

Summary
Scaffold proteins play pivotal roles in the regulation of signal transduction pathways by connecting upstream receptors to downstream effector molecules. During last decade, many scaffold proteins that contain caspase-recruitment domain (CARD) have been identified. Investigating the roles of CARD proteins has revealed that many play crucial roles in signaling cascades that lead to activation of nuclear factor-κB (NF-κB). In this review, we discuss the contributions of CARD proteins to NF-κB activation in various signaling cascades. In particular, we share some of our personal experiences during the initial investigation of the functions of the CARMA family of CARD proteins and then summarize the roles of these proteins in signaling pathways induced by antigen receptors, G protein-coupled receptors, receptor tyrosine kinase, and C-type lectin receptors in the context of recent progress in the field.
Keywords: signal transduction, adapter proteins, signaling proteins
Introduction: overview of CARD proteins
Intracellular signal transduction pathways contain many scaffold/adapter and effector proteins, which ensure the transmission of extracellular stimulation signals to specific multi-protein complexes or subcellular compartments in a timely manner. Scaffold proteins are defined as molecules that do not have any enzymatic activity but have ability to bind to at least two other signaling proteins and assemble various multi-protein complexes that are necessary for integrating and or transmitting signals from cell surface receptors (1). In most cases, scaffold proteins help to localize signaling molecules to specific subcellular compartments and serve as platforms for co-localizing enzymes and their substrates, preventing the non-specific access of enzymes to unwanted substrates, and protecting from undesirable cellular effects.
CARD(caspase-recruitment domain)-containing scaffold/adapter proteins, also known as CARD proteins, play very important roles in the regulation of signaling cascades, the best characterized being the activation of apoptosis. The name ‘CARD’ is based on the alignment of a structural domain originally found in many proteins such as caspase-1, -2, -8, -9, cIAP-1, cIAP-2, EHV-E10, Ced-3, and Ced-4, which were found to be involved in apoptotic signaling (2). The CARD domain consists of six α-helices in the predicted secondary structure, which is evolutionally conserved among different species and is involved in protein-protein interaction through homo- or hetero-dimerization, thereby transmitting or amplifying intracellular signals to the downstream effector molecules.
Many CARD proteins were initially identified through bioinformatics approaches (3–13). As more and more CARD proteins were identified, however, it was found that CARD proteins are not only involved in apoptotic signaling but also function as scaffold molecules in other signaling pathways, including pathways that activate nuclear factor-κB (NF-κB) and induce inflammatory responses (3–13). Based on the year identified, tissue distribution, binding partners, and functions, a list of CARD proteins that regulate NF-κB signaling is summarized in Table 1, and the schematic structure of these CARD proteins is presented in Fig. 1.
Table 1.
CARD proteins that regulate NF-κB activation
| CARD proteins | Year identified | Binding partners | Tissue distribution | Functions |
|---|---|---|---|---|
| Carma1 | 2001 | Bcl10, Malt1, NEMO, | Hematopoietic cells | TCR and BCR induced NF-κB activation |
| Carma3 | 2001 | Bcl10, Malt1, NEMO, β-arrestin | Non-hematopoietic cells | GPCRs and RTKs induced NF-kB activation |
| CARD9 | 2000 | Bcl10, Malt1, NEMO | Myeloid phagocytes | Dectin-1/2-induced NF-kB activation |
| Bcl10 | 1999 | CARMA1/3, CARD9, Malt1 | Ubiquitous | TCR, BCR, GPCR, RTKs, Dectin induced NF-kB activation |
| cIAP-1 | 1995 | TRAF2, TNFR, RIP2 | Ubiquitous | NOD1/2 or TNFα-induced NF-kB activation |
| cIAP-2 | 1995 | TRAF2, TNFR, RIP2 | Ubiquitous | NOD1/2 or TNFα-induced NF-kB activation |
| RIP-2 | 1998 | cIAP-1, cIAP-2, TRAF-1, -5, -6, | Ubiquitous | NOD1/2 induced NF-kB activation |
| NOD-1 | 1999 | RIP2 | Ubiquitous | NOD1 induced NF-kB activation |
| NOD-2 | 2001 | RIP2 | Antigen-presenting cells and epithelial cells | NOD2 induced NF-κactivation |
| RIG-I | 1997 | MAVS, TRIM25 | Inducible | Viral infection induced NF-κB activation |
| MDA-5 | 1999 | MAVS | Inducible | Viral infection induced NF-κB activation |
| MAVS | 2005 | RIG-I, MDA-5, FADD, RIP1 | Ubiquitous | Viral infection induced NF-κB activation |
Fig. 1. Schematic representation of the structural domains of CARD proteins.
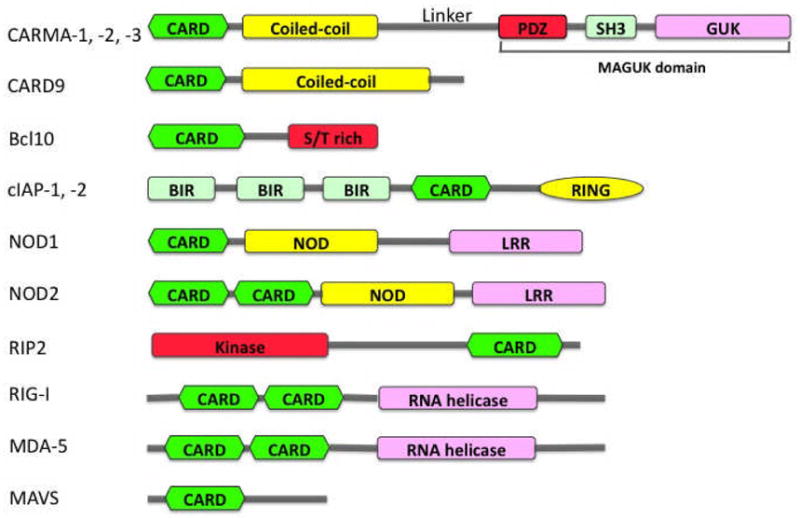
CARD - caspase recruitment domain; MAGUK - membrane-associated guanylate kinase-like domain; BIR - baculovirus IAP repeat domain; NOD - nucleotide-binding oligomerization domain; LRR – Leucine-rich repeat domain; RING – Really-Interesting-New-Gene domain; S/T rich – Ser/Thr rich domain.
CARMA1, CARMA2, and CARMA3 were initially identified based on their CARD domain by bioinformatics approaches and named CARD11 (known as CARMA1), CARD14 (known as CARMA2 or Bimp2), and CARD10 (known as CARMA3 or Bimp1) (12–14). They all contain an N-terminal CARD domain, following with a coiled-coil (CC) domain, a linker, a PDZ domain, a SH3 domain, and a C-terminal guanylate kinase-like (GUK) domain (Fig. 1). The structural module of PDZ-SH3-GUK is also known as membrane-associated GUK (MAGUK) domain. CARMA1, CARMA2, and CARMA3 share a high degree of sequence and structural homology but are expressed in different tissues: CARMA1 is primarily expressed in the hematopoietic system, CARMA2 is expressed in placenta and mucosal tissues, and CARMA3 is expressed in a broad range of tissues but not in hematopoietic cells (15). The distinct tissue distribution indicates that CARMA family members mediate different signaling pathways but use a similar mechanism to activate downstream effector molecules. Consistent with this hypothesis, CARMA proteins, through their CARD domain, form a complex with two downstream signaling molecules, B-cell lymphoma 10 (Bcl10)(16, 17), another CARD-containing scaffold protein, and caspase-like protein MALT1 (mucosa-associated lymphoid tissue lymphoma translocation protein 1)(18–20), and the formation of CARMA-Bcl10-MALT1 complex (commonly known as CBM complex) recruits the downstream IKK complex, leading to activation of NF-κB (21–24).
CARD9, another CARD protein, is structurally similar to CARMA family members (9). It has a N-terminal CARD and a CC domain like CARMA family proteins but lacks the C-terminal MAGUK domain (Fig. 1). Its CARD domain is involved in the association with Bcl10 (9), but the role of CC domain remains to be determined. Unlike CARMA family proteins, CARD9 is mainly expressed in myeloid cells such as dendritic cells (DCs) and macrophages. Recent studies suggest that, in response to fungal stimulation, CARD9 also inducibly forms a complex with Bcl10 and MALT1 in C-type lectin receptor (CLR)-induced signaling pathways (25, 26) and activates the IKK complex, leading to activation of NF-κB (25, 26). More recent studies further suggest that CARD9 also functions downstream of RIG-I and mediates viral infection-induced NF-κB activation (27).
