

Background: The Hippo-Yap signaling pathway is one of the critical pathways regulating cell proliferation, differentiation, and apoptosis.
Results: Knockdown of endogenous Yap1 impairs VSMC proliferation and enhances VSMC contractile phenotype by promoting the association of the myocardin-SRF-CArG complex.
Conclusion: The Yap1 signaling pathway is a central regulator of the phenotypic switch of VSMCs.
Significance: The phenotypic switch of VSMCs and vessel injury response can be controlled by modulation of Hippo-Yap signaling.
Keywords: Atherosclerosis, Cardiovascular Disease, Gene Regulation, Vascular Biology, Vascular Smooth Muscle Cells, Yes-associated Protein 1 (Yap1), Smooth Muscle Cells
Abstract
The Hippo-Yap (Yes-associated protein) signaling pathway has emerged as one of the critical pathways regulating cell proliferation, differentiation, and apoptosis in response to environmental and developmental cues. However, Yap1 roles in vascular smooth muscle cell (VSMC) biology have not been investigated. VSMCs undergo phenotypic switch, a process characterized by decreased gene expression of VSMC contractile markers and increased proliferation, migration, and matrix synthesis. The goals of the present studies were to investigate the relationship between Yap1 and VSMC phenotypic switch and to determine the molecular mechanisms by which Yap1 affects this essential process in VSMC biology. Results demonstrated that the expression of Yap1 was rapidly up-regulated by stimulation with PDGF-BB (a known inducer of phenotypic switch in VSMCs) and in the injured vessel wall. Knockdown of Yap1 impaired VSMC proliferation in vitro and enhanced the expression of VSMC contractile genes as well by increasing serum response factor binding to CArG-containing regions of VSMC-specific contractile genes within intact chromatin. Conversely, the interaction between serum response factor and its co-activator myocardin was reduced by overexpression of Yap1 in a dose-dependent manner. Taken together, these results indicate that down-regulation of Yap1 promotes VSMC contractile phenotype by both up-regulating myocardin expression and promoting the association of the serum response factor-myocardin complex with VSMC contractile gene promoters and suggest that the Yap1 signaling pathway is a central regulator of phenotypic switch of VSMCs.
Introduction
Vascular smooth muscle cells (VSMCs)3 within the vessels retain remarkable plasticity and are characterized in part by their ability to modulate their phenotypes in response to the environmental stimuli through a process characterized by decreased gene expression of VSMC contractile markers and increased proliferation, migration, and matrix synthesis (1). As a result, VSMCs proliferate and synthesize collagens and matrix metalloproteinases (1, 2). Phenotypic switch, thus, is one of the major cellular events underlying many VSMC-related pathological conditions, such as atherosclerosis, postangioplasty restenosis, hypertension, and tumor angiogenesis (1). Unraveling the mechanisms involved in VSMC phenotypic switch is an important step toward better understanding the pathology of these diseases and ultimately designing therapeutic agents for their treatment and prevention.
The switch between the contractile and synthetic VSMC phenotypes is tightly controlled through a synergistic and coordinated molecular regulatory network (3–8). Within these molecules, serum response factor (SRF), a ubiquitously expressed transcription factor that executes multiple functions through binding to evolutionarily conserved cis-elements, the CArG box (CC(A/T)6GG), plays a central role (9, 10). Nearly all of the VSMC-specific contractile protein genes and many other genes involved in migration, proliferation, and extracellular matrix production during the process of VSMC phenotypic switch contain CArG boxes within their promoters (1, 9, 10). The communication between SRF and CArG boxes in response to environmental changes is mostly controlled by the interaction of SRF with additional transcription factors: co-activators, including myocardin (11), myocardin-related transcription factors (12), PRX1 (13), and GATA factors (14, 15), and co-repressors, including ELK-1 (6, 16), KLF4 (7), YY1 (17), and Foxo factors (18), among others. The balance between those positive and negative SRF cofactors instructs the dynamic VSMC gene expression in response to environmental cues (1, 19). Among these regulatory components, myocardin is perhaps the most potent transcriptional co-activator of SRF identified in nature so far and strongly transactivates VSMC-specific gene expression by physically interacting with SRF (11, 20).
In the past decade, the Hippo-Yap signaling pathway has emerged as one of the critical conserved pathways that regulate cell proliferation and apoptosis in response to environmental and developmental signals (21–23). This signaling pathway was discovered in Drosophila (22) with many genes involved in the Hippo pathway evolutionarily conserved in Drosophila and vertebrates. The activation of macrophage-stimulating 1/2 (Mst1/2) kinases by certain environmental cues phosphorylates and subsequently activates large tumor suppressor, homolog 1/2 (Lats1/2) kinase, which in turn directly phosphorylates Yap1. Phosphorylated Yap1 is retained in the cytoplasm, and, as a result, Yap1 function is inhibited (24, 25). Analysis of Hippo-Yap signaling in mammalian organisms has revealed its role in the regulation of maintenance of stem cell pluripotency, tumorigenesis, and organ size (21, 22, 25–31).
However, the function of the Hippo-Yap pathway in VSMC biology remains largely unexplored. In particular, the involvement of Yap1 in VSMC phenotypic switch and the molecular mechanisms underlying such a potential link are unknown. Here, we uncover for the first time a role for Yap1 in VSMC phenotypic switch. We found up-regulation of Yap1 expression during VSMC phenotypic switch induced by PDGF-BB in vitro and during balloon injury-induced vessel lesion formation in vivo. Conversely, the knockdown of Yap1 expression not only led to impaired proliferation of VSMCs but also enhanced expression of SMC contractile phenotype-specific markers. Furthermore, Yap1 affects VSMC phenotypic switch via interference with myocardin-SRF-CArG box tertiary complex formation. This study provides new insights into the mechanisms controlling phenotypic switch of VSMC and identifies a new potential therapeutic target for ameliorating VSMC-related diseases and a potential tool for accelerating viable approaches to vascular engineering.
