

Abstract
Basket axon collaterals synapse onto the Purkinje soma/axon initial segment (AIS) area to form specialized structures, the pinceau, which are critical for normal cerebellar function. Mechanistic details of how the pinceau become organized during cerebellar development are poorly understood. Loss of cytoskeletal adaptor protein Ankyrin G (AnkG) results in mislocalization of the cell adhesion molecule Neurofascin (Nfasc) at the Purkinje AIS and abnormal organization of the pinceau. Loss of Nfasc in adult Purkinje neurons leads to slow disorganization of the Purkinje AIS and pinceau morphology. Here, we used mouse conditional knock-out techniques to show that selective loss of Nfasc, specifically in Purkinje neurons during early development, prevented maturation of the AIS and resulted in loss of Purkinje neuron spontaneous activity and pinceau disorganization. Loss of Nfasc in both Purkinje and basket neurons caused abnormal basket axon collateral branching and targeting to Purkinje soma/AIS, leading to extensive pinceau disorganization, Purkinje neuron degeneration, and severe ataxia. Our studies reveal that the Purkinje Nfasc is required for AIS maturation and for maintaining stable contacts between basket axon terminals and the Purkinje AIS during pinceau organization, while the basket neuron Nfasc in combination with Purkinje Nfasc is required for proper basket axon collateral outgrowth and targeting to Purkinje soma/AIS. Thus, cerebellar pinceau organization requires coordinated mechanisms involving specific Nfasc functions in both Purkinje and basket neurons.
Introduction
Development of neuronal circuits is controlled by signaling mechanisms that coordinate precise targeting of presynaptic axons to their postsynaptic targets (Lu et al., 2009; Kolodkin and Tessier-Lavigne, 2011). The GABAergic interneurons frequently target inhibitory synapses to the soma or axon initial segment (AIS) of principal cells (Freund and Buzsáki, 1996; Somogyi et al., 1998; Ango et al., 2004). In the cerebellum, basket interneurons target the Purkinje soma and AIS (Somogyi and Hámori, 1976; Somogyi et al., 1983; Li et al., 1992; Ango et al., 2004). Since Purkinje neurons are the sole cerebellar output source, their activity must be strictly regulated. Genetic mutations resulting in Purkinje neuron dysfunction lead to cerebellar dysfunction and ataxia (Hamilton et al., 1996; Wang and Zoghbi, 2001; Gold et al., 2003; Levin et al., 2006; Perkins et al., 2010; Rinaldo and Hansel, 2010). Cerebellar basket axons branch into many axonal collaterals that form the basket-shaped pinceau at the Purkinje soma/AIS (Ramon y Cajal, 1911). The pinceau inhibitory synapses modulate the frequency of Purkinje neuron action potential spikes (Sakaba, 2008). However, the mechanisms responsible for the organization of the pinceau and targeting of basket axon collaterals to Purkinje soma/AIS are not well understood.
The Purkinje AIS is enriched in Ig-like domain-containing cell adhesion molecules (CAMs), including Neurofascin (Nfasc) (Brümmendorf et al., 1998; Huang, 2006), which bind to Ankyrin G (AnkG), an adaptor protein that links membrane proteins to the spectrin/actin cytoskeleton (Zhou et al., 1998; Bennett and Baines, 2001; Boiko et al., 2007). Other components of the Purkinje AIS required for action potential initiation are the voltage-gated ion channels, including sodium (NaV) channels (Catterall, 1981; Raman and Bean, 1999; Khaliq and Raman, 2006; Benton and Raman, 2009). The mechanisms that underlie the enrichment and maintenance of ion channels at the AIS are still poorly understood.
Previous studies showed that loss of AnkG from the Purkinje AIS resulted in mislocalization of Nfasc, altered basket axon targeting, and disrupted pinceau synapses (Ango et al., 2004). Purkinje neuron expression of dominant-negative Nfasc also disrupted pinceau synapses, implicating a role for Nfasc in the formation of GABAergic pinceau synapses (Ango et al., 2004). Recent studies used an inducible Cre recombinase to ablate Nfasc in adult neurons and revealed slow destabilization of the Purkinje AIS, causing deficits in Purkinje physiology (Zonta et al., 2011). However, these studies did not address the role of Nfasc in the developmental organization of the Purkinje AIS or basket axon targeting to form the pinceau.
Here, we show that Purkinje neuron-specific ablation of Nfasc results in failure of the Purkinje AIS to mature, and basket axon collaterals fail to establish proper pinceau at the Purkinje soma/AIS. Consequently, basket neuron inhibitory input to Purkinje neurons is significantly reduced. Furthermore, Nfasc ablation in both Purkinje and basket neurons revealed Nfasc expression in basket neurons and caused abnormal basket axon collateral branching and targeting to the Purkinje soma/AIS, resulting in more severe pinceau disorganization. Together, our results establish that Nfasc performs distinct functions in Purkinje and basket neurons to coordinate cerebellar pinceau organization during postnatal development.
Materials and Methods
Animals.
All animal experiments were performed according to University of North Carolina–Institutional Animal Care and Use Committee approved guidelines for ethical treatment of laboratory animals. Parv-Cre and TaumGFP/LacZ mice (data not shown) (Hippenmeyer et al., 2005) were generously provided by Dr. William Snider (University of North Carolina). Pcp2-Cre mice (Barski et al., 2000) and β-Actin-Cre mice (Thaxton et al., 2011) were obtained from Jackson Laboratories (Maine). Gad67-GFP transgenic mice (Ango et al., 2004) were generously provided by Dr. Patricia Maness (University of North Carolina).
Tissue-specific deletion of the Nfasc gene.
The NfascFlox mice used in this study were previously described (Pillai et al., 2009; Thaxton et al., 2011). Genomic DNA was extracted from tail and cerebellar samples using the REDExtract-N-Amp Tissue PCR kit (Sigma-Aldrich). The primer sequences that were used to identify specific genotypes were as follows: Pcp2-Cre 5′- GGCCAATGTCTGACCAAATAC-3′ and 5′-CTCCCACCGTCAGTACGTGAGAT-3′; Parv-Cre 5′-CAGCCTCTGTTCCACATACACTCC-3′ and 5′-TCACTCGAGAGTACCAAGCAGGCAGGAGATATC-3′; TaumGFP/LacZ 5′-ACAACGTCGTGAGTGGGAAAA-3′ and 5′-ATCAACATTAAATGTGAGCGAG-3′.
Antibodies.
The following antisera were described previously: Guinea pig anti-NF186 and rat anti-pan Nfasc (Pillai et al., 2009; Thaxton et al., 2010), rat anti-AnkG (Thaxton et al., 2010), and rabbit anti-Brevican (John et al., 2006). Other primary antibodies used here include: rabbit anti-Calbindin and mouse anti-Calbindin (Sigma), mouse anti-Parvalbumin (Millipore), and mouse anti-phospho-neurofilaments (Covance, SMI 31). The monoclonal antibodies mouse anti-NaV1.6 (K87A,10), mouse anti-KV1.1 (K20/78), mouse anti-KV1.2 (K14/16), and mouse anti-PSD95 (K28/43) were obtained from University of California, Davis/NIH NeuroMab Facility. Secondary antibodies used for immunofluorescence were as follows: Alexa Fluor conjugated -488, -568, and -647 (Invitrogen).
Tissue preparation and immunostaining.
Tissues used for immunostaining were prepared essentially as previously described (Buttermore et al., 2011). Briefly, for cerebellar sections, wild-type and mutant mice of either gender were deeply anesthetized and transcardially perfused with saline buffer followed by ice-cold 4% paraformaldehyde in PBS. The cerebellum was dissected out and postfixed in 4% paraformaldehyde overnight at 4°C. The tissues were rinsed several times in PBS and sectioned to 30 μm using a Vibratome (Leica). The cerebellar sections were then immediately immunostained, beginning with permeabilization in ice-cold acetone for 20 min followed by washing in PBS and blocking in buffer (5% BSA, 1% NGS, and 0.2% Triton X-100, in PBS) for 1 h. Cerebellar sections were then incubated with primary antibodies overnight in blocking buffer, followed by washing in PBS, incubation with secondary antibodies for 1 h at room temperature (RT), and another round of washing in PBS. Sections were mounted in VectaShield (Vector Laboratories) before imaging.
For NaV antibodies, wild-type and mutant mice, of either gender, were deeply anesthetized and transcardially perfused with saline buffer followed by ice-cold 2% paraformaldehyde in PBS. The cerebellum was dissected out and postfixed in 2% paraformaldehyde for 2 h on ice. The tissues were rinsed several times in PBS before being placed in 30% sucrose overnight for two nights. The tissues were then blocked and frozen in M-1 Embedding Matrix (Shandon, Thermo Scientific) and sectioned to 20 μm using a cryostat (Bright Instrument Company LTD). The cerebellar sections were then immediately immunostained as described above.
Quantification and analysis.
Quantification of basket axon collaterals at the Purkinje soma and AIS at P20 was completed using three independent wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox age-matched mice of either gender. Cerebellar tissue was processed as described above and immunostaining with phosphorylated neurofilaments (pNfl) and calbindin (Calb) allowed visualization of basket axon collaterals surrounding the Purkinje soma and AIS. Purkinje neurons from each mouse were selected at random and the number of basket axon collaterals surrounding the Purkinje soma and AIS were counted. The average number of basket collaterals was calculated for each genotype, age, and location (soma vs AIS).
Quantification of compact versus broken/noncompact pinceau, immunostained by KV1.2 channels at P16, P20, and P30 was completed using three independent wild-type, Pcp2-Cre;NfascFlox, and Parv-Cre;NfascFlox age-matched mice of either gender. Cerebellar tissue sections were immunostained with KV1.2 and Calb, and Purkinje neurons were randomly selected to score and count the pinceau phenotype. In addition, the presence of ectopic KV1.2 clusters around the Purkinje soma was also quantified for each genotype.
Neurons were selected randomly for both pNfl and KV1.2 quantification by looking at the immunostaining in the Calb channel alone and at the Purkinje AISs. If a full Purkinje AIS was visible, a confocal image was taken of both Calb and the other channel for counting (pNfl or KV1.2). Images were then used for quantitation.
Quantification of synapse formation on the bottom half of the Purkinje soma closest to the granule-cell layer of one-month-old wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella was completed using three separate EM grids from one mouse of each genotype and of either gender. The average number of synapses per Purkinje soma-base was calculated.
Quantification of Purkinje neuron death in 5-month-old Parv-Cre;NfascFlox compared with wild-type cerebella was completed by immunostaining cerebellar sections with Calb to elucidate Purkinje neurons. The total number of Purkinje neurons present in each of the following cerebellar loops: I, II, III, IV, V, VIa, VIb, and VIII were counted in five independent mice of either gender per genotype. The average number of Purkinje neurons per loop was calculated for each genotype.
In the quantification of pNfl (see Fig. 4) and EM synapses (see Fig. 6), “n” refers to the total number of Purkinje cells/pinceau counted. In the quantification of pinceau Kv1.2 channels (see Fig. 4), “n” refers to the number of pinceau counted per animal in each of three independent animals. The cells were counted from 3–4 separate animals for each genotype. In counting Purkinje neuron death (see Fig. 7), “n” refers to the number of cerebellar loops that were counted from five separate animals for each of the three genotypes. For statistical analysis, we used the Student's t test (unpaired) and ANOVA test (single factor). Data are presented as averages ± SE measurement (SEM).
Figure 4.
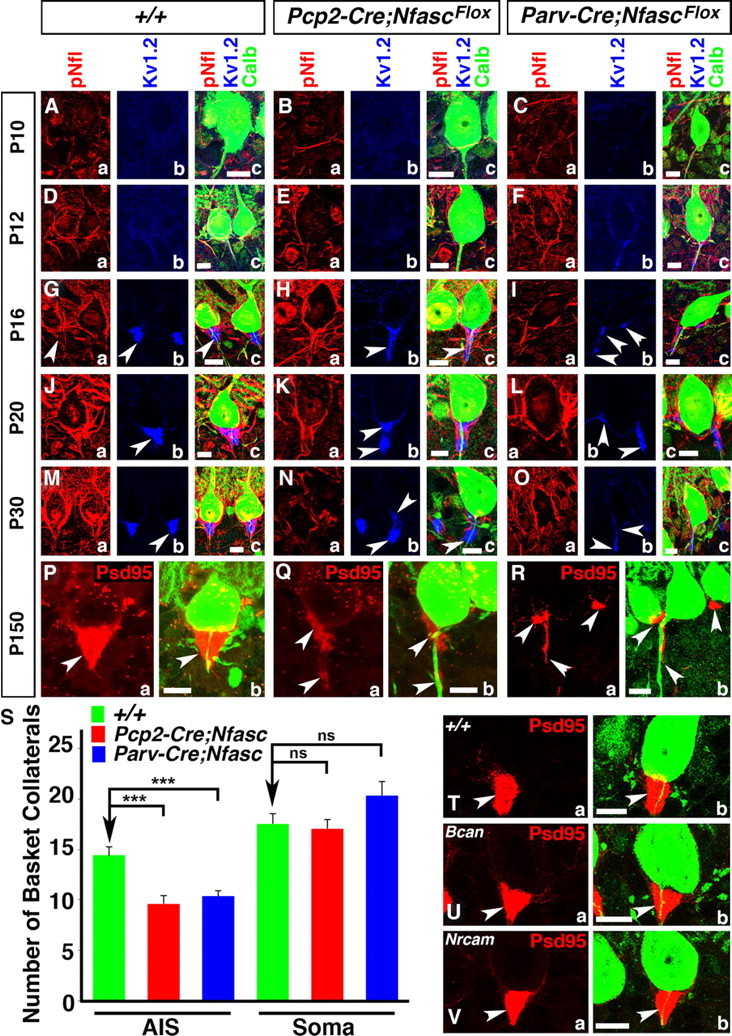
Ablation of Nfasc disrupts basket axon targeting and pinceau organization. Wild-type (A, D, G, J, M), Pcp2-Cre;NfascFlox (B, E, H, K, N), and Parv-Cre;NfascFlox (C, F, I, L, O) cerebellar sections from ages P10 (A–C), P12 (D–F), P16 (G–I), P20 (J–L), and P30 (M–O) immunostained against pNfl (a, red), KV1.2 (b, blue), and Calb (c, green, merged). Pinceau disruption becomes evident in Pcp2-Cre;NfascFlox cerebella at P16, when fewer collaterals appear to reach the Purkinje AIS (Ha, c), and KV1.2 does not become as enriched as at the wild-type pinceau (Hb, c). This trend continues at P20 and P30, where fewer collaterals reach the Purkinje AIS (Ka, Na) and potassium channels fail to cluster properly and appear as broken clusters compared with wild-type (Kb, Nb). In Parv-Cre;NfascFlox cerebella at P16, basket axon targeting is disrupted (Ia, c) and KV1.2 clusters ectopically around the soma and at random places along the Purkinje AIS (Ib, c). This disruption continues at P20 and P30 in Parv-Cre;NfascFlox cerebella with altered basket axon targeting (La, Oa) and ectopic KV1.2 clusters (Lb, Ob). Wild-type (P, T), Pcp2-Cre;NfascFlox (Q), Parv-Cre;NfascFlox (R), Brevican knock-out (Bcan, U), and NrCAM knock-out (V) cerebella immunostained against PSD95 (a, red) and Calb (b, green, merged). At P150, the wild-type pinceau maintains its cone-shaped PSD95 localization (P, T), while it is completely disrupted in both Pcp2-Cre;NfascFlox (Q) and Parv-Cre;NfascFlox cerebella (R). S, Quantification of basket axon collateral targeting to the Purkinje AIS and soma in Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella. The pinceau remains intact in both Bcan (U) and NrCAM (V) mutants. Scale bars: 10 μm. ns, Not significant. ***p < 0.001.
