

Abstract
A dearth of protein isoform-based clinical diagnostics currently hinders advances in personalized medicine. A well-organized protein biomarker validation process that includes facile measurement of protein isoforms would accelerate development of effective protein-based diagnostics. Toward scalable protein isoform analysis, we introduce a microfluidic “single-channel, multistage” immunoblotting strategy. The multistep assay performs all immunoblotting steps: separation, immobilization of resolved proteins, antibody probing of immobilized proteins, and all interim wash steps. Programmable, low-dispersion electrophoretic transport obviates the need for pumps and valves. A three-dimensional bulk photoreactive hydrogel eliminates manual blotting. In addition to simplified operation and interfacing, directed electrophoretic transport through our 3D nanoporous reactive hydrogel yields superior performance over the state-of-the-art in enhanced capture efficiency (on par with membrane electroblotting) and sparing consumption of reagents (ca. 1 ng antibody), as supported by empirical and by scaling analyses. We apply our fully integrated microfluidic assay to protein measurements of endogenous prostate specific antigen isoforms in (i) minimally processed human prostate cancer cell lysate (1.1 pg limit of detection) and (ii) crude sera from metastatic prostate cancer patients. The single-instrument functionality establishes a scalable microfluidic framework for high-throughput targeted proteomics, as is relevant to personalized medicine through robust protein biomarker verification, systematic characterization of new antibody probes for functional proteomics, and, more broadly, to characterization of human biospecimen repositories.
Keywords: isoelectric focusing, nanoporous reactive polymers, lab-on-a-chip, Western blotting, antibody selection
In this postgenomic period, personalized medicine is poised to benefit from proteomics (1). Proteins are key functional components of living organisms and, thus, offer the potential for high-utility disease diagnostics. Nevertheless, the vast majority of protein biomarker candidates stall at the discovery phase, never making it through validation scrutiny and to the clinic (2). Over the last 15 y an average of just 1–2 new protein biomarkers per year have been approved by the Food and Drug Administration for clinical translation. Compounding concerns, consider two ostensibly accepted protein biomarkers: total prostate specific antigen (PSA) and CA-125. Each protein has been used in diagnostics for screening of prostate and ovarian cancer, respectively. Recently, these cancer screening diagnostics have met with limited success and even controversy (3–5). Consequently, the lackluster progress in protein-based diagnostics highlights important gaps in our approach to defining protein biomarkers. Taken together, a pressing need for innovation exists to expedite translation of informative biomarkers into clinical decision making.
In one important example, the mediocre ability of total and free PSA assays to distinguish between malignant and benign prostatic pathology has spurred study of free PSA isoforms (6, 7). Proteomic studies suggest a promising link between prostate cancer incidence and differential isoform expression in healthy and cancer patient sera (6, 8, 9). Although promising, rigorous validation studies are needed to translate the potential of protein isoforms to the clinic. Immunoassay formats including ELISA and microarray formats offer powerful multiplexing and high-sensitivity performance. Recent ELISA-based formats offer notable gains in analytical sensitivity (10, 11). Nevertheless, mounting evidence suggests that protein isoform “fingerprinting” could advance diagnostic performance (6, 9, 12). Unfortunately, ELISA is severely limited for isoform discrimination because antibodies specific to protein isoforms often do not exist (12). Combining protein separations with antibody interrogation (immunoblotting) allows measurement of protein isoforms. High-performance immunoblotting assays—particularly those with scalable frameworks—would bridge the gap between biomarker discovery and translation to the clinic (2, 13).
Despite their analytical power, conventional benchtop immunoprobing assays consume tremendous time, labor, reagents, and sample resources. Further, performance and implementation characteristics limit scalability, including disjointed workflows requiring manual intervention across multiple instruments, transfer between platforms, and limited quantitation (2, 14). To surmount these shortcomings, analytical technologies based on slab-gel and capillary separations are being introduced (15, 16). A capillary-based separation with surface reaction approach has been commercialized (17, 18). However, the proprietary photoactive capillary surface exhibited low target capture efficiency (ca. 0.01%); necessitated complex interfacing involving pumps, valves, and high voltage control; and availability of characterization data is limited. Consequently, continued advances in targeted proteomics technology are needed, including minimized reagent consumption, reduced complexity including interfacing, and automation.
Here we describe microfluidic integration to realize a streamlined, compact assay platform for high-performance protein isoform measurement (14, 19). Microfluidic integration allows us to harness the favorable scaling of electrokinetic transport and reactions, as well as limit consumption of precious diagnostic samples and costly immunoprobing reagents. By utilizing purely electrophoretic transport through our 3D photoreactive hydrogels, we minimize diffusion distances and maximize binding site densities. We demonstrate 100x gains in analyte capture efficiency, rapid high-resolution protein isoform separations, vanishingly small reagent consumption (< 1 ng of each antibody probe is required, as compared to approximately 1 μg necessary for macroscale immunoblotting), and a “single-channel, single-instrument” design that requires no bulky pumps or valves for device actuation. This rational engineering design strategy advances analytical technology for automated, scalable scrutiny of protein isoforms in complex diagnostic fluids as part of a pipeline to realize personalized proteomics in medicine.
