

Abstract
Advanced glycation end products (AGEs) are produced through the non enzymatic glycation and oxidation of proteins, lipids and nucleic acids. Enhanced formation of AGEs occurs particularly in conditions associated with hyperglycaemia such as diabetes mellitus (DM). AGEs are believed to have a key role in the development and progression of cardiovascular disease in patients with DM through the modification of the structure, function and mechanical properties of tissues through crosslinking intracellular as well as extracellular matrix proteins and through modulating cellular processes through binding to cell surface receptors [receptor for AGEs (RAGE)]. A number of studies have shown a correlation between serum AGE levels and the development and severity of heart failure (HF). Moreover, some studies have suggested that therapies targeted against AGEs may have therapeutic potential in patients with HF. The purpose of this review is to discuss the role of AGEs in cardiovascular disease and in particular in heart failure, focussing on both cellular mechanisms of action as well as highlighting how targeting AGEs may represent a novel therapeutic strategy in the treatment of HF.
Keywords: Advanced glycation end products, Diabetes, Cardiovascular disease, Atherosclerosis, Heart failure
INTRODUCTION
Advanced glycation end products (AGEs) are a heterogenous group of molecules that are generated through non-enzymatic glycation and oxidation of proteins, lipids and nucleic acids[1]. They alter tissue function and mechanical properties through crosslinking intracellular as well as extracellular matrix proteins[2-6] and through binding to their cell surface receptor, receptor for AGEs (RAGE), they are capable of modulating multiple cellular processes[7-9]. Enhanced formation and accumulation of AGEs has been reported to occur in conditions such as diabetes mellitus (DM) as well as in natural aging, renal failure and chronic inflammation[10-12].
In vivo detectable AGEs include three main groups: (1) Fluorescent cross-linking AGEs such as pentosidine and crossline[13]; (2) Non-fluorescent cross-linking AGEs such as imidazolium dilysine cross-links named either glyoxal lysine dimer or methylglyoxal lysine dimer result from reactions taking place between glyoxal derivatives and lysine residues[14] ; (3) Non-cross-linking AGEs such as N-carboxymethyllysine (CML)[15].
In patients with diabetes, cardiovascular complications are the principal cause of morbidity and mortality and account for up to 65% of diabetic fatalities[16]. It has been reported that 33% of diabetic patients on insulin therapy will have died from cardiovascular disease by the age of 50 years[17]. It is thought that AGEs have a central role in the pathophysiological processes that lead to the development of such cardiovascular complications observed in diabetes. This review highlights how AGEs are formed, summarises the evidence for the role of AGEs in the development of cardiovascular complications in diabetic patients, as well as their underlying mechanisms of action and finally we review the potential of anti-AGE therapies for their use in clinical practice.
MAILLARD REACTION AND AGE SYNTHESIS
AGE are a spectrum of heterogeneous compounds that are derived from proteins, lipids and nucleic acids that are glycated or oxidized non-enzymatically in a process termed the “Maillard reaction”[18]. D-glucose plays a primary role in glycation of proteins in vivo due to its high concentration in human plasma[19]. The Maillard reaction starts as a reaction between the carbonyl group of a reducing sugar such as glucose and the amino acid of proteins, lipids or nucleic acids leading to the production of an unstable compound known as a “Schiff base”. This step is reversible and usually takes a few hours to occur. Over weeks the Schiff base turns into a more stable compound called the “Amadori” product through various molecular rearrangements. Over months and years, the Amadori products undergo further structural changes through a series of reactions such as oxidation, dehydration and degradation to finally yield highly stable AGE compounds[20,21] (Figure 1).
Figure 1.

Maillard reaction. Schematic illustration of the Maillard process that starts by a non-enzymatic reaction of a protein lysine residue (as an example of the most common residues that get glycated) and glucose with consequent loss of a water molecule. This is a reversible reaction that takes place over a few hours leading to the formation of an unstable compound (Schiff base). The latter undergoes molecular rearrangements over a period of days yielding a more stable compound known as (the Amadori product). Over months/years, the Amadori product turns to a very stable compound known as advanced glycation end product (AGE) through a series of reactions such as oxidation. Therefore AGE molecules acquire more negative charges. AGEs are irreversible once they are formed. The Maillard reaction leads to denaturation and browning of the modified proteins.
Highly reactive dicarbonyl compounds are generated during the conversion of Amadori products to AGEs. Methylglyoxal and 3-deoxyglucosone are the best known AGE dicarbonyl precursors[22-24]. In vivo, CML is the most abundant form of AGEs and is characterized by its highly antigenic nature[25,26].
It has been reported that non-enzymatic glycation of proteins and formation of AGEs affects the protein physiological functions and is capable of inducing enzyme inactivation[27]. The influence of such modifications on protein structure has been far less investigated. Recently, using nuclear magnetic resonance (NMR), Howard et al[28] have showed that glycation of a model helical peptide from human serum albumin (HSA) by glucose caused distortion of the helical structure of the protein at the point of glycation. Another protein structural study for the changes induced by non-enzymatic glycation in the secondary and tertiary protein structure of HSA showed that the in general protein structure was unaltered. Instead the change was only limited to local regions after glycation. This was revealed using fluorescence-dependant methods[29].
AGES AND CARDIOVASCULAR DISEASE
AGEs are produced normally in the body and they accumulate by age and are considered not only as biomarkers of senescence but they correlate inversely with left ventricular ejection fraction (EF) and can predict the outcome of cardiac surgery in elderly patients. A direct linear correlation between CML levels in the pericardial fluid and the age of the patients was shown where mean CML level of 260.8 ± 19.7 ng/mL was detected in patients with mean age 52.2 ± 1.3 years, mean CML level of 293.4 ± 18.8 ng/mL was identified in patients with mean age 65.8 ± 0.6 years and mean CML level of 357.0 ± 28.3 ng/mL was recorded at mean age 75.1 ± 0.6 years[30].