Some CARD proteins play critical roles as pattern recognition receptors (PRRs). RIG-I and MDA-5 are novel PRRs. They are structurally related RNA helicases, critical for host innate antiviral responses. RIG-I and MDA-5 contain two N-terminal CARD domains and a C-terminal DEX(D/H)-box helicase domain, which recognizes single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), or replication intermediates of RNA viruses, leading to activation of NF-κB and IRF family of transcription factors (28–31). MAVS (also known as IPS-1, VISA, or CARDIF), another CARD-containing protein, is recruited by RIG-I/MDA5 through their CARD-CARD interaction, leading to activation of NF-κB and IRF3 (32–35).
NOD1 and NOD2 also contain CARD domains in their N-terminus, and they belong to the nucleotide-binding domain and leucine-rich repeat containing gene (NLR) family (7, 11, 36) (Fig. 1). They also function as PRR for bacterial peptides. Their C-terminal leucine-rich repeat (LRR) is required for specific recognition of muramyl peptides from both Gram-positive and Gram-negative bacteria, whereas CARD domain(s) mediate association with the downstream signaling intermediate RIP2 leading to activation of NF-κB. RIP2 contains a N-terminal Ser/Thr kinase catalytic domain and a C-terminal CARD domain (37, 38). The CARD domain mediates the association of RIP2 with other CARD-containing proteins, including NODs and cIAPs (7, 39, 40).
cIAP1 and cIAP2 contains three characteristic baculovirus IAP repeat (BIR) domains at N-terminus, followed by an ubiquitin-binding domain, a CARD domain, and a C-terminal RING domain (41, 42) (Fig. 1). The BIR domains mediate their interaction with tumor necrosis factor receptor (TNFR)-associated factor 2 (TRAF2) (42, 43). The CARD domain may mediate its association with TNFR2 (44, 45). The RNG domain designates them as E3 ubiquitin ligases, important for promoting ubiquitination of RIP1 and RIP2 (40, 46). cIAP1 and cIAP2 are important players in TNFR and NLRs-induced NF-κB activity(43, 44).
The functional roles for RIG-I/MAD5, NOD1/NOD2, and cIAP1/cIAP2 have been extensively described in various review articles (47–51). Therefore, we do not summarize the roles of these CARD proteins in this review. Instead, we discuss the roles of CARMA family members and their related proteins in signaling pathways that lead to activation of NF-κB.
Personal and historical narrative
Our group has a long-standing interest in revealing the functional role of CARD proteins. This interest stems from our interest in the characterization of T-cell receptor (TCR)-induced signaling. Stimulation of T cells by antigen-presenting cells (APCs) induces the formation of a large multi-component complex at the contact area between the T cell and the APC, termed the supramolecular activation complex (SMAC) or immunological synapse (IS) (52). The SMAC/IS of T cells is highly enriched in cholesterol and glycosphingolipids, also termed lipid rafts or microdomains (53). Some signaling molecules are constitutively associated with lipid rafts, while others are recruited into lipid rafts following the stimulation of the TCR/CD3 complex and CD28 costimulatory receptor (also known as CD3/CD28 costimulation) (54). CD3/CD28 costimulation induces a potent NF-κB activation, which contributes to T-cell activation, survival, and proliferation.
Through our efforts to delineate the signaling pathway mediating TCR-induced NF-κB activation, we found that CD3/CD28 costimulation induced a potent NF-κB activation through the inhibitor of NF-κB (IκB) kinase (IKK) complex, and this activation is sensitive to protein kinase C (PKC) inhibitors (55). Since earlier studies indicate that PKCθ, a novel PKC isoform (56, reviewed in Diaz-Meco & Moscat, this volume), is selectively recruited into the SMAC/IS following the stimulation by APCs (57), we had hypothesized that PKCθ might be the key enzyme regulating TCR-induced NF-κB activation. By expressing dominant-negative mutants of PKCθ, we and others showed that PKCθ is involved in TCR-induced NF-κB activation (55, 58). Indeed, gene knockout studies provided the genetic evidence that PKCθ is required for TCR-induced NF-κB activation (59).
One of important questions at the time was how PKCθ regulates the IKK complex in the TCR signaling pathway. To address this question, we decided to take a somatic genetics approach to identify signaling molecules that connect PKCθ to IKK. We generated a Jurkat T-cell line (JGFP1), in which green-fluorescent protein (GFP) is under the control of a NF-κB-dependent promoter, thereby expressing GFP in response to various NF-κB stimuli. The JGFP1 cells were then subjected to multiple rounds of treatment with acridine mutagen ICR-191 that generates random frame-shift mutations in genome. The resulting cells were subjected to the selection for GFP-negative cells following the stimulation of phorbol myristate acetate (PMA), which can effectively activate PKCθ. We hypothesized that this selecting procedure would be able to obtain the cells that are defective in the signaling components that regulate NF-κB activation downstream of PKCθ. This pool of GFP-negative cells was then subjected to the selection for GFP-positive cells following TNF stimulation. The resulting cells were subjected to additional rounds of the selection for GFP-negative but GFP-positive following PMA or TNF stimulation, respectively. Since previous studies had suggested that both TCR and TNFR signaling pathways could lead to activation of IKK, we predicted that the above ping-pong-selecting procedure would be able to isolate cells containing mutations in signaling components downstream of PKCθ but upstream of IKK in the TCR pathway. Using this selection procedure, we cloned a mutant cell line, JPM50.6, which is defective in PMA/CD28 costimulation- but not TNF-induced NF-κB activation (22), suggesting that JPM50.6 cells contain a mutation in the signaling component(s) downstream of PKCθ but upstream of IKK in TCR signaling pathway.
To identify the signaling molecule(s) that are defective in JPM50.6 cells, we initially tried to use cDNA expression libraries to perform a genetic complementation experiment with the hypothesis that the reconstitution of JPM50.6 cells with the wildtype version of the mutated gene(s) would rescue the defect in PKCθ-mediated IKK activation. However, we encountered a series of difficulties and realized that most of cDNA expression libraries at the time did not encode full-length coding sequences for all genes, especially coding sequences over 3 kb. Therefore, it would be very difficult to rescue the defect in JPM50.6 cells through cDNA complementation, if the mutated gene encodes a large protein.
Our effort to identify the mutation in JPM50.6 cells was significantly accelerated with the finding by Ruland et al. (60) that the CARD protein Bcl10 is required for antigen receptor-induced NF-κB activation. Bcl10 was originally identified from MALT B-cell lymphomas with t(1;14)(p22;q32) (16, 17) and from data mining using bioinformatics approaches (4–6, 8). Since lymphocytes from Bcl10 knockout mice were defective in PMA/ionomycin-induced NF-κB but not affect TNF signaling (60), we hypothesized that JPM50.6 cells might have a mutation in either Bcl10 or its related signaling components. After we found out that JPM50.6 cells did not have a mutation in Bcl10, we investigated whether Bcl10-associated proteins were mutated in JPM50.6 cells. Because Bcl10 contains a CARD domain that can associate with other CARD proteins in signaling pathways, we obtained expression vectors encoding several CARD proteins and expressed them in JPM50.6 cells. Using this approach, we found that JPM50.6 cells are defective in the expression of the CARMA1 protein, and expression of CARMA1 could effectively rescue the defect of CD3/CD28-induced NF-κB activation (22). Together, our studies provided the genetic evidence that CARMA1 is required for TCR-mediated NF-κB activation.
Consistent with our findings, two laboratories independently found that CARMA1 plays a critical role in TCR-induced NF-κB activation by expressing dominant-negative mutants of CARMA1 (21) or using CARMA1 RNA interference approach (61). Subsequently, the mouse gene knockout studies (62–64) and mouse ENU mutagenesis studies (65) further demonstrated that CARMA1 is required for both B-cell receptor (BCR)- and TCR-induced NF-κB activation. Consistent with the defect of antigen receptor-induced NF-κB activation, antigen-induced proliferation of T and B cells from carma1-null mice is defective (62–64). In addition, CARMA1 deficiency also selectively impairs the development of a subset of B cells, which results in reduced marginal-zone B cells and absence of the peritoneal B1 subpopulation (63, 66). Altogether, these studies demonstrate that CARMA1 is an essential component mediating antigen receptor-induced NF-κB activation (Fig. 2).
