

Abstract
Grainyhead genes are involved in wound healing and developmental neural tube closure. In light of the high degree of similarity between the epithelial-mesenchymal transitions (EMT) occurring in wound healing processes and the cancer stem cell-like compartment of tumors, including TGF-β-dependence, we investigated the role of the Grainyhead gene, Grainyhead-Like-2 (GRHL2) in oncogenic EMT. GRHL2 was down-regulated specifically in the claudin-low subclass breast tumors and in basal-B subclass breast cancer cell lines. GRHL2 suppressed TGF-β-induced, Twist-induced or spontaneous EMT, enhanced anoikis-sensitivity, and suppressed mammosphere generation in mammary epithelial cells. These effects were mediated in part by suppression of ZEB1 expression via direct repression of the ZEB1 promoter. GRHL2 also inhibited Smad-mediated transcription and it upregulated mir200b/c as well as the TGF-β receptor antagonist, BMP2. Lastly, ectopic expression of GRHL2 in MDA-MB-231 breast cancer cells triggered a mesenchymal-to-epithelial transition and restored sensitivity to anoikis. Taken together, our findings define a major role for GRHL2 in the suppression of oncogenic EMT in breast cancer cells.
Keywords: Epithelial-mesenchymal transition, EMT, Grainyhead, anoikis, ZEB1, TGF-β, breast cancer
INTRODUCTION
The oncogenic EMT is a transcriptional re-programming event that endows restricted subclasses of tumor cells with enhanced metastatic and stem cell-like properties. These properties include increased migration/invasion, chemo- and radiation-resistance, anoikis-resistance and extraordinary tumor initiation frequency, a cancer stem cell-like capability (1, 2). In breast cancer, two subclasses of tumors, “claudin-low” and “metaplastic”, exhibit frank EMT-like gene expression signatures. These are among the most aggressive and least treatment-responsive tumors (3).
In general, TGF-β signaling can either promote or suppress tumorigenicity and progression (4, 5). It can suppress tumors through Smad-mediated induction of cyclin dependent kinase-inhibitor genes such as p15, and inactivating mutations in this pathway occur in certain tumors. In other contexts, however, TGF-β can promote tumor progression by supporting oncogenic EMT induction, through both transcriptional and non-transcriptional mechanisms. For example, in claudin-low and metaplastic mammary tumors -- as well as in the stem cell-like CD44high/CD24low tumor cell subpopulation fractionated from breast tumors of other subclasses -- TGF-β pathway components are strikingly up-regulated and, indeed, TGF–β receptor kinase inhibitors partially revert the EMT-related gene expression profile (3, 6). This indicates a pro-tumorigenic role of the TGF-β pathway in this context.
Cell culture models confirm these conclusions. An extensively characterized SV40 large T/hTERT-immortalized mammary epithelial cell line, HMLE, exhibits spontaneous EMT in a subpopulation of CD44high/CD24low cells (7). This EMT is accompanied by up-regulation of autocrine TGF-β signaling, mainly through down-regulation of antagonists such as BMP2/4. Reversion to an epithelial phenotype could be achieved by inhibition of this signaling pathway. Moreover, even Twist-induced EMT in this system was partially dependent upon autocrine TGF-β signaling, demonstrating its functional significance in multiple contexts.
The similarities between oncogenic and wound-healing-related EMT, including TGF-β-dependence, have been documented (8–10). In this light, we hypothesized that perhaps other “dedicated” wound-healing genes might prove important in oncogenic EMT. Grainyhead family genes have been shown to play an important role in wound-healing, epidermal integrity and the mechanistically related process of embryonic neural tube closure (11–14). Relevant, albeit limited, Grainyhead family target genes identified so far include E-cadherin, claudin-4, desmoglein-1, transglutaminase-1, rho-GEF19, several Zelda-target genes expressed during the maternal-zygotic transition in Drosophila, and telomerase (11, 12, 14–17). With regard to cancer, GRHL2 gene amplification has been noted in several tumor types, including breast cancer, and suppressed death receptor expression, conferring resistance to apoptosis mediated by the corresponding ligands, indicating that GRHL2 was a potential oncogene (18). On the other hand, GRHL3 was recently shown to suppress squamous cell carcinoma, due to its activation of PTEN expression (19).
Inspired by its role in wound-healing, we hypothesized and report here that GRHL2 suppressed EMT mediated by the TGF-β signaling pathway. Consistent with this effect, GRHL2 was down-regulated specifically in EMT-dependent mammary tumors (claudin-low, metaplastic) and cell lines (basal B). ZEB1 was found to be required for EMT and was a direct target for repression by GRHL2. GRHL2 also enhanced anoikis-sensitivity. These data suggest an EMT-suppressive function of GRHL2 that is down-regulated in the context of TGF-β/EMT-driven tumor types.
MATERIALS AND METHODS
Cell lines
HMLE, HMLE+twist-ER, and HMLE+Ras cells (HMLER) were generous contributions from R. Weinberg (The Whitehead Institute). HMLE and HMLE+Twist-ER were grown in a 1:1 mixture of MEGM (Lonza) and [DME/F12(Gibco)+5% horse serum+1X penicillin-streptomycin-glutamine (PSG) +10 μg/ml insulin, 10 ng/ml EGF, 0.5 μg/ml hydrocortisone], where indicated, 4-hydroxytamixofen (10ng/ml) was added to the HMLE+Twist-ER cells to activate the Twist-ER protein. HMLER cells were grown in MEGM. MDA-MB-231LN were provided by E. Pugacheva (West Virginia University) and were grown in Advanced DMEM (Invitrogen)+10% fetal bovine serum+1X penicillin-streptomycin-glutamine (PSG).
Generation of stable cell lines by retroviral transduction
Human GRHL2 was amplified from a template purchased from Open Biosystems (MHS4426-99625903) and subcloned by standard molecular biology methods into the pMIG or MSCV-IRES-puro retrovirus (Addgene; XhoI site). Retroviruses were packaged and amplified in GP2+293T cells by transfection of 4.5 μg of retroviral plasmid and 2.5 μg of pCMV-VSV-G per 60mm2 dish of cells, using Mirus TransIT reagent. Viral supernatants were collected forty-eight hours later, filtered through 0.45 micron filters (Whatman) and 0.6 ml of supernatant was used to infect one well of a 6-well dish of target cells by centrifugation at 1400 rpm for one hour followed by 6 hour to overnight incubation. Infected cells were either selected for puromycin (2 μg/ml for HMLE, 0.5 μg/ml for MDA-MB-231) or flow-sorted for GFP, followed by western blot analysis to verify expression.
Generation of stable cell knockdown cell lines by lentiviral transduction, and transient knockdown (siRNA)
GRHL2, ZEB1 and scrambled control shRNAs were purchased from Open Biosystems in the pGIPZ vector. (Catalogue numbers: shGRHL2a: RHS4430-99291384; shGRHL2b: RHS4430-99614394. shZEB1a: V3LHS_356186 and shZEB1b: 3 V3LHS_356187; the latter were also sub-cloned into vector pTRIPz using Mlu1/Xho fragments). SiZEB1 Smartpool was from Dharmacon catalogue number (L-006564-01-0005) and was transfected transiently using Lipofectamine RNAi-max (Invitrogen).
