

Enantiomerically pure homopropargylic alcohols are highly useful intermediates, with broad synthetic utility. The terminal alkyne functionality serves as a synthetic handle for cross-coupling, metathesis, and heterocycle synthesis.[1] The addition of allenic or propargylic reagents to carbonyl compounds is mechanistically similar to the analogous reaction with allylic reagents. Though many useful and innovative methods exist for the synthesis of homoallylic alcohols,[2] the enantio-selective synthesis of homopropargylic alcohols remains arduous. Two main complications are 1) the lower reactivity of the allenylic and propargylic substrates in comparison to allylic substrates, and 2) the difficulties associated with controlling the reaction regioselectivity.[3] Herein, we describe a highly enantioselective catalytic method for the preparation of homopropargylic alcohols. Computational studies of the reaction provide insight into the catalysis and stereochemistry of the reaction.
Many current methods for enantioselective propargylation reactions rely upon the use of chiral reagents.[4] Alternative catalytic methods have been developed, but are limited to the use of allenylic or propargylic metal-based reagents or intermediates.[2a,5] Despite notable work, many of these methods are restricted by one or more limitations. Among them are 1) the use of reagents that are relatively difficult to prepare or are unstable to air and/or moisture, 2) the use of undesirable metal reagents or catalysts, and 3) regioselectivity concerns.
In the past decade, Lewis and Brønsted acid-catalyzed allylboration reactions have fascinated the synthetic community.[6,7] However, this methodology remains relatively undeveloped for the more challenging allenylboration of aldehydes. Following our recent report on the development of a chiral phosphoric acid-catalyzed allylboration,[7] we examined the extension of our methodology to the enantioselective propargylation of aldehydes. We began our investigation with the reaction of benzaldehyde and allenyl boronic acid pinacol ester. Boronate 2 is a relatively stable, non-toxic and commercially available reagent. The C–C bond formation proceeded smoothly in the presence of various chiral acid catalysts,[8] with complete control over the regioselectivity (Table 1). PA5[9] afforded product 3 with the highest enantio-selectivity, when toluene was used as the reaction solvent. An increase to 87% ee was seen with the use of higher catalyst loading, in the presence of 4Å M.S. (entry 13). The enantio-selectivity could be further increased, when the reaction was conducted at lower reaction temperatures of 0°C (entry 14) and −20°C (entry 15), albeit with longer reaction times.
Table 1.
Catalyst screening and optimization for the propargylation of benzaldehyde.[a]
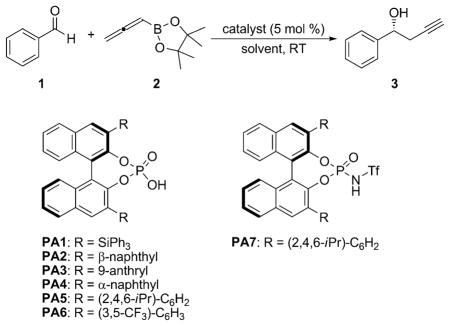
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst[b] | Solvent | t [h] | Yield [%][c] | ee [%][d] |
| 1 | PA1 | toluene | 40 | 94 | 0 |
| 2 | PA2 | toluene | 40 | 93 | 7 |
| 3 | PA3 | toluene | 40 | 92 | 20 |
| 4 | PA4 | toluene | 40 | 95 | 9 |
| 5 | PA5 | toluene | 40 | 91 | 74 |
| 6 | PA6 | toluene | 40 | 93 | 4 |
| 7 | PA7 | toluene | 40 | 94 | 16 |
| 8 | PA5 | benzene | 40 | 89 | 62 |
| 9 | PA5 | DCM | 40 | 87 | 43 |
| 10 | PA5 | PhCF3 | 40 | 94 | 68 |
| 11 | PA5 | p-xylene[e] | 40 | 92 | 75 |
| 12 | PA5 | toluene[e] | 40 | 92 | 77 |
| 13 | PA5 | toluene[e],[f] | 24 | 93 | 87 |
| 14 | PA5 | toluene[e],[f],[g] | 64 | 96 | 90 |
| 15 | PA5 | toluene[e],[f],[h] | 72 | 94 | 91 |
Reaction conditions: 1 (0.10 mmol), 2 (0.12 mmol), catalyst (5 mol%), unless otherwise specified.
All catalysts were washed with 6M HCl after purification by column chromatography.
Yields of isolated product.
Determined by chiral HPLC analysis.
Reaction conducted in presence of 4Å M.S.
20 Mol% catalyst used.
Reaction conducted at 0°C.