Fig. 2. NF-κB activation induced by CARMA family member-mediated signaling pathways.
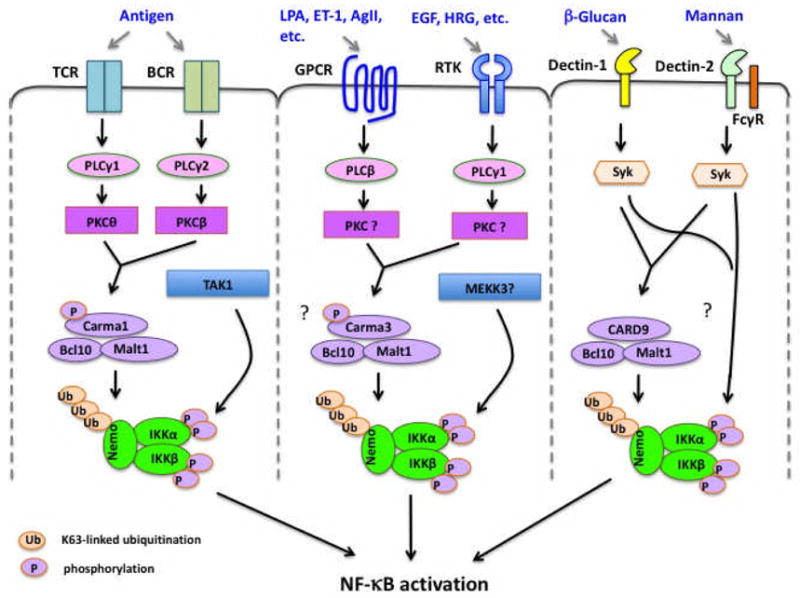
TCR, T-cell receptor; BCR, B-cell receptor; GPCR, G protein-coupled receptor; RTK, receptor tyrosine kinase.
CARD proteins in antigen receptor signaling
Antigen receptors, including TCR and BCR, play essential roles for regulation of lymphocyte activation, proliferation, and survival, leading to proper immune responses. CARD proteins are best characterized for their involvement in antigen receptor signaling (67–70). The first genetic evidence that CARD proteins are involved in antigen receptor signaling was provided by Ruland et al (60), in which they demonstrate that Bcl10 plays an essential role for antigen receptor-induced NF-κB activation. Bcl10 was originally identified by functional cloning from MALT lymphoma cells (16, 17) and by bioinformatics approaches as a CARD-containing protein (4, 5, 7, 71). Studies on Bcl10-deficient mice revealed that Bcl10 is required for lymphocyte activation and proliferation (60) as well as the development of certain subset of B cells (72).
Bcl10 contains a N-terminal CARD domain and a C-terminal Ser/Thr-rich domain (Fig. 1). The CARD domain of Bcl10 is responsible for its association with CARMA1, a CARD protein functioned upstream of Bcl10 in antigen receptor signaling cascades. CARMA1 contains an N-terminal CARD domain, followed by a C-C domain and a linker region, and its C-terminal half encodes a structural module of PDZ-SH3-GUK domain, which is also known as the MAGUK domain (Fig. 1). As described above, studies from our laboratory and others demonstrated that CARMA1 is required for antigen receptor-induced NF-κB activation, leading to lymphocyte activation and proliferation (Fig. 2). During the last several years, the mechanism by which antigen receptor signaling leads to activation of CARMA1 and Bcl10 has been intensively studied (15).
CARMA1 recruits Bcl10 and MALT1 following the stimulation of antigen receptor. Genetic inactivation of the Malt1 gene in mice impairs TCR-induced NF-κB activation (73, 74). However, there are some discrepancies regarding the role of MALT1 in BCR-induced NF-κB in the existing two mouse models. One study suggested that total NF-κB activity is significantly reduced in MALT1-deficient B cells (73). Although another gene-targeting study showed that NF-κB activation is minimally affected by MALT1 deficiency upon BCR stimulation (74). More recent studies using the latter strain of MALT1-deficient mice revealed that the activation of the c-Rel isoform of NF-κB was more severely impaired than was activation of the other isoforms of NF-κB (75). Thus, these studies indicate that MALT1 is a key component of the antigen receptor signaling pathway. However, it remains to be determined why MALT1 deficiency has a more significant impact for the activation of c-Rel than other isoforms of NF-κB in B cells.
Several laboratories including our own showed that the signal-induced phosphorylation of CARMA1 by PKCs (76–78) and other kinases (79, 80) is required for activation of CARMA1. The linker region in CARMA1 contains multiple phosphorylation sites (Fig. 3), which control a conformational switch from an inactive to an active state (76, 77) (Fig. 3). Therefore, phosphorylation on the linker region of CARMA1 may expose the binding site for recruiting Bcl10 and MALT1 (Fig. 3), leading to activation of NF-κB (77, 81–83). The CC domain of CARMA1 likely mediates oligomerization (84, 85), which may amplify signaling from CARMA1 to downstream components. Consistent with this model, mutations that result in the constitutive oligomerization of CARMA1 induce NF-κB activation and contribute to lymphomagenesis (85).
Fig. 3. Schematic presentation of the phosphorylation-dependent conformational change of CARMA1.
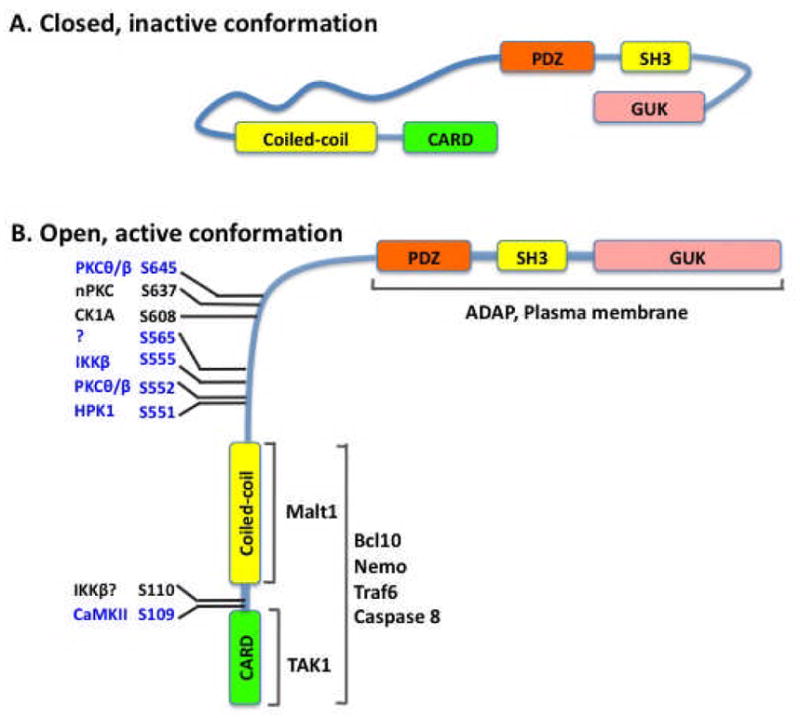
(A) The proposed CARMA1 structure in the inactive status. (B) The proposed CARMA1 structure in the active status. Ser residues that are phosphorylated are shown.
The CARD domain of Bcl10 is also involved in its oligomerized form following the stimulation of antigen receptors (86, 87), and the oligomerization of Bcl10 was found to be polyubiquitinated through a Lys63 (K63) linkage. The K63-linked polyubiquitin chain on Bcl10 serves as the docking site for binding NEMO, a subunit in the IKK complex, thereby recruiting the IKK complex to the CARMA1-Bcl10-MALT1 (CBM) complex (88). Interestingly, another study argued that MALT1 was polyubiquitinated through K63 linkage and served as a docking site for recruiting IKK (89). The relative contribution of MALT1 and Bcl10 polyubiquitination to IKK recruitment is unclear. One mechanism that could explain this seemingly redundant function of MALT1 and Bcl10 is that K63-linked polyubiquitination of both proteins may increase the specificity and affinity with which the CBM complex recruits IKK. In addition to the recruitment of IKK, Bcl10 also binds to TRAF6 and recruits TAK1 to the CBM complex (90). Therefore, the formation of this multiprotein complex brings TAK1 into proximity with IKK and then phosphorylates and activates IKK, leading to activation of NF-κB (90, 91). Interestingly, although CARMA1 deficiency does not affect TCR-induced phosphorylation of IKK, IKK kinase activity is completely defective (91), suggesting that CARMA1 may control other TCR-induced modifications on the IKK complex, which are required for activation of IKK. An earlier study (92) suggested that Bcl10 may contribute to a K63-linked polyubiquitination of NEMO. Consistent with this study, the signal-induced polyubiquitination of IKK is defective in CARMA1-deficient T cells (91). However, it remains to be determined whether the CBM complex recruits an additional E3 ubiquitin ligase to ubiquitinate the IKK complex, since TCR-induced NF-κB activation is still intact in TRAF6-deficient T cells(93).
