

Abstract
Inhibition of soluble epoxide hydrolase (sEH) has been proposed as a new pharmaceutical approach for treating hypertension and vascular inflammation. The most potent sEH inhibitors reported in literature to date are urea derivatives. However, these compounds have limited pharmacokinetic profiles. We investigated non-urea amide derivatives as sEH inhibitors and identified a potent human sEH inhibitor 14–34 having potency comparable to urea-based inhibitors.
Keywords: Soluble epoxide hydrolase, sEH, Non-urea inhibitors, Hypertension, Anti-inflamatory
Soluble epoxide hydrolase (sEH) is an enzyme that is involved in the metabolism of lipid epoxides.1 This enzyme is found in various mammalian tissues and is mostly located in liver, kidneys and vascular tissues.2 It converts endogenous substrate—epoxyeicosarienoic acids (EETs)—to dihydroxy eicosatrienoic acids (DHETs), which show abolished or diminished or changed biological activity. 3 EETs act as vasodilators in various arteries (renal, cerebral, and coronary),4 protect from ischemic injury5 and manifest anti-inflammatory properties.6 sEH inhibition increases cellular EETs levels and promote this activity. Several preclinical studies suggest that inhibition of sEH may represent a novel approach to treat hypertension, organ protection and inflammation.7–10 Furthermore, it has been shown that sEH inhibition reduces pain in several animal models.11
Initial sEH inhibitors with IC50 in the lower nanomolar range included N,N′-disubstituted ureas, N,N′-dicyclohexylurea (DCU) and N-adamantyl-N′-cyclohexylurea (ACU) (Fig. 1).12 However, poor water solubility and limited in vivo studies13 led to a design of the second generation of sEH inhibitors, 12-(3-adamantan-1-ylureido) dodecanoic acid (AUDA) and 1-adamantan-1-yl-3-{5-[2-(ethoxyethoxy) ethoxy]pentyl}urea (AEPU) with improved water solubility and maintained inhibition.14,15 However, these inhibitors suffered from rapid metabolism in vivo.16 Recent studies have focused on piperidine-based di- and trisubstituted ureas, such as N-(1-(2,2,2-trifluoroethanoyl)piperidin-4-yl)-N′-(adamant-1-yl)urea (TPAU) and N-(1-acetylpiperidin-4-yl)-N′-(adamant-1-yl)urea (APAU).17,18 Some of these piperidine-based compounds possess low nano to picomolar activity and good pharmacokinetic properties in different animal models.17–21
Figure 1.

Chemical structures of known sEH inhibitors.
These extensive structure–activity relationship (SAR) studies suggested that the pharmacophore for sEH inhibitors should include a central urea moiety for hydrogen bonding to two tyrosine residues (Tyr381 and Tyr465) and one Asp333 residue—all three located in the hydrolase catalytic pocket of sEH.22
Several common urea modifications such as thiourea, sulfonyl urea, amide and aminomethylene amide have been incorporated into central pharmacophore in order to maintain similar binding profile to that of urea but improve potential therapeutic applications.23
We sought to identify potent, selective non-urea sEH inhibitors effective in vivo with good metabolic stability and pharmacokinetic and distribution properties, to test as an anti-hypertensive and anti-inflammatory drug candidate. A sensitive fluorescent based assay29 was employed to determine IC50 values. Using high throughput screening we have previously reported a series of non-urea sEH inhibitors with low micromolar to nanomolar potency. 24 From the compound collection provided by the NIH Road-map project we identified sulfonyl isonipecotamide 1, a potent sEH inhibitor (IC50 = 20 nM).25 In addition, we synthesized a secondary library of compounds based on 1 and SAR revealed the most potent non-urea sEH inhibitor in that study, compound 2 with IC50 = 7.9 nM (Fig. 2).24 Herein we report design, synthesis and biological evaluation of non-urea sEH inhibitors based on compound 2.
Figure 2.

Chemical structures of non-urea sEH inhibitors.