Mammosphere assay
Mammospheres were seeded at 1×104 cells/well of a 6 well Ultra Low Cluster Plate (Corning) and grown for 7 to 10 days in the appropriate growth media + 0.5% methylcellulose (MC). Wells were fed every third day with 1ml media+MC. Total mammospheres/well were counted and the size cutoff was set at 150mm in diameter; the same cells were plated at 2×105 cells/well of a 6-well plate and the number of cells was counted each day for 4 days, to measure normal growth rate. Error bars represent the standard deviation of replicates.
Anoikis assays
Cells were dissociated using TrypLE Express (Invitrogen), counted and a fixed amount (1×105 to 2×105 cells/well of a 6-well Ultra-Low Attachment Cluster Dish (Costar) were suspended in normal growth media +0.5%MC for the indicated for 6 to 24 hours to induce anoikis. For Cell Death ELISA (CDE) analysis of apoptotic DNA fragmentation, the cells were collected in 3 volumes of media and then spun down at 1500rpm for 3min. The pellet was then washed with D-PBS (Invitrogen) transferred to a microfuge tube, pelleted at 7,000 rpm for 15 seconds and lysed in PBS+0.5% Triton X 100+10mM EDTA (Note: the lysis buffer with the Cell Death ELISA kit from Roche was found to lyse cells inadequately). Lysates were incubated on ice for fifteen minutes with occasional mixing, and then centrifuged at 13,000rpm for 12 minutes. The supernatants were subjected to the CDE according to the manual provided with the kit (Roche). Alternatively, percentage cell death was determined by a trypan blue exclusion assay, wherein cells were suspended in same manner but collected and re-suspended in Accumax (Innovative Cell Technology) to ensure a single cell suspension. After brief incubation (2–3min) trypan blue was added to this solution and the % dead cells were immediately counted on a hemacytometer. All samples were analyzed in either duplicate or triplicate, and time-zero cell death values were subtracted from the data presented here.
Three dimensional culture
3D Matrigel culture methods were adapted from :(20). To summarize, 2.5×103 cells/well were seeded onto 8 well chamber slides (Labtek) where 45ul Matrigel(Trevigen) had been evenly distributed. The cells were overlaid with the appropriate growth media (see above) +2.5% matrigel. After 6 days in culture 3D migration/invasion was quantified by counting the number of structures which had formed protrusions vs. those which grew as lobular structures defined by their contact with matrix. (see images for clarification). At least 200 structures were counted per experiment; error bars represent the standard deviation across three samples.
Microarray methods
RNA isolated by RNeasy Plus kit (Qiagen) was quantified using Nanodrop (Fisher Scientific). The RNA quality was check on Bioanalyzer (Agilent). Two hundred fifty nanograms of each RNA sample with an RIN value greater than seven was processed using the Ambion WT Expression Kit according to the manufacturer’s instructions. The typical yield from the reaction was 6–9 micrograms of cDNA. The required amount of cDNA (5.5 micrograms) was processed for fragmentation and biotin labeling using the Gene Chip WT Terminal Labeling Kit (Affymetrix). The efficiency of fragmentation reaction was checked via Agilent Bioanalyzer. The entire reaction of fragmented and biotin-labeled cDNA (50 microliters) with added hybridization controls was hybridized to the human GeneChip 1.0 ST Exon Arrays (Affymetrix) at 45°C for 17 hours in GeneChip Hybridization Oven 640 (Affymetrix). Human GeneChip 1.0 ST Exon Arrays were stained using FS 450_0001 protocol in Affymetrix GeneChip Fluidics Station 450. Briefly: Biotin-labeled cDNA was reacted using two rounds of washes with a solution containing a streptavidin-phycoerythrin complex, with an intermediate treatment of biotin-labeled anti-streptadvidin antibody to amplify the signal. Phycoerythrin labeling was detected within the Affymetrix GeneChip Scanner 3000 7G plus using 532 nm light and detected by a photomultiplier tube. Expression Consol software (Affymetrix) was used to check quality controls of hybridized chips. All chips that passed quality controls were RMA normalized using Expression Console software. The microarray data were deposited into the NCBI GEO database as accession number GSE36081. To examine the extent to which GRHL2 affects the propensity to undergo Epithelial to Mesenchymal Transition (EMT), we compared the relative expression of genes in an identified EMT signature (21) to the relative expression of genes in cells with constitutive GRHL2 expression. Specifically, we obtained the expression profile of the 251 Core EMT signature genes from table S1 of (21) and computed the mean log ratio of the relative expression. We restricted the genes to those which appeared on our array platform and computed the Pearson Correlation coefficient of those genes to the log expression ratio of GRHL2-regulated genes compared to the control.
Reporter assays
HMLE were transiently transfected using Lipofectamine 2000 (Invitrogen) at a 1μg DNA:2ul Lipofectamine ratio. 1.5 μg DNA/well of a 12 well was the maximum amount of DNA found to be tolerable. Transfection mixtures were incubated for 20 minutes in 200 μl Opti-MEM (Invitrogen) and then added to the cells in normal growth media lacking antibiotics. Cells were incubated for 4h then re-fed with normal growth media. Lysates were made by washing the cells once with PBS then lysing in 1x Cell Culture Lysis Buffer (Promega). Lysates were centrifuged at 13, 000 rpm for 10 minutes and the supernatants were assayed for luciferase and β-galactosidase activity as internal control. Luciferase assay reagent was obtained from Promega and the β-galactosidase 2X assay reagent was 200 mM sodium phosphate (pH 7.3), 2mM MgCl2, 100 mM 2-mercaptoethanol, and 1.33 mg/ml o-nitrophenylgalactoside. The Smad reporter construct 3TP-Lux was from Addgene. The ZEB1 promoter-luciferase construct in pGL3 (22) was kindly provided by Antonio Garcia de Herreros (Barcelona, Spain). CMV-LacZ or TK-LacZ were used as internal controls. The GRHL2 clone was purchased from Open biosystems, cat # MHS4426-99625903, and the coding sequence was cloned into the XhoI site of pcDNA3.1+. Sub-fragments of the ZEB1 promoter were generated and cloned into pGL3-promoter (MluI-BglII) using the following primers: Fragment 1: ZPfr1-f:Ttaat ACGCGT CCTTAAGGTCCTGCACGGCG, ZPfr1-r: tatat agatct AAGTTCCGCTTGCCAGCAGC; Fragment 2: ZPfr2-f: TtaatACGCGT CTAGCCTCTCTTTCAATCCA, ZPfr2-r: tatatagatctTCCGCCCCCCGCACCCCGGGGC; Fragment 3: ZPfr3-f: TtaatACGCGTCACGCGAGGCGTGGGACTGA, ZPfr3-r: tatat agatct GGATGCCGGGAAACCGTAGG; Fragment 4: ZPfr4-f: Ttaat ACGCGT GCCTCCCTCTCCCCACCACA, ZPfr4-r: tatat agatct ACCGCACCTGGTTTACGACA; Fragment 5: ZPfr5-f: TtaatACGCGT GCGGACCGGGTGTGGGAGGC, ZPfr5-r: tatat agatct TACCTGACCCGCGCAGCCCG; Fragment 6: ZPfr6-f: Ttaat ACGCGT GCCTCTCTCCGGTCGCCGCG, ZPfr6-r: tatatagatct GGGGCAGGGAGGGATCTGGC, Fragment 7: ZPfr7-f: Ttaat ACGCGT GGGCAGCCGCGGCGGGTGTG, ZPfr7-r: tatat agatct ACCGTGGGCACTGCTGAATT.