Design Principles
Design of Assay and Microdevice.
Our targeted proteomics platform is a self-contained microfluidic device (Fig. 1 A and B) that executes all protein isoform analysis steps, namely isoelectric focusing (IEF) for separation of protein isoforms, immobilization of separated proteins, probing of immobilized proteins with affinity reagents, and all washing steps. Underpinning integration is a microfluidic strategy comprised of simple straight microchannels, programmable electrophoretic transport, and adaptable 3D hydrogels that switch from molecular sieving matrices to analyte capture scaffolds upon brief exposure to UV. A distinguishing contribution is use of electrophoretic transport for all assay stages with a 3D photoclickable hydrogel matrix, termed a light-activated, volume-accessible gel (LAVAgel) strategy (Fig. 1C).
Fig. 1.

Design and operation of the microfluidic LAVAgel assay for high-specificity protein isoform analysis. (A) Glass microfluidic device with microchannels linking two fluid reservoirs (dye added for clarity). (Scale bar: 2 mm.) (B) The 80-min five-stage immunoprobing assay is completed in a single microchannel. (C) Schematic of microchannel cross-section depicting principle of the LAVAgel: Analytes are electrophoresed through the reactive nanoporous hydrogel, exposed to UV, and covalently immobilized. (Scale bar: 5 μm.) (D) Schematic of reaction between polypeptide backbone and pendant LAVAgel benzophenone groups. Ph denotes phenyl group. For clarity, the electrophilic triplet state of benzophenone, hydrogen abstraction, and radical intermediates are omitted.
Our single-channel, multistage microfluidic assay offers several advantages for protein studies in minimally processed samples. Chiefly, the use of microfluidic technology enables automation and workflow completion in one unified instrument. Five additional advantages arise compared to competing approaches: (i) unification of all assay stages into a single microchannel for minimal operator intervention, (ii) realization of rapid protein separations (< 20 min) owing to miniaturization, (iii) reduced consumption of sparingly available biospecimens and costly affinity reagents, (iv) programmable electrokinetic control to eliminate pumps and valves, thus simplifying external hardware complexity, and (v) no blocking steps prior to antibody probing.
Materials and Transport.
Two major operational advantages underpin microfluidic LAVAgel design and performance (Fig. 1C). Firstly, use of a channel-filling (3D) photoactive hydrogel maximizes protein immobilization efficiency by offering an increased number of available reactive sites, compared to capillary surface capture approaches. For comparison, consider a capillary of inner radius r of approximately 50 μm with a reactive inner surface. The 3D LAVAgel reactive surface area can be approximated as a simple cubic arrangement of ca. 5 × 105 cylindrical nanopores (r = 120 nm, mean pore radius of a 4%T, 2.6%C polyacrylamide gel; ref. 20) packed into a 50 μm radius channel. Because capture efficiency, η, scales with surface area, comparison of surface immobilization to 3D LAVAgel immobilization yields ηgel/ηcap ∼ Agel/Acap ∼ 300 (see SI Text). Thus the LAVAgel offers an approximately two to three orders-of-magnitude increase in capture efficiency over a reactive capillary inner surface. Empirically, we observe an approximately 180-fold improvement in η over that measured for capillary surface photoimmobilization (see Results and Discussion) (17).
Secondly, directed electrokinetic transport through the nanoporous LAVAgel minimizes diffusion distances, yielding efficient mass transport to reaction sites. Short diffusion distances eliminate the need for mixing and reduce the overall reaction time (21), as is important for both the photoimmobilization and the immunoprobing stages. Electromigration of a protein through reactive gel pores can be framed as a homogeneous reaction occurring between two crossing reactant bands (see SI Text) (21). In contrast, boundary layer characteristics can dominate open-surface reactions, limiting analyte transport. The 3D distribution of captured analyte in the LAVAgel approach removes this extra mass transfer resistance term for both analyte capture and antibody probe reactions. Consequently, the appropriate mass transfer timescale for the pseudohomogeneous LAVAgel system can be estimated as tcross = w/urel ∼ 2s, where w is the width and urel is the velocity of a given mobile analyte zone, respectively. As is advantageous to performance, the LAVAgel system is reaction-limited (see SI Text).
Design of Volume-Accessible Photoclickable Hydrogel.