Our evaluation of the SAR for 2 started with replacement of the sulfonamide moiety with both an amide and methylene group, in order to better understand what structural feature of the ‘secondary’ pharmacophore is important for sEH inhibition. Furthermore, amides have in general better properties for formulation. Additionally, we designed a compound in which the sulfonamide group was retained, but the central amide moiety was reversed. These three compounds were prepared by standard synthetic methods from commercially available starting materials (Scheme 1). Thus, per Scheme 1, cycloheptylamine 3 was condensed with Boc-isonipecotic acid 4 under standard peptide coupling conditions. The Boc group of the amide 5 was removed with TFA to afford amine 6, which was reacted with 2,4,6-trimethylbenzoyl chloride to yield amide analog 7. In order to synthesize the methylene analog of compound 2, amine 6 was treated with mesitaldehyde under reductive amination conditions, which gave the final product 8. The reversed amide analog was synthesized in similar fashion. Commercially available cycloheptanecarboxylic acid 9 and 4-amino- 1-Boc-piperidine 10 were condensed under EDC coupling conditions to afford amide 11, which was subsequently deprotected to furnish amine 12. The amine 12 was sulfonylated with 2-mesitylenesulfonyl chloride to afford analog 13 (Scheme 2). This first set of compounds demonstrated that the sulfonamide group is important for recognition by sEH, since replacement with an amide group, as in compound 7, resulted in a sixfold decrease in binding affinity (IC50 = 46.1 nM). Compound 8, which contains a methylene group, suffered a 15-fold loss of inhibitory potency (IC50 = 120 nM). The effect of reversing amide group in compound 13 resulted in a small, 2.5-fold decrease in the binding affinity for sEH (IC50 = 22 nM), supporting previous studies that the proton in NH (both in urea and amide sEH inhibitors) is important in sEH inhibition, ostensibly forming a salt bridge with the catalytic nucleophile Asp333.26
Scheme 1.

Reagents and conditions: (a) EDC, CH2Cl2, rt, 24 h, 68%; (b) TFA, CH2Cl2, rt, 24 h, 89%; (c) 2,4,6-trimethylbenzoyl chloride, Et3N, CH2Cl2, rt, 24 h, 85%; (d) mesitaldehyde, NaBH(OAc)3, CH2Cl2, rt, 12 h, 82%; (e) R-SO2Cl, Et3N, CH2Cl2, rt, 24 h, 70–91%; (f) R′OCl, Et3N, CH2Cl2, rt, 24 h, 85–92%.
Scheme 2.

Reagents and conditions: (a) EDC, CH2Cl2, rt, 24 h, 65%; (b) TFA, CH2Cl2, rt, 24 h, 90%; (c) 2,4,6-trimethylbenzoylsulfonyl chloride, Et3N, CH2Cl2, rt, 24 h, 85%.
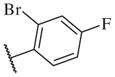
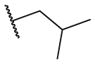
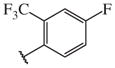
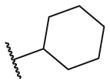
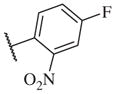
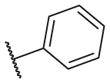
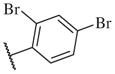
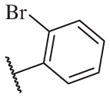
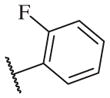
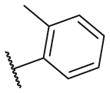
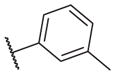
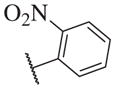
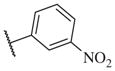
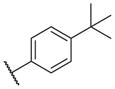
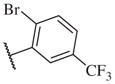
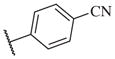
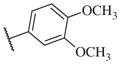
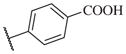
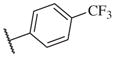
These results returned our attention to the SAR of the right-hand side of the sulfonamide group of the original lead compound 2. Thus, amine 6 was reacted with a variety of sulfonyl chlorides to yield products 14–1 to 14–51. This group of analogs will allow us to evaluate the role of different substituent on this part of the molecule. We introduced as well several different polar groups on the aromatic ring in order to obtain compounds that will be easier to formulate and administer. The results are summarized in Table 1. As illustrated in Table 1, the diverse right-hand side modifications led to improved or similar potency (14–7, 14–27, 14–31 and 14–34) over the original sulfonamide analog 2 and several structure–activity relationships can be discerned in this series of analogs. First, the nonpolar group in the ortho-position is important for potent inhibition. Great loss of activities was observed if ortho-group is deleted (14–14, 14–16, 14–19, 14–20, 14–22, 14–24, 14–25, etc.). Para-substitution is generally tolerated, but placement of any polar group at this position significantly diminished potency of the compounds (14–11, 14–12, 14–15, 14–21, etc).