EXPERIMENTAL PROCEDURES
Rat Aortic SMC (RASMC) Culture
The culture of RASMCs was performed following a previous report (32). Briefly, primary RASMCs were cultured in DMEM/F-12 supplemented with 10% (v/v) FBS in a 5% CO2 humidified atmosphere at 37 °C.
Infection with shRNA Lentivirus
Knockdown of Yap1 was achieved by infection with a lentivirus expressing the scrambled control or Yap1 shRNA (generous gift from Dr. Kunliang Guan, University of California, San Diego, CA) (21, 25, 33). Cultured passage 3 RASMCs were infected with these viruses and selected with 5 μg/ml puromycin in culture medium for 1 week before using them in the experimental procedures. For simplicity, the resulting cells will be referred to as “scrambled” VSMCs and “shYap1” VSMCs throughout this work.
RNA Extraction and Quantitative Real-time Polymerase Chain Reaction
Total RNA from cultured cells was extracted by using the RNeasy minikit (Qiagen), and cDNA was synthesized with the Superscript III first-strand synthesis system (Invitrogen). Quantitative RT-PCR amplification using custom primers (listed in supplemental Table S1) was conducted on a Bio-Rad MyIQ detection system (Bio-Rad), as in our previous reports (34, 35). 18S RNA served as an internal standard.
VSMC Proliferation Assays
Cell proliferation was determined by growth curves of VSMCs derived from cell counting as described before (36). Passage-matched scrambled and shYap1 VSMCs were seeded at ∼6 × 105 cells/ml in growth medium. Cell numbers were determined for the indicated periods with the hemocytometer measurement method in a blinded unbiased fashion. Each count was an average of three repeats, whereas each data point was the average of four experiments.
Western Blot Analysis and Co-immunoprecipitation
Western blot analysis was carried out as we described previously (35, 37). Whole cell lysate samples were prepared using the mammalian protein extraction reagent M-Per (Promega) supplemented with a protease inhibitor mixture (Roche Applied Science). Antibodies against GAPDH (1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), β-tubulin (1:500; Santa Cruz Biotechnology, Inc.), SM α-actin (SMA) (1:5000; Millipore), SM-myosin heavy chain (SMMHC) (1:2000; BTI), SM22α (1:2000; Abcam), p27 (1:1000; Santa Cruz Biotechnology, Inc.), cyclin D1 (CCND1) (1:1000; Santa Cruz Biotechnology, Inc.), cyclin-dependent kinase 4 (CDK4) (1:1000; Santa Cruz Biotechnology, Inc.), proliferating cell nuclear antigen (PCNA) (1:1000; Santa Cruz Biotechnology, Inc.) and Yap1 (1:1000; Santa Cruz Biotechnology, Inc.) were used to examine individual protein expression. For the co-immunoprecipitation (co-IP) assay, pcDNA4-Yap1-Myc (Addgene) and pCGN-SRF (Addgene) were cotransfected into HEK293 cells with either pcDNA3.1 vehicle or pcDNA3.1FLAG-myocardin (generous gift from Dr. Li Li, Wayne State University), and cells were harvested 48 h post-transfection. The co-IP assay was performed as described in a previous report (38). Briefly, cells were lysed in lysis buffer (50 mm Tris-HCl, pH 7.8, 137 mm NaCl, 1 mm EDTA, 0.1% Triton X-100 with a protease inhibitor mixture (Roche Applied Science), and the supernatants were collected after centrifugation and precleared with protein G-agarose for 1 h at 4 °C and then incubated with antibodies as indicated overnight at 4 °C. Normal IgG was used for a negative control. Immunoreactivity and band density were visualized by the Odyssey system (LI-COR Biosciences, Lincoln, NE), according to the manufacturer's instructions.
Luciferase Activity Assay
HEK293 cells seeded in 96-well plates for 24 h were transiently transfected with pGL3-SMMHC-luciferase reporter (generous gift from Dr. G. K. Owens, University of Virginia), thymidine kinase Renilla luciferase control reporter vector and pcDNA4-Yap1 plasmids in the presence of pcDNA3.1 vector to keep DNA constant or pcDNA3.1FLAG-myocardin using Lipofectamine 2000 (Invitrogen). Forty-eight hours post-transfection, cells were lysed, and luciferase activity was detected with the Dual-Luciferase reporter assay system (Promega) in a Wallac 1420 work station. The activity of thymidine kinase Renilla served as an internal standard.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as we described previously (34). Briefly, RASMCs carrying either lentiviral scrambled or shYap1 were cultured to 70–80% confluence. Cellular proteins were cross-linked, and ChIP assay was performed by using the EZ-ChIP assay kit (Millipore). Purified chromatin was immunoprecipitated using anti-SRF antibody (Santa Cruz Biotechnology, Inc.). Eluted DNA fragments were purified to serve as templates for the PCR amplification. Rabbit IgG control (Santa Cruz Biotechnology, Inc.) serves as a negative control to the SRF antibody. The primer sets used to amplify the area containing SRF binding sites on the CrAG boxes of promoters of SMC-specific markers were described before (8) and are included in supplemental Table S1. Data represent the relative enrichment of IP DNA samples as compared with input DNA.
Rat Carotid Artery Injury Model
The rat carotid artery balloon injury model was performed in male Sprague-Dawley rats (230–300 g) following a previous report (32). The arteries were harvested at day 14 after operation and ground for protein analysis (32).
Statistical Analysis
All data, expressed as the mean ± S.D., was analyzed for statistical significance by Student's t test with values of p < 0.05 considered to be significant. All experiments were independently repeated at least three times.