Activated Bcl10 also functions as a scaffold molecule to recruit c-Jun N-terminal kinase (JNK) leading to activation of JNK (94). In this case, oligomerized Bcl10 specifically associates with the C-terminal tail of JNK2 and its upstream kinases, MKK7 and TAK1, in TCR-stimulated T cells, leading to the specific activation of JNK2 (94). Interestingly, the high molecular weight complex of oligomerized Bcl10 was also found to regulate TCR-induced actin polymerization (87). However, it remains to be determined whether the Bcl10-dependent actin polymerization has a significant impact on TCR-induced IKK and JNK activation. Interestingly, Bcl10-dependent actin polymerization has a significant impact on phagocytosis in monocytes, but CARMA1 and MALT1 are not involved in this process (95).
It has been shown that PKCθ is inducibly recruited into the IS (57, 96). Consistent with the findings that PKCθ associates with and phosphorylates CARMA1(76, 83), it has been found that the CBM complex is also recruited into the IS (81, 83). Therefore, an important question is how antigen receptor recruits CBM complex. CARMA1 contains a MAGUK domain, which is typically anchored to the cytoplasmic face of the membranes by various membrane proteins (71). Therefore, the C-terminal MAGUK domain of CARMA1 is likely involved in membrane localization. Consistent with this hypothesis, we have found that a point mutation in the MAGUK domain of CARMA1 (Leu808 replaced with Pro) impairs its membrane localization and recruitment to the IS (83).. Currently, the protein that links CARMA1 to cytoplasmic membrane remains unknown. Although it remains to be determined how CARMA1 is linked to the cytoplasmic membrane, TCR-induced recruitment of CARMA1 into the IS is dependent on its inducible interaction with an adapter protein ADAP (adhesion- and degranulation-promoting adapter protein)(97). Upon TCR engagement, ADAP is inducibly associated with CARMA1 and recruited into the IS, and ADAP deficiency results in impaired CARMA1 translocation to the IS and consequently reduced NF-κB activation (97). However, ADAP is not a membrane protein, and it is unlikely that ADAP anchors the MAGUK domain of CARMA1 to the cytoplasmic membrane. Therefore, another unknown protein is required for anchoring CARMA1 to the cytoplasmic membrane.
CARD proteins are also subjected to negative regulation in antigen receptor signaling. Besides the CARD domain, Bcl10 has a C-terminal Ser/Thr-rich domain. Although the function of the C-terminal Ser/Thr-rich domain of Bcl10 is not fully understood, several studies suggest that signal-dependent phosphorylation of these Ser and Thr residues may mediate degradation of Bcl10 (98–101), thereby terminating NF-κB activation (99). Indeed, Bcl10-deficient T cells reconstituted with Bcl10-S138A mutant have prolonged NF-κB activation and enhanced IL-2 production (100). However, the mechanism by which signal-induced phosphorylation of Bcl10 contributes to its stability remains to be defined.
Posttranslational modifications also contribute to the expression level of CARMA1. Recent studies suggest that phosphorylation of certain residues in CARMA1 may also suppress CARMA1 function (102, 103). One study suggests that the phosphorylation of CARMA1 by casein kinase 1α (CK1α) leads to the attenuation of CBM-mediated NF-κB activation (103). Ser608 in the CARMA1 linker region has been identified as a CK1a phosphorylation site (103); however, the mechanism by which Ser608 phosphorylation suppresses CARMA1 activity remains to be determined. Another study indicates that Ser637 is phosphorylated by undefined PKC isoforms, and mutation of Ser637 to Ala significantly enhances CARMA1-mediated NF-κB activation(102), suggesting that Ser637 phosphorylation may negatively regulate CARMA1 function. Together, these studies indicate that phosphorylation of CARMA1 not only activates CARMA1 but also suppresses its function.
Recent studies indicate that ubiquitination may also regulate the function of CBM complex. It has been shown that the formation of CBM complex appears to be negatively regulated by Cbl-b, an ubiquitin E3 ligase (104). Consistent with this finding, Cbl-b induces mono-ubiquitination of CARMA1, and this mono-ubiquitination disrupts the interaction between CARMA1 and Bcl10 but does not induce CARMA1 degradation (105). Another study suggests that CARMA1 undergoes Lys48 (K48)-linked polyubiquitination following TCR stimulation, which leads to the proteasome-mediated degradation of CARMA1 (106). Mutagenesis studies indicate that the C-terminal MAGUK domain of CARMA1 is involved in this regulation (106). In vitro experiments suggest that cIAP, a CARD-containing E3 ligase, may be involved in the K48-linked polyubiquitination of CARMA1 (106). Finally, recent studies show that CARMA1 and MALT1 recruit CSN5, a component of the COP9 signalosome following TCR stimulation (107), and this recruitment of CSN to CBM complex may be required for maintaining Bcl10 stability in response to T-cell activation, which suggests that COP9 regulates IKK activity through protecting Bcl10 from degradation and stabilizing CBM complex (107).
Somatic mutations in the components of antigen receptor signaling pathways are often associated with hematopoietic malignancy (Reviewed in Lim et al., this volume). Consistent with this notion, previous studies indicate that chromosome translocation of Bcl10 and MALT1 contribute to lymphomagenesis (16–18). Although CARMA1 gene is not commonly rearranged in B- or T-cell lymphomas, mutations and elevated expression of CARMA1 gene was found in adult T-cell leukemia (108), primary gastric B-cell lymphoma (109), and diffuse large B-cell lymphoma (DLBCL) (110). CARMA1 is also mutated in about 10% of systemic ABC-DLBCL and 16% of primary central nervous system DLBCL (85, 111, 112). The oncogenic mutants of CARMA1 constitutively recruit downstream signaling components (113), leading to activation of NF-κB (85). The activation of NF-κB through the alternation of these oncogenic proteins is believed to be one of the hallmarks of lymphoma. Previous studies have shown that NF-κB activity is critical for the survival of malignant cells in ABC-DLBCL (114), and IKK inhibitors (115) or CARMA1 shRNA (110) are toxic for these cells. Therefore, the CBM complex could be a potential therapeutic target for some types of lymphoma.
CARD proteins and NF-κB activation induced by GPCRs and RTKs
CARMA1 is only expressed in the hematopoietic system, whereas Bcl10 and MALT1 are expressed in all tissues. This expression suggests that other CARMA family members may function upstream of Bcl10 and MALT1 to induce NF-κB activation in certain receptor signaling pathways in different cells. Although the role of CARMA1 in antigen receptor signaling had been intensively investigated (67–69, 116), the role of other CARMA proteins was neglected for many years. One exception was CARMA3, which was shown to bind to NEMO and activate NF-κB when overexpressed in mammalian cells (12, 13, 117). To determine the functional role of CARMA3 in signaling pathways, we generated CARMA3-deficient mice. About half of CARMA3−/− mice showed defects in the neural tube closure (NTD), resulting in perinatal mortality (24). Similarly, this phenotpe was found in about 40% of Bcl10−/− mice (60). The similar phenotype of CARMA3- and Bcl10-deficient mice suggests that CARMA3 and Bcl10 function in the same signaling pathway and control the development of neural tube cells. Since CARMA1 and Bcl10 function downstream of PKC in TCR-induced NF-κB acitivation, we predicted that CARMA3 might play a similar role downstream of PKC in undefined signaling pathways.
