol/ml and 89.13 pmol/ml, respectively.
Figure 5.
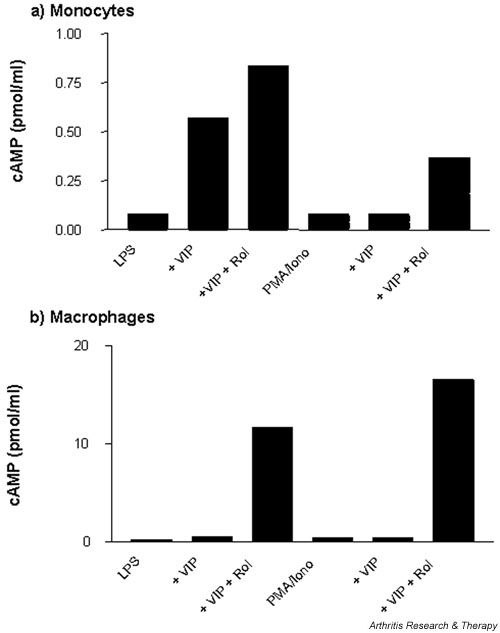
Macrophages have a high endogenous PDE IV activity: VIP induction of cAMP is augmented by rolipram. Human monocytes (a) and monocyte-derived macrophages (b) were plated out at 5 × 105cells per well in a flat-bottomed 24-well plate and simultaneously treated with 10-6 M VIP, or VIP in the presence of 10 μM rolipram, and stimulated with 50 ng/ml PMA and 0.5 μg/ml ionomycin or 1 ng/ml LPS and incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time cell lysates were harvested and assayed for cAMP by immunoassay. Data are mean cAMP levels in pmol/ml of duplicate culture supernatants, showing a representative of n = 2 replicate experiments. Iono, ionomycin; LPS, lipopolysaccharide; PDE, phosphodiesterase; PMA, phorbol 12-myristate 13-acetate; Rol, rolipram; VIP, vasoactive intestinal peptide.
Rolipram and VIP augment IL-10 production in a stimulus- and cell-specific manner
Elevation of intracellular cAMP by the phosphodiesterase-resistant dibutyryl cAMP augments production of IL-10 by PMA/ionomycin-stimulated macrophages. The lack of augmentation of IL-10 production by VIP is suggested by a short-lived elevation in cAMP as a result of high endogenous PDE activity. Here, we have investigated VIP modulation of cytokine production in the presence of rolipram, an inhibitor of PDE IV activity. Our results demonstrate both stimulus- and cell-type-specific responses to VIP in the presence of rolipram. The addition of VIP and rolipram on their own or in combination in the absence of an activating stimulus failed to induce cytokine production. VIP augmented macrophage IL-10 production when stimulated by PMA/ionomycin in the presence of rolipram (Fig. 6a). This was not the case, however, when macrophages were stimulated with LPS; LPS-induced IL-10 production was unaffected by rolipram alone or rolipram+VIP (Fig. 6b). In addition, stimulated T cells also failed to show an augmentation of IL-10 production upon treatment by VIP and rolipram (Fig. 6c). On the other hand, rolipram augmented VIP suppression of TNF-α production in a cell-nonspecific and stimulation-nonspecific manner, as observed for PMA/ionomycin- and LPS-stimulated macrophages and concanavalin-A-stimulated T cells (data not shown).
VIP fails to modulate spontaneous IL-10 and TNF-α production by RA-SMCs
To investigate the role of VIP as a modulator of cytokine production in RA, VIP was added to dissociated, cultured RA-SMCs and spontaneous cytokine production was assessed. In this study, VIP failed to modulate the spontaneous production of IL-10 and TNF-α (Fig. 7a,7b). At the maximal concentration used, VIP suppressed IL-10 by 4% and TNF-α by 18.9%. In comparison, the effect of the PDE-resistant dibutyryl cAMP was also investigated, mimicking the effect of both VIP and rolipram and resulting in stable PDE-resistant and prolonged cAMP. Elevation of cAMP effectively suppressed spontaneous TNF-α production with relatively little effect on IL-10 production by RA-SMCs (Fig. 7c,7d). Dibutyryl cAMP suppressed spontaneous TNF-α production by 36% and 46% at concentrations of 100 and 1000 μM, respectively (IC50 = 20 μM). Spontaneous IL-10 production was partially suppressed by 8% and 15% at 10 μM and 100 μM, respectively. The lack of responsiveness to VIP and effects of cAMP were reproducible between patient samples; however, patient variability exists for spontaneous cytokine production: mean TNF-α production 486 pg/ml (range 70 to 1047 pg/ml), mean IL-10 production 529 pg/ml (range 199 to 1064 μpg/ml).