RESULTS
Yap1 Expression Correlates with VSMC Synthetic Phenotype
Previous publications established that PDGF-BB is a potent mediator of the VSMC phenotype switch from a contractile to a synthetic state by repressing VSMC marker gene expression as well as promoting VSMC proliferation (1, 4). Our data uncovered that the level of Yap1 mRNA in RASMCs treated with 10 ng/ml PDGF-BB was significantly elevated, 2–4-fold, compared with the vehicle-treated control (Fig. 1A). Remarkably, Yap1 expression correlated with down-regulation in either the mRNA or protein levels of VSMC-specific genes associated with a contractile phenotype (including myocardin, SMA, SMMHC, and SM22α) and enhanced pro-proliferation gene expression (e.g. cyclin D1 and cyclin D2) (Fig. 1, A and B), which is consistent with the effects of PDGF-BB on VSMC phenotypic switch and proliferation (1, 4). In accordance with the in vitro results, Yap1 expression is up-regulated in the well established balloon injury-induced vessel lesion formation for 14 days, when phenotypic switch is extensive in vivo (39). This correlates with decreased expression of VSMC contractile apparatus SMA and SM22α and increased proliferation marker PCNA (Fig. 1C). These data suggest that up-regulation of the Hippo-Yap signaling pathway components is positively correlated with the synthetic VSMC phenotype both in vitro and in vivo.
FIGURE 1.

Yap1 expression is increased during VSMC phenotypic switch induced by PDGF-BB and in vessel injury. A, RASMCs grown to 60–70% confluence underwent mitogenic quiescence by serum starvation (DMEM + 0.5% FBS) for 48 h and were subsequently stimulated with 10 ng/ml PDGF-BB for the indicated periods. Time point expression of Yap1 and markers of VSMC proliferation and contractile phenotype were analyzed by quantitative RT-PCR. 18S RNA served as internal control. CCND1, cyclin D1; CCND2, cyclin D2; SMA, SM α-actin; MyoCD, myocardin; SMMHC, smooth muscle myosin heavy chain. n = 3. Data are expressed as mean ± S.D. *, p < 0.05. B, representative Western blot analysis of total protein lysates of cultured RASMCs. RASMCs were treated with 10 ng/ml PDGF-BB for 24 h after serum starvation for 48 h and harvested for Western blot. C, representative Western blot analysis of total protein lysates of carotid arteries. Rat carotid arteries subjected to balloon injury and sham surgery controls were harvested 14 days after surgery and ground for Western blot analysis as indicated in the figure. n = 3.
Yap1 Promotes SMC Proliferation
Yap1 is known to enhance cell proliferation in other systems (26, 30, 31, 40–43). We next tested whether knockdown of Yap1 is able to impair VSMC proliferation. Lentiviruses containing two different target shYap1 sets were tested for their ability to knock down endogenous Yap1, and all subsequent experiments were performed with set 2, showing dramatic down-regulation of Yap1 protein (supplemental Fig. S1, A (set 2) and B). Knockdown of Yap1 in RASMCs reduced cell proliferation compared with scrambled controls as shown in Fig. 2A. Furthermore, consistent with impaired RASMC proliferation, protein levels of the proliferative marker cyclin D1 decreased, whereas p27 protein, an inhibitor of cyclin-dependent kinases, was up-regulated in shYap1 RASMCs (Fig. 2B). Similarly, cell proliferation-associated cyclin D1 mRNA was also down-regulated in these conditions (supplemental Fig. S1B). The proliferation response to PDGF-BB is impaired by Yap1 knockdown, as evidenced by failure in up-regulation of the proliferation marker PCNA, compared with scrambled RASMCs, although no dramatic changes were observed on CDK4 (supplemental Fig. S1B). Thus, these data indicate that proliferation of RASMCs is Yap1-dependent and without the impairment of CDK4.
FIGURE 2.

Knockdown of Yap1 attenuates VSMC proliferation. A, RASMCs infected with lentiviruses encoding Yap1 shRNA (shYap1) or control scrambled shRNA were seeded in 6-well plates, and cell proliferation was measured by counting cell numbers at each time point as described under “Experimental Procedures.” n = 4. Data are expressed as mean ± S.D. (error bars). *, p < 0.05. B, representative Western blot analysis of total protein lysates from RASMCs infected with lentiviruses encoding Yap1 shRNA (shYap1) or control scrambled shRNA actively growing in 10% FBS-containing growth medium using the indicated antibodies. GAPDH served as internal control.
Yap1 Suppresses Expression of VSMC-specific Contractile Protein Genes
To investigate the effect of loss of function of Yap1 on VSMC contractile phenotype-specific gene expression, RASMCs were infected with lentiviral shYap1 or scrambled control as above, and total protein or mRNAs were harvested to measure SMC gene expression by Western blot or quantitative RT-PCR (supplemental Fig. S1B and Fig. 3). Data from these experiments demonstrated that all examined VSMC-specific genes associated with a contractile phenotype (including myocardin, SMA, SM22α, and SMMHC) were significantly up-regulated by the knockdown of Yap1 at both protein and mRNA levels (supplemental Fig. S1B and Fig. 3, A and B). Interestingly, the mRNA expression level of SRF in RASMCs was not affected by knockdown of Yap, indicating that SRF mRNA expression is Yap1-independent. In addition, the mRNA expression of smoothelin B, which is one of the CArG-independent SMC contractile markers (44), was not significantly changed by knockdown of Yap1. Moreover, overexpression of Yap1 in HEK 293 cells attenuated the myocardin-driven activation of an SMMHC-promoter luciferase reporter (Fig. 3C). Together, these data demonstrate that endogenous Yap1 is a repressor of CArG-dependent VSMC contractile phenotype-specific genes probably by antagonizing myocardin-mediated activation.
FIGURE 3.