The acrylamide-based LAVAgel copolymer is functionalized using a benzophenone methacrylamide monomer (N-[3-[(4-benzoylphenyl)formamido]propyl] methacrylamide or BPMAC) (Fig. 1D). Free radical polymerization forms the sieving gel in the microchannel. Upon brief exposure to UV light (350–365 nm, ca. 10 s), the gel switches from a molecular sieve to an immobilization scaffold. Exposure to UV promotes the carbonyl groups of the BPMAC monomer termini to an electrophilic triplet state (22). Subsequent hydrogen abstraction is preferential toward C–H bonds in target polypeptides and other buffer constituents (22), leading to formation of stable covalent linkages to the gel matrix. Importantly, the use of polyacrylamide gels with strong resistance to nonspecific adsorption and this UV-initiated covalent attachment mechanism eliminates the need for separate and time-consuming blocking steps common with conventional blotting materials (e.g., PVDF, nitrocellulose).
Results and Discussion
Integrated Protein Isoform Assay Operation.
We designed our multistage assay to reduce instrument complexity. Consequently, we employ a single microchannel and programmable electrophoretic transport for all stages, including washing. With performance on par with conventional slab-gel and capillary methods, the total assay duration was < 120 min, with hands-on time of < 15 min. To facilitate technical and biological replicates, the prototype glass device houses four separation channels per fluid reservoir pair (Fig. 1A). With four reservoir pairs on each chip in this prototype, 16 channels can be run in parallel. Results from a complete assay are shown in Fig. 2 and detailed here.
Fig. 2.

Characterization of protein isoforms using the single-microchannel 80-min LAVAgel immunoblot. Fluorescence micrographs show (A) loading and IEF of a CE540-labeled protein ladder with 617-nM green WT GFP, and (B) IEF readout via UV excitation. (C) After UV gel photoactivation, the pH gradient is washed out with retention of a portion of each WT GFP isoform. (D and E) Antibody probing of WT GFP with 100 nM Texas red-labeled polyclonal antibody (pAb*) demonstrates specificity and low-background. RFU, relative fluorescence units; CytC, cytochrome C; LCL, lentil lectin; Mb, myoglobin; CA, carbonic anhydrase; BLG, β-lactoglobulin; GOx, glucose oxidase.
During the first assay stage (Fig. 2 A and B), IEF is used to separate proteins based on differences in pI. IEF is an ideal separation mechanism for resolving protein isoforms which may have only slight differences in molecular mass. IEF is achieved by establishing a pH gradient along the channel length using a commercially available mixture of polyprotic amino carboxylic acids (carrier ampholytes) that buffer at their pI values (23). Analysis of a cocktail of fluorescent pI marker peptides revealed that a linear broad range pH gradient (pH 4–8.7) was established in < 20 min (within-chip % relative SD in slope = 6.5%, R2 > 0.99). Focusing of a fluorescent model protein (WT GFP) yielded clear resolution of three well-characterized isoforms (Fig. 2B), with baseline resolution of the two closest neighbors. A resolution of ΔpI = 0.15 pH units was achieved with broad pH range ampholytes (see SI Text). We estimate a peak capacity of 110 ± 22 (n = 3), on par with conventional IEF (23, 24). Using a starting volume of 3 μL and a detectable concentration minimum of approximately 0.1 nM (2.7 ng/mL), we estimate that approximately 8 pg of starting material is needed for detection of WT GFP during IEF, with just 15 fg injected into the 5.8-nL microchannel.
After IEF, the second assay stage is a transition to in situ immunoprobing of the IEF resolved species (Fig. 2 B and C). As discussed in Design Principles, the IEF-focused species and the microfluidic LAVAgel are exposed to UV light to induce photoimmobilization of species to the light-activatable copolymer. The IEF pH gradient is then exchanged to uniform pH buffer conditions using a 20 min chemical mobilization step (Fig. 2C). Gradient “washout” by chemical mobilization eliminates the need for pumps and valves.
Finally, during the third stage (Fig. 2 D and E), immunoaffinity probes are electrophoresed through the protein-decorated 3D LAVAgel. In addition to yielding efficient mass transfer, as described in Design Principles, electrophoretic transport simplifies hardware interfacing and, importantly, requires approximately 1 ng of antibody. Electrophoretic washout of unbound probe reveals the target protein isoform pattern (Fig. 2E). Two color fluorescence imaging shows the resolved WT GFP isoforms (green) and the resultant signal from a red-labeled polyclonal antibody for WT GFP. Comparison of the blot signal to the protein signal reveals specificity for GFP and low off-target background signal, even amidst an approximately 20-fold excess of off-target ladder proteins. A fourth GFP isoform is apparent at pI 5.33 only after immunoprobing, an intriguing consequence hypothesized to arise from a protein charge photoswitching process that is currently under study.
Characterization of Microfluidic LAVAgel Photoimmobilization.
We sought to quantitatively assess performance of our volume-accessible microfluidic LAVAgel material. The LAVAgel capture efficiency is a critical performance metric, as previous reports of analyte photocapture on capillary surfaces (in both research and commercial instruments) report strikingly low capture efficiencies of approximately 0.01% (17). Characterization requires assessment of analyte capture efficiency in the nonuniform pH conditions of IEF, as has not been previously considered to our knowledge. Using a two-pronged approach, we measured capture efficiency for both: (i) a well-characterized three isoform model protein (WT GFP) and (ii) fluorescently labeled ampholytes distributed across a broad pH range.