In addition, AGEs accumulate at a much higher rate in diabetics than in normal population. AGEs serum levels are much higher in diabetic patients when compared to normal non diabetic population. AGE serum levels have been reported in a number of clinical studies involving diabetic patients compared to healthy individuals. AGE levels were recorded as 7.4 U/mL in diabetic patients vs 4.2 U/mL in normal population, whereas CML levels reached 15.6 U/mL in diabetics vs 8.6 U/mL in nondiabetics. Serum AGEs were found to be significantly elevated in diabetic patients with coronary heart disease (CHD) (8.1 U/mL) vs diabetics without CHD (7.1 U/mL)[31]. In another study concerned with AGE tissue levels and the severity of diabetic nephropathy, the average AGE content in arterial wall collagen of diabetics was significantly higher than that of samples obtained from nondiabetic patients (14.5 ± 5.2 U/mg vs 3.6 ± 1.5 AGE U/mg). Furthermore, it was revealed that AGE content in renal tissues obtained from diabetic patients with end-stage renal disease is as twice as AGE content in tissue of diabetic patients without renal disease (21.3 ± 2.8 U/mg vs 11.5 ± 1.9 AGE U/mg)[32]. Serum levels of pentosidine were found to be significantly higher in patients with diabetes than in nondiabetic normal controls (64.4 ± 21.0 μg/L vs 22.8 ± 7.0 μg/L). Moreover, serum pentosidine levels were significantly higher in diabetic patients with cardiovascular disease than in those without (72.3 ± 23.7 μg/L vs 62.3 ± 19.8 μg/L) and they were found to correlate with increased arterial wall stiffness in diabetic patients[33].
AGES AND CARDIOVASCULAR COMPLICATIONS OF DIABETES
As mentioned above, several studies have shown a positive association between serum and tissue AGEs and macro and microvascular complications of diabetes[31,34-39]. The following sections overview how serum AGE levels correlate with the development and the progression of several cardiovascular complications of diabetes.
AGEs, coronary heart disease and diabetic heart failure
Elevated serum levels of AGEs such as CML have been documented in type 2 diabetic patients with coronary heart disease[31]. AGEs have been detected in atheromatous lesions in coronary arteries of diabetic patients, suggesting a role of AGEs in the accelerated development of atherosclerosis reported in diabetics[40]. Moreover, serum AGE levels have been shown to be a biomarker for the severity of coronary artery atherosclerosis in type 2 diabetic patients[27] independent from other well known risk factors such as hypertension, hyperlipidaemia and smoking. Higher serum AGE levels were detected in type 2 diabetic patients with obstructive coronary artery disease in comparison to diabetics with non-obstructive coronary disease and AGE serum levels were found to correlate with the degree of the coronary atherosclerosis in those with obstructive coronary disease[41]. AGE levels have also been reported to influence the success rate of coronary artery revascularization in patients with diabetes. Elevated serum AGE levels in patients undergoing percutaneous coronary intervention has been shown to be an independent risk factor for restenosis in diabetic patients[42]. Following cardiac surgery, serum AGE levels also inversely correlate with left ventricular ejection fraction and are associated with prolonged ventilation time and prolonged stays on the Intensive Care Unit[43,44].
Other studies have demonstrated that elevated serum AGE levels have been shown to predict mortality due to coronary heart disease in female patients with type 2 diabetes followed up for 18 years[31]. Similarly, increased serum AGE levels in type 1 diabetes have been shown to correlate with incident fatal as well as nonfatal cardiovascular events independent from other known cardiovascular risk factors such as age, body mass index, smoking, hypertension and hyperlipidaemia[45].
The risk of development of cardiovascular disease is increased by a factor of 2-4 fold in diabetic patients[46]. DM is considered to be an independent risk factor for incident heart failure (HF)[47-49]. In the Framingham study (between the ages 45-74), the risk of developing HF was higher in diabetic males (2.4:1) and diabetic females (5:1) independent of age, obesity, hypertension, hyperlipidaemia and coronary artery disease[50].
Elevated serum levels of AGEs in patients with diabetes accelerate the development and progression of heart failure both indirectly through their vascular effects (coronary dysfunction, atherosclerosis and thrombosis) and directly through direct actions on the myocardium. In the former, AGEs binding to their cell surface receptor (RAGE) on endothelial cells, smooth muscle cells and monocytes thereby inducing a wide range of signaling pathways that trigger inflammation, atherogenesis and vasoconstriction leading to coronary dysfunction, atherosclerosis and thrombosis[14]. On the other hand, AGEs have been reported to have a direct effect on the myocardium independent of effects on the vascular tree, mediated in part via cross linking of extracellular cardiac proteins and through actions mediated by AGE receptors expressed on the myocardium[5,6,51]. AGEs have been implicated in the development of both systolic and diastolic cardiac dysfunction in diabetics which may explain the increased prevalence of heart failure in diabetic patients[14,52]. It was shown by Steine et al[53] that serum AGE levels and duration of diabetes can predict systolic strain (as evaluated by doppler tissue imaging) in patients of type 1 diabetes. Moreover, Kilhovd et al[31] showed that parameters of left ventricular diastolic dysfunction, e.g., delayed isovolumetric relaxation time and reduced LV end-diastolic diameter correlate with elevated plasma AGE levels in type 2 diabetic patients. Similarly, Willemsen et al[54] showed that increased tissue levels of AGEs (as detected by increased skin autofluorescence) in diabetic heart failure patients are independently associated with diastolic dysfunction and consequently reduced exercise capacity in those patients in comparison to non-diabetic heart failure patients.
Serum AGE levels have been demonstrated as independent predictors of both the severity and prognosis in heart failure patients[14,55]. Plasma pentosidine levels, one of the crosslinking AGEs, has been shown to be an independent predictor of both re-hospitalization and mortality in heart failure patients independent from other known risk factors such as brain natriuretic peptide (BNP), age, renal function and New York Heart Association (NYHA) functional class[55]. Similarly, plasma levels of N-CML, one of the most prevalent antigenic AGEs in sera of diabetic patients, have been shown to correlate with NYHA functional class and predict outcomes in patients with systolic heart failure[14].