Knockdown of Yap1 enhances contractile phenotype-specific gene expression in VSMCs. A and B, RASMCs infected with shYap1 were cultured in 10% FBS-containing DMEM and harvested for quantitative RT-PCR (A) or Western blotting (B) to detect gene expression as indicated. Cells infected with lentiviral scrambled vector served as a control. C, a luciferase reporter for the SMMHC gene promoter was co-transfected with myocardin (MyoCD) and/or Yap1 expression plasmids in HEK293 cells, and the luciferase activity was measured 48 h after transfection as described under “Experimental Procedures.” Thymidine kinase Renilla served as internal control. The basal activity of pcDNA3.1 empty vector on SMMHC promoter activity was normalized to 1. Data are expressed as mean ± S.D. (error bars). n = 3. *, p < 0.05.
Yap1 Modulates SMC Phenotype through Interaction with Myocardin-SRF Complex
To assess the possible mechanism underlying this effect, a series of co-IP assays were performed. Results showed that Yap1 specifically interacts with myocardin in HEK 293 cells transiently transfected with plasmids expressing FLAG-myocardin and Yap1 (Fig. 4A). Thus, IP of myocardin with anti-FLAG pulls down Yap1 as evidenced by Western blot with anti-Myc. In addition, overexpression of Yap1 attenuated the interaction between myocardin and SRF in co-IP experiments in a dose-dependent manner in HEK 293 cells (Fig. 4B) as evidenced by reduced SRF in the complexes pulled down with anti-FLAG.
FIGURE 4.
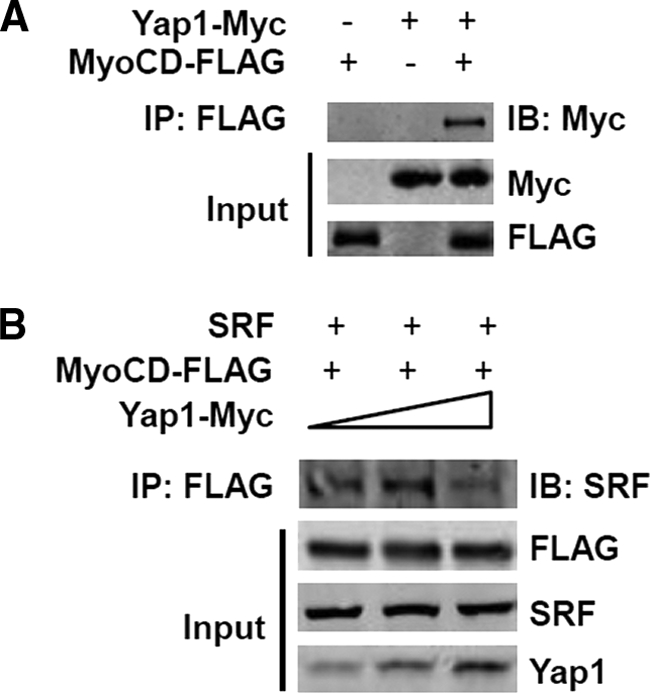
Yap1 binds myocardin. A, HEK 293 cells were transfected with pcDNA-myocardin-FLAG and/or pcDNA4-Yap1-Myc expression plasmids. The specificity of binding between Yap1 and myocardin was confirmed by precipitation with FLAG antibody and further immunoblot with anti-Myc antibody. B, HEK 293 cells were transfected with pcDNA-myocardin-FLAG and SRF while pcDNA4-Yap1-Myc expression plasmid was introduced with increased dosage. Total cell lysates were precipitated with FLAG antibody and further immunoblot with anti-SRF antibodies, FLAG, and Yap1 as indicated. IB, immunoblot.
In order to address the functional relevance of these interactions, we conducted ChIP assays on the SMMHC and SMA promoters. Significantly, results of quantitative ChIP assays showed that Yap1 knockdown in RASMCs cultured in regular conditions was associated with marked enhancement in SRF binding to CArG-containing regions of the promoter of SMA and SMMHC within intact chromatin (Fig. 5). Importantly, these effects were selective in that SRF binding to the c-fos CArG was unchanged by Yap1 knockdown, which is consistent with previous studies showing that SRF binding to the c-fos promoter is independent of myocardin or phenotypic switch (8, 45, 46) (Fig. 5). Taken together, these results provide compelling evidence that Yap1 may interact with both myocardin to mediate the down-regulation of contractile phenotype-specific genes in VSMC and promote proliferation.
FIGURE 5.

Yap1 attenuates the SRF-dependent transcriptional activation of CArG box containing contractile VSMC-specific genes. ChIP assay were performed on crossed-linked chromatin from RASMCs with Yap1 knockdown (shYap1) immunoprecipitated with anti-SRF antibody. The precipitated DNA was amplified by real-time PCR with VSMC gene-specific primers spanning the CArG region. Data represent the relative enrichment of IP DNA samples as compared with input DNA and are expressed as mean ± S.D. (error bars). n = 3. *, p < 0.05.
In summary, these studies support a functional positive role of an inducible Hippo-Yap pathway in coordinating phenotypic switch changes associated with a VSMC proliferative phenotype in response to vascular injury through direct interaction with myocardin and negative regulation of its transcriptional activity on promoters of genes specific to the contractile phenotype of VMSC (Fig. 6).
FIGURE 6.

Working hypothesis whereby Yap1 regulates VSMC phenotypic switch via myocardin. Upon stimuli driving VSMC phenotypic switch from contractile to synthetic, like PDGF-BB, Yap1 interferes with myocardin activity, resulting in reduced expression of VSMC contractile genes and, through an as yet undefined mechanism, increased proliferation markers in the VSMCs.