The primers used to mutate the fragment 4 construct (using Stratagene Quickchange II XL kit) were: Mut-f:gtaaagccgggagtgtcgtcccacaggtgcggtagatctgcg;Mut-r: cgcagatctaccgcacctgtgggacgacactcccggctttac
Immunofluorescence
For Smad2 localization, TGF-β (5 ng/ml) was added for 6h and the coverslips were fixed with 4% paraformaldehyde(PFA) in PBS for 10minutes. PFA was quenched with 100mM glycine in PBS. Cells were permeabilized with 0.2% TX100 in PBS at 4 degrees for 10minutes, washed twice with PBS, and blocked for one hour in: PBS+10% goat serum+0.1% Tween-20+0.1%BSA. Primary and secondary antibodies were diluted in blocking buffer. Primary antibodies were as follows: SMAD2, Cell Signaling, rb, 1:200. Secondary: rb Alexa 555(red), Molecular Probes, 1:1000. Mounting media: Prolong Gold w/DAPI (Invitrogen).
For E-Cadherin, vimentin and GRHL2 the cells were fixed in 100% Methanol at −20°C for at least 1 hour. They were then washed twice with PBS and blocked as above. Ecadherin, ms, BD, 1:200; Vimentin, rb, Cell Signalling, 1:200; GRHL2, rb, Sigma, 1:200. Secondaries used were anti-mouse Alexa 555 (red) or anti-rabbit Alexa 488(green) or A555 (red), (Molecular Probes), diluted 1:1000. Coverslips were mounted in Prolong Gold as above. Images were produced using the Axiovert 200M microscope, AxioCam MRM camera, and Axio Vision 4.3.1 software (Zeiss).
CHIP
5 x100mm dishes of 4-OHT-induced HMLE+twist-ER (+GRHL2, in some experiments) were each fixed in 1.2ml 10% electron microscopy grade paraformaldehyde for 10 minutes. Following quenching with glycine, CHIP was performed exactly as described previously (23) with the following antibodies: (3.3 μg each): GRHL2 (Sigma); Histone H3 (Cell Signaling), or non-immune rabbit IgG (Pierce). CHIP-derived DNA was analyzed by PCR using the following primer sets: ZEBf6: GCGAGGCGTGGGACTGATGG; ZEBr6: AAAGTTGGAGGCTCGGCGGC; ZEBf10: CTGCACGGCGATGACCGCT; ZEBr10: TTCCGCTTGCCAGCAGCCTC; GAPDH-f: ATGGTTGCCACTGGGGATCT; GAPDH-r: TGCCAAAGCCTAGGGGAAGA. For qPCR analysis, CHIP DNAs were analyzed using 2X Power-Sybr Green Master Mix (Applied Biosystems) and signals were calculated relative to input DNA using the “ΔΔ-Ct” method; a correction of 1.5X was applied to adjust for the higher efficiency of GAPDH amplification compared with ZEB1 amplification obtained in a control reaction using lysates from GRHL2-null cells. The primer set used was F8: GCCGCCGAGCCTCCAACTTT; R8: TGCTAGGGACCGGGCGGTTT.
Western blotting
SDS-PAGE was conducted using 4–20% gradient Tris Glycine gels, (Invitrogen). Proteins were immobilized by electophoretically transferring them to a PVDF filter (Immobilon FL, Millipore) in 5% MeOH containing Tris-Glycine transfer buffer. Filters were blocked in PBS+5% non-fat milk, primary antibodies were incubated in PBS+0.1% Tween20+5% non-fat milk, secondaries were incubated in PBS+0.1% Tween20+5%milk+0.01% SDS. Primaries were typically incubated for 2h to overnight, secondaries were incubated for 1h. Primaries used were: Ecadherin, ms, BD Biosciences(BD); Vimentin, ms, Santa Cruz Bio-Tech(SCBT); N-Cadherin, ms, BD; CD44(HCAM), ms, SCBT; ESRP1/2, ms, (Contributed by Russ Carstens, University of Pennsylvania, Philadelphia, Pa); Actin, ms, Millipore; Akt, rb or ms, Cell Signaling(CS); GRHL2, rb, Sigma; Zeb1, rb, Sigma or rb, CS; Ankyrin-G, rb, S.M.F. custom generated (23); total-Smad2/3,ms, BD; phospho-Smad2/3, rb, CS; NF2(merlin), rb, SCBT. Secondary antibodies for chemi-luminescence were either anti-mouse or anti-rabbit, conjugated to horseradish peroxidase (Biorad) and were used at 1:3000 dilution; western blots were developed using (ECL-West Pico, Pierce). Fluorescently tagged secondary antibodies were mouse IRDye 800CW or rabbit IRDye 680LT (Li-Cor) used at 10:000 and detected using the Odyssey Infrared Imaging System (Li-Cor). Fluorescent images were converted to gray scale.
Other bioinformatics methods
We obtained GRHL2 expression data from NCBI Gene Expression Omnibus (accession number GSE10885 (3)). We compared the log expression of GRHL2 among the tumor types Basal, Claudin, Her2, Luminal, and Normal. We performed a Tukey HSD test to determine which comparisons among these groups demonstrated statistically significant differences; family-wise 95% confidence intervals for the log expression were plotted. Statistical analyses were performed using R version 2.13.1 (www.r-project.org).
RESULTS
Loss of GRHL2 Expression is Associated with a Mesenchymal Phenotype
Based on the specific expression of GRHL2 in mouse epithelia (12–14, 24), we investigated its potential regulation during EMT. Using HMLE cells that express a twist-ER fusion -- a previously characterized model of inducible EMT (21) -- we analyzed the levels of GRHL2 over a time-course of twist induction by tamoxifen and found that, indeed, GRHL2 protein was down-regulated during EMT with kinetics similar to the loss of E-cadherin and gain of N-cadherin (Figure 1a). Consistent with this finding, spontaneous, stable EMT in a subpopulation of HMLE cells (“Mesenchymal Subpopulation”) obtained by sorting for CD44high phenotype, described previously (7), also displayed low GRHL2 expression (figure 1b).
Figure 1. GRHL2 is down-regulated in cells and tumors that have undergone EMT.