Earlier studies suggest that G protein-coupled receptors (GPCRs) (118–120), integrins (120), and receptor tyrosine kinases (RTKs) (121) can activate NF-κB through PKC. We decided to test GPCRs, since GPCR is the largest family of cell surface receptors. Indeed, we found that CARMA3 deficiency in mouse embryonic fibroblasts (MEFs) resulted in impaired IKK activation and NF-κB activation upon stimulation of lysophosphatidic acid (LPA) and endothelin-1 (ET-1), which are ligands for two different GPCRs (24). Importantly, CARMA3 is specifically required for GPCR-induced IKK and NF-κB activation, because CARMA3 deficiency does not affect this activation by other stimuli such as TNF and lipopolysaccharide (LPS) (24). Similar to the function of CARMA1 in antigen receptor signaling, CARMA3 deficiency does not affect signal-induced IKK phosphorylation but alters the polyubiquitination of the IKK complex (24). The signal-induced phosphorylation is likely dependent on MEKK3 in GPCR signaling pathways (122) (Fig. 2). Consistent with the similar NTD phenotype found in CARMA3-deficient and Bcl10-deficient mice (24, 60), we and others found that GPCR-induced NF-κB activation is also defective in Bcl10-deficient cells (123–125). More recent studies have further confirmed that the CARMA3-Bcl10-MALT1 complex is required for other GPCRs-induced NF-κB activation in distinct cellular context (23, 124, 126–129). Together, these studies demonstrate that GPCRs-induced NF-κB is dependent on the CARMA3-Bcl10-MALT1 complex (Fig. 2).
RTKs are a family of cell surface receptors that recognize growth factors, cytokines, and polypeptide hormones. They are the key players in mediating multiple cellular responses. Mutations and aberrant expression of these receptors or their downstream signaling components contribute to the development of many cancers. Several growth factors, including epidermal growth factor (EGF) (121, 130, 131), insulin-like growth factor (IGF) (132–134), platelet-derived growth factor (PDGF) (135, 136), and fibroblast growth factor (bFGF) (137, 138), can induce weak but notable NF-κB activation through their cognate RTKs. Although RTK signaling pathways have been intensively studied, the mechanism by which RTKs activate NF-κB is not fully defined, and the functional significance of RTK-induced NF-κB activation in cell proliferation and survival has not been fully appreciated. Since EGFR-induced NF-κB is likely requires the activation of PKC (121), we predicted that EGFR-induced NF-κB activation involves the CBM complex. Consistent with this hypothesis, we found that CARMA3 and Bcl10 are required for EGFR-induced NF-κB activation in cancer cell lines and mouse embryonic fibroblasts, and this NF-κB activation significantly impacts on tumor progression (139). Altogether, these lines of investigation reveal a new aspect to signaling pathways induced by GPCRs and RTKs (Fig. 2). However, it remains to be determined how the CBM complex is linked to GPCRs and RTK receptors.
CARD proteins and NF-κB activation by C-type lectin receptor
CARD9 is another CARD-containing protein with some similarity to CARMA family members. CARD9 was identified through a bioinformatics approach (9). It contains an N-terminal CARD domain and a C-terminal CC domain but lacks the C-terminal MAGUK domain (Fig. 1). Overexpressed CARD9 can associate with Bcl10 and weakly activate NF-κB in mammalian cells (9). Based on these structural features and function, we originally speculated that CARD9 might function as a negative regulator by competing with CARMA family proteins for binding to Bcl10. However, gene-targeting experiments by other investigators and us indicate that CARD9 deficiency does not affect antigen receptor-induced NF-κB activation and lymphocyte activation and proliferation (25, 140, 141). Instead, CARD9 deficiency has significant impacts on innate immunity (25, 140, 141).
Initial characterization of CARD9-deficient mice indicates that CARD9 is required for anti-fungal immune responses (25). This study showed that CARD9 is required for NF-κB activation induced by zymosan, a β-glucan component from yeast cell wall. CARD9-deficient mice are highly susceptible to fungal infections (25). Since β-glucan can activate Dectin-1, a C-type lectin receptor, it has been proposed that CARD9 mediates Dectin-1-induced NF-κB activation in response to fungal infection (25) (Fig. 2). When we characterized another CARD9-deficient mouse strain (141), we surprisingly found that zymosan could still effectively activate NF-κB in macrophages from independently generated CARD9-deficient mouse strain (26), although this strain of CARD9-deficient mice is also highly susceptible to fungal infection(26). These results suggesting that zymosan may activate NF-κB through a CARD9-independent pathway (26). Therefore, the requirement of CARD9 in Dectin-1-induced NF-κB needs to be further confirmed using more specific Dectin-1 ligands such as pure β-Glucan.
Besides the Dectin-1 pathway, it has been demonstrated that CARD9 also mediates the signaling induced by other C-type lectin receptors (Fig. 2), including Dectin-2 (26, 142) and Mincle (143). These receptors have been implicated as the PRRs for different types of fungus (144, 145) or damage-associated molecular pattern (DAMP) signals from necrotic cells (143). In the case of fungal infection, Mannan from the hyphal form of C. albicans may function as the direct ligand for Dectin-2 (146). Recently, a human family with a homozygous germline CARD9 mutation has been identified. Consistent with the role of CARD9 in mouse, human patients with CARD9 mutation have persistent or recurrent infections of the mucosa or the skin with Candida species (147).
The characterization of the third CARD9-deficient mouse strain has revealed that CARD9 also mediates NF-κB activation induced by several ITAM-associated receptors, including FcRγ and DAP12 signaling pathway in myeloid cells (140). Interestingly, it was found that the CARD9-containing complex mediates NF-κB activation induced by the ITAM-associated receptors in myeloid cells, whereas the CARMA1-containing complex mediates NF-κB activation induced by ITAM-associated receptors in lymphoid lineage cells (148, 149). However, because CARMA1 is also expressed in myeloid cells, it remains to be determined why ITAM-associated receptors utilize the CARD9-dependent, but not CARMA1-dependent, pathway to activate NF-κB in myeloid cells.
It has been suggested that CARD9 functions downstream of Syk and recruits Bcl10/MALT1 to activate NF-κB (26, 140, 142, 150, 151). Similar to the defect of CARMA1 deficiency in lymphocytes, our data showed that CARD9 deficiency did not affect the signal-induced phosphorylation of IKK, instead, it caused a defect on the signal-induced polyubiquitination of IKK, which results in a defect in IKK activation (26). Therefore, CARD9-deficient macrophages and dendritic cells are impaired in the expression of TNF, IL-6, and IL-12 in response to the stimulation of fungal particles (25, 26, 142, 151), and CARD9-deficient mice are more susceptible to infection with fungus C. albicans (25, 26, 142, 146, 150).
In addition to the effect on anti-fungal immunity, CARD9 deficiency also affects the innate immune response to intracellular bacteria such as Listeria monocytogenes (140, 141, 152) and Mycobacteruim tuberculosis (153). Later studies from our laboratory indicate that CARD9 inducibly associates with the LyGDI-Rac complex, a protein complex that regulates NADPH oxidase and induces reactive oxygen species (ROS). Therefore, CARD9 deficiency results in the defect of ROS production in macrophage, which plays a critical role for killing the phagocytosed bacteria in macrophages (152). In addition, CARD9 is found to be involved in anti-viral responses (27, 141), in which the CARD9-Bcl10 module appears to be an essential component of the RNA helicase RIG-I-dependent pro-inflammatory response to certain RNA viruses and IL-1β production (27).
Inactivation of the VHL gene is often observed in renal cell carcinoma and may lead to increased NF-κB activity. Another study found that VHL associate with CARD9, and promoted the phosphorylation of CARD9 by casein kinase 2 (CK2) (154). Therefore, it was proposed that the VHL-induced phosphorylation of CARD9 inhibits CARD9, thereby suppressing NF-κB activation, whereas inactivation of VHL gene expression leads to activation of NF-κB (154). Consistent with this hypothesis, ectopic expression of CARD9 mutants that can not be phosphorylated resulted in an increased NF-κB activation and decreased apoptosis in VHL-defective renal carcinoma cells, whereas knockdown of CARD9 suppresses NF-κB activity in these cells (154). Although the role of CARD9 phosphorylation by CK2 is not clear, these authors speculate that this modification may promote an inhibitory intra- or inter-molecular interaction or prevent the binding of CARD9 to NEMO or other protein required for its activity (154). One discrepancy for this proposed regulatory mechanism of posttranslational modification of CARD9 and previous results is that CARD9 is mainly expressed in myeloid cells and the expression level of CARD9 in renal cells is very low (9, 141). Therefore, future studies are needed to investigate the role of CARD9 in non-myeloid cells.