DISCUSSION
Previous studies demonstrated that Yap1 upstream regulators Mst1/2 and Lats1/2 act as tumor suppressors by restricting cell proliferation and promoting cell apoptosis (47). Recent studies reported that cardiac-specific knockdown of Yap1 causes hyperplasic hearts as a result of elevated cardiomyocyte proliferation and with the ensuing impaired cardiac function (31, 43). In addition, Yap1-null mice die at embryonic day 8.5 with defects in yolk sac vasculogenesis, suggesting that Yap1 may play distinct roles during vasculogenesis (48). However, the specific involvement of Yap1 in vascular biology and disease remains unexplored.
In the present study, the first report examining the function of Yap1 on VSMC phenotypic switch, we show that the expression of Yap1 could be enhanced by PDGF-BB, a well accepted driver of this phenomenon in vitro. More importantly, the in vivo expression of Yap1 is up-regulated in the injured vessel walls induced by balloon angioplasty (Fig. 1), which indicates a potential active involvement of Yap1 on VSMC phenotypic switch in vivo. The knockdown of endogenous Yap1 in VSMCs results in up-regulation of contractile phenotype-specific VSMC gene expression but attenuates VSMC proliferation in vitro (Figs. 2 and 3 and supplemental Fig. S1), indicating that endogenous Yap1 is required for both proliferation and the concomitant repression of the contractile phenotype. Furthermore, the effect of Yap1 on VSMC phenotypic switch involves interference with myocardin-SRF-CArG ternary complex, one of the essential transcriptional regulatory effectors in this VSMC response (1). These results clearly suggest that Yap1 acts as an effector for PDGF-BB-induced proliferation and repression of VSMC contractile phenotype-specific genes, thus operating as a central molecular switch for the phenotypic modulation of VSMC, and, therefore, suggest that Yap1 is causally involved in SMC phenotypic switch associated with vascular injury and other vascular pathologies.
Our observations on Yap1 roles are consistent with a recent publication (49) suggesting a relevant role for the Hippo-Yap signaling pathway in VSMC, in that overexpression of Mst1, a negative regulator of Yap1 function, enhanced VSMC apoptosis in vitro and in vivo and suppressed neointimal formation in arteries in a balloon injury model. Although these results are of extensive interest, the precise mechanism for Hippo-Yap signaling in vascular remodeling remained poorly understood. With vascular remodeling being highly regulated by mitogenic factors and involving phenotypic switch, further studies, such as those presented here, are necessary to uncover the mechanisms underlying the effects of Hippo-Yap signaling in mitogen-treated VSMCs.
Intriguingly, knockdown of endogenous Yap1 in VSMCs selectively enhanced expression of an entire subset of VSMC-specific genes associated with the contractile phenotype, as shown in Fig. 3. It is well established that virtually almost all VSMC-selective contractile marker genes characterized to date are CArG-SRF-dependent (1, 2). As such, it is likely that results of the present studies are relevant to mechanisms that induce the coordinated down-regulation of multiple VSMC contractile marker genes in response to PDGF-BB treatment and the ensuing proliferation, thus defining Yap1 as a potential molecular switch controlling SMC phenotypic modulation.
Yap1 has been identified as a Ty1 enhancer activity domain family member (TEAD)-interacting protein (50). Moreover, the interaction of Yap1 and TEAD proteins is essential for Yap1 to modulate cell activities in other cell types (33, 51, 52). Previous studies have shown the presence of functional muscle-specific cytidine-adenosine-thymidine sequence elements within the SMA promoter that bind TEAD proteins and repress the activity of the promoter during VSMC differentiation throughout embryonic development. However, this machinery is not required for SMA promoter activity in adult mature VSMCs (53, 54), suggesting that the effects we observe in markers like SMA in the context of VSMCs may not be mediated by Yap1 interaction with TEAD proteins.
It has been shown that SRF plays a central role on the expression of many different VSMC-specific genes (9). SRF is a multifunctional protein that not only binds a highly conserved CArG box that can be found in most SMC-specific contractile genes but also provides a docking surface for interaction with a wide variety of accessory cofactors (9). Of the SRF-associated proteins, myocardin is the most potent natural activator for CArG box-containing VSMC-specific contractile genes and is a strong inhibitor for VSMC migration and proliferation (11, 55–57). Interestingly, our data here strongly indicate that Yap1 binds myocardin in a complex, whereas the binding of SRF to myocardin is decreased by Yap1 overexpression in a dose-dependent manner (Fig. 4). Although the nature of these complexes, the dynamics of the interaction, and the domains in Yap1 responsible for these effects remain to be explored, these data in conjunction with our ChIP results (Fig. 5) support that, in adult SMC, Yap1 suppresses expression of contractile phenotype genes via inhibition of the recruitment of the essential SRF-myocardin-CArG ternary complex to the promoter of the co-regulated genes. This is probably facilitated through Yap1 decreasing expression of myocardin itself (Fig. 3) because previous studies have shown that myocardin is necessary and sufficient for contractile phenotype-specific VSMC gene expression (58, 59). As shown in Fig. 2, our data also suggest that Yap1 is necessary for VSMC proliferation. Myocardin is a strong inhibitor of VSMC migration and proliferation (11, 55–57) when VSMC cells are in the contractile phenotype. Although the detailed molecular mechanism for Yap1 effects on VSMC proliferation is still unclear and deserves in depth investigation (Fig. 6), the interaction between myocardin and Yap1 is likely to be a primary and essential component within this network to regulate VSMC proliferation by facilitating the phenotypic switch from contractile to synthetic phenotype by interfering with the myocardin-dependent activation of contractile phenotype-specific genes.
