

Abstract
Functional divergence is the process by which new genes and functions originate through the modification of existing ones. Both genetic and environmental factors influence the evolution of new functions, including gene duplication or changes in the ecological requirements of an organism. Novel functions emerge at the expense of ancestral ones and are generally accompanied by changes in the selective forces at constrained protein regions. We present software capable of analyzing whole proteomes, identifying putative amino acid replacements leading to functional change in each protein and performing statistical tests on all tabulated data. We apply this method to 750 complete bacterial proteomes to identify high-level patterns of functional divergence and link these patterns to ecological adaptations. Proteome-wide analyses of functional divergence in bacteria with different ecologies reveal a separation between proteins involved in information processing (Ribosome biogenesis etc.) and those which are dependent on the environment (energy metabolism, defense etc.). We show that the evolution of pathogenic and symbiotic bacteria is constrained by their association with the host, and also identify unusual events of functional divergence even in well-studied bacteria such as Escherichia coli. We present a description of the roles of phylogeny and ecology in functional divergence at the level of entire proteomes in bacteria.
Introduction
Most new genes, functions, and activities originate through the modification of existing ones. The evolutionary process that gives rise to functional differences between related genes is called functional divergence [1], [2]. At the species level, functional diversification is primarily associated with adaptive radiations, when a single ancestor differentiates into multiple descendant species, each adapting by natural selection to one of a new set of ecological niches (Schluter 2000) [3]. Following this theory, environmental variation triggers divergent natural selection, leading to the emergence of niche specialists. In many cases, species under the same ecological conditions differ in their ability to adapt to new niches, even when they stem from the same ancestor [4], [5]. Therefore, other factors such as genetic constraints also play an important role in the process of functional divergence.
The process of functional divergence, or departure of a gene from its ancestral function, is constrained by the requirement to maintain the original function: mutations that confer a new function are likely to interfere with the ancestral function and therefore are eliminated by negative selection. This constraint can be relaxed when selection for the ancestral function is weakened, either through gene duplication (and therefore redundancy), or through changes to the environment inhabited by the organism or a combination of both these factors. After gene duplication, one copy of the gene can be free to evolve in a new direction if the other continues to perform the ancestral function (neofunctionalization). Alternatively, ancestral functions can be partitioned between the two gene copies, potentially leading to later specialization or subfunctionalization [1], [2], [6], [7], [8]. Major changes in the environment or ecological niche can also lead to a relaxation of selective constraints on ancestral functions, although this process is less well characterized. For example, endosymbiotic bacteria have lost many of the genes their free-living relatives need to obtain nutrients from the environment [9], but have also experienced functional divergence in certain genes [10], [11].
Prokaryotes are extraordinarily rich in biological diversity, whether measured in terms of number of species [12], [13], habitat range [14], or the breadth of energy sources and biochemical pathways they can exploit in order to survive [15]. Even photosynthesis and oxidative phosphorylation – the mainstays of eukaryotic energy metabolism – are bacterial inventions acquired by endosymbiosis during early eukaryote evolution [16]. How did this prokaryotic diversity evolve, particularly when the fixation of gene duplications appears to be somewhat more frequent in eukaryotes [17]?
Adaptive evolution in prokaryotes is promoted by at least three main factors: first, a high strength of selection relative to eukaryotes, on account of their generally large population sizes [18]; second, their ability to obtain genes by horizontal gene transfer (HGT), which enables the sharing of niche-relevant functions between distantly-related microbes living in the same environment [19]; and third, their use of stress-induced hypermutation [20], which may increase the production of adaptive variants as a “last gasp” response to a challenging environment.
Although we know that these processes can drive ecological adaptation in prokaryotes, identifying the fraction of genetic variation that is associated with these functional changes remains a challenging problem. In the case of bacteria, whole-genome analyses must take into account widespread HGT, which means that different genes often disagree on the overall species tree [21]. This is a considerable problem for analyses of functional divergence, which require a tree in order to determine the branch upon which a particular trait arose.
The rationale for previous methods to identify functional divergence, and indeed the new approach described here, derives from the neutral theory of Kimura [22] (1983), which predicts that residues important for the function of a protein will be under strong functional constraint and therefore evolve slowly. These considerations have motivated the development of a number of methods for identifying changes in selective constraints on protein-coding genes and on single amino acid sites and lineages in a phylogenetic tree [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33]. At the protein level, Gu [34], [35], [36] developed a Bayesian approach to identify functional divergence, which has become the most widely used. Comparisons of amino acid site-specific evolutionary rate or residue conservation between two homologous clades can therefore be used to identify amino acid sites at which selective constraints have changed, potentially indicating functional divergence.
Recently, we have developed a new distance-based method which explores a bifurcating phylogenetic tree, testing for functional divergence at each node by comparing the two downstream clades to an outgroup in order to identify sites at which substitution rates per amino acid sites have shifted [11], [37]. Similar to other methods, our approach was limited to tests of one gene at a time, unless the phylogeny of all genes could be fixed in advance.
Because general patterns of functional divergence and their link to ecological changes cannot be understood by the analysis of single genes, in the present study, we have optimised our method to (i) handle analyses of functional divergence that include hundreds of complete proteomes, (ii) address the fact that the phylogenies of individual proteins do not necessarily agree with the true phylogeny, as is often the case with organisms that acquire genes through HGT, (iii) provide an intuitive probability assignment for each test which takes the underlying phylogeny of the sequences into account and (iv) explore all levels of each gene tree, testing for functional divergence at each node.
Using this novel method to detect functional divergence, we infer patterns of radical change for each protein individually, and then cluster species according to the functional categories (derived from COG [38]) in which they exhibit significant evidence of functional divergence. We provide a fast, open source implementation of our method in the C++ program CAFS (Clustering analysis of functional shifts). We perform an analysis of functional divergence on 750 bacterial proteomes. This set includes bacteria from various different ecological niches and therefore provides a good dataset for identifying ecology-related functional divergence. Our approach (i) reveals striking patterns of convergent evolution in phylogenetically distinct but ecologically related groups of bacteria, including pathogens, endosymbionts, and thermophiles, (ii) provides additional support for the view that bacteria have a conserved set of core functions, with a more variable metabolic layer and (iii) provides a detailed picture of how individual species of unusual bacteria have diverged from their closest relatives.
Results and Discussion
A conserved functional core and variable crust in the evolution of bacterial proteomes
An obvious sign of functional divergence (also understood here as changes in substitution rates per amino acid site in proteins) would be a set of homologs that spans multiple COG categories. In this study we focus only on those alignments where all sequences have the same COG annotation. This represents the majority of homologs and is a reflection of the relatively broad character of the COG categories.
The kinds of functional shifts that we detect on the basis of conserved, radical amino acid substitutions are therefore subtler and not noticeable from simply comparing the COG classifications across homologous sequences. We used chi-squared tests to evaluate the differences in functional divergence between COG gene categories in our dataset (see Figure 1). We compared the proportion of positive tests for functional divergence within each of the 19 COG categories to the background expectation, which was calculated by combining all categories. If genes in different functional categories have similar propensities to undergo functional divergence, we would expect the proportion of positive tests in each category to be similar to the mean, resulting in few significant cases of enrichment. However, eighteen of the nineteen categories were either enriched or impoverished for functional divergence, while only one category failed to deviate significantly from the background expectation.
Figure 1. Different categories of genes experience different levels of functional divergence.