We first define capture efficiency, η, as the ratio of fluorescence signal measured after photocapture (Iimmobilized—i.e., after IEF and pH gradient washout) to the fluorescence signal measured during IEF (IIEF) or η = IImmobilized/IIEF·ε·100%. Recall that the pH of the washout buffer differs from the local pH during IEF (pHwashout = 9.9, whereas 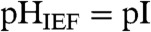 ), thus correction for the anticipated influence of pH on the fluorescence signal of each species is needed. We employ an empirical correction factor, ε, determined to be ε ≈ 0.75 for WT GFP and ε ≈ 1.0 for all fluorescently labeled ampholytes (see SI Text).
), thus correction for the anticipated influence of pH on the fluorescence signal of each species is needed. We employ an empirical correction factor, ε, determined to be ε ≈ 0.75 for WT GFP and ε ≈ 1.0 for all fluorescently labeled ampholytes (see SI Text).
In the WT GFP portion of the study, we observed a maximum photoimmobilization efficiency of 1.8% with photoimmobilization well-described by a first-order model relating η to the UV exposure time (Fig. 3A). Importantly, the capture efficiency is a more than 100-fold improvement over reported surface photoimmobilization values (η ∼ 0.01%) (17). As discussed, we attribute the substantial increase in capture efficiency measured here in the 3D LAVAgel to the high surface area offered by the reactive nanoporous matrix (25). The reaction time constant is 5.5 s. The small time constant suggests that the photocapture dynamics of the microfluidic LAVAgel system are compatible with IEF, but likely also compatible with a wide range of on-chip and nonequilibrium separation methods, including protein sizing by SDS-PAGE as is currently under study in our group.
Fig. 3.

Characterization of LAVAgel photoimmobilization kinetics, capture efficiency, and pH dependence. (A) LAVAgel capture efficiency and resolution losses are optimized by tuning UV exposure duration. Photoactive LAVAgel (BPMAC+, 15 μM WT GFP, ± SD, n = 4, black solid circles) is compared to a nonphotoactive negative control (BPMAC−, red squares). (Inset) Fluorescence micrographs show captured WT GFP fluorescence. Blue open circles and inset images (i–iii) show separation resolution loss for WT GFP isoforms during defocusing. GFP concentration is 617 nM, resolution measured between the pI 5.00 and 5.19 isoforms. (B) Reporter ampholytes (ampholyte*) allow measurement of capture efficiency under focusing conditions for a broad pH range. (Left) Fluorescence micrographs show pI ladder and photocaptured reporter ampholytes after pH gradient washout. (Right) Reporter ampholyte capture efficiency versus pH in BPMA+ and BPMA− LAVAgels, black arrows indicate artifact peaks caused by enhanced local photobleaching of reporter ampholytes in the vicinity of pI marker bands (see SI Text; [ampholyte ∗ ] = 0.025% wt/vol, gray envelopes are ± SD, n = 4).
Dispersion (band broadening) added during the photoimmobilization process will reduce the information content in the immobilized separation, thus reducing the overall performance of the integrated assay. Thus, an assay design trade-off exists between the dominant transport processes and the duration of UV exposure after IEF. Although IEF is an equilibrium separation method, we observed nonnegligible electroosmotic drift during IEF (1.0–3.3 μm/s for EIEF = 300 V/cm at IEF completion). Drift is attributed to the slight negative charge of polyacrylamide gels (23). A 10-s UV exposure yielded a drift distance of 10–33 μm for a focused protein band, on par with the average peak width of focused GFP isoforms (100 μm). Thus, photoimmobilization under IEF conditions should adversely impact overall assay resolution and total peak capacity. Consequently, we limited captured analyte dispersion by performing photocapture of IEF bands under zero-field conditions (E = 0 V/cm or floating). Under zero-field conditions, the drift and the focusing force of IEF go to zero, making molecular diffusion the dominant transport process. Using empirically determined diffusion-associated resolution losses and capture efficiencies for two neighbor peaks (two isoforms of GFP) in the microfluidic LAVAgel system, we found that a 10-s UV exposure under zero-field conditions confers 84% of the achievable capture efficiency for a loss in separation resolution of just 22% from that in the focused state (Fig. 3A).