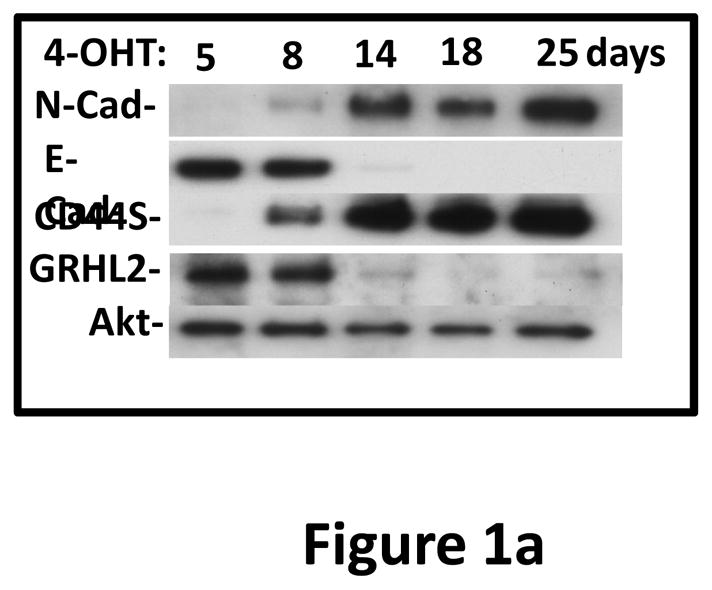
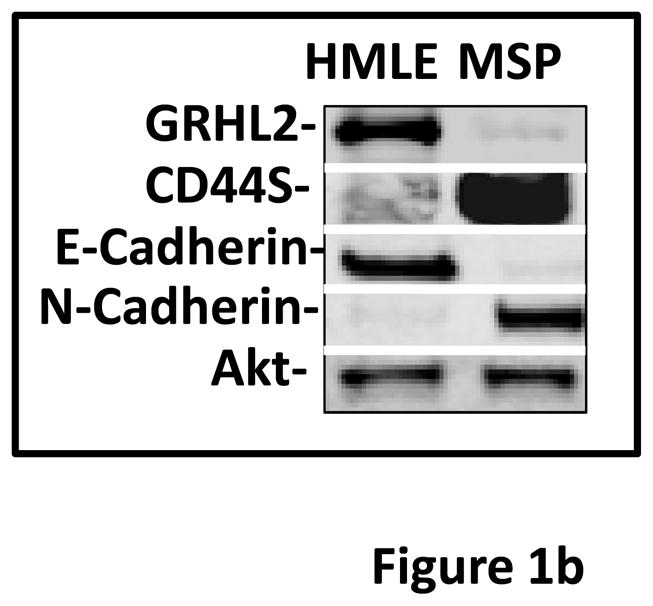


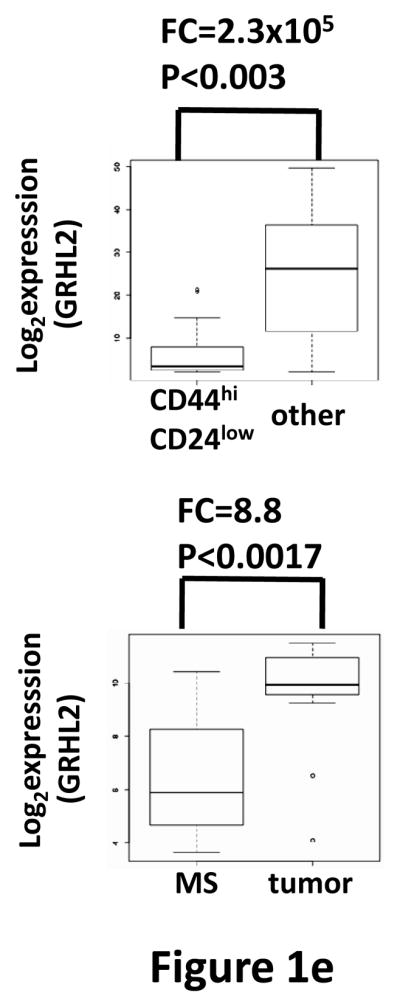
(a) Twist down-regulates GRHL2. HMLE cells with tamoxifen-inducible twist (twist-ER) were induced with 4-hydroxytamoxifen for the indicated periods of time and analyzed for epithelial, mesenchymal and tumor intiating cell (CD44) marker genes. (b) GRHL2 is down-regulated in the Mesenchymal Sub-Population (MSP) cells relative to parental HMLE cells. (c) GRHL2 is down-regulated specifically in “basal B” subclass of breast cancer cell lines characterized by Neve, et al. (42). (d) GRHL2 is down-regulated specifically in the “claudin-low” subclass of breast cancer cell lines characterized by Hennessy, et al. (3). (e) GRHL2 is down-regulated specifically in the tumor initiating/mesenchymal cell subpopulation characterized by Creighton, et al (25).
Breast cancer cell lines have been classified by gene expression profiles. One particular subclass, basal B, was characterized by a mesenchymal profile. GRHL2 was dramatically down-regulated, specifically in this subclass (figure 1c). Analogously, the “claudin-low” subclass of mammary tumor samples – also characterized by a mesenchymal gene expression profile and poor prognosis—expressed substantially lower levels of GRHL2 than other subclasses, which, in fact, showed a modest up-regulation, compared to normal breast tissue (figure 1d, and see Discussion). GRHL2 was also down-regulated dramatically in a different tumor type characterized by EMT, clear cell renal carcinoma (figure S1). Moreover, chemoresistant subpopulations of primary breast tumor cells obtained after chemotherapy of patients by sorting for CD44high/CD24low marker expression or mammosphere generation (25) expressed decreased levels of GRHL2 as well (figure 1e).
These data suggested that GRHL2 loss is a widespread characteristic of both primary and cultured tumor cells that have undergone EMT and acquired a tumor-initiating phenotype, informed the hypothesis that GRHL2 down-regulation was functionally important for EMT.
GRHL2 is an EMT suppressor
The spontaneously occurring, CD44highCD24low MSP cells within the heterogeneous HMLE cell line are characterized by EMT, attributed to autocrine signaling pathway activation (7). Infection of HMLE cells with a GRHL2 expression construct and selection for the infected cells using a GFP marker caused the disappearance of the CD44high subpopulation (figure 2a) within a few days after infection, suggesting a conversion effect rather than selective growth (as demonstrated below).
Figure 2. GRHL2 suppresses EMT.