Figure 6.

Cerebellar pinceau disorganization and infiltration of glial processes in Nfasc-deficient Purkinje and basket neuron cerebella. A–C, Electron micrographs of the pinceau region of cerebellar sections from one-month-old wild-type (+/+) pinceau show compact, electron-dense basket axon collaterals (B) surrounding the Purkinje soma/AIS highlighted by red line (A). Note the presence of synapses between Purkinje (P) AIS and basket collaterals (B) (A, arrowheads). In 1-month-old Pcp2-Cre;NfascFlox cerebella, the pinceau appears slightly disorganized, with spaces beginning to form between basket axon terminals (B) (B, arrows). Note the presence of a synapse between the basket terminal and the Purkinje soma (B, arrowhead). In 1-month-old Parv-Cre;NfascFlox cerebella, the pinceau appears more disorganized with glial processes infiltrating spaces between basket axon terminals (C, arrows). D–F, Electron micrographs at higher magnification showing synapses (black arrowheads) with presynaptic (basket terminal) and postsynaptic (Purkinje AIS) areas in the wild-type (D), Pcp2-Cre;NfascFlox (E), and Parv-Cre;NfascFlox (F) cerebella. G–I, Electron micrographs from cerebellar sections from 3-month-old wild-type cerebella show highly compact basket terminals surrounding the Purkinje AIS (G). Pcp2-Cre;NfascFlox pinceau at 3 months shows increased infiltration of glial processes (H, black arrows). A 3-month-old Parv-Cre;NfascFlox pinceau shows increased disorganization of basket terminals surrounding the Purkinje soma/AIS (I, black arrows). J–O, Electron micrographs of the pinceau from 4-month-old wild-type (J, M), Pcp2-Cre;NfascFlox (K, N), and Parv-Cre;NfascFlox (L, O) cerebella. Wild-type pinceau show compact basket axon terminals surrounding the Purkinje soma/AIS. The Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella show severely disorganized basket terminals with increased infiltrating glial processes (black arrows). In some instances, in Parv-Cre;NfascFlox cerebella the entire pinceau areas are completely disrupted (O, black arrows). Scale bars: 0.5 μm.
Figure 7.

Purkinje neuron-specific ablation of Nfasc abolishes Purkinje neuron spontaneous activity and reduces inhibitory inputs to Purkinje neurons. A, Representative traces of spontaneous Purkinje neuron firing in wild-type (+/+) and Pcp2-Cre;NfascFlox Purkinje neurons. B, Plot of the spontaneous firing rates of wild-type (black circles) and Pcp2-Cre;NfascFlox mutant (white circles) Purkinje neurons. Larger diamonds show average spontaneous firing rates of Purkinje neurons in wild-type (black diamond) and Pcp2-Cre;NfascFlox mutants (white diamond). C, Representative traces of the action potential train response of Purkinje neurons to injected current at increasing depolarizing steps. D–F, Output responses from wild-type (black circles) and Pcp2-Cre;NfascFlox (white circles) Purkinje neurons to increasing levels of injected current. G, Representative traces of mIPSC recordings from wild-type and Pcp2-Cre;NfascFlox Purkinje neurons. H, Plot of mIPSC frequency reveals decreased frequency in Pcp2-Cre;NfascFlox mutant Purkinje neurons. I, Plot of mIPSC amplitude revealing no change between the wild-type and Pcp2-Cre;NfascFlox mutant Purkinje neurons.
Transmission electron microscopy.
Transmission electron microscopy (TEM) of age-matched wild-type, Pcp-2-Cre;NfascFlox and Parv-Cre;NfascFlox mice of either gender was performed essentially as described previously (Garcia-Fresco et al., 2006; Pillai et al., 2009; Thaxton et al., 2010).
Image analysis.
Confocal images were captured with a Bio-Rad Radiance 2000 laser-scanning system attached to a Zeiss Axioplan2 microscope. Scanning parameters were optimized for wild-type tissues and maintained for scanning the mutant tissues. The immunofluorescence images shown are Z stacks of 4–8 sections with a scan step of 0.25 μm. Adobe Photoshop software was used for processing and assembling of all figures.
Electrophysiology
Slice preparation.
Experiments were performed on both wild-type and Pcp2-Cre;NfascFlox mutant littermate mice, aged between 3 months and 6 months. At these developmental time points, Purkinje neuron degeneration is not observed in Pcp2-Cre;NfascFlox mutants. Animals were first anesthetized with halothane and then rapidly decapitated. The cerebellar vermis was immediately removed and cooled to 4°C in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 5 KCl, 1.25 Na2HPO4, 1 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 d-glucose, bubbled with 95% O2 and 5% CO2. Parasagittal slices of the cerebellar vermis (190 μm) were prepared with a vibratome (VT-1000S; Leica). Slices were then incubated at least 1 h at RT in oxygenated ACSF.
Somatic whole-cell patch-clamp recordings.
Throughout recording, slices were continuously perfused with ACSF, at room temperature, to improve the quality of clamp. Patch-clamp recordings from the Purkinje soma were performed using an EPC-10 amplifier (HEKA Electronics). Currents were filtered at 3 kHz, digitized at 5–10 kHz, and acquired using Patchmaster software (HEKA). For recording action potentials, patch pipettes (2–5 MΩ) were filled with a solution containing the following (in mm): 120 K-gluconate, 9 KCl, 10 KOH, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, and 17.5 sucrose (pH 7.25–7.35). For spontaneous and miniature IPSC (sIPSC and mIPSC) recordings, the internal solution contained the following (in mm): 150 CsCl, 4.6 MgCl2, 10 HEPES, 1 EGTA, 0.1 CaCl2, 4 Na2ATP, 0.4 Na3GTP. Purkinje neurons were voltage-clamped at holding potentials in the range of −65 to −70 mV. At this potential and with the CsCl-based internal solution, EPSC and IPSCs were recorded as inward currents. To specifically isolate the excitatory inward currents from inhibitory inward current, NBQX (10 μm) and APV (50 μm) were added to the ACSF throughout the sIPSCs and mIPSCs recordings to block AMPA and NMDA receptors. mIPSCs, corresponding to responses to single synaptic release, were measured under action potential suppression by 1 μm TTX, a voltage-dependent Na+ channel blocker. For EPSPs/EPSCs recordings with the K-Gluconate based internal solution, Picrotoxin (200 μm) was added in the ACSF throughout recordings. To activate PFs, glass electrodes filled with ACSF were placed in the upper molecular layer. The input and series resistances were monitored throughout experiments by applying hyperpolarizing voltage steps (−10 mV) at the end of each sweep. Recordings were excluded if series or input resistances varied by >15% over the course of the experiments.
Analysis.
Data were analyzed with Excel (Microsoft) or Igor (Wavemetrics). Spontaneous IPSCs and miniature IPSCs were analyzed using Patcher's Power Tool program with a threshold of 10 pA for events detection. All data are expressed as the mean ± SEM. For statistical analysis, we used the Student's t test (paired/unpaired) and the Mann–Whitney U test, when appropriate.
Behavioral testing
Ten wild-type and 10 Pcp2-Cre;NfascFlox mice of either gender were tested at one month, two months, three months, and four months in the following tests.
Locomotor activity.
Exploratory activity in a novel environment was assessed by 1 h trials in an open field chamber (40 × 40 × 30 cm) crossed by a grid of photobeams (VersaMax system, AccuScan Instruments). Counts were taken of the number of photobeams broken during the trial in 5 min intervals, with separate measures for ambulation (total distance traveled) and rearing movements.
Rotarod.
Mice were tested for motor coordination and learning on an accelerating rotarod (Ugo Basile, Stoelting). For each test session, animals were given two trials, with 45 s between each trial. Rpm (revolutions per minute) was set at an initial value of 3, with a progressive increase to a maximum of 30 rpm across 5 min (the maximum trial length). Measures were taken for latency to fall from the top of the rotating barrel.
Basket cell dye-injections
Parasagittal slice preparation.
Mice were anesthetized with pentobarbital (40 mg/kg) and decapitated after disappearance of corneal reflexes, in compliance with University of North Carolina guidelines. Brains were rapidly removed and immersed in ice-cold dissection buffer (in mm: 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 75 sucrose, 10 dextrose, 1.3 ascorbic acid, 7 MgCl2, and 0.5 CaCl2) bubbled with 95% O2-5% CO2. The cerebellum was dissected and 250 μm parasagittal slices were prepared using a vibrating microtome (Leica VT1000S). Slices were allowed to recover for 20 min in a 35°C submersion chamber filled with oxygenated ACSF (in mm; 124 NaCl, 3 KCl, 1.25 Na2PO4, 26 NaHCO3, 1 MgCl2 2 CaCl2 and 20 glucose) and then kept at room temperature for >40 min until use.
Biocytin fills of cerebellar basket neurons.
Slices were placed in a submersion chamber, maintained at 30°C, and perfused at 2 ml/min with oxygenated ACSF (as described above). Cells were visualized using a Ziess Axioskop microscope equipped with infrared differential interference contrast (IR-DIC) optics. Patch pipettes were pulled from thick-walled borosilicate glass (P2000, Sutter Instruments). Open tip resistances were between 2.5–5 MΩ when pipettes were filled with the internal solution containing the following (in mm): 100 K-Gluconate, 20 KCl, 0.2 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 10 HEPES, 10 Na-phosphocreatine, 0.5% neurobiotin (Vector Laboratories), and 0.025 Alexa-488 or -568 with pH adjusted to 7.25 with 1 m KOH and osmolarity adjusted to ∼295 mOsm by addition of sucrose. Voltage-clamp recordings were performed in the whole-cell configuration using patch-clamp amplifier (Multiclamp 700A, Molecular Devices). Pipette seal resistances were >1 GΩ, and pipette capacitive transients were minimized before breakthrough. Basket neurons were filled for 15–20 min before the slice was removed and placed in 4% paraformaldehyde (in 0.1 m PBS) overnight. The cerebellar sections were processed for immunostaining as described above.
Quantification and analysis.
Two independent wild-type, Pcp2-Cre;NfascFlox, and Parv-Cre;NfascFlox mice of either gender at P10 and P20 were used for biocytin dye-injection experiments. A minimum of three basket cells were injected in three cerebellar sections from each genotype. Healthy looking basket neurons were randomly selected for injections based on their location above the Purkinje neurons. All clearly stained basket cells were imaged and z-stacks merged. The number of primary branches forming off the main axon for each basket neuron were counted and averaged for each genotype and age, and “n” refers to the number of basket cells counted for each genotype. Averages are also presented with their SEM. For statistical analysis, we used the Student's t test (unpaired) and ANOVA test (single factor).