Proportion (FD) is the proportion of tested branches with at least one functionally divergent site across all gene trees in a particular functional category. Categories are labeled according to the COG ontology system [38], [73]. Eighteen of the nineteen categories fall into two groups: significantly enriched or impoverished. Most information processing genes (K, J, L, A) fall into the latter group, while metabolic functions (E,F,G,H,P,Q) and genes involved in defense (V) or found on the cell surface (M) are enriched for radical change. Description of COG tags can be found in Table S5.
To test whether this polarization of our dataset was simply due to an artifact – for instance, the use of a non-conservative enrichment test – we performed simulations in which the genes in our original dataset were randomly assigned to one of the 19 COG categories before testing for enrichment. In these simulations, events of functional divergence were much more evenly distributed among the categories, so that 93% of categories were neither enriched nor impoverished for functional divergence relative to the background level. This result indicates that the probability of functional change is not evenly distributed among the real categories: there is a stark division between enriched and impoverished categories. This supports the idea that bacterial proteomes comprise a relatively unchanging core (that is, genes in impoverished categories) coupled with a set of more variable functions (enriched categories), as previously noticed [39], [40], [41].
The impoverished categories are almost exclusively those involved with information storage and processing, including DNA replication, recombination, and repair (L); transcription (K), ribosome biogenesis (J); and cell division (D). Metabolic genes were among those enriched for functional divergence, including genes involved in the metabolism of coenzymes (H), secondary metabolites (Q), carbohydrates (G), amino acids (E) and nucleotides (F). Along with these metabolic categories, cell wall and envelope genes (M) and cellular defense mechanisms (V) were among the most enriched categories in our analysis, highlighting the critical role of the environment in directing lineage-specific episodes of functional change. Taken together, our results agree with a number of previous reports indicating that proteins involved in information processing are more conserved across large evolutionary distances than those involved in metabolism [39], [40], [41], [42].
An additional point bears emphasizing here: since our method controls for the level of conservation at each node on the tree, the significance of a particular substitution pattern depends on the background evolutionary rate so that in slow-evolving proteins, relatively conservative substitutions are detected as significant events of functional divergence, whereas only very unusual substitution patterns will attain significance in fast-evolving proteins. Therefore, our results indicate that information processing genes are not only more conserved than others purely in terms of evolutionary rate, but that they also experience less functional change even taking this low rate of sequence evolution into account.
Why are informational genes under greater functional constraint than the rest of the proteome? One possibility, which follows Crick's concept of the “frozen accident” [43], is that too many other genes depend on the basic functions of translation, transcription, and repair: functional changes in these genes would disrupt many other systems in the cell. This hypothesis is supported by the observation that the COG category containing protein trafficking and chaperones (O) is also impoverished: the core activities of generalist chaperones such as GroEL and DnaK are required for the proper folding of many different proteins in bacterial cells [44].
Host interactions constrain functional change in pathogenic and symbiotic bacteria
Does the ecological niche of an organism influence the pattern of functional change it experiences? To answer this question, we evaluated the enrichment of functional divergence in each species relative to the others in our dataset. To calculate the enrichment status of each species, we used the same statistical strategy as employed for enrichment by functional category: we calculated a background proportion of successful tests for functional divergence over all species, and then compared this to the proportion for each species individually using chi-squared tests (a full table of these results can be found in Table S1). We also used chi-squared tests to identify associations between these three enrichment patterns (enrichment, impoverishment, or neither) and organism lifestyle, as is summarized in Table 1. While there was no statistically significant difference between psychrophiles and mesophiles in terms of functional divergence (chi-squared = 0.9762, P = 0.6138), nor indeed was there significance when comparing thermophiles to free living bacteria.
Table 1. Effect of organism lifestyle on functional divergence.
| Lifestyle | Comparison | Enriched | Neither | Impoverished | Significance |
| Psychrophile | Mesophile | 2/61 | 6/433 | 1/66 | N.S. |
| Thermophile | Mesophile | 7/61 | 22/433 | 1/66 | N.S. |
| Pathogen | Non-pathogen | 22/77 | 272/294 | 47/38 | *** (−) |
| Intracellular pathogen | Other pathogen | 0/22 | 26/246 | 4/43 | N.S. |
| Symbiont | Non-symbiont | 4/95 | 36/530 | 13/72 | * |
| *Intracellular endosymbiont | All others | 4/224 | 14/360 | 15/133 | *** (−) |
| All interactors | Free-living | 44/55 | 410/156 | 70/15 | *** (−) |
Associations between lifestyle and enrichment for functional divergence: the numbers of genomes in each category are given in the form Lifestyle/Comparison. Significance was assessed with Yates-corrected chi-squared tests, or Fisher tests when the expected count was lower than 5 for any one cell in the contingency table. Significance codes: N.S. = P>0.05;
= P<0.05,
** = P<0.01,
= P<0.001.
If an association was significant, “+” or “−” denote the direction of the shift associated with the lifestyle being tested. For instance, interactors are significantly impoverished (−) compared to free-living bacteria.
Thermophiles did however present a contrasting pattern of functional divergence in comparison to the general pattern, with two COG categories being enriched for functional divergence in thermophiles while being impoverished in general. These categories are directly related with the survival of cells under heat stress: category K, which comprises mostly transcription factors, and category L, which is involved in DNA replication, recombination and repair. Above certain temperature threshold, molecular pathways undergo dramatic temperature induced alterations that drive to cytotoxicity, radiosensitization and thermotolerance [45], [46]. Among all the responses that take place in the cell under high temperatures, inhibition of DNA, RNA and protein synthesis is the response that involves a complex and fine-tuning of regulation mechanisms, mainly orchestrated by transcription factors [46]. One such important regulated mechanism is the induction of heat-shock proteins, particularly involved in mitigating the cytotoxic effects due to the non-specific aggregation of unfolded and denatured proteins [47], [48].
Interestingly, we found that all bacteria that interact with a host as an integral part of their lifestyle (including pathogens, parasites, symbionts and commensals) were significantly impoverished for functional divergence in comparison to their free-living relatives (see Table 1). This result is somewhat surprising because pathogens and symbionts generally experience higher rates of evolution than free-living bacteria, although much of the increase can be attributed to heightened genetic drift [9]. Our results suggest that once the overall conservation level of proteins is accounted for, these bacteria have undergone less functional change than their free-living relatives. This result can be explained by greater ecological constraints on host-associated bacteria, which must adapt to the highly specific environment of their host. In particular, pathogenic and symbiotic bacteria preferentially lose metabolic genes as they no longer require the capacity to exploit as wide a range of nutrient sources as free-living bacteria [9].
Since these are precisely the kind of genes that are most amenable to functional change (Figure 1), their loss from host-associated bacteria explains the relative impoverishment of functional divergence in these proteomes. The remaining genes are also under strong constraints imposed by the specialized environment they are in, limiting therefore any opportunity for functional divergence (Toft and Fares 2008, 2009). However, variability in genome size is a complicating factor in this analysis because host-associated bacteria tend to have smaller genomes than their free-living relatives. For instance, endosymbiotic bacteria of insects underwent substantial reduction in the gene content, with genomes sizes ranging between 144 kb and 792 kb depending on the host (in comparison, E. coli K12 has a genome size of 4.639 Mb) (See for example [49], [50], [51], [52], [53], [54], [55]. Since functional divergence often follows gene duplication [1], it might be expected that larger genomes would be enriched for new functions in comparison to smaller ones.