Table 1.
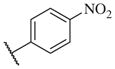
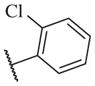
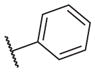
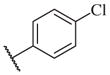
The biological results for sulfonamide analogs 14–1 to 14–52
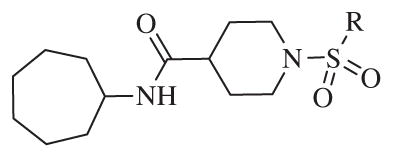
| |||||
|---|---|---|---|---|---|
| Compound | R | IC50a,b (nM) | Compound | R | IC50 (nM) |
| 14–1 |
|
340 | 14–27 |
|
5.4 |
| 14–2 |
|
960 | 14–28 |
|
15 |
| 14–3 |
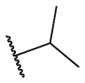
|
490 | 14–29 |
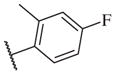
|
18 |
| 14–4 |
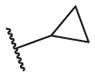
|
450 | 14–30 |
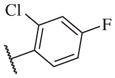
|
27 |
| 14–5 |
|
950 | 14–31 |
|
7.7 |
| 14–6 |
|
510 | 14–32 |
|
12 |
| 14–7 |
|
6.7 | 14–33 |
|
56 |
| 14–8 |
|
78 | 14–34 |
|
1.6 |
| 14–9 |
|
24 | 14–35 |
|
1700 |
| 14–10 |
|
12 | 14–36 |
|
120 |
| 14–11 |
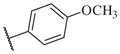
|
450 | 14–37 |
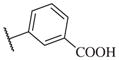
|
150 |
| 14–12 |
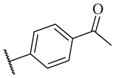
|
340 | 14–38 |
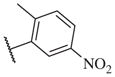
|
32 |
| 14–13 |
|
62 | 14–39 |
|
17 |
| 14–14 |
|
2100 | 14–40 |
|
360 |
| 14–15 |
|
240 | 14–41 |
|
2000 |
| 14–16 |
|
2100 | 14–42 |
|
16 |
| 14–17 |
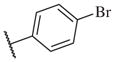
|
560 | 14–43 |
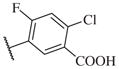
|
6000 |
| 14–18 |
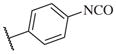
|
150 | 14–44 |
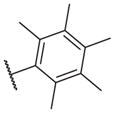
|
9.6 |
| 14–19 |
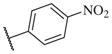
|
50000 | 14–45 |
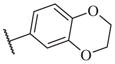
|
224 |
| 14–20 |
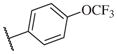
|
1100 | 14–46 |
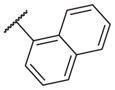
|
35 |
| 14–21 |
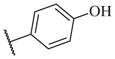
|
140 | 14–47 |
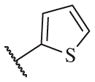
|
870 |
| 14–22 |
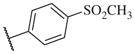
|
13000 | 14–48 |
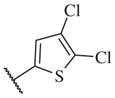
|
4200 |
| 14–23 |
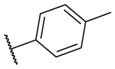
|
570 | 14–49 |
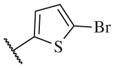
|
710 |
| 14–24 |
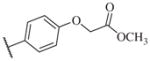
|
1300 | 14–50 |
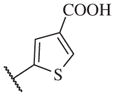
|
6000 |
| 14–25 |
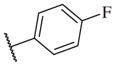
|
1900 | 14–51 |
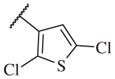
|
21 |
| 14–26 |
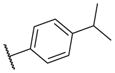
|
470 | |||
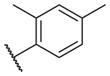
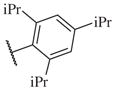
On the other hand, hydrophobic alkyl groups or halogen substitution at the ortho-position enhanced low nanomolar potency of the para- and meta- substituted analogs, suggesting that some of the substituents on the aromatic ring act synergistically (14–27, 14–28, 14–32 and 14–34).