Results
Developmental organization of the cerebellar pinceau
The cerebellar interneurons migrate through the white matter tracks of the cerebellum before moving in the vicinity of the Purkinje neuron layer and differentiating into basket and stellate interneurons in the molecular layer (Palay and Palay, 1974; Zhang and Goldman, 1996; Ango et al., 2004; Sudarov et al., 2011). Axon collaterals from 5–7 basket neurons contribute to the formation of the pinceau, and each basket neuron can innervate several Purkinje neurons, allowing for precise gating of Purkinje neuron output. To establish the developmental organization and localization of various molecular components at the pinceau, we used a combination of molecular markers to follow the developmental timeline of pinceau organization, including the unique enrichment of potassium channels at the core of the pinceau. We performed immunostaining of wild-type cerebellar sections against parvalbumin (Parv) to label both Purkinje neurons and molecular layer interneurons, including basket neurons (Bastianelli, 2003). We also immunostained for Calb as a specific marker for Purkinje neurons (Nordquist et al., 1988), pNfl as a marker for basket neuron collaterals, and potassium channels (KV1.2) to label the core of the pinceau formed by basket axon terminals that target the Purkinje AIS. As shown in Figure 1, coimmunostaining against Parv and Calb at P10 shows Parv expression in basket neurons (b) (Fig. 1Aa,b) in the vicinity of the Purkinje neurons. Note that Parv is also expressed in Purkinje neurons (Fig. 1Ab, merged yellow color), but Calb is not expressed in basket neurons. At this stage, Parv immunoreactivity at the developing pinceaux is not expanded (Fig. 1Ab, arrow). Triple immunostaining of P10 cerebellar sections using antibodies against Calb, pNfl, and KV1.2 showed that basket neuron collaterals are beginning to target the Purkinje AIS (Fig. 1Bb; Bd, merged image, arrow), but the KV1.2 immunoreactivity is not yet detectable at the developing pinceaux (Fig. 1Bc, arrowhead; Bd, merged image). At P12, basket neuron collaterals reach the Purkinje AIS, as visualized by both Parv (Fig. 1Ca) and pNfl (Fig. 1Db, arrowhead). At P12, KV1.2 begins to label the developing pinceau (Fig. 1Dc, arrowhead) around the Purkinje AIS (Fig. 1Dd, merged image). At P16, the structure of the pinceaux begins to take shape and Parv localization broadens around the Purkinje AIS (Fig. 1Ea; Eb, merged image, arrow), and more basket axon collaterals begin to target the Purkinje AIS, as revealed by immunostaining against pNfl (Fig. 1Fb, arrowhead). KV1.2 also begins to show increasing enrichment at the pinceaux (Fig. 1Fc, arrowhead). By P20, the pinceaux takes the characteristic cone-shape and its development is almost complete, as revealed by Parv and pNfl localization (Fig. 1Ga,b, arrow; Hb, arrowhead) and the unique enrichment of KV1.2 channels (Fig. 1Hc, arrowhead; Hd, merged image, arrow). By P30, the general developmental organization of the pinceaux is complete (Fig. 1Ia,b; Ja–d), but the pinceaux becomes larger and the density of KV channels increases by P30 to P90 (Fig. 1Ka,b; La–d) (Sotelo, 2008). We also analyzed BAC transgenic Gad67-GFP mice that express GFP in cerebellar interneurons, including basket neurons and some Purkinje neurons (Ango et al., 2004). Immunostaining of Gad67-GFP cerebella at various postnatal developmental stages using anti-GFP to label basket axon collaterals and anti-Calb further supported the developmental timeline of the pinceau organization, as revealed by various antibody markers in the wild-type cerebella (Fig. 1M–Q). Based on the above immunohistochemical analysis, we have schematized the organization of the cerebellar pinceaux during postnatal development (Fig. 1R). Together, our data reveal distinct features of pinceau development in which basket axon collaterals target the Purkinje soma and the AIS, and only the inner core of the pinceaux becomes enriched with potassium channels, which surrounds the Purkinje AIS.
Figure 1.

Molecular organization of the cerebellar pinceau during postnatal development. Wild-type cerebellar sections from ages P10–P90 (indicated on left) and immunostained against Parv, Calb, pNfl, and KV1.2. A, B, At P10, very few basket neuron (b) collaterals have reached Purkinje neuron (P) AISs (arrows). C, D, At P12, a few basket neuron (b) collaterals have arrived at the Purkinje AIS (arrowhead) and faint traces of KV1.2 channels can be seen around the Purkinje AIS. E, F, By P16, more basket neuron (b) collaterals surround the Purkinje (P) AIS, and KV1.2 channels are enriched at the basket neuron terminals. G, H, At P20, the cone-shape of the pinceau is formed, as evident by Parv (Gb, arrow), pNfl (Hb, arrowhead; Hd, arrow), and KV1.2 clustering (Hc, arrowhead). I, J, At P30, the pinceau is fully mature (Ib, Jd, arrows; Jb, Jc arrowheads). K, L, At P90, the structure of the pinceau does not display major anatomical changes compared withP30 (Kb, Ld, arrows; Lb, Lc, arrowheads). M–Q, Cerebellar sections from Gad67-GFP BAC transgenic mice at ages P10 (M), P12 (N), P16 (O), P20 (P), and P30 (Q) immunostained against GFP (green) and Calb (red). The main basket axons (arrows) give off collaterals (arrowheads) that extend toward the Purkinje AIS begining at P10 (M, arrowhead) and form the cone-shape pinceau at P30 (Q, arrowhead). R, Representative drawings of each stage of pinceau development. Purkinje neurons (P) in green, basket neurons (b) in red and orange and potassium channels in blue. Scale bars: 10 μm.
Nfasc is expressed in cerebellar basket axon terminals at the pinceau
The anatomical features and the organization of the cerebellar pinceau between basket axon collaterals and the Purkinje soma and AIS have raised fundamental questions about what precisely dictates the establishment of the pinceaux. Earlier studies showed that loss of AnkG at the Purkinje AIS led to mislocalization of Nfasc at the Purkinje AIS and disruption in the organization of the pinceau, thereby implicating Nfasc in pinceau organization (Ango et al., 2004). Recently, it was reported that loss of Nfasc in adult mice, using inducible Thy1-Cre, led to progressive disorganization of the Purkinje AIS and the pinceau (Zonta et al., 2011). While both of these studies highlighted the role of Nfasc in the Purkinje neurons for pinceau formation and AIS maintenance, respectively, it remains to be determined whether there is a functional requirement for Nfasc in basket neurons during pinceau assembly. We addressed this issue by specifically ablating Nfasc in Purkinje, and in both Purkinje and basket neurons using cell type-specific Cre recombinase.
To analyze the role of Purkinje neuron Nfasc in pinceau formation, we conditionally ablated Nfasc in Purkinje neurons using Purkinje-cell protein 2 (Pcp2)-Cre (Berrebi et al., 1991; Barski et al., 2000) and previously generated NfascFlox mice (Pillai et al., 2009; Thaxton et al., 2011). We first confirmed the specificity of the Nfasc antibody by immunostaining of cerebellar tissues from Actin-Cre;NfascFlox mice, which are null for Nfasc. This immunostaining showed that Nfasc is absent in Actin-Cre;NfascFlox mice at P5, the age at which it clearly localizes to the Purkinje AIS in the wild-type cerebella (data not shown). We also confirmed that ablation of Nfasc in the cerebellum started from P0 using PCR analysis of Pcp2-Cre;NfascFlox mice during postnatal development (data not shown). To ensure that Nfasc was specifically ablated in Purkinje neurons, we performed immunostaining against Nfasc and Calb in wild-type (+/+) and Pcp2-Cre;NfascFlox mice during postnatal development. At P10, in wild-type cerebella, Nfasc is enriched at the Purkinje AIS and slightly at the base of the Purkinje soma (Fig. 2Aa, arrow), as well as at the basket neuron (b) AIS (Fig. 2Aa, arrowhead; Ab, merged image). In P10 Pcp2-Cre;NfascFlox cerebella, Nfasc is undetectable at the Purkinje cell AIS and soma (Fig. 2Ba, arrow), but is present at the basket (b) AIS (Fig. 2Ba,b, arrowheads). In P12 and P16 wild-type cerebella, Nfasc becomes enriched in the Purkinje soma and AIS (Fig. 2Da,b; Ga,b), as well as at the basket (b) AIS (Fig. 2Da,b, arrowheads). In P12 and P16 Pcp2-Cre;NfascFlox cerebella, Nfasc is absent from the Purkinje soma and AIS (Fig. 2Ea,b; Ha,b, arrows), but is present at the basket (b) AIS (Fig. 2Ea,b; Ha,b, arrowheads). Interestingly, as the basket axon collaterals reach the Purkinje AIS and pinceau begins to take shape, Nfasc labels the basket axon collaterals at the pinceau area that is distinct from its localization at the Purkinje AIS (Fig. 2Da,b; Ga,b; Ja,b; Ma,b; Pa,b, green arrowheads). As the pinceau matures from P20, P30, and into adulthood (P150), Nfasc is not only localized to the Purkinje soma and AIS, but is also found at the pinceau, indicating that it is expressed in the basket axon terminals (Fig. 2Ja,b; Ma,b; Pa,b, green arrowheads). Most importantly, Nfasc localization in the basket axon terminals is not affected in Pcp2-Cre;NfascFlox cerebella at any of the developmental time points analyzed (Fig. 2Ka,b; Na,b; Qa,b, green arrowheads). Together, these data show that Nfasc is expressed in both Purkinje and basket neurons, and that in addition to its localization at the Purkinje soma/AIS, Nfasc also localizes at the basket AIS and basket axon terminals.
Figure 2.
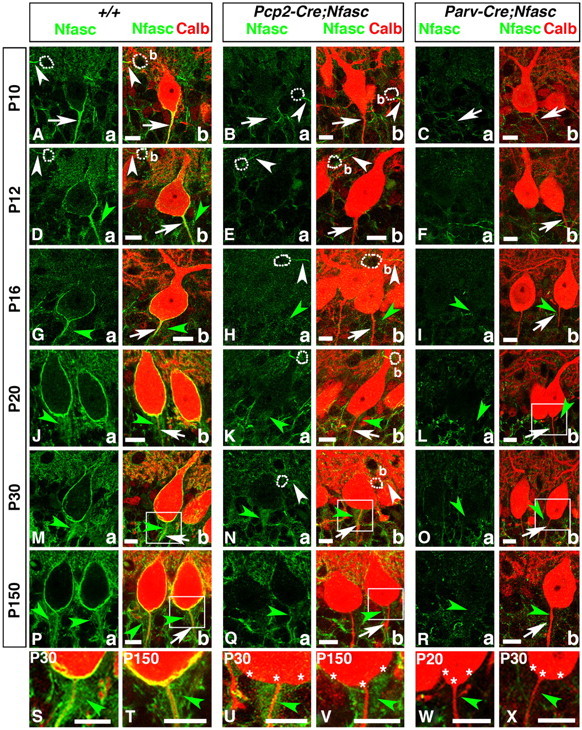
Purkinje and basket-specific ablation of Neurofascin reveals its localization to both Purkinje and basket neurons. Wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebellar sections from ages P10–P150 as indicated on left (A–R) or within panel (S–X), and immunostained against Nfasc (a, green) and Calb (b, red). Note that in P10 wild-type cerebella, Nfasc is localized to the Purkinje AIS (A, arrows) and basket (b) AIS (A, white arrowheads). At later stages, in wild-type cerebella, Nfasc is localized around the entire Purkinje soma and AIS (D, G, J, M, P, arrows), as well as the basket AIS (D, white arrowhead). Nfasc is also localized to basket axon terminals as they form the pinceau in wild-type cerebella (D, G, J, M, P, green arrowheads). In Pcp2-Cre;NfascFlox cerebella, Nfasc is ablated from the Purkinje soma/AIS (B, E, H, K, N, Q, arrows) but remains at the basket AIS (B, E, H, N, white arrowhead) and pinceau (H, K, N, Q, green, arrowheads). In Parv-Cre;NfascFlox cerebella, Nfasc is lost from the Purkinje soma/AIS (C, F, I, L, O, R, arrows), basket AIS, and pinceau (I, L, O, R, green arrowheads). Higher magnification shows that Nfasc is present at the Purkinje soma/AIS and pinceau in wild-type (S, T, green arrowheads), is missing from the Purkinje soma/AIS in Pcp2-Cre;NfascFlox (U, V, stars), while remaining at the pinceau in Pcp2-Cre;NfascFlox (U, V, green arrowheads), and is missing from both the Purkinje soma/AIS (stars) and the pinceau (green arrowheads) in Parv-Cre;NfascFlox cerebella (W, X). Scale bars: 10 μm.
To verify that Nfasc is expressed in basket neurons and localizes to basket axon terminals at the pinceau, we used a Parvalbumin-Cre (Parv-Cre) mouse strain (Hippenmeyer et al., 2005) that expresses Cre in all Parv-expressing interneurons in the nervous system and generated Parv-Cre;NfascFlox mice. Similar to Pcp2-Cre;NfascFlox mice, we confirmed loss of Nfasc in Purkinje and basket neurons at P0 (data not shown). We performed immunostaining for Nfasc to confirm ablation of Nfasc in Parv-Cre;NfascFlox mice (Fig. 2C,F,I,L,O,R). Analysis of P10 (Fig. 2Ca,b), P12 (Fig. 2Fa,b), P16 (Fig. 2Ia,b), P20 (Fig. 2La,b), P30 (Fig. 2Oa,b), and P150 (Fig. 2Ra,b) Parv-Cre;NfascFlox cerebella showed loss of Nfasc in both Purkinje and basket neurons. Most importantly, Nfasc-positive immunostaining observed in wild-type and Pcp2-Cre;NfascFlox cerebella was absent from the basket axon terminals surrounding the Purkinje AIS (Fig. 2Ib, Lb,Ob,Rb, green arrowheads). Higher magnification images of the Purkinje AIS/pinceau area from wild-type (Fig. 2M,P), Pcp2-Cre;NfascFlox (Fig. 2N,Q), and Parv-Cre;NfascFlox (Fig. 2L,O) cerebella show that Nfasc is absent from Purkinje neurons in both Pcp2-Cre;NfascFlox (Fig. 2U,V, asterisks) and Parv-Cre;NfascFlox (Fig. 2W,X, asterisks) cerebella. They also show that Nfasc is present in basket axon terminals surrounding the Purkinje AIS in wild-type (Fig. 2S,T, green arrowheads), and that this staining is present in Pcp2-Cre;NfascFlox (Fig. 2U,V, green arrowheads) and absent in Parv-Cre;NfascFlox (Fig. 2W,X, green arrowheads) cerebella. These data demonstrate that Nfasc is expressed in both Purkinje and basket neurons, and that Nfasc localizes to basket axon collaterals, which form the pinceau at the Purkinje AIS.