Figure 7.
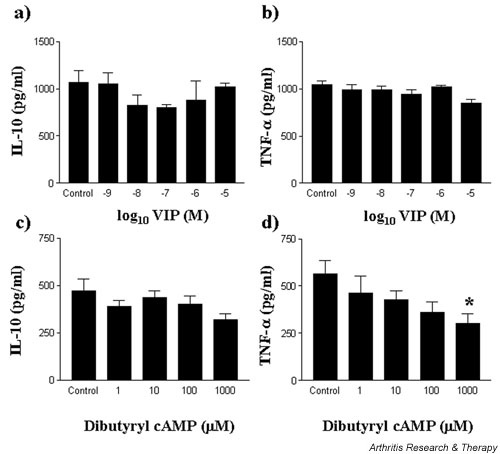
VIP fails to modulate spontaneous IL-10 and TNF-α production by RA-SMCs. RA-SMCs were plated out at 2 × 105 cells per well in a flat-bottomed 96-well plate and treated with VIP (a,b) or PDE-resistant dibutyryl cAMP (c,d) for 24 hours at 37°C in a 5% CO2 humidified atmosphere, after which time supernatants were harvested and assayed for spontaneous production of IL-10 (a,c) and TNF-α (b,d) by ELISA. Data are mean cytokine levels in pg/ml of triplicate culture supernatants ± SD, showing a representative (one patient) of n = 3 replicate experiments for a total of four patient samples. *P < 0.05. PDE, phosphodiesterase; RA-SMC, rheumatoid arthritis synovial membrane cell; VIP, vasoactive intestinal peptide.
Discussion
In a murine model of arthritis (CIA), VIP has been described as a potent anti-inflammatory mediator effectively reducing paw swelling, clinical score, and histological severity of disease [6,7]. This neuropeptide downregulates macrophage and T-cell function as well as modulating T-cell phenotype by altering Th1/Th2 balance in favour of Th2-like cells. There are no such compelling data for the efficacy of VIP in human tissues. The data presented in this paper would argue against VIP alone being a useful therapeutic agent in the treatment of human chronic inflammatory disorders such as RA, because the peptide failed to significantly modulate in vitro TNF-α IL-10 expression by human cells. The lack of effect of VIP in monocyte/macrophage cultures stimulated with LPS may have been due to a high level of endogenous intrinsic phosphodiesterase activity, resulting in a short-lived cAMP in these cell types. This question was addressed by the use of rolipram to inhibit PDE IV and dibutyryl cAMP, which is resistant to PDEs. The treatment of macrophages with VIP in the presence of rolipram facilitated activation of CREB but did not augment IL-10 cytokine production. In addition, this lack of sensitivity of cells to VIP is not as a result of a window of opportunity of action. VIP was added 1 hour prior to stimulation. However, some reports have described VIP to be a more effective anti-inflammatory agent if it is administered at the same time as or after stimulation; addition of VIP to cultures 1 hour before, simultaneously with, or 2 hours after stimulation showed no significant differences to TNF-α/IL-10 ratios in this study.