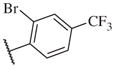
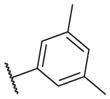
Overall, polar groups were not tolerated in any position on the aromatic ring (14–37, 14–40), even if a hydrophobic group was present in ortho-position (14–38, 14–43). Furthermore, aromatic analogs appeared to be more favorable compared to the alkyl-and cyclo-sulfonamides (14–1, 14–2, 14–3, 14–4, and 14–5). Only the thiophene analog attached to the sulfonamide moiety via position 3 (14–51) had low nanomolar potency compared with other thiophene analogs that are linked via position 2 (14–47, 14–48, 14–49 and 14–50).
Although potency of amide analog 7 showed a sixfold decrease compared to lead compound 2, the SAR on the right-hand side of the molecule was obvious and since the amide has better pharmacokinetic properties28 compared to the corresponding sulfonamide, we decided to design a small library of amide analogs, 15–1 to 15–12, as it is outlined in Scheme 1. However, the various modifications of the aromatic ring did not improve potency further (Table 2).
Table 2.
The biological results for amide analogs 15–1 to 15–12
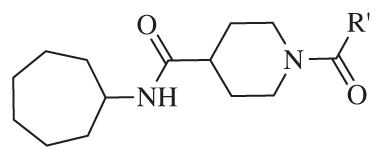
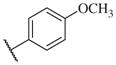
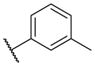
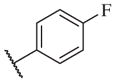
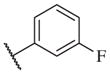
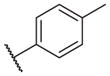
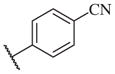
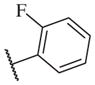
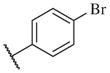
In conclusion, we have successfully improved the potency of non-urea sulfonamide analogs through SAR-guided modification. Compound 14–34,30 with an IC50 of 1.6 nM, represents the most potent non-urea sEH inhibitor reported to date. Pharmacokinetic evaluation and pre-clinical studies of selected potent inhibitors are planned.
Acknowledgments
This work was supported in part by NIEHS R01 ES002710. B.D.H. is a George and Judy Senior fellow of the American Asthma Foundation.
References and notes
- 1.Capdevila JH, Falck JR, Harris RC. J Lipid Res. 2000;41:163. [PubMed] [Google Scholar]
- 2.Hammock BD, Grant D, Storms D. In: Comprehensive Toxicology. Sipes I, McQueen C, Gandolfi A, editors. Pergamon; Oxford: 1997. p. 283. [Google Scholar]
- 3.Newman JW, Morisseau C, Hammock BD. Prog Lipid Res. 2005;44:1. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Behm DJ, Ogbonna A, Wu C, Burns-Kurtis CL, Douglas SA. J Pharmacol Exp Ther. 2009;328:231. doi: 10.1124/jpet.108.145102. [DOI] [PubMed] [Google Scholar]
- 5.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Circ Res. 1996;78:415. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 6.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Science. 1999;285:1276. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loch D, Hoey A, Morisseau C, Hammock BO, Brown L. Cell Biochem Biophys. 2007;47:87. doi: 10.1385/cbb:47:1:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Natl Acad Sci USA. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. J Cardiovasc Pharmacol. 2005;46:842. doi: 10.1097/01.fjc.0000189600.74157.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Proc Natl Acad Sci USA. 2006;103:18733. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Life Sci. 2006;79:2311. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Proc Natl Acad Sci USA. 1999;96:8849. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. J Med Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Biochem Pharmacol. 