Nfasc is required for the maturation and maintenance of the Purkinje axon initial segment
Studies examining the organization of the AIS in vivo and in vitro have led to a debate as to which proteins are critical in the initial organization of the AIS (Grubb and Burrone, 2010). Loss of Nfasc globally does not affect initial clustering of AIS components AnkG, NaV channels, and βIV-Spectrin, but severely disrupts NrCAM localization at the AIS (Zonta et al., 2011). However, loss of Nfasc later in the adult life progressively leads to AIS disorganization (Zonta et al., 2011). RNAi knockdown of AnkG in in vitro hippocampal neuron cultures led to disorganization of the AIS and change in neuronal polarity, but AIS organization was relatively unaffected when Nfasc was knocked down (Hedstrom et al., 2008). These observations have suggested that initial AIS organization is dependent on AnkG, but independent of Nfasc, and that the maintenance of the AIS requires Nfasc. We wanted to address this question using the Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mice to determine the consequences of loss of Nfasc in Purkinje neurons and how that affected the developmental organization of the AIS. At P10 in the wild-type Purkinje neurons, AnkG localizes at the AIS and its expression extends distally from the AIS (Fig. 3Aa,b, arrowheads). This initial distribution of AnkG at the AIS is unaffected when Nfasc is lost in Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mice (Fig. 3Ba,b, Ca,b, arrowheads). By P20, AnkG gets highly enriched at the AIS in the wild-type (Fig. 3Da,b,arrowheads). In P20 Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella, AnkG fails to localize at the AIS and becomes mislocalized to the distal region of the AIS (Fig. 3Ea,b; Fa,b, arrowheads). By P30, in wild-type cerebella, AnkG is concentrated at the AIS (Fig. 3Ga,b, arrowheads), whereas no AnkG is detectable in Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutant AIS (Fig. 3H,a,b; Ia,b, arrows). These data suggest that Nfasc is not essential for initial localization of AnkG at the AIS, but is for its proper retention and stabilization at the AIS.
Figure 3.
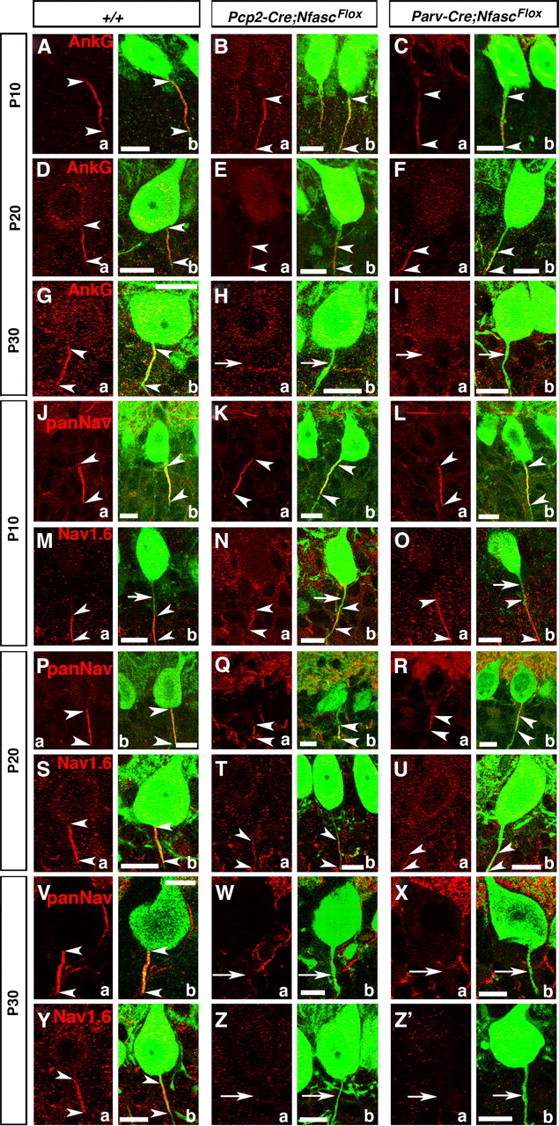
Early postnatal loss of Nfasc disrupts Purkinje AIS maturation and stabilization. Wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebellar sections from mice ages P10–P30 immunostained against AnkG (A–Ia, red), panNaV (J–L; P–R, V–Xa, red), NaV1.6 (M–O; S–U, Y–Z'a, red), and Calb (A–Z'b, green). AnkG is localized (between arrowheads) to the wild-type Purkinje AIS at P10, P20, and P30. In both mutants, AnkG remains (between arrowheads) at the Purkinje AIS at P10, moves distally down the axon at P20 (E, F, between arrowheads), and is undetectable from the Purkinje axon by P30 (H, I, arrows). At P10 (J, between arrowheads), P20 (P, between arrowheads), and P30 (V, between arrowheads), panNaV shows AIS localization in wild-type Purkinje AIS. Localization of panNaV remains the same as in the wild-type in both mutants at P10 (K, L, between arrowheads), but at P20 panNaV shows severely reduced immunostaining at the mutant AISs (Q, R, between arrowheads), and is completely missing from the P30 mutant AISs (W, X, arrows). Localization of NaV1.6 at the wild-type AIS at P10 shows that NaV1.6 is located distally in the AIS (M, between arrowheads) and moves to fill the entire AIS at P20 and P30 (S, Y, between arrowheads). Localization of NaV1.6 remains the same as wild-type in both mutants at P10 (N, O, between arrowheads) and remains distally localized at P20 (T, U, between arrowheads), and is undetectable at the AIS at P30 (Z, Z', arrows). Scale bars: 10 μm.
We next determined the consequences of loss of Nfasc on the developmental localization of NaV channels at the Purkinje AIS. It has been previously reported that the Purkinje AIS initially contains only NaV1.1, and, later, NaV1.6 becomes enriched at the AIS (Van Wart and Matthews, 2006). We performed immunostaining using panNaV and NaV1.6-specific antibodies. At P10, panNaV staining showed proper clustering of NaV channels at the Purkinje AIS in Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants (Fig. 3Ka,b; La,b, arrowheads), which was indistinguishable from that observed in the wild-type (Fig. 3Ja,b, arrowheads). Interestingly, immunostaining against NaV1.6 at P10 showed localization of NaV1.6 distal to the AIS in the wild-type cerebella (Fig. 3Ma,b,arrowheads; arrows point to AIS) and similar localization was observed in Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants (Fig. 3Na,b; Oa,b, arrowheads). These data indicate that NaV1.6 clustering initially does not occur at the AIS, but in the immediate distal region of the AIS. By P20, panNaV immunostaining showed localization of NaV channels at the AIS in the wild-type cerebella (Fig. 3Pa,b, arrowheads), but in Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants, NaV channel localization was already severely affected with localization toward the distal regions from the AIS (Fig. 3Qa,b; Ra,b, arrowheads). Similarly, NaV1.6 immunostaining at P20 revealed that NaV1.6 becomes highly enriched at the wild-type Purkinje AIS (Fig. 3Sa,b, arrowheads). In Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants, by P20, NaV1.6 remained mislocalized in the distal axonal region away from the AIS (Fig. 3Ta,b; Ua,b, arrowheads). At P30, both panNaV and NaV1.6 are enriched at the wild-type Purkinje AIS (Fig. 3Va,b; Ya,b, arrowheads), but are undetectable at the AIS as well as at the distal region of the AIS in P30 Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants (Fig. 3Wa,b; Xa,b; Za,b; Z'a,b, arrows). Together, these data clearly show that the initial phase of AIS organization is independent of Nfasc, and that Nfasc is critically required for the second phase, i.e., the maturation of the AIS with the localization of NaV1.6, and the maintenance of the molecular components of the AIS.
Nfasc is required in both basket neurons and Purkinje neurons for the organization of the pinceau
A hallmark of the Purkinje neuron AIS is that the basket neuron collaterals establish a unique structure, the pinceau, at the AIS (Fig. 1). Our finding, that in addition to Nfasc localization at the Purkinje AIS, Nfasc is also expressed in the basket axon collaterals, has raised an interesting question whether Nfasc is bidirectionally required for proper pinceau organization. Since Pcp2-Cre caused loss of Nfasc in Purkinje neurons and Parv-Cre resulted in the loss of Nfasc in both Purkinje and basket neurons, we compared the development of the pinceau and the phenotypes thereof between Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants. We performed triple immunostaining against pNfl, KV1.2, and Calb (Fig. 1) to determine the extent of pinceau disorganization in these mutants (Fig. 4). As noted earlier (Fig. 1) at P10 and P12, very few basket axon collaterals have reached the Purkinje AIS by these developmental stages, and no significant differences were observed at the Purkinje AISs between wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants (Fig. 4Ba–c, Ca–c, compare with Aa–c; Ea–c, Fa–c, compare with Da–c). At P16, in wild-type cerebella, many basket axon collaterals reach the Purkinje AIS (Fig. 1Ga,c) and KV1.2 channels become enriched at the pinceau (Fig. 1Gb,c, arrowhead). In P16 Pcp2-Cre;NfascFlox cerebella, KV1.2 channels get enriched at the basket axon terminals that have reached the Purkinje AIS, but fail to get properly organized to form a compact structure compared with the wild-type (Fig. 4Hb,c, arrowheads). In P16 Parv-Cre;NfascFlox cerebella, KV1.2 channels fail to get enriched at the Purkinje AIS and instead show small ectopic clusters near the Purkinje soma and regions distal to Purkinje AIS (Fig. 4Ib,c, arrowheads). Quantification of the percentage of noncompact pinceau, based on KV1.2 localization, revealed that at P16, 33.33 ± 3.61% of wild-type pinceau were not compact (n = 16 pinceau from each of 3 independent animals), while this number increased to 84.62 ± 4.27% in Pcp2-Cre;NfascFlox cerebella (n = 13 pinceau from each of 3 independent animals) and 76.19 ± 1.10% in Parv-Cre;NfascFlox cerebella (n = 14 pinceau from each of 3 independent animals). Statistical analyses revealed a significant difference across all three genotypes (F(2,6) = 14.73, p = 0.0048), as well as between each mutant and wild-type (wild-type to Pcp2-Cre;NfascFlox p = 0.014; wild-type to Parv-Cre;NfascFlox p = 0.028). In addition, quantification of ectopic KV1.2 clustering around the Purkinje soma at P16 revealed that 52.38 ± 1.10% of Purkinje neurons in Parv-Cre;NfascFlox cerebella had ectopic clusters, compared with 35.90 ± 3.26% in Pcp2-Cre;NfascFlox and 8.33 ± 0.90% in wild-type cerebella at this age. Statistical analyses again revealed a significant difference across all three genotypes (F(2,6) = 26.52, p = 0.0011), and between each mutant compared with wild-type (wild-type to Pcp2-Cre;NfascFlox p = 0.045; wild-type to Parv-Cre;NfascFlox p = 0.00017). In P20 Pcp2-Cre;NfascFlox cerebella, KV1.2 channels get enriched at the basket axon terminals that have reached the Purkinje AIS area, but begin to form small unstructured KV1.2 clusters (Fig. 4Kb,c, arrowheads; compare with wild-type Jb,c, arrowheads). In P20 Parv-Cre;NfascFlox cerebella, KV1.2 channels either form tiny clusters or small stretched clusters near the Purkinje soma and regions distal to Purkinje AIS (Fig. 4Lb,c, arrowheads), or completely fail to form any KV1.2 clusters at the Purkinje AIS (data not shown). At P20, quantification of the percentage of noncompact KV1.2 pinceau showed that 41.67 ± 4.17% of pinceau were disorganized in Pcp2-Cre;NfascFlox cerebella (n = 12 pinceau from each of 3 independent animals) and 56.15 ± 3.17% were disorganized in Parv-Cre;NfascFlox cerebella (n = 15 pinceau from each of 3 independent animals), compared with14.13 ± 1.97% in wild-type cerebella (n = 14 pinceau from each of 3 independent animals). Statistical analyses revealed a significant difference across all three genotypes (F(2,6) = 10.06, p = 0.012), and between wild-type and Parv-Cre;NfascFlox (p = 0.012), but not between wild-type and Pcp2-Cre;NfascFlox (p = 0.061). At P20, there were also ectopic KV1.2 clusters for 52.38 ± 4.43% of Parv-Cre;NfascFlox Purkinje neurons, 25.0 ± 2.41% of Pcp2-Cre;NfascFlox Purkinje neurons, and 18.73 ± 1.25% of wild-type Purkinje neurons. Statistical analyses revealed a significant difference across all three genotypes (F(2,6) = 7.47, p = 0.024), but not between either mutant independently with wild-type (wild-type to Pcp2-Cre;NfascFlox p = 0.34; wild-type to Parv-Cre;NfascFlox p = 0.068). By P30, Pcp2-Cre;NfascFlox cerebella show basket axon collaterals that are mistargeted to the distal areas of the Purkinje AIS and form broken clusters of KV1.2 channels at the Purkinje AIS area (Fig. 4Nb,c, arrowheads; compare with wild-type Mb,c, arrowhead). By P30, Parv-Cre;NfascFlox cerebella show a total failure of KV1.2 channel clustering at the pinceau (Fig. 4Ob,c, arrowheads; compare with wild-type Mb,c, arrowhead). In P30 Parv-Cre;NfascFlox cerebella, 73.32 ± 1.28% pinceau were disorganized (n = 29 pinceau from each of 3 independent animals), based on KV1.2 localization, compared with 70.76 ± 2.12% in Pcp2-Cre;NfascFlox cerebella (n = 18 pinceau from each of 3 independent animals) and 19.38 ± 1.46% in wild-type cerebella (n = 26 pinceau from each of 3 independent animals). Statistical analyses revealed a significant difference across all three genotypes (F(2,6) = 45.21, p = 0.00024), as well as between each mutant and wild-type (wild-type to Pcp2-Cre;NfascFlox p = 0.0018; wild-type to Parv-Cre;NfascFlox p = 0.0008). Interestingly, ectopic clusters of KV1.2 around the Purkinje soma were not as high in P30 mutant cerebella, compared with P16, with ectopic KV1.2 clusters surrounding 24.67 ± 3.50% of Parv-Cre;NfascFlox Purkinje neurons, 14.62 ± 1.55% of Pcp2-Cre;NfascFlox Purkinje neurons, and 2.56 ± 0.44% of wild-type Purkinje neurons. Statistical analyses did not reveal any significant difference across the three genotypes (F(2,6) = 2.73, p = 0.143), or between each mutant compared with wild-type individually (wild-type to Pcp2-Cre;NfascFlox p = 0.074; wild-type to Parv-Cre;NfascFlox p = 0.18). Together, these data show that the organization of pinceau is more severely disrupted in Parv-Cre;NfascFlox cerebella than in the Pcp2-Cre;NfascFlox cerebella.