The effect of VIP in modulating T-cell function was observed by Delgado and colleagues and by Williams and colleagues, in studies in which murine lymph node cells from CIA mice demonstrated a shift in ratio of IFN-γ/IL-5, from Th1 in favour of a Th2 profile [6,7]. We wished to investigate this modulation of a Th1-driven response (IFN-γ production) in the context of stimulated human T cells. Indeed, VIP failed to modulate IFN-γ. However, elevation of intracellular cAMP by rolipram, dibutyryl cAMP, and forskolin dose-dependently suppressed IFN-γ and TNF-α production. This would suggest that VIP activation of the cAMP pathway is not involved in T-cell IFN-γ production or that the cAMP is rapidly degraded by an active phosphodiesterase present in the cell. This T-cell unresponsiveness to VIP with respect to production of TNF-α, IL-10, and IFN-γ is not a consequence of PMA/ionomycin stimulation, as PHA and concanavalin A also failed to exhibit VIP responsiveness. Alternatively, reports thus far describing modulation of T-cell activity have used PBMCs and lymph node cells, whereas our present studies used purified T cells, which appear relatively insensitive to VIP. We suggest that the modulatory effect of VIP on T-cell cytokine production is indirect, through the regulation of effector functions of antigen-presenting cells (APCs). The role of Th differentiation is likely to play a role where VIP has been described to bias the Th1/Th2 balance in favour of Th2, thus indirectly modulating T-cell cytokine production [17]. The data presented in this paper focus on mature human T-cell modulation by VIP, which has no direct effect on cytokine production; the confirmation of an effect of VIP on T-cell differentiation warrants further investigation in the human system using naïve T cells from cord blood.
Unlike its effect in the murine system, VIP has little effect in modulating IL-10 production by human peripheral blood derived monocytes, macrophages, and T cells. It suppresses monocyte TNF-α production upon stimulation with LPS and is less potent in M-CSF differentiated macrophages. Results obtained with the PDE IV inhibitor rolipram and the PDE-resistant dibutyryl cAMP suggest that the cAMP generated is a potent inhibitor of LPS-induced TNF-α, whereas IL-10 is relatively unaffected. The slight inhibition of IL-10 by elevation of cAMP is thought to be a consequence of the potent inhibition of TNF-α. The failure of VIP to augment macrophage IL-10 production, unlike the murine system, is likely to result from the lack of activation/phosphorylation of CREB, a transcription factor that is readily activated in the murine system by VIP [10]. This is likely to be due to instability of cAMP that results from PDE activation. The combined treatment with VIP and rolipram both activated CREB and augmented IL-10 production. In addition, VIP failed to modulate spontaneous IL-10 or TNF-α production by RA-SMCs. However, spontaneous TNF-α production was suppressed by the PDE-resistant dibutyryl cAMP. This would again suggest that cAMP is failing to activate CREB by a mechanism which involves high PDE activity.
Thus, VIP inhibition of TNF-α was less effective in macrophages than in monocytes and was completely ineffective in RA-SMCs, which suggests that there is an increase in PDE activity during differentiation. One point of note regarding the responsiveness of macrophages and RA-SMCs to VIP is that the effective doses are higher than in earlier reports. In addition to the PDE IV activity, this might be explained by modulation of expression of the VIP receptors (VPAC1 and VPAC2) on these cells. Indeed, in the case of RA-SMCs, it is possible that the method of isolation from synovial membrane tissue might downregulate VIP-receptor expression. Additionally, this could be accounted for by the drug regimen encountered by the patient, where patient tissue obtained by surgery results from the failure to respond to treatments given, decreasing sensitivity to VIP through downmodulation of the receptors. Monocytes, however, were more sensitive to VIP than macrophages, and as such would also suggest that maturation might influence VIP responsiveness through modulation of receptor expression. The relative expression of these receptors is currently under investigation.
Alternatively, this slight discrepancy between effective doses of VIP (IC50) in our data and data already published may result from both different methods of isolation and different cell populations. Our studies use highly purified cells obtained by the centrifugal elutriation of PBMCs, resulting in >90–95% monocytes and T cells that are not prestimulated in any way as a consequence of the purification protocol. Reports in the literature on human cells describe VIP to potently suppress TNF-α (IC50 approximately 20 nM) in whole blood cultures and purified monocytes, where monocytes were separated by clumping and adherence, activating stimuli which may prime VIP responses [24]. In addition, VIP suppresses LPS-induced monocyte IL-8 production (IC50 approximately 0.1 M) [14] and LPS-induced peripheral blood mononuclear cell TNF-α production, its potency and overall effect being dependent on the age of the subject where VIP inhibited LPS-induced TNF-α in young patients but stimulated TNF-α in older subjects [25]. Thus, responsiveness to VIP can be regulated by many factors, including cell type and differentiation status, method of purification, activation stimulus encountered, age of subject, and drug regimens encountered by donors.