Reaction conducted at −20°C.
With the optimized conditions in hand,[10] a variety of aldehydes with different electronic and steric properties were tested to study the scope and limitation of the developed methodology (Table 2). The reaction proved tolerant to electron-donating and electron-withdrawing groups (1a–1j), giving excellent yields and enantioselectivities (92–96% ee). The methodology was extended to aliphatic aldehydes (1k–1m), furnishing the corresponding homopropargylic alcohol products 3k–m in 77–82% ee.
Table 2.
Enantioselective propargylation of aldehydes.[a]
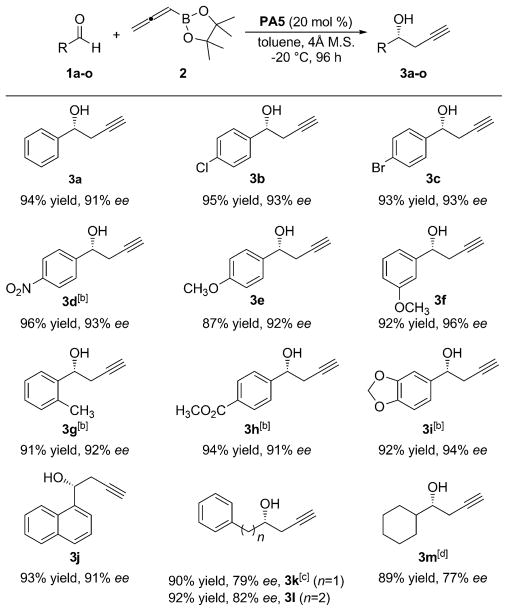
|
Reaction conditions: 1 (0.20 mmol), 2 (0.30 mmol), PA5 (20 mol%). Yields are of isolated product. Enantioselectivity was determined by chiral HPLC. The product configurations were determined to be (R) based on comparison to chiral HPLC analysis and optical rotation data reported in the literature.
The product configurations were determined to be (R) by analogy.
(S) isomer was obtained.
Enantio-selectivity was determined by 1H NMR spectroscopy after conversion to the corresponding Mosher ester.
We prepared several important synthetic scaffolds, previously unavailable from enantioenriched homopropargylic alcohols (Scheme 1). Chiral dihydrofuran-3-ones, such as 4, are important building blocks[11] for the synthesis of biologically active compounds. Despite their importance, a general enantioselective synthesis for this class of molecule has yet to be reported. We successfully transformed 3a[12] into dihydrofuran-3-one 4, by employing gold-catalyzed reaction methodology developed by Zhang and co-workers,[13] with complete preservation of the enantiomeric excess. Crabbé homologation of 3a provided optically active 3,4-allenol 5, which has the potential to serve as a substrate in natural product synthesis.[14] Chiral dihydrofuran 6, currently dependent on the Heck reaction for its synthesis,[15] was obtained through a molybdenum-mediated cycloisomerization of 3a, based on methodology developed by McDonald and co-workers.[16]
Scheme 1.

Synthesis of important chiral moieties.
It is our belief that the propargylation proceeds through a six-membered cyclic transition state, where catalyst activation operates by hydrogen-bonding of the boronate oxygen. To further understand the mechanism and stereoselectivity of this phosphoric acid-catalyzed propargylation reaction, we performed theoretical calculations. Calculated energies of different pathways for allylboration[17] and propargylation showed that Brønsted acids form a strong hydrogen bond with the pseudo-equatorial oxygen of the allenyl boronate.[18] A computed transition state structure involving protonation is shown in Figure 1.
Figure 1.

Transition state structure for the Brønsted acid-catalyzed propargylation reaction.
To explore the origins of the enantioselectivity, we studied the transition state structures for the propargylation reaction, where the phosphoric acid catalyst activates the pseudo-equatorial oxygen of the allenyl boronate. Biphenol(bipol)-derived phosphoric acid was used as the model, in place of the fully derived binol phosphoric acid, to reduce the computational time. Catalyst PA5, bearing a 2,4,6-triisopropylphenyl group at the 3,3′-positions, provides high experimental enantioselectivity. Thus, the diastereomeric transition states of the re-face and si-face attack involving the bipol model of PA5 were compared. Transition states TSr1 and TSs1 are represented in Figure 2. Re-face attack (TSr1) is predicted to be more favored than si-face attack (TSs1) by 1.3 kcalmol−1. This is in agreement with the 74% ee obtained experimentally.
Figure 2.

Optimized structures of TSr1 and TSs1. Relative energies (kcal mol−1) are shown in parentheses.