We also analyzed 5-month-old wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutant cerebella to determine the extent of pinceau defects at the Purkinje AIS. While the wild-type Purkinje AIS showed proper targeting of basket axon collaterals and cone-shaped pinceau (Fig. 4Pa,b, arrowhead), Pcp2-Cre;NfascFlox (Fig. 4Qa,b, arrowheads) and Parv-Cre;NfascFlox (Fig. 4Ra,b, arrowheads) mutant cerebella revealed severely disorganized pinceau. In Parv-Cre;NfascFlox mutants, PSD95 clustering at the pinceau was either completely absent or formed small patchy clusters at ectopic locations at the Purkinje soma or somewhere around the Purkinje AIS. Quantification analyses of the basket axon collaterals that targeted the Purkinje AIS and soma area in P20 Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutant cerebella are shown in Figure 4S. This revealed that fewer basket axon collaterals surround the Purkinje AIS in both mutants, compared with wild-type (WT: 14.42 ± 0.82 axons per AIS, n = 31 Purkinje cells from 3 separate animals; Pcp2-Cre;NfascFlox: 9.64 ± 0.76 axons per AIS, n = 22 Purkinje cells from 3 separate animals, p = 0.00008; Parv-Cre;NfascFlox: 10.35 ± 0.56 axons per AIS, n = 34 Purkinje cells from 3 separate animals, p = 0.00014). However, no significant differences were found in the number of collaterals surrounding the soma in each genotype compared with wild-type (WT: 17.55 ± 1.01 axons per soma, n = 31 Purkinje cells from 3 separate animals; Pcp2-Cre;NfascFlox: 17.09 ± 0.84 axons per soma, n = 22 Purkinje cells from 3 separate animals, p = 0.73; Parv-Cre;NfascFlox: 20.35 ± 1.40 axons per AIS, n = 34 Purkinje cells from 3 separate animals, p = 0.11). A one-way ANOVA was also used to test for statistical difference in the mean number of basket axon terminals at the Purkinje AIS and at the Purkinje soma between the wild-type, Pcp2-Cre;NfascFlox, and Parv-Cre;NfascFlox cerebella. The number of basket axon terminals at the Purkinje AIS differed significantly across the three genotypes, F(2,84) = 13.05, p = 0.000012. However, the number of basket axon terminals at the Purkinje soma did not differ significantly across the three genotypes, F(2,84) = 2.27, p = 0.109. These data show that loss of Nfasc from either the Purkinje neuron or Purkinje and basket neurons results in decreased basket axon targeting to the Purkinje AIS. However, it is not clear from these results whether decreased collaterals are due to destabilization defects or targeting defects.
Next, we determined whether the pinceau disorganization defects observed in Nfasc mutants are also seen in Brevican (Bcan) and Nrcam mutants. Bcan encodes a proteoglycan, which localizes to Purkinje AIS and has been shown to interact with the neuronal Nfasc (Hedstrom et al., 2007). Nrcam encodes a neuron glia-cell adhesion molecule (NrCAM) that is enriched at the Purkinje AIS and gets mislocalized in Nfasc mutants (Zonta et al., 2011). Both Bcan (Fig. 4Ua,b, arrowhead) and Nrcam (Fig. 4Va,b, arrowhead) null mutants revealed normal organization of the pinceau, as seen in the wild-type cerebellum (Fig. 4Ta,b, arrowhead), indicating that Bcan and NrCAM are not critically required for the organization of the cerebellar pinceau. Together, our data suggest that loss of Nfasc in both basket and Purkinje neurons leads to more severe disorganization of the cerebellar pinceau than when Nfasc is lost only in the Purkinje neurons.
Nfasc is required in basket neurons for proper axon collateral outgrowth and targeting during pinceau formation
Our immunohistochemical analyses indicated that loss of Nfasc in Purkinje and basket neurons led to a decrease in the number of basket axon collaterals that targeted to the Purkinje AIS, and that pinceau formation was severely compromised. This phenotype could be caused by defects in basket axon collateral guidance and/or outgrowth during cerebellar development, or due to inability of basket collaterals to form stable contacts at the Purkinje AIS. Earlier studies using chicks have suggested that Nfasc interacts homophilically to induce neurite outgrowth (Pruss et al., 2004), and that alternative splicing of Nfasc allows for expression of multiple Nfasc isoforms, which either promote or inhibit neurite extension and cell adhesion (Koticha et al., 2005; Pruss et al., 2006). However, the in vivo role of Nfasc in neurite outgrowth and adhesion has not been examined. Since Nfasc is localized to the basket axon terminals and loss of Nfasc in Purkinje neurons (Pcp2-Cre;NfascFlox), or Purkinje and basket neurons (Parv-Cre;NfascFlox) caused different pinceau phenotypes, we hypothesized that Nfasc may play a unique role in basket axon terminals for pinceau formation. To address whether loss of Nfasc in basket neurons had any consequences on the basket axon collateral outgrowth and/or stabilization of basket axon collaterals, we performed single-cell fills of individual basket cells with biocytin in wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutants to follow basket axon collateral targeting. Biocytin filled basket neurons were then immunostained to detect biocytin (see Materials and Methods). At P10, as previously observed by pNfl immunostaining (Fig. 1), few basket axon collaterals reach the Purkinje neuron layer and begin to extend toward the Purkinje soma and AIS (Fig. 5A, arrow). In P10 Pcp2-Cre;NfascFlox cerebella, the main basket axon extends through the molecular layer toward the Purkinje cell layer (Fig. 5B, arrow), but branches extensively into collaterals (Fig. 5B, arrowheads), indicating that Purkinje soma/AIS-localized Nfasc is required for terminating basket axon extension and forming stable contacts at the Purkinje AIS. In P10 Parv-Cre;NfascFlox cerebella, the basket axons branched excessively, and failed to extend normally around the Purkinje soma (Fig. 5C, arrowheads), indicating that basket axon/collateral outgrowth fails to occur properly in the absence of Nfasc in basket cells. As development continues, wild-type basket axons extend more branches toward the Purkinje cell layer (Fig. 5D, arrowheads). Similar to the phenotype seen in P10 Pcp2-Cre;NfascFlox cerebella, at P20, the basket axons appear to target correctly to the Purkinje cell layer areas, but branch excessively once they arrive at the Purkinje cell layer (Fig. 5E,G, arrowheads), indicating that Nfasc at the Purkinje soma/AIS may be needed to terminate collateral extensions and to form stable contacts at the AIS. Remarkably, in P20 Parv-Cre;NFascFlox cerebella, the basket axons appear completely lost, branching in multiple directions and seeming to circle around the molecular layer, with no clear targeting pattern (Fig. 5F,H, arrowheads). In many instances, the collaterals fail to target toward the Purkinje soma/AIS (Fig. 5H, asterisks). A quantitative analysis of the basket axon collateral outgrowth and targeting revealed that loss of Nfasc in basket neurons causes more primary axon collateral branches than when Nfasc is only absent in Purkinje neurons (Fig. 5I). (P10 WT: 9.55 ± 1.15 branches per axon, n = 11 basket neurons from 2 separate animals; P10 Pcp2-Cre;NfascFlox: 10.17 ± 1.70 branches per axon, n = 6 basket neurons from 2 separate animals, p = 0.77 Pcp2-Cre;NfascFlox compared with wild-type; P10 Parv-Cre;NfascFlox: 16.71 ± 2.63 branches per axon, n = 7 basket neurons from 2 separate animals, p = 0.036 Parv-Cre;NfascFlox compared with wild-type; P20 WT: 12.5 ± 0.92 branches per axon, n = 6 basket neurons from 2 separate animals; P20 Pcp2-Cre;NfascFlox: 13.91 ± 1.36 branches per axon, n = 11 basket neurons from 2 separate animals, p = 0.42 Pcp2-Cre;NfascFlox compared with wild-type; P20 Parv-Cre;NfascFlox: 20.13 ± 1.53 branches per axon, n = 8 basket neurons from 2 separate animals, p = 0.0014 Parv-Cre;NfascFlox compared with wild-type). A one-way ANOVA was used to test for statistical difference in the mean number of branches off the primary basket axon in both P10 and P20 cerebella from the three genotypes. The number of branches coming off the basket axon differed significantly at both P10 (F(2,21) = 4.86, p = 0.018) and at P20 (F(2,22) = 7.61, p = 0.003). A schematic of the pinceau organization in wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mutant cerebella reflects the phenotypes that are observed when Nfasc is lost in Purkinje, and Purkinje and basket neurons (Fig. 5J). Together, our results suggest that Nfasc function is required in both Purkinje and basket neurons for proper axon collateral extension and targeting to the Purkinje AIS and for their stabilization at the AIS to allow for the formation and organization of the pinceau during cerebellar development.
Figure 5.
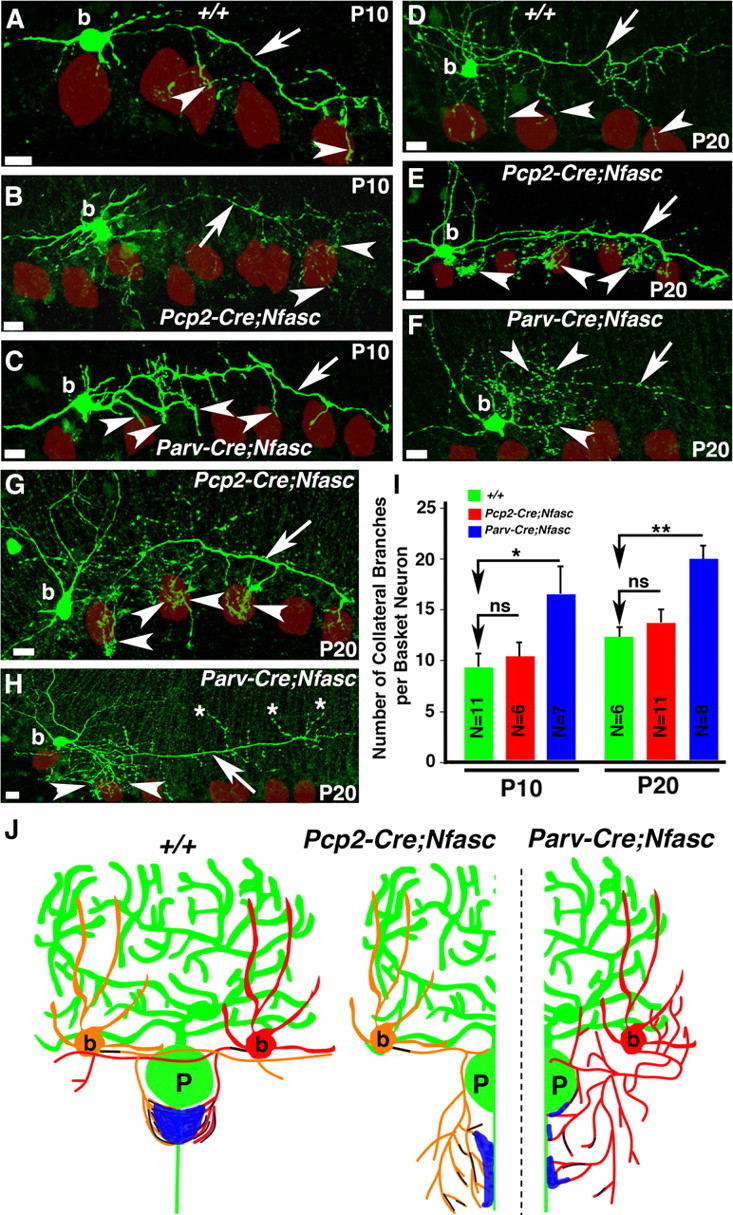
Loss of Nfasc in basket neurons causes mistargeting of basket axon collaterals. Wild-type (A, D), Pcp2-Cre;NfascFlox, (B, E, G), and Parv-Cre;NfascFlox (C, F, H) cerebellar sections from ages P10 (A–C) and P20 (D–H) in which basket cells were injected with biocytin and immunostained with Alexa-conjugated Streptavidin (green). Note that basket axons project toward the Purkinje neuron layer in Pcp2-Cre;NfascFlox mutants, but branch excessively at their terminals (B, E, G, arrowheads). In Parv-Cre;NfascFlox mutants, basket axons branch excessively from the beginning of the extension (C, F, H, arrowheads) and sometimes point in opposite directions (H, asterisks). Purkinje somas are outlined as red circles. I, Quantification of the number of primary basket terminal branches coming off the main basket axon branch at each time point reveal an increase in branching along the length of the Parv-Cre;NfascFlox basket axon compared with wild-type and Pcp2-Cre;NfascFlox basket neurons. J, Representative drawings summarizing the pinceau structures observed in wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella. Scale bars: 10 μm. ns, Not significant. *p < 0.05; **p < 0.01.