In the second portion of the photocapture efficiency study, we characterized photoimmobilization performance across a broad pH range (Fig. 3B). Given that the ampholytes themselves are excellent structural analogs to polypeptides, we imaged fluorescently labeled ampholytes (reporter ampholytes) to map η across pH 5–7.5, as this range encompasses a preponderance of protein isoforms (26). To create reporter ampholytes with a broad continuum in pI, we fluorescently labeled the amine termini of the ampholytes using a CE540 fluorophore that offers a charge-compensating reaction mechanism. This charge-compensation mechanism is thought to avoid the charge heterogeneity that makes other reactive dyes largely incompatible with IEF (27). As shown in Fig. 3B, the immobilized reporter ampholytes distributed along the microfluidic LAVAgel reveal an approximately twofold monotonic rise in the capture efficiency from the acidic to basic end of the pH range studied. In absolute terms, the capture efficiency for the reporter ampholytes ranges from 7.2 ± 2.0% (near pH 5, n = 4) to 13.3 ± 1.7% (near pH 7.5, n = 4) in the LAVAgel. From an assay design perspective, the pH response of η is suitable for protein isoform analyses, given the absence of a strong bias toward any particular pH zone and the fact that protein isoforms are typically clustered over a relatively tight pI range (26). We hypothesize that the increase in η with pH stems from a change in the chemical properties of the ampholyte species, which are also graded along the pH axis (28).
A corollary investigation was undertaken to understand the high capture efficiencies observed using reporter ampholytes, as compared to the WT GFP single protein analysis. We hypothesize that the hydrophobic structure of CE540 (27) may contribute to a higher η by increasing weak “precovalent” interactions of labeled species with the LAVAgel matrix. To elucidate the role of the CE540 dye in photocapture efficiency, we studied two model proteins using the same approach applied to the reporter ampholytes. Both GFP and PSA were labeled with CE540 (GFP*, PSA*) and subjected to IEF and photocapture. Both species exhibited η on par with the reporter ampholytes, here ηGFP∗ = 10.1 ± 1.91% (n = 8) and ηPSA∗ = 9.92 ± 0.86% (n = 3). Interestingly, and with perhaps important implications, we found appreciable effects of the CE540 labeling on the conformational heterogeneity and capture efficiency of WT GFP, see SI Text. Specifically, all CE540 labeled proteins/peptides had capture efficiencies notably higher than unlabeled protein (compare ηGFP∗ = 10% ± 1.91% to ηGFP = 1.30 ± 0.17%, n = 44). Results suggest that precovalent interactions stemming from increased analyte hydrophobicity (affected by both labeling and denaturation state) enhance capture efficiency.
Microfluidic LAVAgel Analysis of PSA Isoforms in Crude Cell Lysate.
We first tested the LAVAgel assay on purified unlabeled PSA as a well-controlled model system (Fig. 4A). PSA was probed after IEF and photoimmobilization using sequential introduction of specific primary and secondary detection antibodies. Two major isoforms with pI values of 6.27 ± 0.02 and 6.77 ± 0.04 (n = 4) are baseline resolved, accompanied by several minor peaks below baseline resolution. Gold-standard comparisons to macroscale slab-gel IEF (see SI Text) and capillary IEF (29) show good agreement between the isoform patterns.
Fig. 4.

LAVAgel assay enables quantitation of PSA isoforms in minimally processed prostate cancer cell lysate and human sera. (A) Fluorescence micrographs and electropherograms for probing of unlabeled PSA purified from human seminal fluid (500 nM): focused pI markers, primary (1°), and secondary (2°) antibody probe signals. Bracketed peak areas used to construct calibration curves. (B) Linear PSA calibration curves for primary (black circles) and secondary (red squares) antibody readouts (RFU, relative fluorescence units; ± SD, n = 4 for all points except 5 nM, n = 2). (C) Primary antibody probing of endogenous PSA isoforms in lysate from a PSA-producing cell line (LAPC-4 cells, +) with negative control lysate (DU145 cells, −). (D) Serum samples from metastatic prostate cancer patients probed with primary antibody to PSA (patients 1 and 2), alongside a low-PSA negative control serum (−).
Linear calibration curves were generated for two scenarios: probing captured PSA with a fluorescently labeled primary antibody and, as is more broadly relevant, probing of the primary antibody with a labeled secondary antibody. In Fig. 4B, the relationship between the spiked PSA concentration and fluorescence readouts for the dominant isoform (pH 6.0–6.5) is linear from approximately 10 to 500 nM. Quantitative capacity is maintained to ca. 5 nM PSA (165 ng mL-1) or ca. 1.1 pg of PSA. Improvement of the absolute lower limit of detection should be feasible through, for example, incorporation of amplified readout approaches to yield a similar sensitivity to conventional benchtop 2D electrophoresis with Western blotting (ca. 0.1 ng mL-1) (6, 19). Such approaches should also increase the dynamic operating range of the present assay. The PSA isoform characterization study allows inference of the stoichiometry of secondary:primary antibody probing from the ratio of the respective fluorescence traces and indicates negligible effects of the gel pore environment on achievable probe-target valency (Fig. 4B and SI Text).