Figure 2 shows a lack of obvious steric differences in the transition states. H–H distances are 2.4 Å or more. However, the distortion of the catalyst is larger in TSs1 than in TSr1 by about 1.2 kcalmol−1. This distortion relieves steric repulsions that would otherwise occur. The preference for re-facial selectivity is therefore the result of the larger distortion of the catalyst–boronate complex in TSs1. The origins of the differences in distortion energies of the catalyst–boronate complex in the two TSs can be visualized from geometries of the catalyst in the TSs. Figure 3a shows the catalyst–boronate complex structure in TSr1. Here, the dioxaborolane ring has no significant steric interaction with the catalyst, and the dihedral angle between the 2,4,6-triisopropylphenyl substituent and the bipol core is 74°, almost the same as the dihedral angle of 72° in the optimized catalyst. Figure 3b shows the catalyst–boronate complex structure in TSs1, with the dioxaborolane ring on the left. The methyl groups (circled in Figure 3b) of the dioxaborolane ring and the isopropyl groups of the catalyst (circled in Figure 3b) are close to each other. In order to minimize such steric repulsions, the 2,4,6-triisopropylphenyl substituent is rotated around the bond to the bipol phenyl core with a dihedral angle of 78°. This is a 6° rotation away from the dihedral angle in the optimized catalyst (72°). The asymmetric induction can be rationalized by differences in distortion energies originating from the steric interactions between the substrates and the bulky 3,3′-substituents on the catalyst.
Figure 3.

a) 3D structure of TSr1 without benzaldehyde. b) 3D structure of TSs1 without benzaldehyde.
For other catalysts screened experimentally, calculations showed the absence of an energy difference between re- and si-attack diastereomeric transition states, suggesting why these catalysts gave low enantioselectivities.
In summary, we have developed the first Brønsted acid-catalyzed propargylation of aldehydes, for the synthesis of chiral homopropargylic alcohols. The reaction is simple and highly efficient, demonstrating broad synthetic utility. Mechanistic studies show the catalyst activating the reaction by forming a strong hydrogen bond with the pseudo-equatorial oxygen of the boronate. The high enantioselectivity obtained with catalyst PA5 originates from steric interactions between the methyl groups of the allenylboronate, the bulky catalyst substituents, and the resulting distortion of the catalyst.
Experimental Section
General procedure for the propargylation of aldehydes: A screw-cap reaction tube, with a stir bar and 4Å MS (100 mg) was evacuated, flame-dried, and back-filled with argon. The tube was charged with (R)-trip-PA catalyst PA5 (20 mol%), freshly distilled aldehyde 1 (0.20 mmol) and 1.5 mL of dry toluene. The reaction mixture was then cooled to −20°C, followed by the addition of allenylboronic acid pinacol ester 2 (0.30 mmol), slowly over 30 s. The mixture was stirred 96 h at this temperature, and then directly purified by silica gel column chromatography (hexanes:EtOAC =9:1) to afford product 3.
Supplementary Material
Footnotes
We thank the National Institutes of Health (NIH GM-36700 and GM-082935) and the National Science Foundation CAREER program (NSF-0847108) for financial support.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201107407.
Contributor Information
Pankaj Jain, Department of Chemistry, University of South Florida, 4202 E. Fowler Avenue, Tampa, FL 33620 (USA).
Hao Wang, Department of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, CA 90095 (USA).
Prof. Dr. Kendall N. Houk, Email: houk@chem.ucla.edu, Department of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, CA 90095 (USA)
Prof. Dr. Jon C. Antilla, Email: jantilla@usf.edu, Department of Chemistry, University of South Florida, 4202 E. Fowler Avenue, Tampa, FL 33620 (USA)
References
- 1.For selected examples, see: Trost BM, Dumas J, Villa M. J Am Chem Soc. 1992;114:9836–9845.McDonald FE, Gleason MM. J Am Chem Soc. 1996;118:6648–6659.Schmidt DR, O’Malley SJ, Leighton JL. J Am Chem Soc. 2003;125:1190–1191. doi: 10.1021/ja0283201.O’Sullivan PT, Buhr W, Fuhry MAA, Harrison JR, Davies JE, Feeder N, Marshall DR, Burton JW, Holmes AB. J Am Chem Soc. 2004;126:2194–2207. doi: 10.1021/ja038353w.Trost BM, Dong G. Nature. 2008;456:485–488. doi: 10.1038/nature07543.Francais A, Leyva A, Etxebarria-Jardi G, Ley SV. Org Lett. 2010;12:340–343. doi: 10.1021/ol902676t.