Nfasc is required for proper basket axon collateral targeting and compaction at the pinceau
Several studies have suggested a role for Nfasc in GABAergic synapse formation at the AIS. It was shown that expression of a dominant-negative form of Nfasc in Purkinje cells disrupted pinceau synapse formation based on GAD65 immunostaining (Ango et al., 2004). More recent studies showed that shRNA-knockdown of Nfasc in granular cells of the rat dentate gyrus resulted in a decrease of presynaptic and postsynaptic components at the synapse (Kriebel et al., 2011). Although synaptic markers clearly define the cone-shape of the cerebellar pinceau, only a few synapses are actually made by basket axons onto the Purkinje AIS at each pinceaux (Somogyi and Hámori, 1976; Sotelo, 2008). No ultrastructural analysis of the pinceau region has been performed either in AnkG mutants or animals that express transgenic dominant-negative Nfasc constructs (Ango et al., 2004) or in Nfasc adult mutants reported in Zonta et al. (2011). To determine the ultrastructural changes that occur at the pinceau as a result of loss of Nfasc in Purkinje, and Purkinje and basket neurons, we performed electron microscopic (EM) analysis. EM analysis of 1-month-old wild-type cerebella at the level of the Purkinje AIS and the pinceau revealed a high density of basket (B) axon collaterals surrounding the Purkinje (P) soma and AIS, and presence of synapses between basket collaterals and the Purkinje AIS (Fig. 6A, arrowheads). The Purkinje soma/AIS membrane is highlighted by the red line to distinguish it from the basket collateral areas. In P30 Pcp2-Cre;NfascFlox cerebella, we observed a decrease in the density of basket axons and clear gaps between the Purkinje soma/AIS and basket collaterals (Fig. 6B, arrows), but basket axons were able to form synapses with the Purkinje soma and AIS (Fig. 6B, arrowhead). In P30 Parv-Cre;NfascFlox cerebella, we observed a further decrease of the basket collaterals at the pinceau and more gaps between the Purkinje soma/AIS and basket axon collaterals (Fig. 6C, arrows). Again, basket collaterals were able to form synapses with the Purkinje soma/AIS, indicating that loss of Nfasc in Purkinje and basket neurons does not abolish synapse formation (Fig. 6E,F, compare with wild-type D). Quantification of the number of synapses formed on the Purkinje soma/AIS at 1 month revealed no significant differences in the total number of synapses between wild-type, Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mice (wild-type: 5.03 ± 0.26 synapses per soma, n = 105 Purkinje neurons; Pcp2-Cre;NfascFlox: 4.48 ± 0.30 synapses per soma, n = 75 Purkinje neurons; Parv-Cre;NfascFlox: 4.13 ± 0.29 synapses per soma, n = 68 Purkinje neurons; F(2,245) = 2.65, p = 0.073). These data suggest that basket axon collaterals retain their ability to form synapses with the Purkinje soma/AIS in the absence of Nfasc, indicating that Nfasc is not required for synapses formation. Next, we analyzed 3-month-old wild-type cerebella, which showed that basket axon collaterals remain tightly compacted surrounding the Purkinje AIS area (Fig. 6G). However, at 3 months, both Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella revealed more severe disorganization of the pinceau area with glial processes infiltrating the gaps that are created by mistargeting of the basket axon collaterals (Fig. 6H,I, arrows). At 4 months, both mutants displayed much more severe disorganization at the pinceau with increased infiltration of glial processes between mislocalized basket axon collaterals (Fig. 6K,L,N,O, arrows). Wild-type cerebella at 4 months maintained a compact array of basket collaterals with the Purkinje soma/AIS (Fig. 6J,M). Together, the ultrastructural analysis of Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox cerebella shows that loss of Nfasc disrupts the targeting and stabilization of the basket axon collaterals at the pinceau and does not affect Purkinje and basket neuron synapse formation.
Nfasc-deficient Purkinje neurons lack spontaneous activity and receive reduced inhibitory input from basket neurons
Since loss of Nfasc resulted in a failure to assemble AIS molecular components, and also caused mistargeting of the basket axon collaterals at the Purkinje AIS, we wanted to address how loss of Nfasc affected the electrophysiological properties of the Purkinje neurons. In voltage-clamp mode, we tested whether the spontaneous activity, defined as firing rate in the absence of any current injection, of Pcp2-Cre;NfascFlox mutant Purkinje neurons was different from the control Purkinje neurons (Fig. 7A,B). In the control group, 13 of 19 wild-type neurons (68.4%) produced action potentials spontaneously, while only 6 of 18 Pcp2-Cre;NfascFlox neurons (33.3%) fired spontaneously, the rest of them remaining silent (Fig. 7A). The mean firing rate across all wild-type neurons was significantly different from the mean firing rate across all Pcp2-Cre;NfascFlox mutant neurons (24.6 ± 5.6 spikes/s, n = 19, and 7.7 ± 3.3 spikes/s, n = 18, respectively, p = 0.02) (Fig. 7B). Comparing only the spontaneously active neurons, wild-type neurons fired at 36.1 ± 5.9 Hz (n = 13) and Pcp2-Cre;NfascFlox mutant cells fired at 23.2 ± 6.1 Hz (n = 6, p = 0.17). No significant difference in the mean amplitude of action potentials was noticeable (control: 60.4 ± 2.5 mV, n = 13, Pcp2-Cre;NfascFlox mutant: 61.0 ± 2.9 mV, n = 6) (p = 0.96). We hypothesized that the silent cells had a more hyperpolarized resting potential, preventing their depolarization to threshold. However, the resting potential of the 12 silent Pcp2-Cre;NfascFlox mutant neurons (−53.3 ± 1.8 mV) was not significantly more hyperpolarized than the resting potential of the 13 spontaneously active control neurons (−50.5 ± 0.6 mV, p = 0.14) (Fig. 7B). Thus, the trend to decreased spontaneous firing rate in the mutant neurons is not caused by a more hyperpolarized resting potential. Furthermore, no significant differences in neuronal input resistance, series resistance, or capacitance was detected between the control and Pcp2-Cre;NfascFlox genotypes (data not shown), indicating that the active membrane properties of mutant and wild-type neurons were similar.
Next, to further test the firing properties of Pcp2-Cre;NfascFlox mutant Purkinje neurons, we recorded action potentials triggered by depolarizing current steps in both control and Pcp2-Cre;NfascFlox mutant Purkinje neurons (Fig. 7C–F). In current-clamp mode, cells were held at −70 mV, and 500 ms steps of 100–800 pA were applied (by increments of 100 pA). Characteristic responses of wild-type and Pcp2-Cre;NfascFlox mutant cells to 200, 400, 600, and 800 pA injections are illustrated in Figure 7C. All cells responded to depolarizing injections with a train of action potentials, a single spike, or a burst. The mean threshold of current injection at which cells started to fire was not significantly higher in mutants than in control cells (control: 268.4 ± 40.5 pA, n = 19, mutant: 427.8 ± 75 pA, n = 18, p = 0.23), indicating that Pcp2-Cre;NfascFlox mutant cells are able to start to fire for similar intensity of depolarization. The number of spikes during each step was counted (Fig. 7D). The sustained firing rates were quantified as the number of spikes divided by the time between the first and the last spike; the firing rate for steps eliciting only a single spike was scored as 0 spikes/s. Although the sustained firing rate was not different in control and mutant cells for every step (Fig. 7E), the number of spikes generated in mutant cells was significantly lower than in control cells for current injections >300 pA. These data show that Pcp2-Cre;NfascFlox mutant cells are able to fire repetitively like the control cells, but they are unable to fire more than ∼10 spikes, resulting in a lower spike number (Fig. 7F).
Because the loss of Nfasc expression results in altered targeting of the basket axon collaterals, we investigated the possibility that inhibitory inputs could be impaired in Pcp2-Cre;NfascFlox mutant Purkinje neurons. In the current-clamp mode, we recorded sIPSCs in Purkinje cells, in the presence of glutamatergic antagonists (NBQX and APV). In Pcp2-Cre;NfascFlox mutants, the frequency of sIPSCs was significantly decreased, compared with control cells (WT: 1.5 ± 0.2 Hz, n = 5; Pcp2-Cre;NfascFlox mutant: 0.2 ± 0.04 Hz, n = 9, p = 0.001). The amplitude of sIPSCs was not significantly changed in Pcp2-Cre;NfascFlox mutant cells compared with control cells (WT: 32.1 ± 3.4 pA, n = 5; Pcp2-Cre;NfascFlox mutant: 28.5 ± 4.9 pA, n = 9, p = 0.52). The reduction in the frequency of inhibition observed in Pcp2-Cre;NfascFlox mutants in the sIPSCs recordings indicates that inhibitory inputs are impaired in mutant cells. To determine whether the decreased sIPSC reflected a decrease in spontaneous action potential firing in basket neurons or a decrease in the number or release probability of inhibitory inputs, we recorded mIPSCs in the presence of TTX (1 μm) and glutamatergic antagonists (Fig. 7G,H). No significant difference was found in the mIPSC amplitude (WT: 27.4 ± 4.4 pA, n = 11; Pcp2-Cre;NfascFlox mutant: 35.5 ± 6.4, n = 13, p = 0.57; Fig. 7H). However, the mIPSC frequency was decreased [1.2 ± 0.3 Hz in WT (n = 11), 0.26 ± 0.05 Hz (n = 13) in mutant cells (p = 0.026] (Fig. 7H). These data indicate that inhibitory inputs onto Pcp2-Cre;NfascFlox mutant Purkinje neurons are decreased in number or have a decreased rate of presynaptic release.
To test whether excitatory inputs were altered in response to the disrupted inhibitory input onto Purkinje neurons, we also tested the parallel fiber (PF) inputs onto Purkinje cells in Pcp2-Cre;NfascFlox cerebella (data not shown). In voltage-clamp mode, PF-EPSCs were recorded in Pcp2-Cre;NfascFlox mutant cells and compared with wild-type PF-EPSCs. No significant difference between genotypes was noticed when the intensity of stimulation was increased, and the paired-pulse facilitation was similar in both groups for different tested delays, suggesting that PF connections are not affected presynaptically or postsynaptically by the abnormal inhibitory inputs and the disrupted AIS in mutant Purkinje neurons. Together, these results indicate that Purkinje neuron physiology is compromised with Nfasc ablation and inhibitory input onto Nfasc mutant Purkinje neurons is reduced.
Loss of Nfasc in Purkinje and basket neurons leads to progressive ataxia and neurodegeneration
The synaptic circuitry of the cerebellum relies on the Purkinje neurons as the sole output source. Thus, Purkinje neuron dysfunction is thought to be the major contributing factor in most ataxias and motor coordination deficits (Zoghbi, 2000; Manto and Marmolino, 2009). Since loss of Nfasc in Purkinje neurons leads to Purkinje AIS and pinceau disorganization, we wanted to determine whether Pcp2-Cre;NfascFlox and Parv-Cre;NfascFlox mice displayed any signs of motor incoordination and ataxia and Purkinje neuron degeneration. We first performed rotarod tests as a measure of motor coordination on Pcp2-Cre;NfascFlox mice. These mice became progressively more ataxic and showed decreased latency to fall from the accelerating rotarod as they aged (data not shown). The Parv-Cre;NfascFlox mice were not used for rotarod tests as they displayed extremely severe motor deficits, because of loss of Nfasc in other areas of the nervous system. Visually, at ∼P90, the Pcp2-Cre;NfascFlox mice begin to display motor deficits, including a wide-based gait and stumbling in the cage. As these mice age, these phenotypes become more severe. To determine whether older Pcp2-Cre;NfascFlox mice undergo neurodegeneration in the cerebellum, we performed triple immunostaining of the cerebella from one-year-old (P365) wild-type and Pcp2-Cre;NfascFlox mutant littermates against Calb, Parv, and KV1.2 to assess the loss of Purkinje and basket neurons, as well as the phenotypes at the pinceau in older mutants. As shown in Figure 8, A and B, the wild-type cerebellum displays a well ordered Purkinje neuron layer (Fig. 8Aa,d; Ba,d, white arrows), presence of basket interneurons (b) (Fig. 8Ab,d; Bb,d, red arrow), and normal shape pinceau (Fig. 8Ac,d; Bc,d, blue arrowhead). In contrast, the Pcp2-Cre;NfascFlox mutant cerebellum has essentially lost all its Purkinje neurons (Fig. 8Ca,d; Da,d, white arrows) and the molecular layer has been reduced in size to almost one third of that of the wild-type (Fig. 8, compare Cd, Ad). The Calb immunoreactivity is essentially absent in Pcp2-Cre;NfascFlox mutant cerebella, except in some background cell nuclei (Fig. 8Da,d, green arrows). Basket neurons (b) are present and their axon collaterals form bundles (Fig. 8Db, red arrowheads) with traces of KV1.2 immunoreactivity reflecting the disorganized pinceau (Fig. 8Dc, blue arrowheads). Similarly, we analyzed cerebella from 4 ½-month-old Parv-Cre;NfascFlox mutant mice, which also revealed Purkinje neuron degeneration (Fig. 8Ga,Ha, white arrows). The basket neurons (b) are still present (Fig. 8Gb,Hb), but their axon collaterals are mistargeted and display severely disorganized pinceau structures (Fig. 8Gc,Hc, blue arrowheads). The Parv-Cre;NfascFlox mutant mice become extremely uncoordinated as they reach ∼6 months of age and rarely survive past this age. At this point they also display more Purkinje neuron degeneration (data not shown). To quantify the levels of Purkinje neuron degeneration that occurs before the Parv-Cre;NfascFlox mice die, we counted the number of Purkinje neurons in cerebellar loops I, II, III, IV, V, VIa, VIb, and VIII from several cerebellar sections from each of five separate mice for both wild-type and Parv-Cre;NfascFlox and averaged the total number of Purkinje cells per loop. We found that there were an average of 159.79 ± 9.63 Purkinje cells per loop in wild-type cerebella at 5 months (n = 43 loops from 5 separate animals) and 97.53 ± 4.82 Purkinje cells per loop in Parv-Cre;NfascFlox cerebella at 5 months (n = 79 loops from 5 separate animals) (p = 0.00000024). This is a 38.96% decrease in Purkinje neurons by the time the Parv-Cre;NfascFlox reach 5 months of age. These data show that loss of Nfasc in Purkinje neurons leads to progressive ataxia and Purkinje neuron degeneration. Together, the data presented in this study reveal critical roles for Nfasc in both Purkinje and basket neurons for proper basket axon outgrowth, organization of the pinceau, maturation of the Purkinje AIS, and thus cerebellar function.