We next assayed a PSA-producing cell line relevant to the study of prostate cancer (Fig. 4C). Here, we quantify endogenous PSA isoforms present in 3 μL of minimally processed lysate from LAPC-4 cells derived from a lymph node of a human prostate cancer patient. The probed LAPC-4 lysate presents a distinctive four peak pattern in the pI 6.9–7.9 range that is similar to slab-gel assays of PSA purified from LAPC-4 cell culture medium (8, 30). As a negative control, lysate from a PSA negative cell line (DU145) was assayed and shows no detectable PSA isoform readout, as expected. The crude cell lysate samples yielded some nonspecific signal near the anodic well, likely due to that channel region being the electrophoretic introduction point for both sample and labeled antibody probe. The total PSA concentration via the microfluidic LAVAgel assay was determined to be 27.8 ± 4.7 nM (n = 4) using the purified PSA calibration curve of Fig. 4B, in reasonable agreement with benchmark ELISA measurements (see Materials and Methods).
To validate the capability of the microfluidic LAVAgel assay to measure immunoreagent isoform specificity, we compared the isoform distribution of IEF-focused CE540-labeled PSA* to the fluorescence readout after capture and probing with both monoclonal and polyclonal PSA antibodies (see SI Text). Ratiometric comparison of the probed and focused PSA* signals suggests uniform probing across the pH region of interest, for both polyclonal and monoclonal detection antibodies. Use of high-throughput LAVAgel IEF assays for isoform-specific immunoreagent selection assays could enable rapid development of next-generation ELISA microplate-based bioassays and clinical diagnostics with isoform resolution.
Microfluidic LAVAgel Analysis of PSA Isoforms in Metastatic Prostate Cancer Patient Sera.
We further demonstrated clinical utility of the LAVAgel assay by separating and probing PSA isoforms in minimally processed sera from advanced metastatic prostate cancer patients (Fig. 4D). The low-volume requirement of the assay (3 μL) is critical for screening of often ephemeral and volume-limited human biospecimen repositories.
Human sera from two patients were assayed in addition to a low-PSA negative control sample, all at 10x dilution. The two PSA+ samples each show three major PSA isoforms falling within the pI 6.4–7.5 range, in good agreement with comparatively laborious slab-gel IEF studies (6, 8). Patient-specific differences in PSA isoform representation and pI are clearly apparent, recapitulating the potential utility of isoform ratio measurements in clinical diagnostics and personalized medicine (6, 7). Ongoing studies are currently in progress to validate the LAVAgel assay for rapid, high-throughput classification of cancer and benign prostate pathology patient groups.
Conclusions
Quantitative, robust protein isoform assays designed for analysis of minimally processed fluids are needed to advance diagnostics for personalized medicine. We demonstrate a quantitative protein isoform assay that harnesses microfluidic integration, fully electrophoretic control, and a photoactivatable 3D hydrogel for automated, pump-free operation. The automated assay reports isoform levels in 80–120 min, a 5- to 15-fold improvement in assay time over 2D electrophoresis with Western blotting and a twofold improvement over capillary immunoblotting (6, 17). Two aspects of our design rationale distinguish the present study and underpin observed performance gains, as compared to currently available immunoblotting assays. Firstly, the 3D LAVAgel significantly boosts protein immobilization and probing efficiency over 2D surface capture approaches owing to the availability of approximately 102–103 more reactive sites per unit channel length and use of directed electrokinetic transport through the nanoporous LAVAgel. The strategy yields capture efficiencies that are two to three orders-of-magnitude higher than competing surface capture approaches (0.01% vs. 1.3–13% demonstrated here).
In contrast to ELISA-based approaches, the microfluidic LAVAgel platform allows quantitation of distinct biomarker isoforms and requires just a single primary antibody (not capture and detection matched pairs) and an optional secondary detection antibody. We demonstrate PSA isoform detection in crude cell lysate and serum repository biospecimens from metastatic prostate cancer patients. Microfluidic integration yields sparing consumption of precious biospecimens (1–5 μL), low consumption of costly probing antibodies (1 ng antibody), and total assay completion in one unified instrument. The measurement operating range of the assay was optimized for clinical relevance to PSA and isoforms in prostate cancer sera. Adaptation and optimization should allow for protein isoform assessment (including quantitation) in other human diagnostic fluids and tissue samples (e.g., from laser capture microdissection). This first report of a self-contained, electronically controlled immunoblotting platform fills an important gap in translation of promising protein biomarkers from as-discovered to validated high-utility biomarkers of disease. We continue to actively develop the platform as a core technology adaptable to protein biomarker scrutiny in a broad range of local and systemic diseases, rapid analysis of promising diagnostic biomarkers from biospecimens only available in minute volumes. Continued innovation is focused on realizing higher throughput through scaleup of electrode and liquid handling architectures for simultaneous analysis of panels of protein biomarkers in larger patient sample sets while maintaining compatibility with existing microplate handling systems.
Materials and Methods
Microfluidic Assay Instrumentation.
Fabrication of microchannels in optical white soda lime glass, high-voltage control, fluorescence microscope, and UV exposure system details are in SI Text.