Although VIP activity has been documented to be regulated in a cAMP-dependent manner, there are additional cAMP-independent mechanisms capable of transducing VIP function. One such mechanism involves the inhibition of NF-κB, a crucial factor for the expression of inflammatory mediators such as TNF-α [26]. Thus, the effects of VIP can be explained not only through the cAMP/PKA pathway. This dichotomy in mechanisms of VIP action may explain differential regulation of proinflammatory and anti-inflammatory cytokines: inhibition of NF-κB suppresses TNF-α production, whereas activation of the cAMP/PKA/ CREB pathway in the presence of low endogenous PDE activity not only suppresses TNF-α but also positively regulates IL-10 production. Further investigation into the cAMP-dependent mechanism led to an interesting observation which described ATF-1 activation by LPS-stimulated macrophages whereas CREB was not activated; the addition of VIP abrogated this activation. The combined treatment of VIP+rolipram, however, resulted in activation of both CREB and ATF-1 upon LPS stimulation. It is possible that, in addition to NF-κB, the suppressive effect of VIP on TNF-α production is achieved through inhibition of ATF-1 activation, whereas activation of CREB by VIP upon PDE inhibition augments IL-10 production in a cell- and stimulus-specific manner.
Treatment of RA by neuropeptides such as VIP alone may not be efficacious but may be useful in combination with a PDE inhibitor. This, however, cannot be explained wholly by a failure to elevate cAMP, as treatment with dibutyryl cAMP and other cAMP-elevating drugs alone failed to induce monocyte, macrophage, and T-cell IL-10 but could, however, downregulate TNF-α upon stimulation and spontaneous TNF-α production by RA-SMCs. In the presence of an exogenous stimulus such as LPS for monocytes and macrophages and PMA/ionomycin for T cells, TNF-α production was downregulated whereas IL-10 was relatively unaffected. Of particular interest is the observation that elevation of cAMP augments macrophage IL-10 production with a concomitant potent inhibition of TNF-α upon PKC activation by PMA/ionomycin. This augmentation was not observed for VIP alone; however, treatment with VIP and simultaneous PDE inhibition by rolipram did augment IL-10 production by PMA/ionomycin-stimulated macrophages. This augmentation response is specific, as it was not reproducible in LPS-stimulated macrophages and stimulated T cells. This suggests that there is a complex relationship between the PKC and cAMP-dependent PKA pathways regulating macrophage IL-10 production and suggests that IL-10 augmentation is specific not only to cell type but also to the stimulus.
Comparing these data with those found in the mouse, the augmentation of IL-10 and downregulation of TNF-α and IFN-γ results in a Th2 cytokine profile that is dependent on cell types present, differentiation status, and the activation stimulus encountered. The effectiveness of VIP as an anti-inflammatory agent in vivo for CIA in the mouse contrasts starkly with in vitro human data. This may be due to differences between murine and human cells but it is also possible that VIP activates regulatory circuits in vivo that are not seen in vitro. The differences in VIP sensitivity between these two systems requires further work and is currently being investigated in our laboratory. Cyclic-AMP levels can be raised by increasing the activity of adenylate cyclase or by decreasing the activity of phosphodiesterases. In the light of the observation that a physiological activator of adenylate cyclase, like VIP, failed to modulate cytokine production by RA synovial cells, probably due to high PDE activity in these cells, our findings suggest that a PDE inhibitor is more likely to be effective in human RA than an activator of adenylate cyclase. However, the possibility of a combined approach using VIP together with a PDE inhibitor merits further investigation.
Conclusion
This report has demonstrated that, in humans, VIP is relatively ineffective as an anti-inflammatory mediator capable of augmenting IL-10 while concurrently inhibiting TNF-α production. This, however, is specific to cell type and to the stimulus and is dependent on endogenous PDE activity. Macrophages stimulated by phorbol ester in the presence of the PDE-resistant dibutyryl cAMP augment IL-10 and strongly suppress TNF-α. No such modulation of anti-inflammatory IL-10 was observed in T cells or macrophages stimulated by LPS. These data suggest efficacy for a combination of VIP and a phosphodiesterase inhibitor.
Competing interests
None declared.