Figure 8.
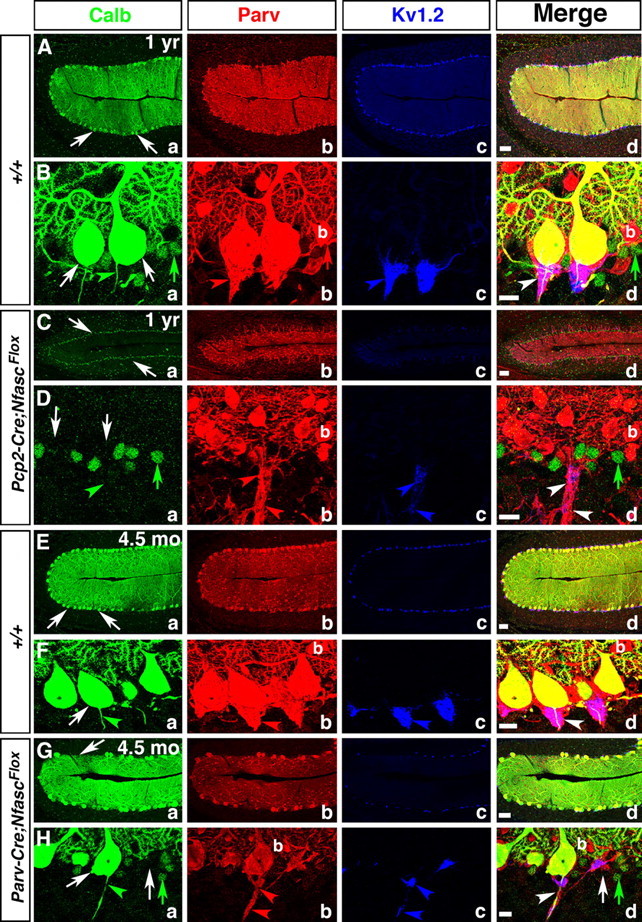
Ablation of Nfasc in Purkinje neurons leads to progressive Purkinje neuron degeneration. One-year old wild-type (A, B), one-year-old Pcp2-Cre;NfascFlox (C, D), 4.5-month-old wild-type (E, F), and 4.5-month-old Parv-Cre;NfascFlox (G, H) cerebellar sections immunostained against Calb (a, green), Parv (b, red), KV1.2 (c, blue), and merged (d). One-year and 4.5 month wild-type cerebella maintain a full complement of healthy Purkinje neuron layer (Aa, Ba, Ea, Fa, arrows), while one-year-old Pcp2-Cre;NfascFlox cerebella show decreased molecular layer size with loss of Purkinje neurons (Ca, Da, arrows). The pinceau is stably maintained in one-year and 4.5-month-old wild-type cerebella (Bb–d, Fb–d, arrows). In Pcp2-Cre;NfascFlox cerebella, the basket axons appear clumped together (Db, d, arrowheads), but do not form a pinceau structure as all Purkinje neurons have died (Da, arrows). In 4.5 month Parv-Cre;NfascFlox cerebella, some Purkinje cells have begun to degenerate (Ga, d; Ha, d, arrows) and the pinceau structure remains disrupted with ectopic clusters of potassium channels (Hb–d, arrowheads). Scale bars: (in d) A, C, E, G, 50 μm; B, D, F, H, 10 μm.
Discussion
The molecular organization of the cerebellar pinceau has intrigued neuroscientists since Ramon y Cajal's (1911, 1995) drawings of the cerebellum first appeared. This intrigue has heightened with observations that normal pinceau organization is required for normal motor behaviors (Bobik et al., 2004; Xie et al., 2010). The physiological function of the pinceau in modulating Purkinje neuron output remains to be understood (Bobik et al., 2004; Huang et al., 2007). Here, we used cell-specific Cre recombinase to ablate Nfasc, revealing that Nfasc is expressed in both Purkinje and basket neurons, and that Nfasc function is required in both neurons for pinceau organization. Importantly, we demonstrate that loss of Nfasc does not abolish pinceau synapse formation. Instead, basket axon collaterals are mistargeted leading to pinceau disorganization. Electrophysiological analyses revealed a decreased probability of spontaneous firing and failure to maintain evoked action potential bursts in Nfasc mutant Purkinje neurons. Together, our studies establish that Nfasc is required in both Purkinje and basket neurons for proper basket axon outgrowth and targeting for pinceau organization.
Neurofascin in adhesive interactions between Purkinje and basket neurons
A key finding of this study is that Nfasc expression in both Purkinje and basket neurons may establish intercellular adhesion, either homophilically or heterophilically, to allow for stable interactions between the Purkinje soma/AIS and basket axon collaterals. While the role of Nfasc in AIS formation and stability is known (Hedstrom et al., 2007, 2008; Zonta et al., 2011), Nfasc function in basket neurons is unknown. Our ultrastructural analysis showed that pinceau synapses formed in Parv-Cre;NfascFlox mice, indicating that Nfasc is not critical for synapse formation (Fig. 6). CAMs similar to Nfasc have been shown to play a critical role in axon guidance (Shiga et al., 1993; Wiencken-Barger et al., 2004) and in establishing intercellular adhesion between extending axons and their target cells (Ushkaryov et al., 1992; Rader et al., 1993). Based on the abnormal branching of basket axon collaterals in Parv-Cre;NfascFlox cerebella, Nfasc is needed for proper outgrowth and guidance of basket axons toward the Purkinje soma/AIS, and similarly, based on the extensive branching of the basket terminals around the Purkinje soma/AIS in Pcp2-Cre;NfascFlox cerebella, Nfasc is involved in bidirectional, mostly heterophilic, interactions between the Purkinje soma/AIS and basket collaterals to stabilize the pinceau. Possible binding partners for Nfasc at the pinceau may include NrCAM, which localizes to the Purkinje soma/AIS. However, NrCAM ablation does not significantly disrupt pinceau organization (Fig. 4), but coordinated loss of NrCAM and Nfasc may lead to pinceau disruption. Another potential binding partner of Nfasc at the AIS/pinceau is the β-subunit of NaV channels, which functions as a CAM, and has been shown to stimulate neurite outgrowth (Isom, 2002; Davis et al., 2004; McEwen and Isom, 2004; Koticha et al., 2006). Nfasc may also interact with ECM proteins to aid in pinceau organization. Elucidating the molecular nature of the Nfasc binding partners at the pinceau will allow for a better understanding of how Nfasc functions in basket and Purkinje neurons during cerebellar pinceau organization.
Loss of Nfasc in basket and/or Purkinje neurons and consequences on pinceau organization
Our comparative analyses revealed that pinceau organization was more severely affected in Parv-Cre;NfascFlox than in Pcp2-Cre;NfascFlox cerebella, leading to several possible interpretations. First, the increased loss of Nfasc could contribute to the more severe phenotype in Parv-Cre;NfascFlox cerebella such that the compounded loss of Nfasc in basket and Purkinje neurons allows for mislocalization of other molecular components critical for pinceau organization. Thus, when Nfasc is lost in Parv-Cre;NfascFlox cerebella, these components are more disrupted than in Pcp2-Cre;NfascFlox cerebella and the pinceau is further disorganized. Alternatively, if Nfasc function was only required at the Purkinje soma/AIS, then loss of Nfasc in Purkinje and Purkinje/basket neurons should lead to an identical phenotype. However, in Pcp2-Cre;NfascFlox cerebella, a smaller pinceau, based on localization of KV channels and PSD95, can form. These molecules are more disrupted in Parv-Cre;NfascFlox cerebella. This suggests Nfasc functions in a trans heterophilic manner during pinceau organization. Furthermore, basket axon collaterals target the Purkinje AIS better in Pcp2-Cre;NfascFlox than in Parv-Cre;NfascFlox cerebella, suggesting that Nfasc in basket axons interacts with another molecule(s) at the Purkinje soma/AIS. In Pcp2-Cre;NfascFlox cerebella, that molecule(s) is lost from the AIS in the absence of Nfasc. Our immunohistochemical results show that NaV1.6 channel, the marker of the mature AIS (Van Wart and Matthews, 2006), fails to become enriched at the Purkinje AIS. Thus, the primary function of Nfasc in Purkinje neurons is to ensure maturation and stabilization of the AIS molecules, including molecules that serve as receptors or ligands for basket neuron-expressed Nfasc. When Nfasc is also absent in basket neurons, the axon collaterals fail to target properly and are unable to stably interact with the Purkinje AIS.
The signaling mechanisms triggered by Nfasc in basket neurons remain to be determined. Alternative splicing and post-translational modifications of CAMs affect their interactions with downstream signaling effectors. For example, spatiotemporal control of functionally distinct isoforms of NCAM and L1 regulate their interactions with FGF tyrosine kinase receptors to modulate axon growth and guidance (Walsh and Doherty, 1997). Furthermore, several CAMs induce neurite outgrowth by interacting with FGF receptors in growth cones (Doherty et al., 2000; Hansen et al., 2008). Importantly, Nfasc interacts with FGF receptors for neurite outgrowth, such that the cytosolic domain of Nfasc is critical for FGF receptor activation, while the extracellular domain is critical for regulating FGF receptor signaling in vitro (Kirschbaum et al., 2009). Our studies provide in vivo evidence that Nfasc is required for basket axon collateral outgrowth and targeting, and this may require receptor-mediated signal transduction pathways.
Basket neuron inhibitory input to Purkinje neurons and the role of pinceau
Nfasc functions in the organization of inhibitory synapses by clustering gephyrin at the future AIS in the developing hippocampus, and, in adult dentate gyrus, stabilizes GABAergic synaptic components (Kriebel et al., 2011). Interestingly, only a few basket axon collaterals synapse with the Purkinje soma/AIS at the pinceau, while the remaining collaterals just surround the AIS (Palay and Palay, 1974; Somogyi and Hámori, 1976; Sotelo, 2008). Our patch-clamp recordings revealed that Pcp2-Cre;NfascFlox Purkinje neurons show reduced spontaneous firing rates and failure to maintain depolarization-evoked high-frequency spike firing. These observations suggest that an intact AIS is not required for action potential initiation, but is essential for maintaining firing in response to current injection and for spontaneous firing. Similar observations were made in mice that lack NaV1.6 channels (Khaliq et al., 2003). Thus, the abnormal spike firing properties in Pcp2-Cre;NfascFlox Purkinje neurons are likely related to the mislocalization of NaV1.6 channels (Fig. 3). Recent studies by Zonta et al. (2011) also showed that ablation of Nfasc in adult Purkinje neurons results in silent Purkinje neurons that fail to maintain spontaneous firing. Together, these data confirm that Purkinje AIS is critical for Purkinje neuron spontaneous activity and maintaining action potentials.
What role does the pinceau play in Purkinje neuron activity? Our studies uncovered a 5–6-fold reduction in mIPSC frequency, but not mIPSC amplitude, suggesting that the number of synaptic release sites, or their release probability, is decreased. This is consistent with our EM analysis, which revealed the presence of synapses between basket axons and Purkinje AIS, but fewer basket axon collaterals around the Purkinje AIS. Interestingly, mutations in Kcna2, which encodes basket axon collateral-specific KV1.2, also resulted in defective inhibitory input to Purkinje neurons (Xie et al., 2010). Together, these data suggest that pinceau plays an important role in modulating Purkinje neuron activity.
Ataxia and neurodegeneration in Nfasc mutants
Many genetic mutations are associated with Purkinje neuron degeneration and cerebellar dysfunction leading to ataxia (Wang and Zoghbi, 2001; Perkins et al., 2010; Rinaldo and Hansel, 2010). In this study, the Pcp2-Cre;NfascFlox mice developed ataxia, consistent with the Purkinje neuron loss found in older Pcp2-Cre;NfascFlox cerebella. What might cause the Purkinje neuron death? Previous studies have shown that ion channels are essential for sustained high-frequency firing by Purkinje neurons (Raman and Bean, 1997, 1999; Sacco et al., 2006; Zagha et al., 2008), and mutant forms of human channels are associated with cerebellar atrophy and spinocerebellar ataxias (Browne et al., 1994; Ophoff et al., 1996; Zhuchenko et al., 1997; Waters et al., 2006). A high density of ion channels at the Purkinje AIS is necessary for proper action potential propagation, and, as discussed above, loss of Nfasc results in their mislocalization from the AIS, leading to Purkinje neuron dysfunction. Interestingly, mutations in Kcna2 also lead to ataxia, suggesting that an intact Purkinje AIS is not sufficient to maintain normal Purkinje neuron firing, further highlighting an equally important role of the pinceau in ensuring proper Purkinje neuron output modulation (Xie et al., 2010). Additionally, Pcp2-Cre;NfascFlox mice show progressive Purkinje neuron degeneration beginning at ∼7–8 months because of secondary consequences. In several instances, Purkinje neuron dysfunction has been attributed to glutamatergic excitotoxicity, so this could also contribute to Purkinje neuron loss (Slemmer et al., 2005; Perkins et al., 2010). Future studies in which Nfasc is only ablated in basket neurons may help differentiate Purkinje AIS versus basket Nfasc functions during pinceau organization, and how inhibitory input to Purkinje neurons would be affected. In summary, we have provided in vivo evidence that cerebellar pinceau organization and function requires coordinated mechanisms involving distinct Nfasc functions in Purkinje and basket neurons.