Glass channels were functionalized with acrylate-terminated self-assembled monolayers (19). Microfluidic LAVAgels were fabricated via introduction of a gel precursor solution by capillary action. The precursor contained 4% wt/vol total acrylamide (4%T) with 2.6% of the total as the cross-linker bisacrylamide (2.6%C), 2% Pharmalyte 3–10 titrated to pH 9.9 with NaOH (17-0456-01; GE Healthcare), 3% CHAPS detergent (C9426; Sigma), 10% sorbitol, 200 mM nondetergent sulfobetaine-256 (17236; Sigma), 4.5 mM BPMAC (see Reagents and Samples). The initiators ammonium persulfate (0.08%, A3678; Sigma) and N,N,N',N'-tetramethylethylenediamine (0.08% vol/vol, T9281; Sigma) were added just before introduction of degassed precursor to channels. Just after visible gelation of the excess precursor, wells were flushed and replaced with gel buffer (details of buffers used and microfluidic LAVAgel chip operation protocol are provided in SI Text).
Reagents and Samples.
BPMAC monomer was synthesized in-house and verified by 1H NMR and mass spectrometry as described in SI Text. The monomer was added to BPMAC+ gel precursor solutions at 4.5 mM (ca. 1 mol % with respect to acrylamide) from a 100 mM stock in DMSO. BPMAC− precursors contained an equivalent volume of DMSO lacking BPMAC. Purified proteins, antibodies, and fluorescence labeling protocols are described in SI Text; antibodies were used at 100 nM. LAPC-4 and DU145 lysates were purified in P-6 Bio-Spin columns (Bio-Rad) and added to samples at 2x dilution; serum samples from Stanford University Medical Center Oncology Clinic and negative control serum were used directly at 10x dilution (see SI Text).
Equal volumes of a set of fluorescent IEF pI markers with absorption maxima in the near UV (pI 4.0, 4.5, 5.5, 6.2, 6.6, 7.2, 7.6, and 8.7) were mixed in a cocktail and added to samples at 20x dilution (89827 and related products; Sigma). WT GFP was included as a loading and immobilization standard along with the pI marker cocktail. Samples in loading buffer were titrated to pH 9.9 with 1M NaOH just prior to electrophoretic loading (see SI Text).
Benchmark Analysis.
ELISA, slab-gel, and microplate experiments are detailed in SI Text. The LAPC-4 cell lysate expressed PSA at a concentration of 19.5 ± 2.7 nM, as quantified by ELISA (n = 8).
Data Acquisition and Analysis.
Whole channel imaging at 10x was conducted via stitching of adjacent, overlapping CCD images in ImageJ (National Institutes of Health) to produce full gel channel images and electropherograms as previously described (19). Imaging scans along both streets required approximately 40 s to complete (see SI Text).
Supplementary Material
Acknowledgments.
The authors gratefully acknowledge O. Lee (Frechet group, UC Berkeley) for NMR assistance and A. Tentori, L. Bugaj, S. Tia, and O. Westesson for helpful discussions. A.J.H. is a Department of Defense National Defense Science and Engineering Graduate Research Fellow. A.E.H. is an Alfred P. Sloan Research Fellow (chemistry). Financial support from New Innovator Grant 1DP2OD007294 (to A.E.H.) from the National Institutes of Health (NIH) Office of the Director is gratefully acknowledged, along with NIH Grant 5R01CA121460 (to D.M.P.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108617109/-/DCSupplemental.
References
- 1.Brennan DJ, O’Connor DP, Rexhepaj E, Ponten F, Gallagher WM. Antibody-based proteomics: Fast-tracking molecular diagnostics in oncology. Nat Rev Cancer. 2010;10:605–617. doi: 10.1038/nrc2902. [DOI] [PubMed] [Google Scholar]
- 2.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 3.Zhu CS, et al. A framework for evaluating biomarkers for early detection: Validation of biomarker panels for ovarian cancer. Cancer Prev Res. 2011;4:375–383. doi: 10.1158/1940-6207.CAPR-10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esserman L, Shieh Y, Thompson I. Rethinking screening for breast cancer and prostate cancer. JAMA. 2009;302:1685–1692. doi: 10.1001/jama.2009.1498. [DOI] [PubMed] [Google Scholar]
- 5.Etzioni R, et al. The case for early detection. Nat Rev Cancer. 2003;3:243–252. doi: 10.1038/nrc1041. [DOI] [PubMed] [Google Scholar]
- 6.Jung K, et al. Analysis of subforms of free prostate-specific antigen in serum by two-dimensional gel electrophoresis: Potential to improve diagnosis of prostate cancer. Clin Chem. 2004;50:2292–2301. doi: 10.1373/clinchem.2004.040469. [DOI] [PubMed] [Google Scholar]
- 7.Sarrats A, et al. Differential percentage of serum prostate-specific antigen subforms suggests a new way to improve prostate cancer diagnosis. Prostate. 2010;70:1–9. doi: 10.1002/pros.21031. [DOI] [PubMed] [Google Scholar]