2002;63:1599. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 15.Kim IH, Morisseau C, Watanabe T, Hammock BD. J Med Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe T, Schulz D, Morisseau C, Hammock BD. Anal Chim Acta. 2006;559:37. doi: 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Bioorg Med Chem Lett. 2006;16:5212. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen HC, Ding FX, Deng Q, Xu S, Chen HS, Tong X, Tong V, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Zhang B, Roy S, Tata JR, Berger JP, Colletti SL. Bioorg Med Chem Lett. 2009;19:5314. doi: 10.1016/j.bmcl.2009.07.138. [DOI] [PubMed] [Google Scholar]
- 19.Shen HC, Ding FX, Wang S, Deng Q, Zhang X, Chen Y, Zhou G, Xu S, Chen HS, Tong X, Tong V, Mitra K, Kumar S, Tsai C, Stevenson AS, Pai LY, Alonso-Galicia M, Chen X, Soisson SM, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. J Med Chem. 2009;52:5009. doi: 10.1021/jm900725r. [DOI] [PubMed] [Google Scholar]
- 20.Anandan SK, Webb HK, Chen D, Wang YX, Aavula BR, Cases S, Cheng Y, Do ZN, Mehra U, Tran V, Vincelette J, Waszczuk J, White K, Wong KR, Zhang LN, Jones PD, Hammock BD, Patel DV, Whitcomb R, MacIntyre DE, Sabry J, Gless R. Bioorg Med Chem Lett. 2011;21:983. doi: 10.1016/j.bmcl.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. J Med Chem. 2010;53:7067. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Protein Sci. 2006;15:58. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anandan SK, Do ZN, Webb HK, Patel DV, Gless RD. Bioorg Med Chem Lett. 2009;19:1066. doi: 10.1016/j.bmcl.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 24.Xie Y, Liu Y, Gong G, Smith DH, Yan F, Rinderspacher A, Feng Y, Zhu Z, Li X, Deng SX, Branden L, Vidovic D, Chung C, Schurer S, Morisseau C, Hammock BD, Landry DW. Bioorg Med Chem Lett. 2009;19:2354. doi: 10.1016/j.bmcl.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.AID:1026; Pubchem 2008.
- 26.Argiriadi MA, Morisseau C, Goodrow MH, Dowdy DL, Hammock BD, Christianson DW. J Biol Chem. 2000;275:15265. doi: 10.1074/jbc.M000278200. [DOI] [PubMed] [Google Scholar]
- 27.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. J Med Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.IC50 Assay Conditions: Cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyloxyran-2-yl) methyl carbonate (CMNPC) was used as the fluorescent substrate. Human sEH (1 nM) or murine sEH (1 nM) was incubated with the inhibitor for 5 min in pH 7.0 Bis–Tris/HCl buffer (25 mM) containing 0.1 mg/mL of BSA at 30 °C prior to substrate introduction ([S] = 5 μM). Activity was determined by monitoring the appearance of 6-methoxy-2-naphthaldehyde over 10 min by fluorescence detection with an excitation wavelength of 330 nm and an emission wavelength of 465 nm. Reported IC50 values are the average of the three replicates with at least two datum points above and at least two below the IC50.
- 30.Analytical data for the compound 14–34: 1H NMR (300 MHz, CDCl3): δ 7.81–7.78 (d, J = 8.1 Hz, 1H) 7.12 (s, 2H), 5.35 (br, 1H), 3.92–3.89 (m, 1H), 3.72–3.68 (d, J = 12 Hz, 2H), 2.74–2.67 (t, J = 21 Hz, 2H), 2.57 (s, 3H), 2.37 (s, 3H), 2.13–2.05 (m, 1H), 1.88–1.84 (m, 2H), 1.78–1.69 (m, 6H), 1.62–1.39 (m, 8H), 13C NMR (75 MHz, CDCl3) δ 172.6, 143.8, 138.1, 133.7, 133.0, 130.7, 126.8, 50.6, 44.8, 43.0, 35.5, 28.8, 28.3, 24.4, 21.6, 20.8; ESI-MS (M++H): 393.