Does genome size alone account for the observed differences between host-associated and free-living bacteria? To test this possibility, we modeled genome enrichment and impoverishment for functional divergence as a function of lifestyle (host-associated vs. free-living) and genome size (in nucleotides) using a generalized linear model, a saturated model was fit using the glm function in R (R Development Core Team, 2010), with enrichment or impoverishment for functional divergence as the response variable (binomial errors), and genome size (continuous, bp) and lifestyle (categorical, free-living or host-associated) as the explanatory variables. This was simplified to a minimal adequate model using the step function. The interaction between genome size and lifestyle was non-significant and was removed during model simplification (see Table S2). Both lifestyle and genome size were significant, with host-associated bacteria significantly more likely to be impoverished (P = 1.28×10−13) and, perhaps surprisingly, a modest tendency towards impoverishment in larger genomes (P = 0.03). Therefore, variation in genome size does not account for the observed differences in functional divergence between host-associated and free-living bacteria.
To better define the effect of lifestyle on functional divergence, we identified the functional categories with the greatest consistent differences in enrichment status between host-associated and free-living bacteria. Interestingly, genes involved in vesicular transport and secretion systems (U) were enriched for functional divergence in host-associated bacteria but neither enriched nor impoverished in free-living bacteria, while signal transduction genes (T) were impoverished in host-associated bacteria but enriched in their free-living relatives (Table S3). This pattern can be readily understood in terms of the lifestyles of host-associated bacteria, as pathogens use elaborate secretion systems for delivering toxins and other virulence factors to their host [56], while symbionts provision their hosts with nutrients as part of their mutually beneficial relationship [57], [58]. In addition, the impoverishment in host-associated signal transduction genes reflects their adaptation to a relatively constant host environment, which is considerably more stable than the fluctuating conditions experienced by their free-living relatives.
What is the relative importance of genome size variation compared to functional divergence in bacterial adaptation? This question is difficult to answer because both of these evolutionary phenomena are at work in the process of ecological bacterial adaptations. Therefore, micro-evolutionary processes, such as functional divergence, necessarily accompany macro-evolutionary processes, such as genome shrinkage or HGT, during bacteria adaptation to different ecological conditions. The timing and relative importance for each of these phenomena is, nevertheless, varied over the different stages of adapting to a new environment. Taking the example of symbiotic bacteria of insects, these bacteria are characterized by a dramatic genome streamlining, high mutation rates and the functional divergence of genes involved in an endosymbiotic lifestyle. When did these processes occur? We predict that HGT was important for the free-living ancestor of these bacteria to acquire pathogenic genes and invade the eukaryotic cells of the host. Dramatic gene loss in these invading bacteria may have become the next important evolutionary leap, making bacteria dependent upon the host. Finally, functional divergence may have contributed importantly to the refinement of the adaptation of these bacteria to the novel ecological conditions.
From proteome-wide to residue-level functional divergence
In order to visualize the results of our functional divergence analysis, we performed two-dimensional hierarchical clustering on the enrichment status (enriched, impoverished, or neither) associated with each species and functional category – that is, we clustered species according to similarities in their enrichment status across the 19 functional categories, resulting in the heatmap and dendrogram in Figure 2a (complete dendogram and heatmap is available in Figure S1). This is a powerful and intuitive way to represent our results because it reveals the overall patterns in the data – such as the extreme conservation among informational genes, particularly those involved in ribosome biogenesis (J) - while also highlighting individual, lineage-specific exceptions to the general trends. In this section, we demonstrate the utility of this approach by using the heatmap to identify species that have undergone major functional shifts.
Figure 2. Visualizing high-level patterns of functional divergence.

We used hierarchical clustering to reveal the main patterns of functional divergence in our dataset of 750 bacterial proteomes. (a) The complete heatmap, with a dendrogram corresponding to category clustering, and species clustering along the left hand side. Visualizing the data in this way reveals the extreme impoverishment of proteins involved in ribosome biogenesis (J), as well as the enrichment of categories involved in interaction with the environment (E, M, G, H, C, P) across all species. (b) Lineage-specific events of functional divergence picked out from the heatmap (dendrogram colors denote the regions expanded upon – a larger version of the complete heatmap is available as Figure S1). Unlike other Bartonella species, B. bacilliformus is impoverished for divergence in cell motility genes (N), and is unique among Bartonella species in using a flagellum to infect erythrocytes. (c) Two strains of E. coli – SMS 3–5 and UMN026 – have phylogenetically atypical patterns of functional divergence: the constraints on cell motility (N) are among those that have relaxed relative to the other strains in SMS 3–5, while UMN026 is uniquely enriched for secretion system (U) genes.
Although top-level bacterial groups (such as the divisions of the proteobacteria, the Firmicutes, Actinobacteria, and so on) are not resolved in our dendrogram of functional divergence (Figure 2), family and genus-level relationships often are, presumably because of close phylogenetic relatedness, shared gene content, and similarity of ecological niche. This allows us to identify individual species with atypical patterns of functional divergence. A particularly striking case is that of the Bartonella genus (Figure 2b), which are a group of intracellular parasites that infect and replicate in erythrocytes [59]. Of the four Bartonella species in our dataset, only one – Bartonella bacilliformus – is enriched for functional divergence in cell motility genes (N), with the others being impoverished (2 species) or neither enriched nor impoverished (1 species).
Remarkably, this is the only member of the genus that possesses flagella [60]. Since erythrocytes lack an active cytoskeleton, they cannot be induced to take up external bacteria by invagination [61]. Instead, erythrocyte invasion by Bartonella species is an active process [62]. The mechanism employed by Bartonella bacilliformus involves the use of its flagella [63] and is more efficient than that of other Bartonella species, with up to 80% of erythrocytes infected [62], [64]. This appears to be a clear case where our approach has identified an interesting, lineage-specific case of adaptation to a specialized ecological niche.
Our heatmap turns up surprises even among relatively well-characterized species (Figure 2c). As expected, closely related E. coli and Shigella strains cluster together at the bottom of the dendrogram (Figure 2a). E. coli SMS 3–5, a multidrug-resistant, heavy-metal tolerant strain isolated from a polluted industrial environment [65] is distinguished from other E. coli strains on the basis of a relaxed functional constraint in the category of cell motility (N); most others are impoverished for functional divergence, while SMS 3–5 is enriched. This profile correlates well with what is known about the biology of this strain, which is unique among sequenced E. coli genomes in possessing a second, intact lateral flagellar system called Flag-2, in addition to the normal peritrichious flagella found in other E. coli strains [65], [66]. This system was originally characterized in a different strain, 042, where it has been rendered nonfunctional by a frameshift mutation in one of the component genes [66], although it appears to be complete in SMS 3–5 [65].
Another E. coli proteome with an unusual pattern of functional divergence is O17:K52:H18 (strain UMN026), a multidrug-resistant strain that causes urinary tract infections [67]. Unique among E. coli and Shigella species, this strain is enriched for functional divergence among genes involved in secretion (U). Investigation of the genes underlying this enrichment revealed functional divergence in the VirB8 and VirB9 genes, which encode core proteins in a Type IV secretion system found only in two E. coli strains – UMN026 and 018 (ED1a), although the latter species is not enriched in this category. In other bacteria, Type IV systems are involved in the exchange of DNA with the environment, as well as the delivery of effector proteins to host cells [68]. Since these two proteins are important components of the Type IV secretion systems of other bacteria, functional divergence in these genes may be involved in adapting the system to an UMN026-specific role (see Figure 3).
Figure 3. Amino acid residues under functional divergence in E. coli UMN026 VirB8.

Right: the structure of a Type IV secretion system found only in two strains of E. coli. CM = cytoplasmic membrane, OM = outer membrane. The complex structure is based on that of Baron (2006) [84]. In UMN026, the central complex proteins VirB8 and VirB9 are under functional divergence. Left: Of the five sites detected by CAFS that could be mapped to the VirB8 crystal structure [69], there is evidence that four are involved in forming the VirB8 homodimer, suggesting that functional divergence at these positions is involved in altering the quaternary structure of the complex.