Perspectives: conclusions and outstanding questions
In last decade, significant progress towards understanding the function of CARD proteins in the NF-κB signaling pathways has been made. These studies demonstrate that CARMA/CARD9-dependent IKK activation is involved in antigen receptor-, GPCR-, RTK-, and CLR-induced NF-κB activation. However, further investigation is needed to define the precise mechanism by which CBM complexes activate IKK and to reveal the mechanisms by which different CBM complexes are linked to receptors. More research is required to define the regulation of CARD protein-mediated signaling and the role of these proteins in cancer and other genetic diseases. For example, it has been shown that human card9 gene contains a germline mutation, which results in a high susceptible to fungal infection. It will be interesting to investigate whether CARMA family members and Bcl10 also have as yet unidentified human germline mutations that contribute to genetic disorders. These lines of investigation will not only reveal the molecular mechanism of CARD proteins but also provide the molecular insight for designing therapeutic agents for cancer and other diseases.
Acknowledgments
This work was partially supported by grants from the National Institutes of Health (RO1GM065899, RO1GM079451, RO1AI050848) to XL.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Shaw AS, Filbert EL. Scaffold proteins and immune-cell signalling. Nat Rev Immunol. 2009;9:47–56. doi: 10.1038/nri2473. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signalling motif. Trends Biochem Sci. 1997;22:155–156. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- 3.Thome M, et al. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–888. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- 4.Thome M, et al. Equine herpesvirus-2 E10 gene product, but not its cellular homologue, activates NF-kappaB transcription factor and c-Jun N-terminal kinase. J Biol Chem. 1999;274:9962–9968. doi: 10.1074/jbc.274.15.9962. [DOI] [PubMed] [Google Scholar]
- 5.Srinivasula SM, et al. CLAP, a novel caspase recruitment domain-containing protein in the tumor necrosis factor receptor pathway, regulates NF-kappaB activation and apoptosis. J Biol Chem. 1999;274:17946–17954. doi: 10.1074/jbc.274.25.17946. [DOI] [PubMed] [Google Scholar]
- 6.Yan M, Lee J, Schilbach S, Goddard A, Dixit V. mE10, a novel caspase recruitment domain-containing proapoptotic molecule. J Biol Chem. 1999;274:10287–10292. doi: 10.1074/jbc.274.15.10287. [DOI] [PubMed] [Google Scholar]
- 7.Inohara N, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 8.Koseki T, et al. CIPER, a novel NF kappaB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J Biol Chem. 1999;274:9955–9961. doi: 10.1074/jbc.274.15.9955. [DOI] [PubMed] [Google Scholar]
- 9.Bertin J, et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. J Biol Chem. 2000;275:41082–41086. doi: 10.1074/jbc.C000726200. [DOI] [PubMed] [Google Scholar]
- 10.Bertin J, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 11.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, et al. Card10 is a novel caspase recruitment domain/membrane-associated guanylate kinase family member that interacts with BCL10 and activates NF-kappa B. J Biol Chem. 2001;276:21405–21409. doi: 10.1074/jbc.M102488200. [DOI] [PubMed] [Google Scholar]
- 13.McAllister-Lucas LM, et al. Bimp1, a MAGUK family member linking protein kinase C activation to Bcl10-mediated NF-kappaB induction. J Biol Chem. 2001;276:30589–30597. doi: 10.1074/jbc.M103824200. [DOI] [PubMed] [Google Scholar]
- 14.Gaide O, Martinon F, Micheau O, Bonnet D, Thome M, Tschopp J. Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-kappaB activation. FEBS Lett. 2001;496:121–127. doi: 10.1016/s0014-5793(01)02414-0. [DOI] [PubMed] [Google Scholar]
- 15.Blonska M, Lin X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011;21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willis TG, et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell. 1999;96:35–45. doi: 10.1016/s0092-8674(00)80957-5. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, et al. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32) Nat Genet. 1999;22:63–68. doi: 10.1038/8767. [DOI] [PubMed] [Google Scholar]
- 18.Akagi T, et al. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene. 1999;18:5785–5794. doi: 10.1038/sj.onc.1203018. [DOI] [PubMed] [Google Scholar]
- 19.Dierlamm J, et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood. 1999;93:3601–3609. [PubMed] [Google Scholar]
- 20.Morgan JA, et al. Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res. 1999;59:6205–6213. [PubMed] [Google Scholar]
- 21.Gaide O, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:836–843. doi: 10.1038/ni830. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, et al. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:830–835. doi: 10.1038/ni824. [DOI] [PubMed] [Google Scholar]
- 23.McAllister-Lucas LM, et al. CARMA3/Bcl10/MALT1-dependent NF-kappaB activation mediates angiotensin II-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci U S A. 2007;104:139–144. doi: 10.1073/pnas.0601947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grabiner BC, et al. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-{kappa}B activation. Genes Dev. 2007;21:984–996. doi: 10.1101/gad.1502507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross O, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 26.Bi L, et al. CARD9 mediates dectin-2-induced IkappaBalpha kinase ubiquitination leading to activation of NF-kappaB in response to stimulation by the hyphal form of Candida albicans. J Biol Chem. 2010;285:25969–25977. doi: 10.1074/jbc.M110.131300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poeck H, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 28.Berghall H, et al. The interferon-inducible RNA helicase, mda-5, is involved in measles virus-induced expression of antiviral cytokines. Microbes Infect. 2006;8:2138–2144. doi: 10.1016/j.micinf.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 30.Hausmann S, Marq JB, Tapparel C, Kolakofsky D, Garcin D. RIG-I and dsRNA-induced IFNbeta activation. PLoS One. 2008;3:e3965. doi: 10.1371/journal.pone.0003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barral PM, et al. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: key regulators of innate immunity. Pharmacol Ther. 2009;124:219–234. doi: 10.1016/j.pharmthera.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 33.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 35.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 36.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 37.McCarthy JV, Ni J, Dixit VM. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem. 1998;273:16968–16975. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- 38.Inohara N, del Peso L, Koseki T, Chen S, Nunez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem. 1998;273:12296–12300. doi: 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- 39.Bertin J, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem. 1999;274:12955–12958. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- 40.Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 42.Mace PD, Smits C, Vaux DL, Silke J, Day CL. Asymmetric recruitment of cIAPs by TRAF2. J Mol Biol. 2010;400:8–15. doi: 10.1016/j.jmb.2010.04.055. [DOI] [PubMed] [Google Scholar]
- 43.Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC. Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem. 2006;281:1080–1090. doi: 10.1074/jbc.M509381200. [DOI] [PubMed] [Google Scholar]
- 44.Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci U S A. 1996;93:13973–13978. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertrand MJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 47.Yoneyama M, Fujita T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity. 2008;29:178–181. doi: 10.1016/j.immuni.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 48.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 49.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 50.Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–398. doi: 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
- 51.Srinivasula SM, Ashwell JD. IAPs: what’s in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dustin ML, et al. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 1998;94:667–677. doi: 10.1016/s0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 53.Xavier R, Seed B. Membrane compartmentation and the response to antigen. Curr Opin Immunol. 1999;11:265–269. doi: 10.1016/s0952-7915(99)80043-0. [DOI] [PubMed] [Google Scholar]
- 54.Bromley SK, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 55.Lin X, O’Mahony A, Mu Y, Geleziunas R, Greene WC. Protein kinase C-theta participates in NF-kappaB activation induced by CD3-CD28 costimulation through selective activation of IkappaB kinase beta. Mol Cell Biol. 2000;20:2933–2940. doi: 10.1128/mcb.20.8.2933-2940.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baier G, et al. Molecular cloning and characterization of PKC theta, a novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J Biol Chem. 1993;268:4997–5004. [PubMed] [Google Scholar]