Footnotes
This work was supported by NIH Grant GM063074 (M.A.B.), NS-062771 (C.H.), and funds from the State of North Carolina (M.A.B.). We are grateful to W. Snider, V. Bautch, and P. Maness for generously sharing the Parv-Cre and TaumGFP/LacZ, R26RLacZ, and Gad67-GFP mouse strains, respectively. We thank C. Seidenbecher for Brevican knock-out cerebella and anti-Brevican antibodies, P. Maness and G. Demyanenko for Nrcam knock-out cerebella, J. Dupree and V. Madden for suggestions and assistance with electron microscopy, B. Taylor-Blake for help with cryosectioning, and S. Moy and R. Nonneman for help with mouse behavioral studies. We also thank H. Bellen, H. Zoghbi, K. Bender, S. Smith, I. Smith, B. Arenkiel, F. Frohlich, and A. Fanning for comments on this manuscript and members of the Bhat laboratory for many insightful and helpful discussions.
The authors declare no competing financial interests.
References
- Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell. 2004;119:257–272. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–98. [PubMed] [Google Scholar]
- Bastianelli E. Distribution of calcium-binding proteins in the cerebellum. Cerebellum. 2003;2:242–262. doi: 10.1080/14734220310022289. [DOI] [PubMed] [Google Scholar]
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- Benton MD, Raman IM. Stabilization of Ca current in Purkinje neurons during high-frequency firing by a balance of Ca-dependent facilitation and inactivation. Channels. 2009;3:393–401. doi: 10.4161/chan.3.6.9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrebi AS, Oberdick J, Sangameswaran L, Christakos S, Morgan JI, Mugnaini E. Cerebellar Purkinje cell markers are expressed in retinal bipolar neurons. J Comp Neurol. 1991;308:630–649. doi: 10.1002/cne.903080409. [DOI] [PubMed] [Google Scholar]
- Bobik M, Ellisman MH, Rudy B, Martone ME. Potassium channel subunit Kv3.2 and the water channel aquaporin-4 are selectively localized to cerebellar pinceau. Brain Res. 2004;1026:168–178. doi: 10.1016/j.brainres.2004.07.088. [DOI] [PubMed] [Google Scholar]
- Boiko T, Vakulenko M, Ewers H, Yap CC, Norden C, Winckler B. Ankyrin-dependent and -independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J Neurosci. 2007;27:590–603. doi: 10.1523/JNEUROSCI.4302-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Brümmendorf T, Kenwrick S, Rathjen FG. Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr Opin Neurobiol. 1998;8:87–97. doi: 10.1016/s0959-4388(98)80012-3. [DOI] [PubMed] [Google Scholar]
- Buttermore ED, Dupree JL, Cheng J, An X, Tessarollo L, Bhat MA. The cytoskeletal adaptor protein band 4.1B is required for the maintenance of paranodal axoglial septate junctions in myelinated axons. J Neurosci. 2011;31:8013–8024. doi: 10.1523/JNEUROSCI.1015-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci. 1981;1:777–783. doi: 10.1523/JNEUROSCI.01-07-00777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TH, Chen C, Isom LL. Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem. 2004;279:51424–51432. doi: 10.1074/jbc.M410830200. [DOI] [PubMed] [Google Scholar]
- Doherty P, Williams G, Williams EJ. CAMs and axonal growth: a critical evaluation of the role of calcium and the MAPK cascade. Mol Cell Neurosci. 2000;16:283–295. doi: 10.1006/mcne.2000.0907. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, Bhat MA. Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc Natl Acad Sci U S A. 2006;103:5137–5142. doi: 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold DA, Baek SH, Schork NJ, Rose DW, Larsen DD, Sachs BD, Rosenfeld MG, Hamilton BA. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40:1119–1131. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Building and maintaining the axon initial segment. Curr Opin Neurobiol. 2010;20:481–488. doi: 10.1016/j.conb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- Hansen SM, Berezin V, Bock E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell Mol Life Sci. 2008;65:3809–3821. doi: 10.1007/s00018-008-8290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178:875–886. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ. Subcellular organization of GABAergic synapses: role of ankyrins and L1 cell adhesion molecules. Nat Neurosci. 2006;9:163–166. doi: 10.1038/nn1638. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Di Cristo G, Ango F. Development of GABA innervation in the cerebral and cerebellar cortices. Nat Rev Neurosci. 2007;8:673–686. doi: 10.1038/nrn2188. [DOI] [PubMed] [Google Scholar]
- Isom LL. The role of sodium channels in cell adhesion. Front Biosci. 2002;7:12–23. doi: 10.2741/isom. [DOI] [PubMed] [Google Scholar]
- John N, Krügel H, Frischknecht R, Smalla KH, Schultz C, Kreutz MR, Gundelfinger ED, Seidenbecher CI. Brevican-containing perineuronal nets of extracellular matrix in dissociated hippocampal primary cultures. Mol Cell Neurosci. 2006;31:774–784. doi: 10.1016/j.mcn.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschbaum K, Kriebel M, Kranz EU, Pötz O, Volkmer H. Analysis of non-canonical fibroblast growth factor receptor 1 (FGFR1) interaction reveals regulatory and activating domains of neurofascin. J Biol Chem. 2009;284:28533–28542. doi: 10.1074/jbc.M109.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Tessier-Lavigne M. Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol. 2011;3. pii:a001727. doi: 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koticha D, Babiarz J, Kane-Goldsmith N, Jacob J, Raju K, Grumet M. Cell adhesion and neurite outgrowth are promoted by neurofascin NF155 and inhibited by NF186. Mol Cell Neurosci. 2005;30:137–148. doi: 10.1016/j.mcn.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Koticha D, Maurel P, Zanazzi G, Kane-Goldsmith N, Basak S, Babiarz J, Salzer J, Grumet M. Neurofascin interactions play a critical role in clustering sodium channels, ankyrin G and beta IV spectrin at peripheral nodes of Ranvier. Dev Biol. 2006;293:1–12. doi: 10.1016/j.ydbio.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Kriebel M, Metzger J, Trinks S, Chugh D, Harvey RJ, Harvey K, Volkmer H. The Cell Adhesion Molecule Neurofascin Stabilizes Axo-axonic GABAergic Terminals at the Axon Initial Segment. J Biol Chem. 2011;286:24385–24393. doi: 10.1074/jbc.M110.212191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin SI, Khaliq ZM, Aman TK, Grieco TM, Kearney JA, Raman IM, Meisler MH. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. J Neurophysiol. 2006;96:785–793. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- Li XG, Somogyi P, Tepper JM, Buzsáki G. Axonal and dendritic arborization of an intracellularly labeled chandelier cell in the CA1 region of rat hippocampus. Experimental brain research Experimentelle Hirnforschung Experimentation cerebrale. 1992;90:519–525. doi: 10.1007/BF00230934. [DOI] [PubMed] [Google Scholar]
- Lu B, Wang KH, Nose A. Molecular mechanisms underlying neural circuit formation. Curr Opin Neurobiol. 2009;19:162–167. doi: 10.1016/j.conb.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manto M, Marmolino D. Cerebellar ataxias. Curr Opin Neurol. 2009;22:419–429. doi: 10.1097/WCO.0b013e32832b9897. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Isom LL. Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem. 2004;279:52744–52752. doi: 10.1074/jbc.M405990200. [DOI] [PubMed] [Google Scholar]
- Nordquist DT, Kozak CA, Orr HT. cDNA cloning and characterization of three genes uniquely expressed in cerebellum by Purkinje neurons. J Neurosci. 1988;8:4780–4789. doi: 10.1523/JNEUROSCI.08-12-04780.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Palay SL, Palay VC. Cerebellar cortex. New York: Springer; 1974. [Google Scholar]
- Perkins EM, Clarkson YL, Sabatier N, Longhurst DM, Millward CP, Jack J, Toraiwa J, Watanabe M, Rothstein JD, Lyndon AR, Wyllie DJ, Dutia MB, Jackson M. Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J Neurosci. 2010;30:4857–4867. doi: 10.1523/JNEUROSCI.6065-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–1793. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruss T, Niere M, Kranz EU, Volkmer H. Homophilic interactions of chick neurofascin in trans are important for neurite induction. Eur J Neurosci. 2004;20:3184–3188. doi: 10.1111/j.1460-9568.2004.03773.x. [DOI] [PubMed] [Google Scholar]
- Pruss T, Kranz EU, Niere M, Volkmer H. A regulated switch of chick neurofascin isoforms modulates ligand recognition and neurite extension. Mol Cell Neurosci. 2006;31:354–365. doi: 10.1016/j.mcn.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Rader C, Stoeckli ET, Ziegler U, Osterwalder T, Kunz B, Sonderegger P. Cell–cell adhesion by homophilic interaction of the neuronal recognition molecule axonin-1. Eur J Biochem. 1993;215:133–141. doi: 10.1111/j.1432-1033.1993.tb18015.x. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Properties of sodium currents and action potential firing in isolated cerebellar Purkinje neurons. Ann N Y Acad Sci. 1999;868:93–96. doi: 10.1111/j.1749-6632.1999.tb11279.x. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S. Histologie du système nerveux de l'hommes et des vertébrés. Paris: Maloine; 1911. [Google Scholar]
- Ramon y Cajal S. Histology of the nervous system of man and vertebrates. New York: Oxford UP; 1995. [Google Scholar]
- Rinaldo L, Hansel C. Ataxias and cerebellar dysfunction: involvement of synaptic plasticity deficits? Funct Neurol. 2010;25:135–139. [PMC free article] [PubMed] [Google Scholar]
- Sacco T, De Luca A, Tempia F. Properties and expression of Kv3 channels in cerebellar Purkinje cells. Mol Cell Neurosci. 2006;33:170–179. doi: 10.1016/j.mcn.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Sakaba T. Two Ca(2+)-dependent steps controlling synaptic vesicle fusion and replenishment at the cerebellar basket cell terminal. Neuron. 2008;57:406–419. doi: 10.1016/j.neuron.2007.11.029. [DOI] [PubMed] [Google Scholar]
- Shiga T, Shirai T, Grumet M, Edelman GM, Oppenheim RW. Differential expression of neuron-glia cell adhesion molecule (Ng-CAM) on developing axons and growth cones of interneurons in the chick embryo spinal cord: an immunoelectron microscopic study. J Comp Neurol. 1993;329:512–518. doi: 10.1002/cne.903290407. [DOI] [PubMed] [Google Scholar]
- Slemmer JE, De Zeeuw CI, Weber JT. Don't get too excited: mechanisms of glutamate-mediated Purkinje cell death. Prog Brain Res. 2005;148:367–390. doi: 10.1016/S0079-6123(04)48029-7. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Hámori J. A quantitative electron microscopic study of the Purkinje cell axon initial segment. Neuroscience. 1976;1:361–365. doi: 10.1016/0306-4522(76)90127-5. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Nunzi MG, Gorio A, Smith AD. A new type of specific interneuron in the monkey hippocampus forming synapses exclusively with the axon initial segments of pyramidal cells. Brain Res. 1983;259:137–142. doi: 10.1016/0006-8993(83)91076-4. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Development of “Pinceaux” formations and dendritic translocation of climbing fibers during the acquisition of the balance between glutamatergic and gamma-aminobutyric acidergic inputs in developing Purkinje cells. J Comp Neurol. 2008;506:240–262. doi: 10.1002/cne.21501. [DOI] [PubMed] [Google Scholar]
- Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, Joyner AL. Ascl1 genetics reveals insights into cerebellum local circuit assembly. J Neurosci. 2011;31:11055–11069. doi: 10.1523/JNEUROSCI.0479-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Labasque M, Dupree JL, Faivre-Sarrailh C, Bhat MA. In vivo deletion of immunoglobulin domains 5 and 6 in neurofascin (Nfasc) reveals domain-specific requirements in myelinated axons. J Neurosci. 2010;30:4868–4876. doi: 10.1523/JNEUROSCI.5951-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA. Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron. 2011;69:244–257. doi: 10.1016/j.neuron.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Südhof TC. Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J Neurosci. 2006;26:7172–7180. doi: 10.1523/JNEUROSCI.1101-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh FS, Doherty P. Neural cell adhesion molecules of the immunoglobulin superfamily: role in axon growth and guidance. Annu Rev Cell Dev Biol. 1997;13:425–456. doi: 10.1146/annurev.cellbio.13.1.425. [DOI] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Müller U, Dürr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Wiencken-Barger AE, Mavity-Hudson J, Bartsch U, Schachner M, Casagrande VA. The role of L1 in axon pathfinding and fasciculation. Cereb Cortex. 2004;14:121–131. doi: 10.1093/cercor/bhg110. [DOI] [PubMed] [Google Scholar]
- Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. 2010;285:32160–32173. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagha E, Lang EJ, Rudy B. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J Neurosci. 2008;28:1291–1300. doi: 10.1523/JNEUROSCI.4358-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Goldman JE. Generation of cerebellar interneurons from dividing progenitors in white matter. Neuron. 1996;16:47–54. doi: 10.1016/s0896-6273(00)80022-7. [DOI] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Spinocerebellar ataxias. Neurobiol Dis. 2000;7:523–527. doi: 10.1006/nbdi.2000.0346. [DOI] [PubMed] [Google Scholar]
- Zonta B, Desmazieres A, Rinaldi A, Tait S, Sherman DL, Nolan MF, Brophy PJ. A critical role for Neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron. 2011;69:945–956. doi: 10.1016/j.neuron.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]