To gain further insight into the possible implications of the UMN026-specific changes in these proteins, we mapped the specific residues under functional divergence in VirB8 (also output by CAFS) onto the Agrobacterium tumefaciens crystal structure [69]. Of the 14 sites under functional divergence (see Table S4), 5 could be mapped onto the crystallized region of the protein. Of these 5, 4 are at or close to positions previously shown to be of functional importance. Thr-196(residues numbered according to the A. tumefaciens sequence), which CAFS detected as being under functional divergence in UMN026, is directly involved in the stabilization of the VirB8 homodimer [69], as is Leu-211, another functionally divergent site. Additionally two sites identified by our approach are at positions that suggest they may have an indirect role in dimerization. Val-218 is located between two other residues (Leu-217 and Val-219) that are involved in dimer formation, while Phe-127 is adjacent to Ser-128, a conserved residue that stabilizes the interaction surface on VirB8. The function of the other site detected under functional divergence, Val-183, is currently unknown. Taken together, these results indicate that functional divergence in E. coli UMN026 VirB8 has occurred at residues important in forming the homodimer, which may have important implications for the overall structure and function of the complex. With no crystal structure available for VirB9, it is more difficult to evaluate the functional significance of the sites detected there. Further, we detected functional divergence on 19 branches of the VirB9 tree, suggesting that this protein experiences a more general pattern of radical change.
Methods
Design and implementation
Our analysis of functional divergence, the individual steps of which are detailed below, is summarized in Figure 4.
Figure 4. CAFS program workflow.

After alignments have been built for each gene in the analysis, the alignments are sorted by functional category. In this case, the COG system was used [38], but any other ontology can be used as well. Trees are built for each gene using BIONJ [76] and the JTT substitution model [77], and sites are scored for functional divergence on each branch. Significance is assessed by simulating a distribution of test scores under a model of neutral evolution, taking the real phylogeny into account and using the False Discovery Rate approach to correct for multiple testing. For each species and functional category, we use chi-squared tests to evaluate whether the species is enriched or impoverished for functional divergence in that category, and then cluster species according to similarities in their profile across all 19 categories. This approach enables us to account for HGT while identifying interesting and atypical patterns of functional change in the data, as discussed in the main text.
Sequences, orthology, and alignment
The first step in a whole-proteome analysis of functional divergence is the grouping of orthologs within the species of interest. We leave orthology assignment for which a number of tools are already in use [70], to the users' choice according to their own needs. For the present analysis, we retrieved pairwise orthology assignments for 750 completely-sequenced bacterial genomes from the OMA database [71], [72], representing all bacterial data in the October 2009 revision of the database.
We chose the OMA project for its very broad phylogenetic coverage, as well as the favourable performance of its algorithm against other current orthology assignment methods [70]. In addition to providing pairwise orthology calls, the OMA algorithm assembles strict orthologous groups in which every member is directly orthologous to every other. The rationale for this strict approach to grouping is the exclusion of paralogs, which is important for a number of potential applications of the OMA database, such as phylogenetic analysis. Unfortunately, these groups are unsuitable for functional divergence analysis across large phylogenetic distances because lineage-specific gene duplications tend to break up genuine orthologs into multiple, overlapping groups (that is, clustering problems arise because pairwise orthologies are not necessarily transitive). Using these groups in our analysis would result in multiple testing of the same clade, each time with overlapping but incomplete sampling of downstream sequences. The inclusion of both orthologs and lineage-specific paralogs in the same group is, however, of no concern in our per-species comparison of divergence between different functional categories of genes, because our method relies on individual gene trees and not a single “species tree” to detect functional divergence (see below).
Therefore, we decided to build our own groups from the pairwise homology assignments in OMA, with the less stringent requirement that any sequence in a group be connected to at least one other sequence by pairwise homology. This strategy produces groups containing all orthologs and paralogs for a given gene, as appropriate for analysis of functional divergence. However, the approach is vulnerable to erroneous homology calls in the original database, because a single false call will cause two unrelated groups of sequences to be merged.
To assess the possible effect of false OMA homology assignments on our dataset, we used the relevant genomic data at NCBI to assign COG ontology tags to each sequence [38], [73]. We then calculated the frequency of the modal COG tag in each group (see Figure 5). The largest group (4,788 alignments) contained only one COG tag each, validating our approach to grouping homologs (since COG categories are relatively broad, related sequences are expected to be annotated with the same tag). To avoid ambiguity in the clustering of functional categories, we only analyzed these single-tag alignments. We then filtered out poorly-characterized groups (annotated with the ambiguous R or S COG categories) and any group containing less than 9 sequences (one outgroup and 4 sequences downstream of the inner nodes), which we chose as the minimum number required for analysis (both as a requirement for stringency and also comparison to similar software Gu 1999). The final dataset comprised 3,813 groups, which were then analyzed with our novel approach (CAFS: Clustering Analysis of Functional Shifts). Other functional classifications such as Gene Ontology (GO) can also be used. However, caution is required because GO contains overlapping categories and alignments with multiple tags can lead to ambiguous results.
Figure 5. Number of COG assignments (tags) for each group of homologous sequences.

We included both orthologs and paralogs in our sequence groups, because we are interested in functional divergence. The majority of groups consisted of sequences that had all been assigned to the same COG category, suggesting our grouping strategy did not lump together unrelated sequences due to spurious orthology calls. Since COG categories are relatively broad, we do not generally expect functional divergence to cause a sequence to shift from one category to another, an hypothesis that is also borne out by the clustering of related sequences within the same category. In our study, to avoid ambiguity we only use alignments in the group with a single COG tag.
Sequence alignments were built for each group with MUSCLE [74], using the default parameters. Data on the ecological niches occupied by the species included in the analysis was retrieved from HAMAP [75] and from the Genome database at NCBI.
A typical alignment of 78 sequences takes 2 minutes and 40 seconds to analyze for functional divergence using CAFS on a standard desktop computer, including NJ tree-building. At the other extreme, the large-scale analysis reported below (44,416 tests of functional divergence/3,813 alignments) took 92 hours on a 40-node cluster.
Building gene trees
When analyzing entire proteomes for functional divergence, the use of a species tree to infer events on each branch is problematic: extensive horizontal gene transfer (HGT), particularly among prokaryotes, means that genomes may not be related in a tree-like way [21]. We therefore calculated a tree for each gene (set of homologous sequences) in the dataset using BIONJ [76] (see Text S1 and Table S6 for a justification of the use of BIONJ and comparison with the use of maximum-likelihood trees), under the JTT model of protein sequence evolution [77] along with a gamma distribution(n = 4, alpha = 1.0) to correct for among sites variation of evolutionary rates(a fixed alpha value was used because of the time constraints involved in assessing alpha and other parameters for all alignments). Calculations for that gene were then made exclusively using the resulting tree.
Scoring functional divergence
We here define functional divergence as the potential departure of the derived protein function from its ancestral one as a result of amino acid changes at important functional sites. Therefore functional divergence is detected on the basis of shifts in substitutution rates per amino acid site in proteins. This analysis of functional divergence can be used to provide a list of candidate genes for further experimental testing. Our method identifies amino acid sites within a protein, which have radical substitutions between clades and are statistically significant. This is carried out in each of the lineages of a tree, with each lineage being a cluster of 4 or more sequences.