(a) Ectopic expression of GRHL2 suppresses the CD44 expression in the spontaneously occurring mesenchymal subpopulation of HMLE cells (“MSP”). HMLE+Twist-ER cells (without 4-OHT) infected with GRHL2 or empty retroviral vector (pMIG) were analyzed for CD44 expression by FACS. (b) GRHL2 reverts MSP cells to an epithelial phenotype. The CD44high mesenchymal sub-population of HMLE, obtained by flow-sorting of HMLE cells, were infected with empty vector or GRHL2 and then (top panel) stained for the indicated proteins by immunofluorescence or (lower panel) probed for epithelial and mesenchymal marker genes by western blotting. (c) GRHL2 expression in MSP cells restores anoikis-sensitivity (left panels: top graph represents a DNA fragmentation ELISA assay, lower graph represents a trypan blue-permeability assay) and reduces mammosphere (middle panels), without affecting growth rate (right panel). (d) GRHL2 suppresses Twist-induced EMT. HMLE+twist-ER expressing either empty vector or GRHL2 were treated with 4-OHT (to activate the twist transgene) for 7 days, and (top panel) photographed to record morphology. (lower panel): Time course of changes in epithelial and mesenchymal marker genes after induction of the twist gene with 4-OHT, in cells with or without ectopic GRHL2 expression; (e) GRHL2 suppresses EMT and anoikis-resistance in MDA-MB-231LN cells. MDA-MB-231LN expressing either empty vector or GRHL2 were analyzed for morphology (phase contrast microscopy, top left), E-cadherin expression and localization (immunofluorescence, top right), expression of epithelial and mesenchymal markers (western blotting, bottom left), anoikis-sensitivity (DNA fragmentation ELISA (middle) and adherent growth rate (lower right).
To further characterize this phenomenon, we isolated MSP cells from the HMLE cell line by flow sorting and infected these with the GRHL2 retrovirus. Based on E-cadherin immunofluorescence and western blotting for epithelial and mesenchymal markers, GRHL2 reverted MSP back to an epithelial phenotype (figure 2b). Anoikis-resistance and the ability to form mammospheres are key characteristics associated with EMT in HMLE cells. GRHL2 expression in the MSP cells restored anoikis-sensitivity and reduced mammosphere formation dramatically, without affecting adherent cell growth (figure 2c).
These data indicated that GRHL2 reverted the spontaneous EMT and accompanying tumor initiating cell characteristics of MSP cells. To test the effect of GRHL2 in other scenarios of EMT, we expressed it constitutively in HMLE+Twist-ER cells and in the prototypical EMT-like/triple-negative breast cancer cell line, MDA-MB-231, where it caused dramatic reversion of EMT and anoikis-resistance in both cases (figures 2d, 2e), indicating a surprisingly broad specificity for this effect.
GRHL2 suppresses TGF-β-induced EMT
MSP cells and Twist-expressing HMLE cells rely on autocrine TGF-β signaling for their maintenance of mesenchymal and tumor-initiating properties (7), suggesting that GRHL2 could be suppressing EMT through this common pathway. We confirmed that twist-mediated EMT and acquisition of anoikis-resistance were TGF-β-dependent by using LY364947, a specific inhibitor of the TGF-βR1 kinase activity (figure S2). Because this inhibitor mimicked the effects of ectopic GRHL2 in some respects, we tested the effect of GRHL2 on TGF-β-induced EMT.
TGF-β alone was previously reported to be insufficient for EMT induction in HMLE, a process requiring activation of multiple pathways (7). When GRHL2 was partially depleted by two distinct shRNAs, TGF-β alone induced EMT more efficiently than in cells with control shRNA (figure 3a). GRHL2 knockdown facilitated several activities of TGF-β: induction of a mesenchymal morphology, down-regulation of epithelial specific genes (E-cadherin, ESRP1 and ankyrin-G – an epithelial cytoskeletal protein that regulates anoikis (23)), and up-regulation of vimentin as well as the tumor initiating cell marker, CD44S; surprisingly, TGF-β induced N-cadherin partly independently of GRHL2 expression. Coincident with this facilitation of EMT, GRHL2 knockdown also permitted TGF-β to confer a mammosphere-generating, anoikis-resistant state (figure 3a,b). GRHL2 also suppressed another feature of EMT, the formation of large protrusive structures during the growth of colonies in three-dimensional matrigel culture, indicative of invasive potential (figure S3).
Figure 3. GRHL2 suppresses TGF-β-induced EMT.





(a) GRHL2 knockdown permits HMLE cells to undergo TGF-β-induced EMT. HMLE+twist-ER cells (no 4-OHT) with two distinct GRHL2 shRNAs or a scrambled control shRNA were exposed to TGF-β for two weeks prior to analysis for cell morphology (top left), E-cadherin expression/localization (immunofluorescence, top right) or mammosphere generation (graph, lower right). Epithelial and mesenchymal markers were also analyzed at the indicated times of TGF-β treatment (western blot, lower left). (b) GRHL2 suppresses the ability of TGF-β to confer anoikis-resistance. The cell lines described above were assayed for anoikis by DNA fragmentation after two weeks incubation with or without TGF-β. (c) GRHL2 inhibits Smad-mediated transcription. (left panel): Effect of stable GRHL2 knockdown on activity of a Smad-responsive reporter construct (3TP-lux) was determined by luciferase assays of transiently transfected HMLE+twist-ER cells (no 4-OHT) expressing either scrambled or GRHL2a shRNA that were treated with TGF-β for sixteen hours prior to lysis; values are luciferase activity normalized to an internal co-transfected β-galactosidase control. (lower panel): Effect of stable GRHL2 knockdown on induction of TGF-β/Smad target genes, CTGF and ZEB1, by exogenous TGF-β, determined by qRT-PCR. (top panel): No effect of GRHL2 on phosphorylation of Smad2/3. MSP cells expressing empty vector or GRHL2 were treated with the indicated concentrations of TGF-β for 24h and analyzed for total and phospho-smad2/3. (right panel): No effect of GRHL2 on nuclear translocation of Smad2. The GRHL2 knockdown or control cells described in part a were treated with TGF-β for six hours and analyzed for Smad2 localization by immunofluorescence. (d) GRHL2 knockdown induces EMT in HMLE cells with Ras (HMLER). HMLER expressing two distinct GRHL2 shRNAs or scrambled shRNA control were imaged using phase contrast microscopy (top), immunoblotted for epithelial and mesenchymal markers (right) or assayed for anoikis (trypan blue exclusion, left panel). (e) GRHL2 up-regulates BMP2. RT-PCR was performed on RNAs from HMLE+Twist-ER cell lines with or without constitutive GRHL2 expression (induced with 4-OHT), using the indicated primer sets.
These results suggested that signaling downstream of the TGF-β receptor might be suppressed by GRHL2. We focused on Smad-mediated transcription, as it plays an important, although not exclusive role in the response to exogenous TGF-β-mediated EMT (4). Using a well-characterized reporter assay (3TP-lux; (4)), GRHL2 knockdown stimulated TGF-β/Smad mediated transcription by ~4.6X relative to control cells (figure 3c). Correspondingly, GRHL2 knockdown promoted the induction of the TGF-β/Smad target genes, CTGF (26) and ZEB1 (27), by exogenous TGF-β. Surprisingly, there was no discernable effect of GRHL2 on either the phosphorylation or nuclear translocation of Smad2, suggesting that other mechanisms of inhibition were operative (see Discussion). These results indicated that GRHL2 inhibited Smad-mediated transcription in response to exogenous TGF-β.