- 57.Monks CR, Kupfer H, Tamir I, Barlow A, Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385:83–86. doi: 10.1038/385083a0. [DOI] [PubMed] [Google Scholar]
- 58.Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci U S A. 2000;97:3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Z, et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- 60.Ruland J, et al. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell. 2001;104:33–42. doi: 10.1016/s0092-8674(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 61.Pomerantz JL, Denny EM, Baltimore D. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J. 2002;21:5184–5194. doi: 10.1093/emboj/cdf505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hara H, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 2003;18:763–775. doi: 10.1016/s1074-7613(03)00148-1. [DOI] [PubMed] [Google Scholar]
- 63.Egawa T, et al. Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Curr Biol. 2003;13:1252–1258. doi: 10.1016/s0960-9822(03)00491-3. [DOI] [PubMed] [Google Scholar]
- 64.Newton K, Dixit VM. Mice lacking the CARD of CARMA1 exhibit defective B lymphocyte development and impaired proliferation of their B and T lymphocytes. Curr Biol. 2003;13:1247–1251. doi: 10.1016/s0960-9822(03)00458-5. [DOI] [PubMed] [Google Scholar]
- 65.Jun J, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 66.Pappu BP, Lin X. Potential role of CARMA1 in CD40-induced splenic B cell proliferation and marginal zone B cell maturation. Eur J Immunol. 2006;36:3033–3043. doi: 10.1002/eji.200535663. [DOI] [PubMed] [Google Scholar]
- 67.Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–359. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- 68.Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor kappa B. Immunity. 2006;25:701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 69.Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol. 2006;6:799–812. doi: 10.1038/nri1944. [DOI] [PubMed] [Google Scholar]
- 70.Blonska M, Lin X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol Rev. 2009;228:199–211. doi: 10.1111/j.1600-065X.2008.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dimitratos SD, Woods DF, Stathakis DG, Bryant PJ. Signaling pathways are focused at specialized regions of the plasma membrane by scaffolding proteins of the MAGUK family. Bioessays. 1999;21:912–921. doi: 10.1002/(SICI)1521-1878(199911)21:11<912::AID-BIES3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 72.Xue L, et al. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol. 2003;4:857–865. doi: 10.1038/ni963. [DOI] [PubMed] [Google Scholar]
- 73.Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. 2003;302:1581–1584. doi: 10.1126/science.1090769. [DOI] [PubMed] [Google Scholar]
- 74.Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–758. doi: 10.1016/s1074-7613(03)00293-0. [DOI] [PubMed] [Google Scholar]
- 75.Ferch U, et al. MALT1 directs B cell receptor-induced canonical nuclear factor-kappaB signaling selectively to the c-Rel subunit. Nat Immunol. 2007;8:984–991. doi: 10.1038/ni1493. [DOI] [PubMed] [Google Scholar]
- 76.Matsumoto R, et al. Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity. 2005;23:575–585. doi: 10.1016/j.immuni.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 77.Sommer K, et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity. 2005;23:561–574. doi: 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 78.Shinohara H, et al. PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J Exp Med. 2005;202:1423–1431. doi: 10.1084/jem.20051591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishiguro K, et al. Ca2+/calmodulin-dependent protein kinase II is a modulator of CARMA1-mediated NF-kappaB activation. Mol Cell Biol. 2006;26:5497–5508. doi: 10.1128/MCB.02469-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brenner D, et al. Phosphorylation of CARMA1 by HPK1 is critical for NF-kappaB activation in T cells. Proc Natl Acad Sci U S A. 2009;106:14508–14513. doi: 10.1073/pnas.0900457106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hara H, et al. The molecular adapter Carma1 controls entry of IkappaB kinase into the central immune synapse. J Exp Med. 2004;200:1167–1177. doi: 10.1084/jem.20032246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Che T, You Y, Wang D, Tanner MJ, Dixit VM, Lin X. MALT1/paracaspase is a signaling component downstream of CARMA1 and mediates T cell receptor-induced NF-kappaB activation. J Biol Chem. 2004;279:15870–15876. doi: 10.1074/jbc.M310599200. [DOI] [PubMed] [Google Scholar]
- 83.Wang D, et al. CD3/CD28 costimulation-induced NF-kappaB activation is mediated by recruitment of protein kinase C-theta, Bcl10, and IkappaB kinase beta to the immunological synapse through CARMA1. Mol Cell Biol. 2004;24:164–171. doi: 10.1128/MCB.24.1.164-171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanner MJ, Hanel W, Gaffen SL, Lin X. CARMA1 coiled-coil domain is involved in the oligomerization and subcellular localization of CARMA1 and is required for T cell receptor-induced NF-kappaB activation. J Biol Chem. 2007;282:17141–17147. doi: 10.1074/jbc.M700169200. [DOI] [PubMed] [Google Scholar]
- 85.Lenz G, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 86.Schaefer BC, Kappler JW, Kupfer A, Marrack P. Complex and dynamic redistribution of NF-kappaB signaling intermediates in response to T cell receptor stimulation. Proc Natl Acad Sci U S A. 2004;101:1004–1009. doi: 10.1073/pnas.0307858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rossman JS, Stoicheva NG, Langel FD, Patterson GH, Lippincott-Schwartz J, Schaefer BC. POLKADOTS are foci of functional interactions in T-Cell receptor-mediated signaling to NF-kappaB. Mol Biol Cell. 2006;17:2166–2176. doi: 10.1091/mbc.E05-10-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu CJ, Ashwell JD. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105:3023–3028. doi: 10.1073/pnas.0712313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oeckinghaus A, et al. Malt1 ubiquitination triggers NF-kappaB signaling upon T- cell activation. EMBO J. 2007;26:4634–4645. doi: 10.1038/sj.emboj.7601897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 91.Shambharkar PB, et al. Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. Embo J. 2007;26:1794–1805. doi: 10.1038/sj.emboj.7601622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou H, et al. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 93.King CG, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. 2006;12:1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- 94.Blonska M, et al. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity. 2007;26:55–66. doi: 10.1016/j.immuni.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rueda D, et al. Bcl10 controls TCR- and FcgammaR-induced actin polymerization. J Immunol. 2007;178:4373–4384. doi: 10.4049/jimmunol.178.7.4373. [DOI] [PubMed] [Google Scholar]
- 96.Bi K, et al. Antigen-induced translocation of PKC-theta to membrane rafts is required for T cell activation. Nat Immunol. 2001;2:556–563. doi: 10.1038/88765. [DOI] [PubMed] [Google Scholar]
- 97.Medeiros RB, et al. Regulation of NF-kappaB activation in T cells via association of the adapter proteins ADAP and CARMA1. Science. 2007;316:754–758. doi: 10.1126/science.1137895. [DOI] [PubMed] [Google Scholar]
- 98.Wegener E, et al. Essential role for IkappaB kinase beta in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol Cell. 2006;23:13–23. doi: 10.1016/j.molcel.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 99.Lobry C, Lopez T, Israel A, Weil R. Negative feedback loop in T cell activation through IkappaB kinase-induced phosphorylation and degradation of Bcl10. Proc Natl Acad Sci U S A. 2007;104:908–913. doi: 10.1073/pnas.0606982104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeng H, et al. Phosphorylation of Bcl10 negatively regulates T-cell receptor-mediated NF-kappaB activation. Mol Cell Biol. 2007;27:5235–5245. doi: 10.1128/MCB.01645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ishiguro K, Ando T, Goto H, Xavier R. Bcl10 is phosphorylated on Ser138 by Ca2+/calmodulin-dependent protein kinase II. Mol Immunol. 2007;44:2095–2100. doi: 10.1016/j.molimm.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 102.Moreno-Garcia ME, Sommer K, Haftmann C, Sontheimer C, Andrews SF, Rawlings DJ. Serine 649 phosphorylation within the protein kinase C-regulated domain down-regulates CARMA1 activity in lymphocytes. J Immunol. 2009;183:7362–7370. doi: 10.4049/jimmunol.0902438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bidere N, et al. Casein kinase 1alpha governs antigen-receptor-induced NF-kappaB activation and human lymphoma cell survival. Nature. 2009;458:92–96. doi: 10.1038/nature07613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qiao G, et al. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. 2008;28:2470–2480. doi: 10.1128/MCB.01505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kojo S, Elly C, Harada Y, Langdon WY, Kronenberg M, Liu YC. Mechanisms of NKT cell anergy induction involve Cbl-b-promoted monoubiquitination of CARMA1. Proc Natl Acad Sci U S A. 2009;106:17847–17851. doi: 10.1073/pnas.0904078106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moreno-Garcia ME, Sommer K, Shinohara H, Bandaranayake AD, Kurosaki T, Rawlings DJ. MAGUK-controlled ubiquitination of CARMA1 modulates lymphocyte NF-kappaB activity. Mol Cell Biol. 2010;30:922–934. doi: 10.1128/MCB.01129-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Welteke V, Eitelhuber A, Duwel M, Schweitzer K, Naumann M, Krappmann D. COP9 signalosome controls the Carma1-Bcl10-Malt1 complex upon T-cell stimulation. EMBO Rep. 2009;10:642–648. doi: 10.1038/embor.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oshiro A, et al. Identification of subtype-specific genomic alterations in aggressive adult T-cell leukemia/lymphoma. Blood. 2006;107:4500–4507. doi: 10.1182/blood-2005-09-3801. [DOI] [PubMed] [Google Scholar]