The method steps through the phylogenetic tree and calculates functional divergence scores at each of the inner nodes. For each site of the protein, our approach compares the amino acid composition between two clades to that of an outgroup. This comparison is performed using BLOSUM62 amino acid substitution matrix [78], indeed any substitution matrix can be used. BLOSUM62 and related matrices provide an empirical measure of the likelihood of the transition of one amino acid to any of the other 20 (including its conservation). Scores of functional divergence (FDscore) for each column are given by:
| (1) |
where 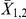 are the mean substitution scores for the transition from clades on either side of the bifurcation in the phylogenetic tree relative to the outgroup and
are the mean substitution scores for the transition from clades on either side of the bifurcation in the phylogenetic tree relative to the outgroup and 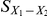 , the standard error for unequal sample sizes with unequal variances, is given by:
, the standard error for unequal sample sizes with unequal variances, is given by:
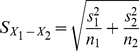 |
(2) |
Significance testing
To test the significance of functional divergence events, we simulated multiple sequence alignments of the same size as the real alignment but in which proteins were evolved under a neutral evolution model. Because functional divergence was tested in protein alignments, the seed ancestral sequence was protein based and this evolved under the JTT model.
For our simulations, we used the gene-specific tree topology and branch lengths calculated above. We built at least 1000 such simulated alignments (more simulations are created if mean and standard deviation have not converged within a difference of 1*10−6 after 1000), in each of which we searched for functional divergence and calculated a score according to equation (1). To allow for the gaps in the sequences we simulated alignments with the number of columns equal to the length of the input alignment minus the average number of gaps in each species. This search resulted in a null distribution of the test score against which P-values for the real data were calculated. These values were then corrected for multiple testing by the False Discovery Rate method [79] using an alpha value of 5% as the threshold of significance. Following this procedure, branches on the tree that still possess at least one significant amino acid site were considered to be under functional divergence for the purposes of enrichment and clustering.
Enrichment analysis
Once all alignments were analyzed, we performed three different enrichment tests to ask three different biological questions. These are based on a chi-squared test:
| (3) |
Where Oi is the observed frequency of genes/alignments under functional divergence, Ei is the expected frequency and n is the number of possible outcomes of each event. We used the enrichment tests to identify (i) species and (ii) categories of genes that experienced significantly more (enriched: Oi – Ei>0) or significantly less (impoverished: Oi – Ei<0) functional divergence when compared to the background level (that is, P<0.05 in a chi-squared test). We then calculated (iii) the enrichment status of each category within each species, in order to identify lineage-specific shifts in the pattern of functional divergence. It should be noted that the chi-squared test used scales for the size of each of the groupings considered in our results.
Hierarchical clustering
We created a heatmap from the enrichment status of functional categories within species to help visualize the structure in our large dataset. To do this we used the heatmap.2 function from the gplots library in R (R Development Core Team, 2010). This function performs two-dimensional hierarchical clustering according to Euclidean distance and outputs a heatmap together with a corresponding dendrogram. Visualizing the results of the analysis in this way allows identifying unusual patterns of functional divergence in particular functional categories or convergent functional divergence among phylogenetically unrelated sequences.
Implementation
CAFS was implemented in C++ and is available under the GNU General Public License v.3 for Linux, Mac and Windows. The code was written using the GNU Scientific Library and the Bio++ libraries [80]. The program is accompanied by full documentation and enables the user to perform several different kinds of analyses, including the identification of lineage-specific functional divergence in a gene-of-interest (such as that reported by Williams et al. (2010) [37]) and the kind of multi-proteome investigation reported here. The latest version of the code and documentation is available at http://bioinf.gen.tcd.ie/~faresm/software/software.html.
Comparison to other applications and assessment of Error
As mentioned previously many programs have been developed to predict putative sites of functional divergence in a multiple sequence alignment. Of those, the most commonly used method is DIVERGE [81], therefore we used this as a benchmark. We found that our method could analyze large alignments, automate proteome scale analyses and performs analyses on tagging systems which DIVERGE does not (Also see Table S7, Text S2, Table S8 and Text S3 for a detailed comparison of our method with Diverge). Our method also performs simulations, which scales the cutoff value for each alignment, this scaling allows us to analyze all homologs and hence is not limited to orthologs. Our program automates the building of trees with a more detailed model of evolution and better tree building algorithm. Given that the tree building algorithm is distance based we performed a comparison to trees built using RaxML [82], 92% of the sites identified using the distance based trees were identical to those using the maximum likelihood method. Given the differences between our software and that of DIVERGE it is very difficult to make comparisons in terms of the sites reported by each program. DIVERGE compares two clades after a duplication event and our software compares two clades and an outgroup. We do so in order to assess events of functional divergence at any testable node on a tree and also because we feel it provides stronger evidence of functional divergence. Another widely used software to identify the strength of selection in protein-coding genes is PAML. This package employs the programs yn00 and codeml for testing the Dn/Ds ratio. This means PAML works on nucleotide level while CAFS uses protein sequences, which gives it the ability to assess divergences of greater magnitude. An additional feature of our program is the ability to easily automate and carry out large analyses, as shown in this study. Another philosophical difference between both approaches is that, to detect positive selection, using programs like PAML a strong signal is required, which would make it difficult to detect episodic positive selection. CAFS only requires an amino acid substitution fixed by positive selection in a functionally important region of the protein followed by strong purifying selection.
Given the difficulty of finding definitive positive controls for an analysis of this nature we felt it important to demonstrate that there would not be a large false positive rate. We simulated 20 alignments under a codon model with neutral evolution (non-synonymous, Ka, to synonymous, Ks, rates ratio ω = Ka/Ks = 1) using the evolver package in PAML [83]. These alignments were converted to amino acids and analysed with CAFS under the default alpha value of 0.05, with this value the expectation would be 5% of sites being reported as functionally divergent. Our software reported an average of 2.3% of sites as functionally divergent. Given this percentage we are confident that our methodology of significance testing and implementation of false discover rate is not susceptible to a large number of false positives. Further details of this section can be found in Text S1 and S2 and Tables S6 and S7.
In conclusion, The identification of functional divergence and ecological adaptation from sequence data is an interesting and important goal in evolutionary biology, with the potential to deepen our understanding of the evolution of individual traits and species, as well as the processes of evolution as a whole. Bacteria display an astonishing capacity for adaptation to different lifestyles and ecological niches, but investigating the evolution of these traits is problematic because their phylogenetic context is often unclear.
Here, we have circumvented this problem by evaluating functional divergence on gene trees and then clustering species by gene functional category. Our approach revealed the overall patterns that have characterized functional change during the evolution of bacteria, including strong constraint on information storage and processing genes and also constraints induced by the host on pathogenic and symbiotic bacteria. It also identified lineage-specific events of atypical functional divergence, such as the use of flagella by Bartonella bacilliformus to invade host erythrocytes and residue-level changes in the VirB8 protein of E. coli UMN026. This is, to our knowledge, the first method that can be used to identify functional divergence at the level of entire proteomes. Although used here to perform a large-scale analysis on bacteria with the use of COG categories, our CAFS software is extremely flexible and can be applied to individual genes, lineages, or groups of proteomes using any ontology system in order to investigate functional divergence at every level of biological organization.
Supporting Information
Visualizing high-level patterns of functional divergence. We used hierarchical clustering to reveal the main patterns of functional divergence in our dataset of 750 bacterial proteomes. The complete heatmap, with a dendrogram corresponding to category clustering, and species clustering along the left hand side. Visualizing the data in this way reveals the extreme impoverishment of proteins involved in ribosome biogenesis (J), as well as the enrichment of categories involved in interaction with the environment (E, M, G, H, C, P) across all species.