In HMLE cells expressing low levels of activated K-ras, (HMLER), GRHL2 knockdown sufficed to induce EMT --i.e., even without exogenous TGF–β--based on the criteria used above (Figure 3d). This EMT was clearly dependent upon autocrine TGF-β signaling, in that LY364947 reversed it (Figure S4). The TGF-β signaling antagonists BMP2 and-4 were previously shown to be down-regulated in MSP cells relative to normal HMLE, which promoted autocrine TGF-β signaling (7). Interestingly, BMP2 expression was activated by GRHL2 (figure 3e), consistent with the idea that GRHL2 suppressed not only TGF-β signaling in response to exogenous ligands but also (by comparison with reported data on the same cell lines (7)) autocrine signaling.
GRHL2 Represses ZEB-1 expression
Suppression of EMT by GRHL2 could occur by a diversity of mechanisms. To elucidate one or more of these in an unbiased manner, we performed a microarray-based gene expression profiling comparing the HMLE-Twist cells with or without GRHL2 expression. This analysis revealed that genes regulated by GRHL2 (GEO database accession number GSE36081) correlated negatively (R=−.7508) with genes regulated during EMT (by multiple transcription factors) in the HMLE system, validating the EMT-suppressive effect of GRHL2 (Figure 4a). The genes regulated by GRHL2 included markers of epithelial vs. mesenchymal phenotypes, several of which were ZEB1 target genes; transcription factors implicated in the control of EMT were also noted.
Figure 4. GRHL2 represses the ZEB1 gene.
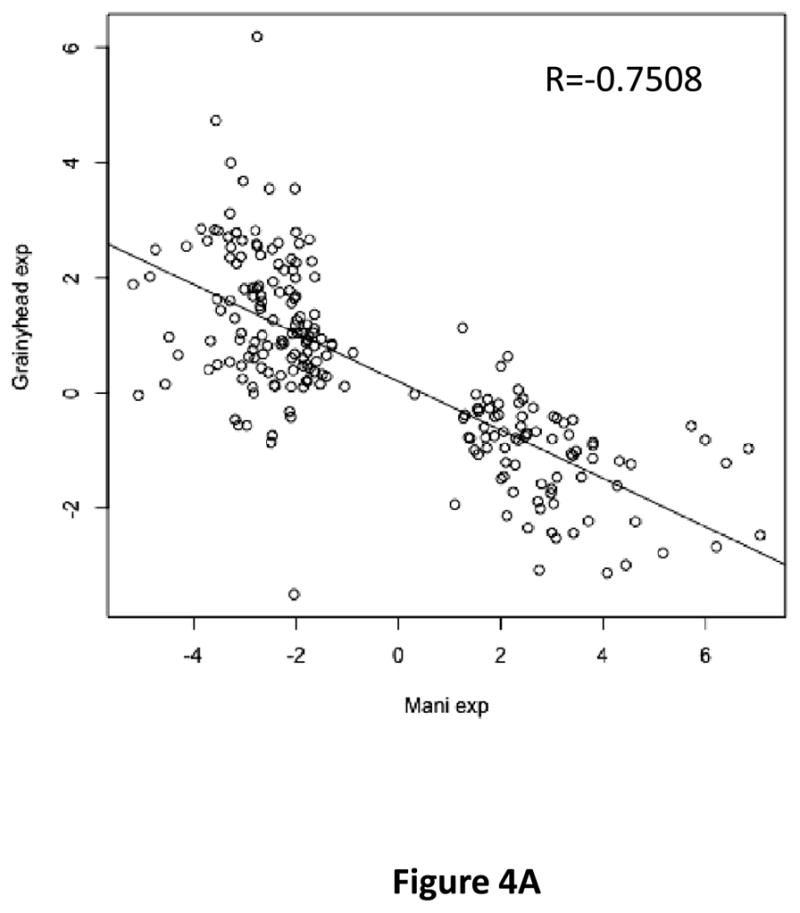




(a) HMLE+Twist-ER expressing either empty vector or GRHL2 were induced with 4-OHT for 17 days; four days following removal, RNAs were isolated and compared by microarray profiling. Gene changes due to GRHL2 were compared to those due to Twist in the same cell line; a regression plot of this comparison is shown. (b) GRHL2 up-regulates mir-200b/c. RNAs from HMLE or HMLER cells expressing GRHL2 or control shRNAs were compared for the indicated mir-200 transcripts by RT-PCR. (c) GRHL2 represses the ZEB1 promoter. (left panel): MSP cells were co-transfected with a ZEB1 promoter-luciferase reporter construct, with the indicated input amounts of GRHL2 expression vector or equal amounts of empty vector. Values represent the relative luciferase activity normalized to an internal β-galactosidase control; (right panel): HMLE+Twist-ER cells (no 4-OHT) with stable GRHL2 knockdown or control (scrambled) knockdown were assayed for luciferase activity after transient transfection of the ZEB1 promoter. (d) Identification of a ZEB1 promoter fragment that is GRHL2-sensitive, and contains a GRHL2 consensus binding site. (left panel): Fragments (~200bp) spanning the 1 kb ZEB1 promoter were assayed for transcriptional activity in the presence or absence of co-transfected GRHL2. The fragment 1–4 sequence and predicted GRHL2 binding site are shown. (right panel): Fragment 4 containing wild-type or mutant versions of the predicted GRHL2 binding site were assayed for repression by co-transfected GRHL2. (e) GRHL2 protein interacts with the ZEB1 promoter. Chromatin from HMLE+Twist-ER+GRHL2 (upper panel) or MDA-MB-231+GRHL2 (lower panel) was immunoprecipitated with GRHL2, histone H3 or non-immune antibody and analyzed by gel-based PCR or qPCR, using the indicated CHIP primers.
Interestingly, one of the major down-regulated GRHL2 target genes was the E-cadherin repressor/EMT inducer ZEB1, as shown by RT-PCR (figure 3e) and Western blotting in MSP cells (figure 2b), MDA-MB-231 cells (figure 2e), HMLE+shGRHL2 cells with TGF-β (figure 3a), HMLER+shGRHL2 cells (figure 3d) and HMLE+Twist-ER cells (figure 2d). Functional consequences of ZEB1 down-regulation (28) such as the up-regulation of mir-200b/c (figure 4b) and ESRP1 (figure 3e) were also evident. The down-regulation of ZEB1 by GRHL2 was investigated further as a potential mechanism for suppression of EMT.
To determine whether the ZEB1 gene could be a direct target for GRHL2, we co-transfected the previously characterized ZEB1 promoter (22) together with GRHL2 into the MSP cells (which express low endogenous GRHL2). This revealed a marked repression of the ZEB1 promoter by GRHL2, as did the converse experiment, transfection of the ZEB1 promoter into cells with or without knockdown of endogenous GRHL2 (figure 4c).