- 8.Tabares G, et al. Different glycan structures in prostate-specific antigen from prostate cancer sera in relation to seminal plasma PSA. Glycobiology. 2005;16:132–145. doi: 10.1093/glycob/cwj042. [DOI] [PubMed] [Google Scholar]
- 9.Kyselova Z, et al. Alterations in the serum glycome due to metastatic prostate cancer. J Proteome Res. 2007;6:1822–1832. doi: 10.1021/pr060664t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaster RS, et al. Matrix-insensitive protein assays push the limits of biosensors in medicine. Nat Med. 2009;15:1327–1333. doi: 10.1038/nm.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rissin DM, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28:595–600. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vickers AJ, et al. A panel of kallikrein markers can reduce unnecessary biopsy for prostate cancer: Data from the European Randomized Study of Prostate Cancer Screening in Göteborg, Sweden. BMC Med. 2008;6:19–28. doi: 10.1186/1741-7015-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zolg W. The proteomic search for diagnostic biomarkers: Lost in translation? Mol Cell Proteomics. 2006;5:1720–1726. doi: 10.1074/mcp.R600001-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.He M, Herr AE. Automated microfluidic protein immunoblotting. Nat Protoc. 2010;5:1844–1856. doi: 10.1038/nprot.2010.142. [DOI] [PubMed] [Google Scholar]
- 15.Ciaccio MF, Wagner JP, Chuu CP, Lauffenburger DA, Jones RB. Systems analysis of EGF receptor signaling dynamics with microwestern arrays. Nat Methods. 2010;7:148–155. doi: 10.1038/nmeth.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson GJ, Cipolla CM, Kennedy RT. Western blotting using capillary electrophoresis. Anal Chem. 2011;83:1350–1355. doi: 10.1021/ac102671n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Neill RA, et al. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc Natl Acad Sci USA. 2006;103:16153–16158. doi: 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan AC, et al. Nanofluidic proteomic assay for serial analysis of oncoprotein activation in clinical specimens. Nat Med. 2009;15:566–571. doi: 10.1038/nm.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes AJ, Herr AE. Quantitative enzyme activity determination with zeptomole sensitivity by microfluidic gradient-gel zymography. Anal Chem. 2010;82:3803–3811. doi: 10.1021/ac100201z. [DOI] [PubMed] [Google Scholar]
- 20.Holmes DL, Stellwagen NC. Estimation of polyacrylamide gel pore size from Ferguson plots of linear DNA fragments II. Comparison of gels with different crosslinker concentrations, added agarose and added linear polyacrylamide. Electrophoresis. 1991;12:612–619. doi: 10.1002/elps.1150120903. [DOI] [PubMed] [Google Scholar]
- 21.Matta A, et al. Computational study of band-crossing reactions. J Microelectromech Syst. 2004;13:310–322. [Google Scholar]
- 22.Dorman G, Prestwich GD. Benzophenone photophores in biochemistry. Biochemistry. 1994;33:5661–5673. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 23.Righetti PG. Isoelectric Focusing: Theory, Methodology and Applications. New York: Elsevier; 1983. [Google Scholar]
- 24.Sommer GJ, Singh AK, Hatch AV. On-chip isoelectric focusing using photopolymerized immobilized pH gradients. Anal Chem. 2008;80:3327–3333. doi: 10.1021/ac702523g. [DOI] [PubMed] [Google Scholar]
- 25.Squires TM, Messinger RJ, Manalis SR. Making it stick: Convection, reaction and diffusion in surface-based biosensors. Nat Biotechnol. 2008;26:417–426. doi: 10.1038/nbt1388. [DOI] [PubMed] [Google Scholar]
- 26.Gianazza E, Righetti PG. Size and charge distribution of macromolecules in living systems. J Chromatogr. 1980;193:1–8. [Google Scholar]
- 27.Craig DB, Wetzl BK, Duerkop A, Wolfbeis OS. Determination of picomolar concentrations of proteins using novel amino reactive chameleon labels and capillary electrophoresis laser-induced fluorescence detection. Electrophoresis. 2005;26:2208–2213. doi: 10.1002/elps.200410332. [DOI] [PubMed] [Google Scholar]
- 28.Righetti PG, Simo C, Sebastiano R, Citterio A. Carrier ampholytes for IEF, on their fortieth anniversary (1967–2007), brought to trial in court: The verdict. Electrophoresis. 2007;28:3799–3810. doi: 10.1002/elps.200700232. [DOI] [PubMed] [Google Scholar]
- 29.Donohue MJ, et al. Capillary electrophoresis for the investigation of prostate-specific antigen heterogeneity. Anal Biochem. 2005;339:318–327. doi: 10.1016/j.ab.2005.01.043. [DOI] [PubMed] [Google Scholar]
- 30.Vaisanen V, et al. Characterization and processing of prostate specific antigen (hK3) and human glandular kallikrein (hK2) secreted by LNCaP cells. Prostate Cancer Prostatic Dis. 1999;2:91–97. doi: 10.1038/sj.pcan.4500289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.