Another study conducted by Neeper et al[56] showed that serum levels of soluble RAGE (sRAGE) are markers of the development and the progression of heart failure in diabetic and non-diabetic patients. The term sRAGE includes both cleaved RAGE (cRAGE) and endogenously secreted RAGE (esRAGE). The former is cleaved RAGE from cell surface by action of metalloproteinases that are increased in HF. This form lacks the V-domain and is therefore unable to neutralize AGEs. The latter is an endogenously secreted RAGE that helps to neutralize serum AGEs. Both higher serum levels of cRAGE and lower serum levels of esRAGE correlate with the severity of cardiac dysfunction, severity of symptoms and clinical outcomes in patients with heart failure[56]. Similar findings have been recorded in a number of other HF studies. For instance, Koyama et al[57] have shown that serum levels of sRAGE correlate with NYHA functional class and that they are particularly higher in HF patients with preserved EF suggesting that they have a role in diastolic dysfunction. Furthermore, Koyama et al[57] reported that sRAGE levels are independent prognostic factors in HF[57]. Similarly, Raposeiras-Roubín et al[58] also demonstrated that sRAGE is a highly sensitive and specific marker of prognosis of HF patients.
Other studies have yielded conflicting observations about the role of AGEs in the development of heart failure whether directly or indirectly. For instance, in another study, the authors demonstrated that increased serum sRAGE levels are associated with increased severity of HF particularly in patients with an ischemic cause of heart failure although this correlation was shown to be independent of AGE serum levels[59]. Similarly, in the study of Campbell et al[60], left ventricular biopsies were taken from pre-diabetic, type 2 diabetic and control subjects undergoing coronary heart bypass surgery to assess myocardial fibrosis and myocardial expression of the AGE N-CML and the RAGE. All the patients had similar degrees of coronary disease with no previous history of myocardial infarction or heart failure although diabetic patients exhibited diastolic dysfunction on echocardiography. The study demonstrated no significant differences in either myocardial AGE or RAGE expression between patient groups or the control group suggesting that myocardial AGE/RAGE expression was not an important factor in diastolic dysfunction development in diabetes. In a similar study in which endomyocardial biopsies of failing hearts of diabetic patients were studied, no increase in myocardial fibrosis or myocardial CML expression was observed in cases with preserved left ventricular ejection fraction although both myocardial fibrosis and CML expression were elevated in cases with significant systolic dysfunction[60].
AGEs and other forms of macrovascular diseases
Serum levels of AGEs were also shown to correlate with the presence of other forms of macrovascular pathology as carotid stenosis and peripheral artery occlusive disease. Serum levels of the esRAGE that can neutralize circulating AGEs were found to be inversely proportionate to carotid artery intima-media thickness in type 1 diabetic patients[61]. The same was reported in type 2 diabetic patients as well as non-diabetic cases[62]. Higher expression of the RAGE was detected in carotid artery plaques and was shown to be associated with enhanced inflammatory reactions[63].
Type 2 diabetic patients with peripheral artery disease exhibited higher levels of serum AGEs vs the non-diabetic subjects[39]. A tight link has been shown between AGE plasma levels and pulse pressure in type 1 diabetic patients as was reported in the EURODIAB Prospective Complications Study[64].
AGEs and microangiopathy
Serum and tissue levels of AGEs are predictors of the development of microvascular complications in diabetes. For example in type 1 diabetes control and complications trial, increased AGE levels in skin biopsies predict the development of diabetic microvascular complications, e.g., retinopathy and nephropathy[13,65]. AGE/RAGE interaction has been suggested as a possible mechanism coupling microangiopathy with diabetic nephropathy, retinopathy and neuropathy[43].
Diabetic nephropathy: Correlation between the levels of skin collagen AGEs particularly CML and the development and worsening of microvascular complications including nephropathy in type 1 diabetic patients was initially reported by Genuth et al[65]. Similarly, in type 2 diabetes skin AGEs (skin autofluorescence) were shown to be potent independent predictors of evolution of microvascular complications including nephropathy[66]. In diabetes, the kidney is an important site for AGE accumulation but also contributes to the increased levels of AGEs in the serum of diabetic patients since the kidney is the main site for AGE clearance[67]. Diabetic animal models exhibit higher levels of AGE deposition in their kidneys[68] which has been linked to renal structural alterations reported in diabetic nephropathy such as glomerular basement membrane thickening, glomerulosclerosis, mesangial expansion and tubulointerstitial fibrosis. Similar histological findings have also been also documented in murine models injected with AGE-albumin[69].
Diabetic retinopathy: Accumulation of AGEs especially CML in retinal blood vessels of type 2 diabetic patients has been reported. It has been also documented that the levels of AGEs correlated with the severity of the retinopathy[70,71]. Infusion of AGE-albumin in non-diabetic animals has shown that AGEs localize both inside and around the pericytes co-localizing with the AGE receptor causing thickening of the basement membrane and destruction of the blood retinal barrier[72,73]. In vitro exposure of retinal cells to AGEs induced upregulation of vascular endothelial growth factor (VEGF) that may contribute to retinal neovascularisation reported in diabetic retinopathy[74].
Diabetic neuropathy: Both peripheral and autonomic neuropathies have been shown to correlate with AGE levels even before neuropathy becomes clinically evident[75]. Skin autofluorescence reflecting AGE bound collagen levels has been reported to have a tight link with the degree of diabetic neuropathic foot ulceration[76]. AGEs have been shown to accumulate in peripheral nerves of diabetic patients and they were shown to co-localize with RAGE receptor in endoneurial, perineurial and epineurial blood vessels[77].
MECHANISMS OF ACTIONS OF AGES
AGEs mediate their tissue effects through three main mechanisms: (1) Cross linking extracellular (matrix) proteins thereby affecting tissue mechanical properties[51]; (2) Cross linking intracellular proteins thus altering their physiological functions[5,6]; and (3) Binding to their cell surface receptor RAGE to inducing multiple intracellular signalling cascades[56]. In the following sections these mechanisms will be discussed.