Inspection of the ~1 kb of promoter sequence that was GRHL2-responsive revealed several potential binding sites for grainyhead proteins. We tested ~200bp nested fragments of the ZEB1 upstream region, in the context of an SV40 promoter, for repression by GRHL2, and identified one fragment (fragment 4) that was highly repressed. This fragment contained a consensus GRHL2 binding site and also carried a strong enhancer; the repression by GRHL2 was completely eliminated by a four-base mutation of this consensus site (figure 4d). To determine whether the ZEB1 promoter was a direct target for repression by GRHL2, CHIP analysis was performed, demonstrating a strong enrichment of PCR signal using GRHL2 antibody, with respect to non-immune IgG or a primer set (using GRHL2 antibody) representing an unrelated region of the genome (figure 4e). These results indicated that GRHL2 repressed ZEB1 expression and interacted directly with the ZEB1 promoter.
Suppression of ZEB1 is critical for the suppression of EMT by GRHL2
ZEB1 plays a critical role in EMT in response to various stimuli including TGF-β (29–33), informing the hypothesis that GRHL2 suppressed EMT, at least in part, by repressing ZEB1 expression. To test this, ZEB1 was expressed ectopically, using a doxycycline-inducible promoter, in the HMLE+twistER+GRHL2 cells. By the criteria of morphology, expression of epithelial and mesenchymal markers, and anoikis-resistance, ZEB1 restored EMT that had previously been blocked by GRHL2 expression (figure 5a). Analogous effects of ZEB1 expression were also observed in MSP cells that had been reverted to an epithelial phenotype by stable GRHL2 expression (figure S5). Conversely, in the HMLE cells where GRHL2 knockdown predisposed the cells toward TGF-β induced EMT, ZEB1 knockdown blocked this induction (Figure 5b). Similarly, EMT that was induced by GRHL2 knockdown in HMLER cells was reversed by ZEB1 knockdown (figure S6). These results indicated that the repression of ZEB1 was a key mechanism by which GRHL2 suppressed EMT.
Figure 5. Suppression of ZEB1 is important for the suppression of EMT by GRHL2.


(a) ZEB1 restores EMT capability and anoikis-resistance in cells that express GRHL2 constitutively. HMLE+twist-ER with or without constitutive GRHL2 expression and with empty vector or ectopic ZEB1 expression vector were treated with 4OHT and then analyzed for anoikis (DNA fragmentation ELISA, top left); E-cadherin expression/localization (immunofluorescence, top right), cell morphology (phase contrast, lower left) and expression of epithelial and mesenchymal markers (western blot, lower right). (b) ZEB1 knockdown prevents TGF-β-induced EMT in GRHL2 knockdown cells. HMLE+twistER (without 4OHT) expressing shGRHL2a were transfected with either non-targeting siRNA or ZEB1 siRNA prior to incubation with or without TGF-β Cells were analyzed for epithelial and mesenchymal markers (western blot, top panel), morphology (phase contrast, lower left panel), or E-cadherin expression/localization (immunoflourescence, lower right).
DISCUSSION
Mammalian GRHL2 is a transcription factor that plays important role in epidermal junctions, in part due to activation of target genes including claudin-4 and E-cadherin. Consistent with this role, the Drosophila Grainyhead gene is among the first transcription factors utilized in the maternal to zygotic transition (MZT) during embryonic development (16), and the three mammalian Grainyhead genes are critical for embryonic and adult wound healing (11–14). In light of the fact that wound-healing is orchestrated in part by TGF-β signaling (9), the suppressive effect of GRHL2 on this pathway suggests that GRHL2 may contribute to the resolution phase of wound-healing, wherein transient EMT-like cell conversions in keratinocytes are instructed to reverse. The suppressive effect of GRHL2 on oncogenic EMT may be understood by analogy to this function, given the similarities between the two contexts of EMT (8).
The significance of mammalian Grainyhead proteins in cancer is emerging. GRHL3 was recently shown to function as a tumor suppressor in squamous cell carcinoma, acting, at least in part, as a direct activator of PTEN expression (19); EMT-related issues were not examined, however. The GRHL2 gene shows frequent amplification in unclassified breast tumor samples, and has been proposed as a potential oncogene in breast cancer, due, in part, to its suppression of death receptor expression (18). Consistent with this, modest up-regulation of GRHL2 mRNA was observed in luminal A, B and HER2-positive tumor types (figure 1d) (although it is unclear whether this is an artifactual result of expansion of the epithelial cell compartment relative to normal mammary gland during tumor outgrowth). By contrast, our results show that GRHL2 is down-regulated in EMT models and EMT-driven tumor subclasses, and that it suppresses TGF-β-induced ZEB1 expression; in light of the established pro-tumorigenic potential of ZEB1, this result predicts that GRHL2 will specifically suppress EMT-like tumors (34, 35).
These results can be reconciled in light of the diametrically opposed, context-dependent effects of TGF-β: growth arrest and tumor suppression in certain tumors vs. tumor promotion in others (4). In breast cancer, fewer than 10% of patients have tumor types (claudin-low, metaplastic) in which EMT/TGF-β contributes critically to tumor progression, while in the majority of tumors (other basal, luminal A,B and HER2-positive subclasses) – which most transgenic mouse models emulate—TGF-β is tumor-suppressive (3, 36). By targeting the TGF-β pathway, GRHL2 is predicted usually to act as an oncogene (i.e., in the most common subclasses of breast cancer) or, less frequently, as a tumor suppressor gene (i.e., in EMT-like subclasses).
The results here indicate that GRHL2 interferes with the response to TGF-β by at least two mechanisms, interference with Smad2/3 mediated transcriptional activation and direct repression of the ZEB1 promoter (diagrammed in figure 6). Consistent with previous observations in other systems (28, 37), ZEB1 was required for EMT in response to Twist, TGF-β and spontaneous conversion. GRHL2 also up-regulated mir-200b/c, consistent with a critical role of the established ZEB1/mir-200 feed-forward regulatory loop in EMT (28). The precise mechanism by which GRHL2 represses the ZEB1 promoter may relate to Grainyhead proteins’ ability to repress transcription, by recruiting polycomb repression complex components or by interfering with the binding of a transactivator (16, 38).
Figure 6. Summary of the proposed model.

EMT is induced by the combination of TGF-β with other micro-environmental factors, and requires the activation of ZEB1 plus other target genes. Microenvironmental factors (Wnt, NF-kb agonists?) up-regulate Twist and Snail genes, which down-regulate GRHL2. This down-regulation alleviates the repression of the ZEB1 promoter, permitting TGF-β (partly, through Twist and Snail themselves) to activate ZEB1 expression. The down-regulation of GRHL2 also enhances Smad-mediated transactivation of TGF-β target genes, which, together with ZEB1, induce EMT and anoikis-resistance.