A TEAD binding element in the myocardin promoter has been identified in cardiomyocytes, and TEAD2 could significantly activate a myocardin promoter reporter gene (60). It is possible that knockdown of Yap1 attenuates the formation of Yap1-TEAD complexes and subsequently enhances TEAD function, resulting in up-regulation of myocardin expression and thereby the potentiated expression of SMC contractile genes in VSMCs. This would require that, unlike what has been reported for similar elements in the SMA promoter, the TEAD element in the myocardin promoter be functional in adult VSMC, a possibility that remains to be investigated. It is also possible that these two mechanisms may co-exist and act coordinately to account for the up-regulation of VSMC contractile phenotype-specific gene expression resulting from the down-regulation of Yap1. The present studies support a mechanism whereby down-regulation of Yap1 selectively increases SMC contractile gene expression by allowing increased expression of the SRF co-activator myocardin (Fig. 3) and simultaneously releasing the direct inhibition on myocardin-SRF-CArG ternary complex with the subsequent activation of contractile phenotype-specific gene expression mediated by this complex (Figs. 4 and 5). Therefore, the results presented here provide evidence for an emerging model revealing a molecular mechanism whereby the up-regulation of Yap1 in the presence of PDGF-BB selectively inhibits VSMC contractile phenotype-specific genes via both inhibition of myocardin expression and concurrent direct inhibition of myocardin-mediated activation of CArG-dependent transcription by selectively attenuating SRF binding to CArG box regions of these genes within the intact chromatin (Fig. 6). Additionally, endogenous Yap1 is clearly required for optimal proliferation of VSMC (Fig. 2, A and B), an effect described previously for other cell types (26, 30, 31, 40–43), which, in the context of the new data presented here, places Yap1 at the critical cross-regulatory junction linking proliferation and synthetic phenotypic switch (1).
This phenotypic switch of VSMC from a quiescent, contractile phenotype to a proliferative state may contribute to the repair of vascular injury. Conversely, in the normal vessels, quiescent, contractile phenotypes are maintained through repression of Yap1, which, in turn results in up-regulation of myocardin expression and synergistic direct potentiation of SRF binding to the CArG box of the co-regulated contractile targets. Here, we have shown the up-regulation of Yap1 in the balloon injury-induced vessel lesion (Fig. 1C). However, there is currently no direct evidence that Yap1 plays this role in vivo because, as mentioned above, Yap1-null mice die at embryonic day 8.5 with, most interestingly, defects in yolk sac vasculogenesis (48). Additional studies involving VSMC-selective conditional knockout of this gene are needed to directly test whether Yap1 contributes to VSMC phenotypic switch in vivo during vascular injury and atherosclerosis pathogenesis as well as to further dissect the mechanisms by which Yap1 suppresses myocardin expression and selectively controls SRF binding to CArG elements within the intact chromatin.
There is extensive evidence indicating that VSMC phenotypic switch by mitogens in response to vascular injury occurs through the interaction of a transcriptional network, which contributes to overall phenotypic switching, including stimulation of VSMC growth (61–63). Among others, KLF4 (7) and ELK1 (6, 16) have been reported to mediate PDGF-BB effects on phenotypic switch. The present studies are the first to provide direct evidence identifying Yap1 as a novel effector of PDGF-BB-induced repression of VSMC differentiation marker genes, yet it is still possible that PDGF-BB may also induce suppression of VSMC contractile genes through Yap1-independent mechanisms or that Hippo-Yap signaling may cross-talk with other PDGF-BB-dependent pathways. For instance, Hippo-Yap signaling may cooperate synergistically with the inhibitory effect of KLF4 and ELK1 on the binding of myocardin to modulate VSMC phenotypic switch in response to environmental stimulation.
The ability to manipulate the phenotypic switch of VSMC in vitro and in vivo will prove essential as a therapeutic intervention to prevent proliferative vascular disorders as well as the complications of atherosclerosis and development of aneurysms (1, 2). This study provides new insights into the mechanisms controlling phenotypic modulation of VSMC and defines Yap1 as a central molecular switch controlling this process, thus identifying a new potential therapeutic target for ameliorating VSMC-related diseases and a potential tool for accelerating viable approaches to stem cell differentiation and vascular engineering.
Supplementary Material
Acknowledgment
We thank Dr. Minerva T. Garcia-Barrio at the Morehouse School of Medicine for critical review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL105114, HL068878, and HL89544 (to Y. E. C.).

This article contains supplemental Fig. S1 and Table S1.
- VSMC
- vascular smooth muscle cell
- Yap
- Yes-associated protein
- SRF
- serum response factor
- SMMHC
- SM-myosin heavy chain
- SMA
- SM α-actin
- TEAD
- Ty1 enhancer activity domain
- SMC
- smooth muscle cell
- RASMC
- rat aortic SMC
- PCNA
- proliferating cell nuclear antigen
- IP
- immunoprecipitation.