Abbreviations
APC = antigen-presenting cell; ATF-1 = activating transcription factor-1; CIA = collagen-induced arthritis; CREB = cAMP response element binding protein; ELISA = enzyme-linked immunosorbent assay; FCS = fetal calf serum; IC50 = median inhibitory concentration; IFN = interferon; IL = interleukin; LPS = lipopolysaccharide; M-CSF = macrophage-colony stimulating factor; NF-κB = nuclear factor κB; PBMC = peripheral blood mononuclear cells; PDE = phosphodiesterase; PKA = protein kinase A; PKC = protein kinase C; PMA = phorbol 12-myristate 13-acetate; RA = rheumatoid arthritis; RA-SMC = rheumatoid arthritis synovial membrane cell; RPMI = Roswell Park Memorial Institute [medium]; Th1/Th2 = T helper cell type 1/2; TNF-α = tumour necrosis factor α; VIP = vasoactive intestinal peptide.
Acknowledgments
Acknowledgements
This work has been funded by a Wellcome Trust project grant and the Kennedy Institute of Rheumatology is supported by a core grant from the Arthritis and Rheumatism Council of Great Britain.
References
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Ann Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Kambayashi T, Jacob CO, Zhou D, Mazurek N, Fong M, Strassmann G. Cyclic nucleotide phosphodiesterase type IV participates in the regulation of IL-10 and in the subsequent inhibition of TNFα and IL-6 release by endotoxin-stimulated macrophages. J Immunol. 1995;155:4909–4916. [PubMed] [Google Scholar]
- Meisel C, Vogt K, Platzer C, Randow F, Liebenthal C, Volk H-D. Differential regulation of monocytic tumour necrosis factor-a and interleukin-10 expression. Eur J Immunology. 1996;26:1580–1586. doi: 10.1002/eji.1830260726. [DOI] [PubMed] [Google Scholar]
- Ross SE, Williams RO, Mason LJ, Mauri C, Marinova-Mutafchieva L, Malfait A-M, Maini RN, Feldmann M. Suppression of TNFα expression, inhibition of Th1 activity, and amelioration of collagen-induced arthritis by rolipram. J Immunol. 1997;159:6253–6259. [PubMed] [Google Scholar]
- Nyman U, Mussener A, Larsson E, Lorentzen J, Klareskog L. Amelioration of collagen II-induced arthritis in rats by the type IV phosphodiesterase inhibitor Rolipram. Clin Exp Immunol. 1997;108:415–419. doi: 10.1046/j.1365-2249.1997.3931291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat Med. 2001;7:563–568. doi: 10.1038/87887. [DOI] [PubMed] [Google Scholar]
- Williams RO. Therapeutic effect of vasoactive intestinal peptide in collagen-induced arthritis. Arthritis Rheum. 2002;46:271–273. doi: 10.1002/1529-0131(200201)46:1<271::AID-ART10039>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Delgado M, Pozo D, Martinez C, Leceta J, Calvo JR, Ganea D, Gomariz RP. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit endotoxin-induced TNFα production by macrophages: in vitro and in vivo studies. J Immunol. 1999;162:2358–2367. [PubMed] [Google Scholar]
- Delgado M, Martinez C, Pozo D, Calvo JR, Leceta J, Ganea D, Gomariz RP. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) protect mice from lethal endotoxemia through the inhibition of TNF-a and IL-6. J Immunol. 1999;162:1200–1205. [PubMed] [Google Scholar]
- Delgado M, Munoz-Elias EJ, Kan Y, Gozes I, Fridkin M, Brenneman DE, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumour necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun. J Biol Chem. 1998;273:31427–31436. doi: 10.1074/jbc.273.47.31427. [DOI] [PubMed] [Google Scholar]
- Martinez C, Delgado M, Pozo D, Leceta J, Calvo JR, Ganea D, Gomariz RP. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide modulate endotoxin-induced IL-6 production by murine peritoneal macrophages. J Leukoc Biol. 1998;63:591–601. doi: 10.1002/jlb.63.5.591. [DOI] [PubMed] [Google Scholar]