Crosslinking tissue proteins
Cross linking extracellular matrix proteins: AGE for-mation is a process of chronicity usually affecting long-lived proteins. Extracellular matrix proteins especially collagen type IV that is involved in basement membrane structure, are more prone to advanced glycation due to their long turnover[20,78]. Advanced glycation and crosslinking of other extracellular matrix proteins, e.g., collagen I and elastin render them stiffer and less susceptible to proteolytic digestion[51] (Figure 2). This may contribute to the observed increase in vascular stiffness reported in diabetes and old age[20,51,79]. In addition, cross linking myocardial collagen with AGEs has been suggested to cause myocardial stiffness and diastolic dysfunction in diabetic patients[51,80].
Figure 2.
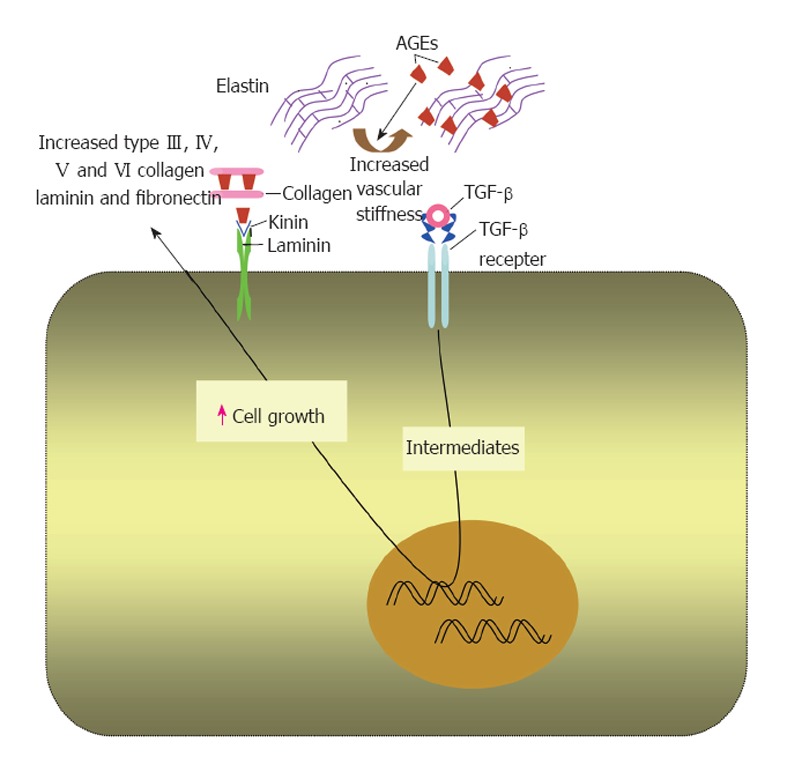
Effects of advanced glycation end products on extracellular matrix proteins. In extracellular matrix, advanced glycation end products (AGEs) form on different molecules as collagen, laminin and elastin. This alters the physiological properties of the matrix and increases its stiffness. AGEs upregulate transforming growth factor (TGF)-β that increases the production of extracellular matrix components by binding to its receptor.
AGEs alter the structure of low density lipoproteins (LDL) through glycation therefore preventing their clearance from the circulation via the normal elimination route, i.e., uptake by the endothelial cells. Instead they are uptaken by the blood monocytes leading to foam cells that contribute to the pathogenesis of atherosclerosis[81,82].
Cross linking intracellular proteins: AGEs have been shown to be implicated in crosslinking of intracellular proteins and hence altering their physiological properties and functions. For instance AGEs were shown to cross link the domains of both the Ryanodine receptor[5] and SERCA2a[6] in cardiomyocytes leading to alterations in calcium homeostasis reported in diabetic cardiomyopathy[83,84].
RAGE dependant effects of AGEs
RAGE receptor: RAGE belongs to the immunoglobulin superfamily of receptors[56]. It has been reported that RAGE gene is located on chromosome 6 in humans between genes coding for class II and class III major histocompatibility complexes[85]. The RAGE promoter has been shown to possess nuclear factor kappa B (NF-κB) binding sites, hence linking RAGE expression to the inflammatory cascade[86].
RAGE is a multiligand receptor with advanced glycation end products being identified as its first known ligands. Hereafter, multiple other RAGE ligands have been revealed including; high mobility group protein box-1[21], some members of the S100 protein family[87] amyloid β[88,89] and fibrillar protein aggregates[90,91]. Therefore, RAGE has an important role in the pathogenesis of the diseases induced by such ligands, e.g., inflammation, tumours, neurodegeneration and amyloidoses[21,88,92,93].
RAGE structure: Full length RAGE comprises three domains, an extracellular domain of 332-amino acids arranged as a single “V”-type immunoglobulin-like (variable) domain with subsequent two “C”-type (constant) domains[89,94]. Modern biochemical techniques have revealed evidence suggesting that both the V and C1 domains of RAGE function together as an incorporated single structural unit for the binding of some ligands. In contrast, C2 RAGE domain is suggested to function completely independent from the VC1 complex while remaining attached to it through a flexible hinge (Figure 3). It has been deduced from experimental work that different RAGE ligands interact with one or more of its domains[19].
Figure 3.
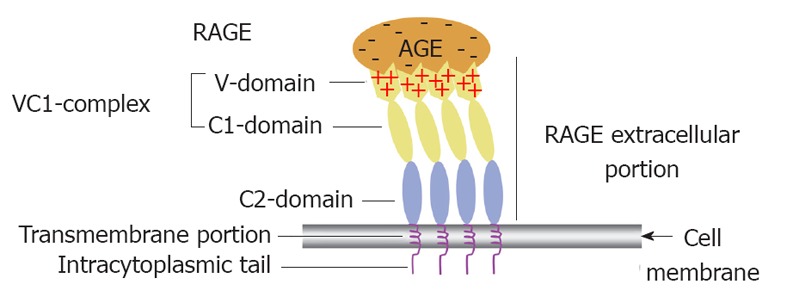
Receptor for advanced glycation end product (i.e., colon) receptor structure and its functional implication in binding different advanced glycation end products. A diagrammatic illustration of the structure of the receptor for advanced glycation end product (RAGE) showing that it is composed of an extracelluar portion, a transmembrane portion and an intracytoplasmic tail. The extracelluar portion comprises three domain V, C1 and C2. The first two are believed to work together as a single functional complex (VC1) whereas the C2 domain remains attached to the VC1 complex but works independently from it. The diagram also illustrates how multiple RAGE receptors polymerise within the cell membrane to facilitate high affinity binding of the positively charged V domain with the negatively charged advanced glycation end products (AGEs) independent of their chemical structure. That is why RAGE is considered one of the pattern recognition receptors.