- 2.For selected examples, see: Denmark SE, Fu J. Chem Rev. 2003;103:2763–2794. doi: 10.1021/cr020050h.Brown HC, Jadhav PK. J Am Chem Soc. 1983;105:2092–2093.Corey EJ, Yu CM, Kim SS. J Am Chem Soc. 1989;111:5495–5496.Keck GE, Tarbet KH, Geraci LS. J Am Chem Soc. 1993;115:8461–8462.Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044–8049. doi: 10.1021/ja043612i.Lachance H, Hall DG. Org React. 2008;73:1.Chen M, Handa M, Roush WR. J Am Chem Soc. 2009;131:14602–14603. doi: 10.1021/ja904599h.Althaus M, Mahmood A, Suarez JR, Thomas SP, Aggarwal VK. J Am Chem Soc. 2010;132:4025–4028. doi: 10.1021/ja910593w.
- 3.Yamamoto H. In: Comprehensive Organic Synthesis: Propargyl and Allenyl Organometallics. Heathcock CH, editor. Vol. 2. Pergamon; Oxford: 1991. pp. 81–98. [Google Scholar]
- 4.a) Ikeda N, Arai I, Yamamoto H. J Am Chem Soc. 1986;108:483–486. doi: 10.1021/ja00263a020. [DOI] [PubMed] [Google Scholar]; b) Haruta R, Ishiguro M, Ikeda N, Yamamoto H. J Am Chem Soc. 1982;104:7667–7669. [Google Scholar]; c) Corey EJ, Yu CM, Lee DH. J Am Chem Soc. 1990;112:878–879. [Google Scholar]; d) Lee KC, Lin MJ, Loh TP. Chem Commun. 2004:2456–2457. doi: 10.1039/b411653d. [DOI] [PubMed] [Google Scholar]; e) Lai C, Soderquist JA. Org Lett. 2005;7:799–802. doi: 10.1021/ol0476164. [DOI] [PubMed] [Google Scholar]
- 5.For reviews on asymmetric propargylation, see: Ding CH, Hou XL. Chem Rev. 2011;111:1914–1937. doi: 10.1021/cr100284m.for selected examples, see: Boldrini GP, Tagliavini E, Trombini C, Umani-Ronchi A. J Chem Soc Chem Commun. 1986:685–686.Minowa N, Mukaiyama T. Bull Chem Soc Jpn. 1987;60:3697–3704.Keck GE, Krishnamurthy D, Chen X. Tetrahedron Lett. 1994;35:8323–8324.Yu CM, Yoon SK, Choi HS, Baek K. Chem Commun. 1997:763–764.Yu CM, Yoon SK, Baek K, Lee JY. Angew Chem. 1998;110:2504–2506.Angew Chem Int Ed. 1998;37:2392–2395. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2392::AID-ANIE2392>3.0.CO;2-D.Iseki K, Kuroki Y, Kobayashi Y. Tetrahedron: Asymmetry. 1998;9:2889–2894.Denmark SE, Wynn T. J Am Chem Soc. 2001;123:6199–6200. doi: 10.1021/ja016017e.Evans DA, Sweeney ZK, Rovis T, Tedrow JS. J Am Chem Soc. 2001;123:12095–12096. doi: 10.1021/ja011983i.Hanawa H, Uraguchi D, Konishi S, Hashimoto T, Maruoka K. Chem Eur J. 2003;9:4405–4413. doi: 10.1002/chem.200305078.Inoue M, Nakada M. Org Lett. 2004;6:2977–2980. doi: 10.1021/ol048826j.Naodovic M, Xia G, Yamamoto H. Org Lett. 2008;10:4053–4055. doi: 10.1021/ol801452c.Fandrick DR, Fandrick KR, Reeves JT, Tan Z, Tang W, Capacci AG, Rodriguez S, Song JJ, Lee H, Yee NK, Senanayake CH. J Am Chem Soc. 2010;132:7600–7601. doi: 10.1021/ja103312x.Usanov DL, Yamamoto H. Angew Chem. 2010;122:8345–8348.Angew Chem Int Ed. 2010;49:8169–8172. doi: 10.1002/anie.201002751.Shi SL, Xu LW, Oisaki K, Shibasaki M. J Am Chem Soc. 2010;132:6638–6639. doi: 10.1021/ja101948s.Chen J, Captain B, Takenaka N. Org Lett. 2011;13:1654–1657. doi: 10.1021/ol200102c.Trost BM, Ngai MY, Dong G. Org Lett. 2011;13:1900–1903. doi: 10.1021/ol200043n.for the lone example of catalytic asymmetric propargylation not involving the use of allenylic or propargylic metal reagents or intermediates, see chiral biphenol-catalyzed allenylboration of ketones: Barnett DS, Schaus SE. Org Lett. 2011;13:4020–4023. doi: 10.1021/ol201535b.