- Delgado M, Munoz-Elias EJ, Gomariz RP, Ganea D. VIP and PACAP inhibit IL-12 production in LPS-stimulated macrophages. Subsequent effect on IFNgamma synthesis by T cells. J Neuroimmunol. 1999;96:167–181. doi: 10.1016/S0165-5728(99)00023-5. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Inhibition of endotoxin-induced macrophage chemokine production by vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide in vitro and in vivo. J Immunol. 2001;167:966–975. doi: 10.4049/jimmunol.167.2.966. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Vasoactive intestinal peptide inhibits IL-8 production in human monocytes. Biochem Biophys Res Commun. 2003;301:825–832. doi: 10.1016/S0006-291X(03)00059-7. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide prevent inducible nitric oxide synthase transcription in macrophages by inhibiting NF-kB and IFN regulatory factor 1 activation. J Immunol. 1991;162:4685–4696. [PubMed] [Google Scholar]
- Delgado M, Munoz-Elias E, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: in vitro and in vivo studies. J Immunol. 1999;162:1707–1716. [PubMed] [Google Scholar]
- Delgado M, Ganea D. Cutting edge: is vasoactive intestinal peptide a type 2 cytokine? J Immunol. 2001;166:2907–2912. doi: 10.4049/jimmunol.166.5.2907. [DOI] [PubMed] [Google Scholar]
- Ganea D. Regulatory effects of vasoactive intestinal peptide on cytokine production in central and peripheral lymphoid organs. Adv Neuroimmunol. 1996;6:61–74. doi: 10.1016/s0960-5428(96)00007-1. [DOI] [PubMed] [Google Scholar]
- Said S. VIP as a modulator of lung inflammation and airway constriction. Am Rev Respir Dis. 1991;143:22–24. doi: 10.1164/ajrccm/143.3_Pt_2.S22. [DOI] [PubMed] [Google Scholar]
- Pozo D, Delgado M, Martinez C, Guerrero JM, Leceta J, Gomariz RP, Calvo JR. Immunobiology of vasoactive intestinal peptide (VIP) Immunol Today. 2000;21:7–11. doi: 10.1016/S0167-5699(99)01525-X. [DOI] [PubMed] [Google Scholar]
- Abrams J, Roncorolo MG, Yssel H, Andersson U, Gleich GJ, Silver J. Strategies and practice of anti-cytokine monoclonal antibody development: immunoassay of IL-10 and IL-5 in clinical samples. Immunol Rev. 1992;127:5–24. doi: 10.1111/j.1600-065x.1992.tb01406.x. [DOI] [PubMed] [Google Scholar]
- Engelberts I, Moller A, Schoen GJ, van der Linden CJ, Buurmann WA. Evaluation of measurement of human TNF in plasma by ELISA. Lymphokine Cytokine Res. 1991;10:69–76. [PubMed] [Google Scholar]
- Foey AD, Parry SL, Williams LM, Feldmann M, Foxwell BMJ, Brennan FM. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNFα: Role of the p38 and p42/44 mitogen-activated protein kinases. J Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- Dewitt D, Gourlet P, Amraoui Z, Vertongen P, Willems F, Robberecht P, Goldman M. The vasoactive intestinal peptide analogue RO25-1553 inhibits the production of TNF and IL-12 by LPS-activated monocytes. Immunol Lett. 1998;60:57–60. doi: 10.1016/S0165-2478(97)00129-6. [DOI] [PubMed] [Google Scholar]
- Hernanz A, Tato E, De la Fuente M, de Miguel E, Arnalich F. Differential effects of gastrin-releasing peptide, neuropeptide Y, somatostatin and vasoactive intestinal peptide on interleukin-1 beta, interleukin-6 and tumour necrosis factor-alpha production by whole blood cells from healthy young and old subjects. J Neuroimmunol. 1996;71:25–30. doi: 10.1016/S0165-5728(96)00118-X. [DOI] [PubMed] [Google Scholar]
- Foxwell B, Brown K, Bondeson J, Clarke C, de Martin R, Brennan F, Feldmann M. Efficient adenoviral infection with IkappaB alpha reveals that macrophage tumour necrosis factor alpha production in rheumatoid arthritis is NF-kappaB dependent. Proc Natl Acad Sci USA. 1998;95:8211–8215. doi: 10.1073/pnas.95.14.8211. [DOI] [PMC free article] [PubMed] [Google Scholar]