(TIFF)
Enrichment of COG gene categories for functional divergence. The annotation for each COG category was retrieved from http://www.ncbi.nlm.nih.gov/COG/. Enrichment was evaluated with Chi-squared tests. “Not enriched” indicates there was no significant association between the number of lineages under functional divergence and the COG category. Impoverishment and Enrichment denotes the direction of a significant association.
(DOCX)
Enrichment status of 750 bacterial species. Enrichment status was calculated with a Chi-squared test based on the number of testable branches on our gene trees where a particular species was under functional divergence.
(DOCX)
Effect of bacterial lifestyle and genome size on functional divergence. We used a generalized linear model with binomial errors to assess the impact of lifestyle and genome size on the enrichment and impoverishment of genomes for functional divergence. The saturated model was fit with the glm function in R, and simplified to a minimal adequate model with the step function, which determined that the interaction was not significant. Both lifestyle and genome size have a significant impact on enrichment status, with host-associated bacteria and bacteria with larger genomes more likely to be impoverished for functional divergence.
(DOCX)
Enrichment status of gene categories in host-associated and free-living bacteria. Categories U and T show different levels of enrichment for functional divergence when the analysis is run on these groups of bacteria independently.
(DOCX)
Sites under functional divergence in VirB8. The left column shows the sites found in E.coli UMN026, the sites on the right show the homologous sites in Agrobacterium tumefaciens. Sites without a value for Agrobacterium tumefaciens represent sites, which have not been crystalised.
(DOCX)
Detecting functional divergence using BioNJ trees. The number of FD sites predicted at the 0.05 p-value level using BioNJ trees is consistently smaller. This indicates a stricter scoring scheme, which potentially reduces false positives. It can be seen from the last column, that the percentage of the FD sites detected through BioNJ trees that are also detected through ML trees increases with larger number of sequences in the alignment and approaches 100%. Only the smaller alignments show noticeable discrepancies but some of these can already be explained by the effect of different tree topologies. Overall, the above results confirm the suitability of BioNJ for tree construction, particularly for alignments with a large number of sequences.
(DOCX)
Comparison of runtime for two methods of functional divergence. The most widely used program for detection of functional divergence is DIVERGE [81]. Even though it is well-suited for individual analyses, it can not be used for a large-scale study such as the one presented here. This is because the size of alignments dealt with exceeds the limits of the data that DIVERGE can handle. It is also not designed to be run automatically.
(DOCX)
Functionalities of CAFS in comparison with DIVERGE.
(DOCX)
Justification for use of BioNJ and Comparison of results performed with Maximum Likelihood trees vs BIONJ trees.
(DOCX)
A Note about testable alignments and computation time for functional divergence detection. It should be noted that since DIVERGE could not run the 4 largest alignments in this subset of our dataset, we predict that at least half of the alignments in our full dataset can not be analysed by the DIVERGE software. Indeed we were unable to run analysis on any alignment over 86 sequences long, however DIVERGE would read alignments up to 100 sequences long. Another further problem about the calculations performed with DIVERGE2.0 is the impossibility to perform analyses collected in Gu2001 which pertains to the method in [81]. This analysis did not work for any of the alignments above. All alignments failed with the error “Please recheck input sequence data and tree information”, for which we could not find documentation. In addition to the times stated above we would like to add that DIVERGE works on an alignment by alignment basis and as such the user must manually chose files to be analysed and also the trees to be input and the nodes to be tested. This makes proteome level analyses prohibitive. The runtimes given in the table above do not include the time it takes for the user to choose nodes for functional divergence testing with DIVERGE.
(DOCX)
Conclusion of Comparisons of the methods to identify functional divergence. We conclude that whilst we understand and acknowledge the value of building maximum likelihood trees we feel that one of the biggest assetts of our program is speed and automation. With this in mind, we wish to demonstrate to the user that whilst BioNJ trees are calculated within the program the results recieved are ∼92% comparable to those returned after the maximum likelihood analysis. Given the high level of similarity between the maximum likelihood built trees and those built with BioNJ we feel that the conclusions drawn in the manuscript would hold in either circumstance. We also feel that analyses on the scale of those detailed in the main text are not possible for many who have limited computational resources. In reference to the DIVERGE comparisons we feel justified in our calculations that DIVERGE is not ideal for large scale analyses and can be troublesome to the user for even relatively small alignments. In addition to the tests run above we would like to say that on large scale analyses all of the information about the sites tested are collected and automatically analysed according to any tagging system applied to the dataset.
(DOCX)
Acknowledgments
We would like to acknowledge all members of Fares Lab and from the Department of Genetics in TCD for their constructive criticism to the manuscript.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This study was supported by a grant from the Spanish Ministerio de Ciencia e Inovación (BFU2009-12022) and a grant of the Research Frontiers Program (10/RFP/GEN2685) from Science Foundation Ireland. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 2.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 3.Schluter D. The ecology of adaptive radiation. New York: Oxford University Press; 2000. [Google Scholar]
- 4.Gillespie JH. Population genetics: a concise guide. Johns Hopkins University Press; 2004. [Google Scholar]
- 5.Pinto G, Mahler DL, Harmon LJ, Losos JB. Testing the island effect in adaptive radiation: rates and patterns of morphological diversification in Caribbean and mainland Anolis lizards. Proc Biol Sci. 2008;275:2749–2757. doi: 10.1098/rspb.2008.0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohno S. Evolution by gene duplication. Springer Verlag; 1970. [Google Scholar]
- 7.Lynch M, Katju V. The altered evolutionary trajectories of gene duplicates. Trends Genet. 2004;20:544–549. doi: 10.1016/j.tig.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nature Reviews Genetics. 2010;11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 9.Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002;108:583–586. doi: 10.1016/s0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- 10.Williams TA, Codoñer FM, Toft C, Fares MA. Two chaperonin systems in bacterial genomes with distinct ecological roles. Trends in Genetics. 2009 doi: 10.1016/j.tig.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Toft C, Williams TA, Fares MA. Genome-wide functional divergence after the symbiosis of proteobacteria with insects unraveled through a novel computational approach. PLoS Comput Biol. 2009;5:e1000344. doi: 10.1371/journal.pcbi.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dykhuizen DE. Santa Rosalia revisited: why are there so many species of bacteria? Antonie Van Leeuwenhoek. 1998;73:25–33. doi: 10.1023/a:1000665216662. [DOI] [PubMed] [Google Scholar]
- 13.Gans J, Wolinsky M, Dunbar J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science. 2005;309:1387–1390. doi: 10.1126/science.1112665. [DOI] [PubMed] [Google Scholar]
- 14.Pikuta EV, Hoover RB, Tang J. Microbial extremophiles at the limits of life. Crit Rev Microbiol. 2007;33:183–209. doi: 10.1080/10408410701451948. [DOI] [PubMed] [Google Scholar]
- 15.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 16.Dyall SD, Brown MT, Johnson PJ. Ancient invasions: from endosymbionts to organelles. Science. 2004;304:253–257. doi: 10.1126/science.1094884. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–298. [Google Scholar]