- 6.For selected examples, see: Kennedy JWJ, Hall DG. J Am Chem Soc. 2002;124:11586–11587. doi: 10.1021/ja027453j.Ishiyama T, Ahiko TA, Miyaura N. J Am Chem Soc. 2002;124:12414–12415. doi: 10.1021/ja0210345.Yu SH, Ferguson MJ, McDonald R, Hall DG. J Am Chem Soc. 2005;127:12808–12809. doi: 10.1021/ja054171l.Rauniyar V, Hall DG. Angew Chem. 2006;118:2486–2488. doi: 10.1002/anie.200504432.Angew Chem Int Ed. 2006;45:2426–2428. doi: 10.1002/anie.200504432.Rauniyar V, Zhai H, Hall DG. J Am Chem Soc. 2008;130:8481–8490. doi: 10.1021/ja8016076.for chiral biphenols-catalyzed allylborations see: Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660–12661. doi: 10.1021/ja0651308.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398–15404. doi: 10.1021/ja075204v.Barnett DS, Moquist PN, Schaus SE. Angew Chem. 2009;121:8835–8838. doi: 10.1002/anie.200904715.Angew Chem Int Ed. 2009;48:8679–8682. doi: 10.1002/anie.200904715.
- 7.Jain P, Antilla JC. J Am Chem Soc. 2010;132:11884–11886. doi: 10.1021/ja104956s.for reduction of ketones using phosphoric acid-derived catalysts, see: Zhang Z, Jain P, Antilla JC. Angew Chem. 2011;123:11153–11156.Angew Chem Int Ed. 2011;50:10961–10964. doi: 10.1002/anie.201103883.
- 8.For reviews on phosphoric acid catalysis, see: Akiyama T. Chem Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j.Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r.Terada M. Chem Commun. 2008:4097–4112. doi: 10.1039/b807577h.
- 9.Both enantiomers of trip-PA (R and S) are commercially available or easily prepared from binol by using a literature procedure: Hoffmann S, Seayad A, List B. Angew Chem. 2005;117:7590–7593. doi: 10.1002/anie.200503062.Angew Chem Int Ed. 2005;44:7424–7427. doi: 10.1002/anie.200503062.
- 10.Though benzaldehyde showed >90% conversion within 48 h at −20°C, the reaction time was not further optimized and all the substrates were allowed to react for 96 h to ensure complete conversion.
- 11.A Scifinder search of compounds with the dihydrofuran-3-one moiety resulted in more than 5000 entries with reported biological activities.
- 12.95% ee was obtained for 3a when the reaction was run on a larger scale (2 mmol, benzaldehyde) when compared to a run on the 0.2 mmol scale (91% ee) under identical conditions.
- 13.Ye L, Cui L, Zhang G, Zhang L. J Am Chem Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Crabbé P, Nassim B, Robert-Lopes MT. Org Synth. 1985;63:203. [Google Scholar]; b) Ma S, Hou H, Zhao S, Wang G. Synthesis. 2002:1643–1645. [Google Scholar]; c) Cheng X, Jiang X, Yu Y, Ma S. J Org Chem. 2008;73:8960–8965. doi: 10.1021/jo8015677. [DOI] [PubMed] [Google Scholar]; d) Coeffard V, Aylward M, Guiry PJ. Angew Chem. 2009;121:9316–9319. doi: 10.1002/anie.200903647. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9152–9155. doi: 10.1002/anie.200903647. [DOI] [PubMed] [Google Scholar]; e) Bates RW, Lu Y. Org Lett. 2010;12:3938–3941. doi: 10.1021/ol1016492. [DOI] [PubMed] [Google Scholar]
- 15.For selected examples, see: Dai WM, Yeung KKY, Wang Y. Tetrahedron. 2004;60:4425–4430.Wu W, Peng Q, Dong D, Hou X, Wu Y. J Am Chem Soc. 2008;130:9717–9725. doi: 10.1021/ja7104174.
- 16.McDonald FE, Connolly CB, Gleason MM, Towne TB, Treiber KD. J Org Chem. 1993;58:6952–6953. [Google Scholar]
- 17.Frisch MJ, et al. Gaussian03, revisionC.02. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 18.Wang H, Jain P, Antilla JC, Houk KN. manuscript in preparation. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.