REFERENCES
- 1. Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- 2. Owens G. K. (1995) Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 75, 487–517 [DOI] [PubMed] [Google Scholar]
- 3. Majesky M. W. (2007) Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 27, 1248–1258 [DOI] [PubMed] [Google Scholar]
- 4. Xie C., Ritchie R. P., Huang H., Zhang J., Chen Y. E. (2011) Smooth muscle cell differentiation in vitro. Models and underlying molecular mechanisms. Arterioscler. Thromb. Vasc. Biol. 31, 1485–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim S., Ip H. S., Lu M. M., Clendenin C., Parmacek M. S. (1997) A serum response factor-dependent transcriptional regulatory program identifies distinct smooth muscle cell sublineages. Mol. Cell Biol. 17, 2266–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Z., Wang D. Z., Hockemeyer D., McAnally J., Nordheim A., Olson E. N. (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428, 185–189 [DOI] [PubMed] [Google Scholar]
- 7. Liu Y., Sinha S., McDonald O. G., Shang Y., Hoofnagle M. H., Owens G. K. (2005) Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 280, 9719–9727 [DOI] [PubMed] [Google Scholar]
- 8. McDonald O. G., Wamhoff B. R., Hoofnagle M. H., Owens G. K. (2006) Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J. Clin. Invest. 116, 36–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miano J. M. (2003) Serum response factor. Toggling between disparate programs of gene expression. J. Mol. Cell Cardiol. 35, 577–593 [DOI] [PubMed] [Google Scholar]
- 10. Sun Q., Chen G., Streb J. W., Long X., Yang Y., Stoeckert C. J., Jr., Miano J. M. (2006) Defining the mammalian CArGome. Genome Res. 16, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A., Olson E. N. (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862 [DOI] [PubMed] [Google Scholar]
- 12. Wang D. Z., Li S., Hockemeyer D., Sutherland L., Wang Z., Schratt G., Richardson J. A., Nordheim A., Olson E. N. (2002) Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. U.S.A. 99, 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshida T., Hoofnagle M. H., Owens G. K. (2004) Myocardin and Prx1 contribute to angiotensin II-induced expression of smooth muscle α-actin. Circ. Res. 94, 1075–1082 [DOI] [PubMed] [Google Scholar]
- 14. Chang D. F., Belaguli N. S., Iyer D., Roberts W. B., Wu S. P., Dong X. R., Marx J. G., Moore M. S., Beckerle M. C., Majesky M. W., Schwartz R. J. (2003) Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev. Cell 4, 107–118 [DOI] [PubMed] [Google Scholar]
- 15. Nishida W., Nakamura M., Mori S., Takahashi M., Ohkawa Y., Tadokoro S., Yoshida K., Hiwada K., Hayashi K., Sobue K. (2002) A triad of serum response factor and the GATA and NK families governs the transcription of smooth and cardiac muscle genes. J. Biol. Chem. 277, 7308–7317 [DOI] [PubMed] [Google Scholar]
- 16. Zhou J., Hu G., Herring B. P. (2005) Smooth muscle-specific genes are differentially sensitive to inhibition by Elk-1. Mol. Cell Biol. 25, 9874–9885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Itoh S., Katoh Y., Konishi H., Takaya N., Kimura T., Periasamy M., Yamaguchi H. (2001) Nitric oxide regulates smooth muscle-specific myosin heavy chain gene expression at the transcriptional level. Possible role of SRF and YY1 through CArG element. J. Mol. Cell Cardiol 33, 95–107 [DOI] [PubMed] [Google Scholar]
- 18. Liu Z. P., Wang Z., Yanagisawa H., Olson E. N. (2005) Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev. Cell 9, 261–270 [DOI] [PubMed] [Google Scholar]
- 19. McDonald O. G., Owens G. K. (2007) Programming smooth muscle plasticity with chromatin dynamics. Circ. Res. 100, 1428–1441 [DOI] [PubMed] [Google Scholar]
- 20. Spiegelman B. M., Heinrich R. (2004) Biological control through regulated transcriptional coactivators. Cell 119, 157–167 [DOI] [PubMed] [Google Scholar]
- 21. Lian I., Kim J., Okazawa H., Zhao J., Zhao B., Yu J., Chinnaiyan A., Israel M. A., Goldstein L. S., Abujarour R., Ding S., Guan K. L. (2010) The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 24, 1106–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao B., Li L., Lei Q., Guan K. L. (2010) The Hippo-YAP pathway in organ size control and tumorigenesis. An updated version. Genes Dev. 24, 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harvey K., Tapon N. (2007) The Salvador-Warts-Hippo pathway. An emerging tumor-suppressor network. Nat. Rev. Cancer 7, 182–191 [DOI] [PubMed] [Google Scholar]
- 24. Hao Y., Chun A., Cheung K., Rashidi B., Yang X. (2008) Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509 [DOI] [PubMed] [Google Scholar]
- 25. Zhao B., Wei X., Li W., Udan R. S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L., Zheng P., Ye K., Chinnaiyan A., Halder G., Lai Z. C., Guan K. L. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Camargo F. D., Gokhale S., Johnnidis J. B., Fu D., Bell G. W., Jaenisch R., Brummelkamp T. R. (2007) YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 [DOI] [PubMed] [Google Scholar]
- 27. Lu L., Li Y., Kim S. M., Bossuyt W., Liu P., Qiu Q., Wang Y., Halder G., Finegold M. J., Lee J. S., Johnson R. L. (2010) Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. U.S.A. 107, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song H., Mak K. K., Topol L., Yun K., Hu J., Garrett L., Chen Y., Park O., Chang J., Simpson R. M., Wang C. Y., Gao B., Jiang J., Yang Y. (2010) Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 107, 1431–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Staley B. K., Irvine K. D. (2010) Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr. Biol. 20, 1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou D., Conrad C., Xia F., Park J. S., Payer B., Yin Y., Lauwers G. Y., Thasler W., Lee J. T., Avruch J., Bardeesy N. (2009) Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16, 425–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heallen T., Zhang M., Wang J., Bonilla-Claudio M., Klysik E., Johnson R. L., Martin J. F. (2011) Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu M., Zhang J., Tseng Y. H., Cui T., Zhu X., Xiao Y., Mou Y., De Leon H., Chang M. M., Hamamori Y., Kahn C. R., Chen Y. E. (2005) Rad GTPase attenuates vascular lesion formation by inhibition of vascular smooth muscle cell migration. Circulation 111, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 33. Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C. Y., Chinnaiyan A. M., Lai Z. C., Guan K. L. (2008) TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang H., Xie C., Sun X., Ritchie R. P., Zhang J., Chen Y. E. (2010) miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. J. Biol. Chem. 285, 9383–9389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie C., Huang H., Sun X., Guo Y., Hamblin M., Ritchie R. P., Garcia-Barrio M. T., Zhang J., Chen Y. E. (2011) MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. 20, 205–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen K. H., Guo X., Ma D., Guo Y., Li Q., Yang D., Li P., Qiu X., Wen S., Xiao R. P., Tang J. (2004) Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 6, 872–883 [DOI] [PubMed] [Google Scholar]