- 109.Nakamura S, et al. Overexpression of caspase recruitment domain (CARD) membrane-associated guanylate kinase 1 (CARMA1) and CARD9 in primary gastric B-cell lymphoma. Cancer. 2005;104:1885–1893. doi: 10.1002/cncr.21421. [DOI] [PubMed] [Google Scholar]
- 110.Ngo VN, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 111.Montesinos-Rongen M, et al. Mutations of CARD11 but not TNFAIP3 may activate the NF-kappaB pathway in primary CNS lymphoma. Acta Neuropathol. 2010;120:529–535. doi: 10.1007/s00401-010-0709-7. [DOI] [PubMed] [Google Scholar]
- 112.Compagno M, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry. 2010;49:8240–8250. doi: 10.1021/bi101052d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lam LT, et al. Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin Cancer Res. 2005;11:28–40. [PubMed] [Google Scholar]
- 116.Lin X, Wang D. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin Immunol. 2004;16:429–435. doi: 10.1016/j.smim.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 117.Stilo R, et al. Physical and functional interaction of CARMA1 and CARMA3 with Ikappa kinase gamma-NFkappaB essential modulator. J Biol Chem. 2004;279:34323–34331. doi: 10.1074/jbc.M402244200. [DOI] [PubMed] [Google Scholar]
- 118.Shahrestanifar M, Fan X, Manning DR. Lysophosphatidic acid activates NF-kappaB in fibroblasts. A requirement for multiple inputs. J Biol Chem. 1999;274:3828–3833. doi: 10.1074/jbc.274.6.3828. [DOI] [PubMed] [Google Scholar]
- 119.Cummings R, et al. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. J Biol Chem. 2004;279:41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 120.Juliano RL. Signal transduction by cell adhesion receptors and the cytoskeleton: functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu Rev Pharmacol Toxicol. 2002;42:283–323. doi: 10.1146/annurev.pharmtox.42.090401.151133. [DOI] [PubMed] [Google Scholar]
- 121.Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:8542–8547. doi: 10.1073/pnas.97.15.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun W, et al. MEKK3 is required for lysophosphatidic acid-induced NF-kappaB activation. Cellular signalling. 2009;21:1488–1494. doi: 10.1016/j.cellsig.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klemm S, Zimmermann S, Peschel C, Mak TW, Ruland J. Bcl10 and Malt1 control lysophosphatidic acid-induced NF-kappaB activation and cytokine production. Proc Natl Acad Sci U S A. 2007;104:134–138. doi: 10.1073/pnas.0608388103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McAllister-Lucas LM, et al. The CARMA3-Bcl10-MALT1 signalosome promotes angiotensin II-dependent vascular inflammation and atherogenesis. J Biol Chem. 2010;285:25880–25884. doi: 10.1074/jbc.C110.109421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang D, et al. Bcl10 plays a critical role in NF-kappaB activation induced by G protein-coupled receptors. Proc Natl Acad Sci U S A. 2007;104:145–150. doi: 10.1073/pnas.0601894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rehman AO, Wang CY. CXCL12/SDF-1 alpha activates NF-kappaB and promotes oral cancer invasion through the Carma3/Bcl10/Malt1 complex. Int J Oral Sci. 2009;1:105–118. doi: 10.4248/IJOS.09059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem. 2009;284:6038–6042. doi: 10.1074/jbc.C800207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Borthakur A, Bhattacharyya S, Alrefai WA, Tobacman JK, Ramaswamy K, Dudeja PK. Platelet-activating factor-induced NF-kappaB activation and IL-8 production in intestinal epithelial cells are Bcl10-dependent. Inflamm Bowel Dis. 2010;16:593–603. doi: 10.1002/ibd.21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Delekta PC, et al. Thrombin-dependent NF-{kappa}B activation and monocyte/endothelial adhesion are mediated by the CARMA3.Bcl10. MALT1 signalosome. J Biol Chem. 2010;285:41432–41442. doi: 10.1074/jbc.M110.158949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun L, Carpenter G. Epidermal growth factor activation of NF-kappaB is mediated through IkappaBalpha degradation and intracellular free calcium. Oncogene. 1998;16:2095–2102. doi: 10.1038/sj.onc.1201731. [DOI] [PubMed] [Google Scholar]
- 131.Biswas DK, Iglehart JD. Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J Cell Physiol. 2006;209:645–652. doi: 10.1002/jcp.20785. [DOI] [PubMed] [Google Scholar]
- 132.Kaliman P, Canicio J, Testar X, Palacin M, Zorzano A. Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear factor-kappaB and inducible nitric-oxide synthase define a common myogenic signaling pathway. J Biol Chem. 1999;274:17437–17444. doi: 10.1074/jbc.274.25.17437. [DOI] [PubMed] [Google Scholar]
- 133.Pons S, Torres-Aleman I. Insulin-like growth factor-I stimulates dephosphorylation of ikappa B through the serine phosphatase calcineurin (protein phosphatase 2B) J Biol Chem. 2000;275:38620–38625. doi: 10.1074/jbc.M004531200. [DOI] [PubMed] [Google Scholar]
- 134.Kim HJ, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27:3165–3175. doi: 10.1128/MCB.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Olashaw NE, Kowalik TF, Huang ES, Pledger WJ. Induction of NF-kappa B-like activity by platelet-derived growth factor in mouse fibroblasts. Mol Biol Cell. 1992;3:1131–1139. doi: 10.1091/mbc.3.10.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 137.Byrd VM, Ballard DW, Miller GG, Thomas JW. Fibroblast growth factor-1 (FGF-1) enhances IL-2 production and nuclear translocation of NF-kappaB in FGF receptor-bearing Jurkat T cells. J Immunol. 1999;162:5853–5859. [PubMed] [Google Scholar]
- 138.Bushdid PB, et al. NF-kappaB mediates FGF signal regulation of msx-1 expression. Dev Biol. 2001;237:107–115. doi: 10.1006/dbio.2001.0356. [DOI] [PubMed] [Google Scholar]
- 139.Jiang T, et al. CARMA3 is crucial for EGFR-Induced activation of NF-kappaB and tumor progression. Cancer Res. 2011;71:2183–2192. doi: 10.1158/0008-5472.CAN-10-3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hara H, et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol. 2007;8:619–629. doi: 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- 141.Hsu YM, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- 142.Robinson MJ, et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med. 2009;206:2037–2051. doi: 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 144.Willment JA, Brown GD. C-type lectin receptors in antifungal immunity. Trends Microbiol. 2008;16:27–32. doi: 10.1016/j.tim.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 145.Graham LM, Brown GD. The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine. 2009;48:148–155. doi: 10.1016/j.cyto.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saijo S, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010;32:681–691. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 147.Glocker EO, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hara H, Saito T. CARD9 versus CARMA1 in innate and adaptive immunity. Trends Immunol. 2009;30:234–242. doi: 10.1016/j.it.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 149.Hara H, et al. Cell type-specific regulation of ITAM-mediated NF-kappaB activation by the adaptors, CARMA1 and CARD9. J Immunol. 2008;181:918–930. doi: 10.4049/jimmunol.181.2.918. [DOI] [PubMed] [Google Scholar]
- 150.LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 151.Goodridge HS, et al. Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J Immunol. 2009;182:1146–1154. doi: 10.4049/jimmunol.182.2.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wu W, Hsu YM, Bi L, Songyang Z, Lin X. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat Immunol. 2009;10:1208–1214. doi: 10.1038/ni.1788. [DOI] [PubMed] [Google Scholar]
- 153.Werninghaus K, et al. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma-Syk-Card9-dependent innate immune activation. J Exp Med. 2009;206:89–97. doi: 10.1084/jem.20081445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang H, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell. 2007;28:15–27. doi: 10.1016/j.molcel.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]