- 18.Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 19.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 20.McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. The SOS response regulates adaptive mutation. Proc Natl Acad Sci U S A. 2000;97:6646–6651. doi: 10.1073/pnas.120161797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dagan T, Martin W. The tree of one percent. Genome Biology. 2006;7:118–118. doi: 10.1186/gb-2006-7-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura M. The neutral theory of molecular evolution. Cambridge University Press; 1983. [Google Scholar]
- 23.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Molecular Biology and Evolution. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki Y, Gojobori T. A method for detecting positive selection at single amino acid sites. Molecular Biology and Evolution. 1999;16:1315–1328. doi: 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Molecular Biology and Evolution. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 28.Fares MA, Elena SF, Ortiz J, Moya A, Barrio E. A sliding window-based method to detect selective constraints in protein-coding genes and its application to RNA viruses. J Mol Evol. 2002;55:509–521. doi: 10.1007/s00239-002-2346-9. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki Y. New methods for detecting positive selection at single amino acid sites. J Mol Evol. 2004;59:11–19. doi: 10.1007/s00239-004-2599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J. Frequent false detection of positive selection by the likelihood method with branch-site models. Molecular Biology and Evolution. 2004;21:1332–1339. doi: 10.1093/molbev/msh117. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki Y. Three-dimensional window analysis for detecting positive selection at structural regions of proteins. Molecular Biology and Evolution. 2004;21:2352–2359. doi: 10.1093/molbev/msh249. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology and Evolution. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 33.Berglund AC, Wallner B, Elofsson A, Liberles DA. Tertiary windowing to detect positive diversifying selection. J Mol Evol. 2005;60:499–504. doi: 10.1007/s00239-004-0223-4. [DOI] [PubMed] [Google Scholar]
- 34.Gu X. Statistical methods for testing functional divergence after gene duplication. Molecular Biology and Evolution. 1999;16:1664–1674. doi: 10.1093/oxfordjournals.molbev.a026080. [DOI] [PubMed] [Google Scholar]
- 35.Gu X. Mathematical modeling for functional divergence after gene duplication. J Comput Biol. 2001;8:221–234. doi: 10.1089/10665270152530827. [DOI] [PubMed] [Google Scholar]
- 36.Gu X. A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Molecular Biology and Evolution. 2006;23:1937–1945. doi: 10.1093/molbev/msl056. [DOI] [PubMed] [Google Scholar]
- 37.Williams TA, Codoner FM, Toft C, Fares MA. Two chaperonin systems in bacterial genomes with distinct ecological roles. Trends Genet. 2010;26:47–51. doi: 10.1016/j.tig.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, et al. Comparative genomics of the Archaea (Euryarchaeota): evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–628. [PubMed] [Google Scholar]
- 40.Lake JA, Jain R, Rivera MC. Mix and match in the tree of life. Science. 1999;283:2027–2028. doi: 10.1126/science.283.5410.2027. [DOI] [PubMed] [Google Scholar]
- 41.Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci U S A. 1996;93:10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azuma Y, Ota M. An evaluation of minimal cellular functions to sustain a bacterial cell. BMC Syst Biol. 2009;3:111. doi: 10.1186/1752-0509-3-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crick FH. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 44.Lund PA. Multiple chaperonins in bacteria–why so many? FEMS Microbiol Rev. 2009;33:785–800. doi: 10.1111/j.1574-6976.2009.00178.x. [DOI] [PubMed] [Google Scholar]
- 45.Kampinga HHDJ, Dikoney E. Mechanism of radiosensitization by hyperthermia (>or = 43 degrees C) as derived from studies with DNA repair defective mutant cell lines. International Journal of Hyperthermia. 2004:131–139. doi: 10.1080/02656730310001627713. [DOI] [PubMed] [Google Scholar]
- 46.Laszlo A. The effects of hyperthermia on mammalian cell structure and function. Cellular Proliferation. 1992:59–87. doi: 10.1111/j.1365-2184.1992.tb01482.x. [DOI] [PubMed] [Google Scholar]
- 47.Kregel K. Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. Journal of Applied Physiology. 2002:2177–2186. doi: 10.1152/japplphysiol.01267.2001. [DOI] [PubMed] [Google Scholar]
- 48.Lepock J. Protein denaturation during heat shock. Advances in Molecular Cell Biology. 1997:223–259. [Google Scholar]
- 49.Degnan PH, Lazarus AB, Wernegreen JJ. Genome sequence of Blochmannia pennsylvanicus indicates parallel evolutionary trends among bacterial mutualists of insects. Genome Res. 2005;15:1023–1033. doi: 10.1101/gr.3771305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gil R, Sabater-Munoz B, Latorre A, Silva FJ, Moya A. Extreme genome reduction in Buchnera spp.: toward the minimal genome needed for symbiotic life. Proc Natl Acad Sci U S A. 2002;99:4454–4458. doi: 10.1073/pnas.062067299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Brocal V, Gil R, Ramos S, Lamelas A, Postigo M, et al. A small microbial genome: the end of a long symbiotic relationship? Science. 2006;314:312–313. doi: 10.1126/science.1130441. [DOI] [PubMed] [Google Scholar]
- 52.Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- 53.Tamas I, Klasson L, Canback B, Naslund AK, Eriksson AS, et al. 50 million years of genomic stasis in endosymbiotic bacteria. Science. 2002;296:2376–2379. doi: 10.1126/science.1071278. [DOI] [PubMed] [Google Scholar]
- 54.van Ham RC, Kamerbeek J, Palacios C, Rausell C, Abascal F, et al. Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci U S A. 2003;100:581–586. doi: 10.1073/pnas.0235981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- 56.Baron C. Antivirulence drugs to target bacterial secretion systems. Curr Opin Microbiol. 2010;13:100–105. doi: 10.1016/j.mib.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 57.Douglas AE. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu Rev Entomol. 1998;43:17–37. doi: 10.1146/annurev.ento.43.1.17. [DOI] [PubMed] [Google Scholar]
- 58.Sandstrom J, Telang A, Moran NA. Nutritional enhancement of host plants by aphids - a comparison of three aphid species on grasses. J Insect Physiol. 2000;46:33–40. doi: 10.1016/s0022-1910(99)00098-0. [DOI] [PubMed] [Google Scholar]
- 59.Anderson BE, Neuman MA. Bartonella spp. as emerging human pathogens. Clin Microbiol Rev. 1997;10:203–219. doi: 10.1128/cmr.10.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brenner DJ, O'Connor SP, Hollis DG, Weaver RE, Steigerwalt AG. Molecular characterization and proposal of a neotype strain for Bartonella bacilliformis. J Clin Microbiol. 1991;29:1299–1302. doi: 10.1128/jcm.29.7.1299-1302.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dramsi S, Cossart P. Intracellular pathogens and the actin cytoskeleton. Annu Rev Cell Dev Biol. 1998;14:137–166. doi: 10.1146/annurev.cellbio.14.1.137. [DOI] [PubMed] [Google Scholar]
- 62.Dehio C. Bartonella interactions with endothelial cells and erythrocytes. Trends Microbiol. 2001;9:279–285. doi: 10.1016/s0966-842x(01)02047-9. [DOI] [PubMed] [Google Scholar]
- 63.Scherer DC, DeBuron-Connors I, Minnick MF. Characterization of Bartonella bacilliformis flagella and effect of antiflagellin antibodies on invasion of human erythrocytes. Infect Immun. 1993;61:4962–4971. doi: 10.1128/iai.61.12.4962-4971.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ihler GM. Bartonella bacilliformis: dangerous pathogen slowly emerging from deep background. FEMS Microbiol Lett. 1996;144:1–11. doi: 10.1111/j.1574-6968.1996.tb08501.x. [DOI] [PubMed] [Google Scholar]
- 65.Fricke WF, Wright MS, Lindell AH, Harkins DM, Baker-Austin C, et al. Insights into the environmental resistance gene pool from the genome sequence of the multidrug-resistant environmental isolate Escherichia coli SMS-3–5. J Bacteriol. 2008;190:6779–6794. doi: 10.1128/JB.00661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren CP, Beatson SA, Parkhill J, Pallen MJ. The Flag-2 locus, an ancestral gene cluster, is potentially associated with a novel flagellar system from Escherichia coli. J Bacteriol. 2005;187:1430–1440. doi: 10.1128/JB.187.4.1430-1440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manges AR, Johnson JR, Foxman B, O'Bryan TT, Fullerton KE, et al. Widespread distribution of urinary tract infections caused by a multidrug-resistant Escherichia coli clonal group. N Engl J Med. 2001;345:1007–1013. doi: 10.1056/NEJMoa011265. [DOI] [PubMed] [Google Scholar]