RAGE has a single transmembrane domain and a highly charged cytosolic tail formed of 43 amino acids (Figure 3). The cytosolic tail is vital for RAGE ligands to activate intracellular signalling cascades. RAGE isoforms in which the cytosolic tail was absent, bind AGEs but fail to elicit intracellular signaling on ligand binding[95]. It has recently been shown that for some RAGE actions, its tail binds directly to a cytoplasmic molecule known as the mammalian diaphanous-1 which is essential for eliciting phosphorylation/activation steps needed for initiation of the signalling cascade[96].
Despite the wide variation in their chemical structure, AGEs bind to the V domain of the RAGE receptor[94]. The fact that RAGE recognizes a class of biochemically heterogeneous ligands such as AGEs categorizes RAGE as one of the pattern recognition receptors[97-99] that identifies common features or patterns rather than a specific ligand. Though AGEs exhibit diverse chemical structure, however, they have some common general characteristics. First, all AGE modified proteins demonstrate a net negative charge that accumulates during their formation by glycation and oxidation[100,101]. The second main feature is that modifications of proteins by AGEs lead to creation of multiple covalent cross-links resulting in higher molecular mass molecules (multimers). This ligand geometry is thought to be important for RAGE activation[100].
Recent molecular structure revealing technologies including X-ray crystallographic, NMR together with conventional biochemical data have illustrated that the unusual ability of the RAGE receptor to bind different AGEs lies in its extracellular portion (VC1 ectodomain) where it has been suggested that the ligand binding is triggered mainly by electrostatic interactions between the positively charged surface of this subunit and negatively charged ligands (Figure 3)[102]. The V domain has been shown by NMR to exhibit three distinct areas for mediating AGE-V domain interactions. Such areas are situated in the positively charged regions of the V domain. The first interaction surface includes strand C and loop CC’, the second interaction surface comprises strand C’, strand F and loop FG, and the third interaction one consists of strand A’ and loop EF[101].
The second main point to be considered here is that studies based on using a fluorescence-labelled receptor demonstrated that RAGE does not float as one molecule in the plasma membrane but instead multiple RAGE receptors aggregate to form receptor assemblies[19,101]. Therefore, RAGE exists in constitutive multimers that usually includes four molecules or more within the plasma membrane[101,103]. RAGE multimers are thought to display a parallel orientation with VC1 subunits exhibiting side to side contacts (Figure 3) as revealed in protein crystals. Therefore it has been speculated that the general multimeric structure adopted by AGE modified proteins together with numerous AGE-modified side chains are essential requirements for preserving the receptor assemblies’ stability needed for activation of the receptor[100] probably in a similar way to that previously described for the receptor tyrosine kinases where the intracellular domains of the receptor that possess intrinsic kinase activity must come very close together in a particular orientation that facilitates cross-phosphorylation of the domains and hence commencement of the signalling cascade[104].
RAGE isoforms: Despite the presence of a single gene coding for RAGE, there are several splice variants of this gene with three main RAGE isoforms having been identified: full length RAGE, dominant-negative RAGE (DN-RAGE) and endogenous soluble RAGE (es-RAGE)[105]. Full length RAGE is composed of the whole three domains; extracellular, trans-membrane and intracellular domains. DN-RAGE has extracellular and transmembrane domains, but no cytosolic tail. Endogenous soluble RAGE possesses the extracellular domain only so it is found free in the circulation. The only RAGE isoform that is capable of eliciting intracellular signalling upon interacting with its ligands is the full length RAGE as this is induced through its cytosolic tail. However, the other two isoforms; DN-RAGE and esRAGE help in clearance and neutralisation of circulating AGEs by competing with full length RAGE in binding them[56,105].
RAGE tissue distribution: RAGE expression has been detected in a number of cells including endothelial cells, smooth muscle cells, monocytes/macrophages, T lymphocytes, cardiomyocytes, glomerular podocytes, dendritic cells, neurons of the central and peripheral nervous systems and transformed cells[106]. Generally, there is a low expression of RAGE in tissues. However, it becomes up-regulated in an environment rich with its ligands as in the case of diabetes or aging. RAGE expression was higher in endothelial cells, monocytes and smooth muscles in diabetic vascular tissue[107].
INTRACELLULAR EFFECTS OF AGE-RAGE BINDING ON CARDIOVASCULAR SYSTEM
RAGE activation by high serum and tissue levels of AGEs induces multiple intracellular signalling pathways (Figure 4) that have been implicated in pathogenesis of serious diabetic complications such as enhanced atherosclerosis, cardiovascular disease, nephropathy and chronic inflammation[9,10,108-110]. It has been documented that circulating AGEs bind with endothelial RAGE resulting in endothelial dysfunction via activation of a number of signaling pathways, e.g., activation of nicotinamide adenine dinucleotide phosphate oxidase that enhances the production of reactive oxygen species (ROS)[108]. ROS have been shown to play a pivotal role in causing major cardiovascular damage in diabetes through modifying the structure of cellular proteins, lipids and nucleic acids and therefore altering their physiological roles[111].
Figure 4.