The mechanism by which GRHL2 inhibits Smad-mediated transcription is unresolved at present. Previous work has shown that ZEB1 protein can bind to the Smad2/3 complex, enhancing transactivation (39); our preliminary observations indicated that this mechanism did not apply in our system. Smad2/3 nuclear vs. cytoplasmic localization is regulated by phosphorylation as well as signaling from the Crumbs polarity complex through Hippo pathway components (26). These mechanisms were, however, unlikely to explain the suppression by GRHL2, because Smad phosphorylation and nuclear translocation were not apparently affected. Other nuclear proteins that affect Smad2/3 transactivation, such as TGIF, Ski, and Sno (4), remain to be tested in the context of GRHL2.
TGF-β-induced EMT is a highly restricted phenomenon in cell culture models, occurring in only a small number of epithelial cell lines (40). In fact, we observed that the mouse mammary epithelial cell line NMuMg, commonly used to study this phenomenon, has undetectable GRHL2 expression, while other mouse mammary lines that are unresponsive do express GRHL2 (figure S7). These results are consistent with the previous finding that additional factors from the tumor microenvironment confer TGF-β–responsiveness upon HMLE cells, suggesting that one or more of these factors could function by down-regulating GRHL2 (7). More generally, the GRHL2 expression profile in breast cancer samples and cell lines indicate that GRHL2 is a general barrier to EMT. Accordingly, GRHL2 prevented TGF-β from conferring anoikis-resistance, mammosphere formation (a tumor initiating cell behavior), and invasive growth in three-dimensional culture, predicting a tumor-suppressive effect in this context.
These results also suggest that GRHL2 may be a useful biomarker for tumors predicted to respond to TGF-β receptor inhibitory drugs currently in clinical trials (41): GRHL2-null tumors, being susceptible to the tumor-promoting effects of TGF-β, are predicted to respond specifically to this class of drugs, an approach that could improve their efficacy substantially.
Supplementary Material
Acknowledgments
The authors wish to thank Kathy Brundage (flow cytometry), Kimmi Alonge, and the laboratories of Peter Stoilov, Mike Ruppert, Laura Gibson, Antonio Garcia de Herreros, Antonio Postigo, Richard Myers and Kai-Schmidt-Ott for help, advice and reagents. We thank Wioletta Szeszel-Fedorowicz for performing the microarray experiment. S.M.F. was supported by NIH grant R01CA123359. The flow cytometry core facility (Mary Babb Randolph Cancer Center) was supported by NIH grants RR020866 and P20 RR16440. J.D. was supported in part by the West Virginia IDEA Network of Biomedical Research Excellence (INBRE) P20 RR 016477-12 (Rankin, Gary, PI). A.V.I. was supported by NIH grant P20 RR16440, American Cancer Society grant 122300-IRG-09-061-04-IRG and Susan G. Komen for the Cure grant KG110350.
References
- 1.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 2.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 15:169–90. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Scheel C, Eaton EN, Li SH, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell States in the breast. Cell. 145:926–40. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–38. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 9.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 11:97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–9. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ting SB, Caddy J, Hislop N, et al. A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science. 2005;308:411–3. doi: 10.1126/science.1107511. [DOI] [PubMed] [Google Scholar]
- 12.Boglev Y, Wilanowski T, Caddy J, et al. The unique and cooperative roles of the Grainy head-like transcription factors in epidermal development reflect unexpected target gene specificity. Dev Biol. 349:512–22. doi: 10.1016/j.ydbio.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Pyrgaki C, Liu A, Niswander L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev Biol. 353:38–49. doi: 10.1016/j.ydbio.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werth M, Walentin K, Aue A, et al. The transcription factor grainyhead-like 2 regulates the molecular composition of the epithelial apical junctional complex. Development. 137:3835–45. doi: 10.1242/dev.055483. [DOI] [PubMed] [Google Scholar]
- 15.Wilanowski T, Caddy J, Ting SB, et al. Perturbed desmosomal cadherin expression in grainy head-like 1-null mice. EMBO J. 2008;27:886–97. doi: 10.1038/emboj.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison MM, Botchan MR, Cline TW. Grainyhead and Zelda compete for binding to the promoters of the earliest-expressed Drosophila genes. Dev Biol. 345:248–55. doi: 10.1016/j.ydbio.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Dong Q, Shin KH, et al. Grainyhead-like 2 enhances the human telomerase reverse transcriptase gene expression by inhibiting DNA methylation at the 5′-CpG island in normal human keratinocytes. J Biol Chem. 285:40852–63. doi: 10.1074/jbc.M110.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dompe N, Rivers CS, Li L, et al. A whole-genome RNAi screen identifies an 8q22 gene cluster that inhibits death receptor-mediated apoptosis. Proc Natl Acad Sci U S A. 108:E943–51. doi: 10.1073/pnas.1100132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darido C, Georgy SR, Wilanowski T, et al. Targeting of the Tumor Suppressor GRHL3 by a miR-21-Dependent Proto-Oncogenic Network Results in PTEN Loss and Tumorigenesis. Cancer Cell. 20:635–48. doi: 10.1016/j.ccr.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol. 2003;163:315–26. doi: 10.1083/jcb.200304159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dave N, Guaita-Esteruelas S, Gutarra S, et al. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 286:12024–32. doi: 10.1074/jbc.M110.168625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Park SH, Cieply B, et al. A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol Cell Biol. 31:4036–51. doi: 10.1128/MCB.01342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rifat Y, Parekh V, Wilanowski T, et al. Regional neural tube closure defined by the Grainy head-like transcription factors. Dev Biol. 345:237–45. doi: 10.1016/j.ydbio.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 25.Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–5. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varelas X, Samavarchi-Tehrani P, Narimatsu M, et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell. 19:831–44. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 22:1686–98. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 11:670–7. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guaita S, Puig I, Franci C, et al. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem. 2002;277:39209–16. doi: 10.1074/jbc.M206400200. [DOI] [PubMed] [Google Scholar]
- 30.Taube JH, Herschkowitz JI, Komurov K, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 107:15449–54. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smit MA, Peeper DS. Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene. doi: 10.1038/onc.2011.96. [DOI] [PubMed] [Google Scholar]
- 32.Aigner K, Dampier B, Descovich L, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26:6979–88. doi: 10.1038/sj.onc.1210508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grooteclaes ML, Frisch SM. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene. 2000;19:3823–8. doi: 10.1038/sj.onc.1203721. [DOI] [PubMed] [Google Scholar]
- 34.Spaderna S, Schmalhofer O, Wahlbuhl M, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68:537–44. doi: 10.1158/0008-5472.CAN-07-5682. [DOI] [PubMed] [Google Scholar]
- 35.Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 36.Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–66. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 38.Tuckfield A, Clouston DR, Wilanowski TM, Zhao LL, Cunningham JM, Jane SM. Binding of the RING polycomb proteins to specific target genes in complex with the grainyhead-like family of developmental transcription factors. Mol Cell Biol. 2002;22:1936–46. doi: 10.1128/MCB.22.6.1936-1946.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003;22:2453–62. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown KA, Aakre ME, Gorska AE, et al. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004;6:R215–31. doi: 10.1186/bcr778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganapathy V, Ge R, Grazioli A, et al. Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 9:122. doi: 10.1186/1476-4598-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.