- 68.Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Microbiol. 2003;1:137–149. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bailey S, Ward D, Middleton R, Grossmann JG, Zambryski PC. Agrobacterium tumefaciens VirB8 structure reveals potential protein-protein interaction sites. Proc Natl Acad Sci U S A. 2006;103:2582–2587. doi: 10.1073/pnas.0511216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Computational Biology. 2009;5:e1000262–e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roth A, Gonnet G, Dessimoz C. Algorithm of OMA for large-scale orthology inference. BMC bioinformatics. 2008;9:518–518. doi: 10.1186/1471-2105-9-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider A, Dessimoz C, Gonnet G. OMA Browser Exploring orthologous relations across 352 complete genomes. Bioinformatics. 2007;23:2180–2182. doi: 10.1093/bioinformatics/btm295. [DOI] [PubMed] [Google Scholar]
- 73.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics. 2004 doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lima T, Auchincloss AH, Coudert E, Keller G, Michoud K, et al. HAMAP: a database of completely sequenced microbial proteome sets and manually curated microbial protein families in UniProtKB/Swiss-Prot. Nucleic Acids Res. 2009;37:D471–478. doi: 10.1093/nar/gkn661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gascuel O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Molecular Biology and Evolution. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 77.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences: CABIOS. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 78.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences. 1992 doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 80.Dutheil J, Gaillard S, Bazin E, Glemin S, Ranwez V, et al. Bio++: a set of C++ libraries for sequence analysis, phylogenetics, molecular evolution and population genetics. BMC Bioinformatics. 2006;7:188–188. doi: 10.1186/1471-2105-7-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gu X, Vander Velden K. DIVERGE: phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics. 2002;18:500–501. doi: 10.1093/bioinformatics/18.3.500. [DOI] [PubMed] [Google Scholar]
- 82.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- 83.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 84.Baron C. VirB8: a conserved type IV secretion system assembly factor and drug target. Biochem Cell Biol. 2006;84:890–899. doi: 10.1139/o06-148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Visualizing high-level patterns of functional divergence. We used hierarchical clustering to reveal the main patterns of functional divergence in our dataset of 750 bacterial proteomes. The complete heatmap, with a dendrogram corresponding to category clustering, and species clustering along the left hand side. Visualizing the data in this way reveals the extreme impoverishment of proteins involved in ribosome biogenesis (J), as well as the enrichment of categories involved in interaction with the environment (E, M, G, H, C, P) across all species.
(TIFF)
Enrichment of COG gene categories for functional divergence. The annotation for each COG category was retrieved from http://www.ncbi.nlm.nih.gov/COG/. Enrichment was evaluated with Chi-squared tests. “Not enriched” indicates there was no significant association between the number of lineages under functional divergence and the COG category. Impoverishment and Enrichment denotes the direction of a significant association.
(DOCX)
Enrichment status of 750 bacterial species. Enrichment status was calculated with a Chi-squared test based on the number of testable branches on our gene trees where a particular species was under functional divergence.
(DOCX)
Effect of bacterial lifestyle and genome size on functional divergence. We used a generalized linear model with binomial errors to assess the impact of lifestyle and genome size on the enrichment and impoverishment of genomes for functional divergence. The saturated model was fit with the glm function in R, and simplified to a minimal adequate model with the step function, which determined that the interaction was not significant. Both lifestyle and genome size have a significant impact on enrichment status, with host-associated bacteria and bacteria with larger genomes more likely to be impoverished for functional divergence.
(DOCX)
Enrichment status of gene categories in host-associated and free-living bacteria. Categories U and T show different levels of enrichment for functional divergence when the analysis is run on these groups of bacteria independently.
(DOCX)
Sites under functional divergence in VirB8. The left column shows the sites found in E.coli UMN026, the sites on the right show the homologous sites in Agrobacterium tumefaciens. Sites without a value for Agrobacterium tumefaciens represent sites, which have not been crystalised.
(DOCX)
Detecting functional divergence using BioNJ trees. The number of FD sites predicted at the 0.05 p-value level using BioNJ trees is consistently smaller. This indicates a stricter scoring scheme, which potentially reduces false positives. It can be seen from the last column, that the percentage of the FD sites detected through BioNJ trees that are also detected through ML trees increases with larger number of sequences in the alignment and approaches 100%. Only the smaller alignments show noticeable discrepancies but some of these can already be explained by the effect of different tree topologies. Overall, the above results confirm the suitability of BioNJ for tree construction, particularly for alignments with a large number of sequences.
(DOCX)
Comparison of runtime for two methods of functional divergence. The most widely used program for detection of functional divergence is DIVERGE [81]. Even though it is well-suited for individual analyses, it can not be used for a large-scale study such as the one presented here. This is because the size of alignments dealt with exceeds the limits of the data that DIVERGE can handle. It is also not designed to be run automatically.
(DOCX)
Functionalities of CAFS in comparison with DIVERGE.
(DOCX)
Justification for use of BioNJ and Comparison of results performed with Maximum Likelihood trees vs BIONJ trees.
(DOCX)
A Note about testable alignments and computation time for functional divergence detection. It should be noted that since DIVERGE could not run the 4 largest alignments in this subset of our dataset, we predict that at least half of the alignments in our full dataset can not be analysed by the DIVERGE software. Indeed we were unable to run analysis on any alignment over 86 sequences long, however DIVERGE would read alignments up to 100 sequences long. Another further problem about the calculations performed with DIVERGE2.0 is the impossibility to perform analyses collected in Gu2001 which pertains to the method in [81]. This analysis did not work for any of the alignments above. All alignments failed with the error “Please recheck input sequence data and tree information”, for which we could not find documentation. In addition to the times stated above we would like to add that DIVERGE works on an alignment by alignment basis and as such the user must manually chose files to be analysed and also the trees to be input and the nodes to be tested. This makes proteome level analyses prohibitive. The runtimes given in the table above do not include the time it takes for the user to choose nodes for functional divergence testing with DIVERGE.
(DOCX)
Conclusion of Comparisons of the methods to identify functional divergence. We conclude that whilst we understand and acknowledge the value of building maximum likelihood trees we feel that one of the biggest assetts of our program is speed and automation. With this in mind, we wish to demonstrate to the user that whilst BioNJ trees are calculated within the program the results recieved are ∼92% comparable to those returned after the maximum likelihood analysis. Given the high level of similarity between the maximum likelihood built trees and those built with BioNJ we feel that the conclusions drawn in the manuscript would hold in either circumstance. We also feel that analyses on the scale of those detailed in the main text are not possible for many who have limited computational resources. In reference to the DIVERGE comparisons we feel justified in our calculations that DIVERGE is not ideal for large scale analyses and can be troublesome to the user for even relatively small alignments. In addition to the tests run above we would like to say that on large scale analyses all of the information about the sites tested are collected and automatically analysed according to any tagging system applied to the dataset.
(DOCX)