Intracellular effects of AGEs after AGE/RAGE binding. Diagrammatic representation of AGE/RAGE interaction on the surface of an endothelial cell leads to transduction of a signalling cascade; activates nicotinamide adenine dinucleotide phosphate oxidase and enhances ROS production and phosphorylate p21 RAS and MAPKs. Moreover, the AGE/RAGE interaction induces signalling through activation of p38 MAPK. A main step in AGE/RAGE signalling is activation of NF-κB and its translocation to the nucleus, where it enhances transcription of target genes as endothelin-1, ICAM-1, E-selectin and tissue factor. Hence, AGE/RAGE binding triggers an inflammatory cascade. AGE may decrease NO availability by reducing eNOS activity and by inactivating NO. AGE: Advanced glycation end-product; RAGE: Receptor for advanced glycation end products; ROS: Reactive oxygen species; MAPKs: Mitogen-activated protein kinases; ERK1/2: Extracellular signal-regulated kinase 1/2; NF-κB: Nuclear factor kappa B; ICAM-1: Intercellular adhesion molecule-1; VCAM-1: Vascular cell adhesion molecule-1; VEGF: Vascular endothelial growth factor; IL-1α: Interleukin-1α; IL-6: Interleukin-6; TNF-α: Tumor necrosis factor-α; eNOS: Endothelial nitric oxide synthase; NO: Nitric oxide.
In addition, it has been reported as well that AGE-RAGE engagement increases phosphorylation of p21ras, the mitogen-activated protein kinases, extracellular signal-regulated kinase 1/2 and p38, and activates GTPases Cdc42 and Rac, ultimately inducing activation and translocation of NF-κB from cytoplasm to the nucleus where it starts transcribing its target set of genes[112]. The latter include, intercellular adhesion molecule-1, vascular cell adhesion molecule-1, VEGF, endothelin-1, tissue factor, E-selectin, thrombomodulin and proinflammatory cytokines, including interleukin (IL)-1α, IL-6 and tumor necrosis factor-α[113,114]. The above mentioned cytokines and adhesion molecules have central roles in both inflammation and atherosclerosis[108,115].
ANTI-AGE THERAPIES
Therapies that target AGEs can be classified into two main categories, the ones that prevent the formation of AGEs and others that breakdown the already formed AGEs.
Prevention of AGEs formation
Aminoguanidine (AG), is a hydrazine compound that inhibits AGE formation via trapping of carbonyl intermediates (early active glycation products) and hence preventing modification of nucleophilic residues in proteins[116,117]. In a placebo-controlled, randomized trial involving type 1 diabetes patients, the use of aminoguanidine hindered reduction in glomerular filtration rate, reduced 24-h urinary proteinuria and prevented the deterioration of retinopathy. However, it did not influence time-to-doubling of serum creatinine[118]. Being an NOS inhibitor, which is an essential renovasodilator, this may offset some of AG benefits as an AGE inhibitor[119].
Pyridoxamine, (form of vitamin B6 that naturally exists) and benfotiamine (a lipid-soluble derivative of thiamine), are known to inhibit AGE formation. In phase 2 trials using pyridoxamine in diabetic patients suffering from obvious diabetic nephropathy, a significant decline in the serum levels of creatinine from baseline was observed although this was not accompanied by a corresponding change in urinary albumin excretion[120]. Using benfotiamine in type 2 diabetic patients prevented both macro- and microvascular endothelial dysfunction and oxidative stress induced by an AGE rich meal[121]. Moreover, the combined use of benfotiamine and alpha-lipoic acid normalized elevated AGE levels and blocked the enhancement hexosamine-modified protein formation in monocytes in type 1 diabetic patients[122].
AGE degradation (AGE cross link breaker, ALT-711)
Cross link breakers such as ALT-711 contain a thiazolium structure that is capable of breaking α-carbonyl compounds by cleaving the carbon-carbon bond between carbonyls. In vitro, incubation of AGE crosslinked collagen with ALT-711 promotes collagen digestibility by metalloproteinases (MMP), a phenomenon used as a key sign of an effective cross link-breaker effect[123].
ALT-711 has been used in a number of small clinical studies to investigate the effects of targeting AGE on cardiovascular complications. A phase II clinical trial demonstrated that ALT-711 (210 mg/d, 8 wk) reduced the arterial pulse pressure and improved the compliance of large arteries in older patients (> 50 years) who exhibited age-dependent stiffening of their large arteries secondary to isolated systolic hypertension[124].
Two more phase 2b clinical trial trials currently being undertaken to assess the safety as well as the efficacy of ALT-711 use in the treatment of isolated systolic hypertension with or without left ventricular hypertrophy (LVH) in elderly patients (> 50 years), termed systolic and pulse pressure hemodynamic improvement by restoring elasticity (SAPPHIRE) and systolic hypertension interaction with left ventricular remodeling (SILVER). SAPPHIRE is designed to use multiple doses (four) of ALT-711 over a period of 6 mo in patients with systolic hypertension without LVH whilst the control group will be treated with placebo. The SILVER study is being conducted on patients with systolic hypertension and LVH, including both diabetics and non-diabetics treated with a single dose of ALT-711 and a placebo group for 6 mo. The primary endpoints of both studies are alterations in both systolic blood pressure and pulse pressure. Secondary endpoints involve extra measurements of arterial blood pressure and modifications in definite urological parameters. The results of both trials are still not yet available[125].
In addition, two small and open label clinical trials suggested an important role of AGEs in the development of cardiac dysfunction and HF. In both trials HF patients were treated with an AGE cross-link breaker Alagebrium (ALT-711). In the Distensibility Improvement and Remodeling in Diastolic Heart Failure (DIAMOND) trial, 23 patients with stable diastolic HF received ALT-711 for 16 wk after which patients were assessed by magnetic resonance imaging and tissue doppler which revealed both a reduction of left ventricular mass and an improvement of left ventricular diastolic function[126]. The Patients with Impaired Ejection Fraction and Diastolic Dysfunction: Efficacy and Safety Trial of Alagebrium trial was conducted on 22 patients suffering from systolic HF and diastolic dysfunction who were treated by ALT-711 (35-420 mg). The results of this trial were in line with those of the DIAMOND trial. Doppler assessment showed improvement in isovolumetric relaxation time and consequently reduction in the left atrial pressure. Furthermore, reduced left ventricular mass and left ventricular end-diastolic volume was also reported in some patients[127].