- 37. Xie C. Q., Huang H., Wei S., Song L. S., Zhang J., Ritchie R. P., Chen L., Zhang M., Chen Y. E. (2009) A comparison of murine smooth muscle cells generated from embryonic versus induced pluripotent stem cells. Stem. Cells Dev. 18, 741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan Y., Guo Y., Hamblin M., Chang L., Zhang J., Chen Y. E. (2011) Inhibition of gluconeogenic genes by calcium-regulated heat-stable protein 1 via repression of peroxisome proliferator-activated receptor α. J. Biol. Chem. 286, 40584–40594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Faxon D. P., Coats W., Currier J. (1997) Remodeling of the coronary artery after vascular injury. Prog. Cardiovasc Dis. 40, 129–140 [DOI] [PubMed] [Google Scholar]
- 40. Fernandez-L A., Northcott P. A., Dalton J., Fraga C., Ellison D., Angers S., Taylor M. D., Kenney A. M. (2009) YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. 23, 2729–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schlegelmilch K., Mohseni M., Kirak O., Pruszak J., Rodriguez J. R., Zhou D., Kreger B. T., Vasioukhin V., Avruch J., Brummelkamp T. R., Camargo F. D. (2011) Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 144, 782–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou D., Zhang Y., Wu H., Barry E., Yin Y., Lawrence E., Dawson D., Willis J. E., Markowitz S. D., Camargo F. D., Avruch J. (2011) Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. U.S.A. 108, E1312–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xin M., Kim Y., Sutherland L. B., Qi X., McAnally J., Schwartz R. J., Richardson J. A., Bassel-Duby R., Olson E. N. (2011) Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 4, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rensen S. S., Niessen P. M., Long X., Doevendans P. A., Miano J. M., van Eys G. J. (2006) Contribution of serum response factor and myocardin to transcriptional regulation of smoothelins. Cardiovasc. Res. 70, 136–145 [DOI] [PubMed] [Google Scholar]
- 45. Hendrix J. A., Wamhoff B. R., McDonald O. G., Sinha S., Yoshida T., Owens G. K. (2005) 5′ CArG degeneracy in smooth muscle α-actin is required for injury-induced gene suppression in vivo. J. Clin. Invest. 115, 418–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawai-Kowase K., Kumar M. S., Hoofnagle M. H., Yoshida T., Owens G. K. (2005) PIAS1 activates the expression of smooth muscle cell differentiation marker genes by interacting with serum response factor and class I basic helix-loop-helix proteins. Mol. Cell Biol. 25, 8009–8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao B., Lei Q. Y., Guan K. L. (2008) The Hippo-YAP pathway. New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 20, 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morin-Kensicki E. M., Boone B. N., Howell M., Stonebraker J. R., Teed J., Alb J. G., Magnuson T. R., O'Neal W., Milgram S. L. (2006) Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell Biol. 26, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ono H., Ichiki T., Ohtsubo H., Fukuyama K., Imayama I., Hashiguchi Y., Sadoshima J., Sunagawa K. (2005) Critical role of Mst1 in vascular remodeling after injury. Arterioscler. Thromb. Vasc. Biol. 25, 1871–1876 [DOI] [PubMed] [Google Scholar]
- 50. Vassilev A., Kaneko K. J., Shu H., Zhao Y., DePamphilis M. L. (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 15, 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen L., Loh P. G., Song H. (2010) Structural and functional insights into the TEAD-YAP complex in the Hippo signaling pathway. Protein Cell 1, 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li Z., Zhao B., Wang P., Chen F., Dong Z., Yang H., Guan K. L., Xu Y. (2010) Structural insights into the YAP and TEAD complex. Genes Dev. 24, 235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gan Q., Yoshida T., Li J., Owens G. K. (2007) Smooth muscle cells and myofibroblasts use distinct transcriptional mechanisms for smooth muscle α-actin expression. Circ. Res. 101, 883–892 [DOI] [PubMed] [Google Scholar]
- 54. Swartz E. A., Johnson A. D., Owens G. K. (1998) Two MCAT elements of the SM α-actin promoter function differentially in SM versus non-SM cells. Am. J. Physiol. 275, C608–C618 [DOI] [PubMed] [Google Scholar]
- 55. Chen J., Yin H., Jiang Y., Radhakrishnan S. K., Huang Z. P., Li J., Shi Z., Kilsdonk E. P., Gui Y., Wang D. Z., Zheng X. L. (2011) Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler. Thromb. Vasc. Biol. 31, 368–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang X., Hu G., Zhou J. (2010) Repression of versican expression by microRNA-143. J. Biol. Chem. 285, 23241–23250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xin M., Small E. M., Sutherland L. B., Qi X., McAnally J., Plato C. F., Richardson J. A., Bassel-Duby R., Olson E. N. (2009) MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 23, 2166–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li S., Wang D. Z., Wang Z., Richardson J. A., Olson E. N. (2003) The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. U.S.A. 100, 9366–9370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Z., Wang D. Z., Pipes G. C., Olson E. N. (2003) Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. U.S.A. 100, 7129–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Creemers E. E., Sutherland L. B., McAnally J., Richardson J. A., Olson E. N. (2006) Myocardin is a direct transcriptional target of Mef2, Tead, and Foxo proteins during cardiovascular development. Development 133, 4245–4256 [DOI] [PubMed] [Google Scholar]
- 61. Holycross B. J., Blank R. S., Thompson M. M., Peach M. J., Owens G. K. (1992) Platelet-derived growth factor-BB-induced suppression of smooth muscle cell differentiation. Circ. Res. 71, 1525–1532 [DOI] [PubMed] [Google Scholar]
- 62. Jawien A., Bowen-Pope D. F., Lindner V., Schwartz S. M., Clowes A. W. (1992) Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J. Clin. Invest. 89, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mack C. P. (2011) Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 31, 1495–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.