However, a more recent clinical trial BENEFICIAL, a prospective, randomized, double-blind, phase II, placebo controlled trial on the effects of the AGE-breaker alagebrium on exercise capacity and diastolic function in 102 patients with systolic HF did not support the findings of the earlier trials. In BENEFICIAL study, patients were assessed after receiving 200 mg alagebrium twice daily or placebo treatment for a period of 36 wk. No improvement was reported in exercise capacity or systolic function of those patients. In addition, no significant change was detectable in a number of other parameters including diastolic dysfunction, NYHA functional class, NT pro-BNP levels and AGE levels (skin autofluorescence). However, the authors highlighted some important points that may explain their findings. Firstly, inclusion into this study did not mandate the presence of diastolic dysfunction as an entry criterion into the trial, and it is this sub group with diastolic dysfunction that may have shown a better response to alagebrium treatment. Secondly, tissue AGE levels were relatively low in the patient cohort, especially since only (18%) of the patients included into this study were diabetic. Finally, the duration of treatment might be to short for alagebrium to produce a therapeutic effect[128].
Anti-RAGE receptor therapies
Recently, therapies targeting the RAGE receptor, e.g., sRAGE have been used in experimental studies and proved to be effective in reducing atherosclerosis in diabetic mice[129,130] as well as microvascular complications of diabetes as diabetic retinopathy[131], nephropathy[132] and neuropathy[98]. The use of the latter in the clinical setting has not yet been approved.
CONCLUSION
Cardiovascular diseases are one of the leading causes of morbidity and mortality in the western world particularly amongst patients with diabetes. AGEs accumulate rapidly in the hyperglycaemic milieu of diabetes and have an important role in the development of the macro and microvascular complications of diabetes.
Several clinical and experimental studies support the view that AGEs might have a significant role in pathogenesis of heart failure by contributing to its development and progression either via indirect mechanisms mediated through enhancing coronary artery disease or directly by inducing myocardial damage independent of vascular effects. AGEs adverse effects on cellular and tissue function arise from their potential to cross link intracellular and extracellular proteins thus altering their function and through binding to their cell surface receptor RAGE activating multiple signalling cascades in many different cells within the cardiovascular system. Preliminary data suggests that targeting AGEs therapeutically may represent a novel treatment strategy in the management of DM and its cardiovascular complications.
Footnotes
Peer reviewers: Cristina Vassalle, PhD, G. Monasterio Foun-dation and Institute of Clinical Physiology, Via Moruzzi 1, I-56124 Pisa, Italy; Sandeep A Saha, MD, Sacred Heart Medical Center, Internal Medicine Faculty Hospitalists, 101 West 8th Avenue, PO Box 2555, Spokane, WA 99220-2555, United States; Dr. Wayne Grant Carter, Department of Biomedical Sciences, School of Biomedical Sciences, University of Nottingham, Queen’s Medical Centre, Nottingham NG7 2UH, United Kingdom
S- Editor Cheng JX L- Editor A E- Editor Li JY
References
- 1.Thorpe SR, Baynes JW. Maillard reaction products in tissue proteins: new products and new perspectives. Amino Acids. 2003;25:275–281. doi: 10.1007/s00726-003-0017-9. [DOI] [PubMed] [Google Scholar]
- 2.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 3.Kent MJ, Light ND, Bailey AJ. Evidence for glucose-mediated covalent cross-linking of collagen after glycosylation in vitro. Biochem J. 1985;225:745–752. doi: 10.1042/bj2250745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sell DR, Lapolla A, Odetti P, Fogarty J, Monnier VM. Pentosidine formation in skin correlates with severity of complications in individuals with long-standing IDDM. Diabetes. 1992;41:1286–1292. doi: 10.2337/diab.41.10.1286. [DOI] [PubMed] [Google Scholar]
- 5.Bidasee KR, Nallani K, Yu Y, Cocklin RR, Zhang Y, Wang M, Dincer UD, Besch HR. Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes. 2003;52:1825–1836. doi: 10.2337/diabetes.52.7.1825. [DOI] [PubMed] [Google Scholar]
- 6.Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, Besch HR. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. 2004;53:463–473. doi: 10.2337/diabetes.53.2.463. [DOI] [PubMed] [Google Scholar]
- 7.Min C, Kang E, Yu SH, Shinn SH, Kim YS. Advanced glycation end products induce apoptosis and procoagulant activity in cultured human umbilical vein endothelial cells. Diabetes Res Clin Pract. 1999;46:197–202. doi: 10.1016/s0168-8227(99)00094-7. [DOI] [PubMed] [Google Scholar]
- 8.Wautier JL, Guillausseau PJ. Diabetes, advanced glycation endproducts and vascular disease. Vasc Med. 1998;3:131–137. doi: 10.1177/1358836X9800300207. [DOI] [PubMed] [Google Scholar]
- 9.Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 10.Yan SF, Ramasamy R, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and cardiovascular disease. Expert Rev Mol Med. 2009;11:e9. doi: 10.1017/S146239940900101X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charney DI, Walton DF, Cheung AK. Atherosclerosis in chronic renal failure. Curr Opin Nephrol Hypertens. 1993;2:876–882. doi: 10.1097/00041552-199311000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Farhey Y, Hess EV. Accelerated atherosclerosis and coronary disease in SLE. Lupus. 1997;6:572–577. doi: 10.1177/096120339700600704. [DOI] [PubMed] [Google Scholar]
- 13.Taneda S, Monnier VM. ELISA of pentosidine, an advanced glycation end product, in biological specimens. Clin Chem. 1994;40:1766–1773. [PubMed] [Google Scholar]
- 14.Hartog JW, Voors AA, Bakker SJ, Smit AJ, van Veldhuisen DJ. Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail. 2007;9:1146–1155. doi: 10.1016/j.ejheart.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Onorato JM, Thorpe SR, Baynes JW. Immunohistochemical and ELISA assays for biomarkers of oxidative stress in aging and disease. Ann N Y Acad Sci. 1998;854:277–290. doi: 10.1111/j.1749-6632.1998.tb09909.x. [DOI] [PubMed] [Google Scholar]
- 16.Bucala R, Makita Z, VegaG , Grundy S, Koschinsky T, Cerami A, Vlassara H. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. 1994;91:9441–9445. doi: 10.1073/pnas.91.20.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timmis AD. Diabetic heart disease: clinical considerations. Heart. 2001;85:463–469. doi: 10.1136/heart.85.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frye EB, Degenhardt TP, Thorpe SR, Baynes JW. Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem. 1998;273:18714–18719. doi: 10.1074/jbc.273.30.18714. [DOI] [PubMed] [Google Scholar]
- 19.Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, Fritz G. Structural basis for ligand recognition and activation of RAGE. Structure. 2010;18:1342–1352. doi: 10.1016/j.str.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 21.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 22.Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki D, Miyata T, Saotome N, Horie K, Inagi R, Yasuda Y, Uchida K, Izuhara Y, Yagame M, Sakai H, et al. Immunohistochemical evidence for an increased oxidative stress and carbonyl modification of proteins in diabetic glomerular lesions. J Am Soc Nephrol. 1999;10:822–832. doi: 10.1681/ASN.V104822. [DOI] [PubMed] [Google Scholar]
- 24.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344 Pt 1:109–116. [PMC free article] [PubMed] [Google Scholar]
- 25.Furth AJ. Glycated proteins in diabetes. Br J Biomed Sci. 1997;54:192–200. [PubMed] [Google Scholar]
- 26.Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeboah FK, Alli I, Yaylayan VA, Yasuo K, Chowdhury SF, Purisima EO. Effect of limited solid-state glycation on the conformation of lysozyme by ESI-MSMS peptide mapping and molecular modeling. Bioconjug Chem. 2004;15:27–34. doi: 10.1021/bc034083v. [DOI] [PubMed] [Google Scholar]
- 28.Howard MJ, Smales CM. NMR analysis of synthetic human serum albumin alpha-helix 28 identifies structural distortion upon amadori modification. J Biol Chem. 2005;280:22582–22589. doi: 10.1074/jbc.M501480200. [DOI] [PubMed] [Google Scholar]
- 29.Mendez DL, Jensen RA, McElroy LA, Pena JM, Esquerra RM. The effect of non-enzymatic glycation on the unfolding of human serum albumin. Arch Biochem Biophys. 2005;444:92–99. doi: 10.1016/j.abb.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 30.Simm A, Wagner J, Gursinsky T, Nass N, Friedrich I, Schinzel R, Czeslik E, Silber RE, Scheubel RJ. Advanced glycation endproducts: a biomarker for age as an outcome predictor after cardiac surgery? Exp Gerontol. 2007;42:668–675. doi: 10.1016/j.exger.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Kilhovd BK, Berg TJ, Birkeland KI, Thorsby P, Hanssen KF. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care. 1999;22:1543–1548. doi: 10.2337/diacare.22.9.1543. [DOI] [PubMed] [Google Scholar]
- 32.Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, Friedman EA, Cerami A, Vlassara H. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325:836–842. doi: 10.1056/NEJM199109193251202. [DOI] [PubMed] [Google Scholar]
- 33.Yoshida N, Okumura K, Aso Y. High serum pentosidine concentrations are associated with increased arterial stiffness and thickness in patients with type 2 diabetes. Metabolism. 2005;54:345–350. doi: 10.1016/j.metabol.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Salman AG, Mansour DE, Swelem AH, Al-Zawahary WM, Radwan AA. Pentosidine - a new biochemical marker in diabetic retinopathy. Ophthalmic Res. 2009;42:96–98. doi: 10.1159/000225661. [DOI] [PubMed] [Google Scholar]
- 35.Norlinah MI, Hamizah R, Md Isa SH, Wan Nazaimoon WM, Khalid BA. The effects of short-term, rapid glycemic control on the peroneal nerve function and serum VCAM-1 and AGE in type 2 diabetic patients in Malaysia. Indian J Med Sci. 2009;63:131–138. [PubMed] [Google Scholar]
- 36.Piarulli F, Sartore G, Ceriello A, Ragazzi E, Reitano R, Nollino L, Cosma C, Fedele D, Lapolla A. Relationship between glyco-oxidation, antioxidant status and microalbuminuria in type 2 diabetic patients. Diabetologia. 2009;52:1419–1425. doi: 10.1007/s00125-009-1367-y. [DOI] [PubMed] [Google Scholar]
- 37.Sugiyama S, Miyata T, Ueda Y, Tanaka H, Maeda K, Kawashima S, Van Ypersele de Strihou C, Kurokawa K. Plasma levels of pentosidine in diabetic patients: an advanced glycation end product. J Am Soc Nephrol. 1998;9:1681–1688. doi: 10.1681/ASN.V991681. [DOI] [PubMed] [Google Scholar]
- 38.Miura J, Yamagishi S, Uchigata Y, Takeuchi M, Yamamoto H, Makita Z, Iwamoto Y. Serum levels of non-carboxymethyllysine advanced glycation endproducts are correlated to severity of microvascular complications in patients with Type 1 diabetes. J Diabetes Complications. 2003;17:16–21. doi: 10.1016/s1056-8727(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 39.Lapolla A, Piarulli F, Sartore G, Ceriello A, Ragazzi E, Reitano R, Baccarin L, Laverda B, Fedele D. Advanced glycation end products and antioxidant status in type 2 diabetic patients with and without peripheral artery disease. Diabetes Care. 2007;30:670–676. doi: 10.2337/dc06-1508. [DOI] [PubMed] [Google Scholar]
- 40.Obayashi H, Nakano K, Shigeta H, Yamaguchi M, Yoshimori K, Fukui M, Fujii M, Kitagawa Y, Nakamura N, Nakamura K, et al. Formation of crossline as a fluorescent advanced glycation end product in vitro and in vivo. Biochem Biophys Res Commun. 1996;226:37–41. doi: 10.1006/bbrc.1996.1308. [DOI] [PubMed] [Google Scholar]
- 41.Kiuchi K, Nejima J, Takano T, Ohta M, Hashimoto H. Increased serum concentrations of advanced glycation end products: a marker of coronary artery disease activity in type 2 diabetic patients. Heart. 2001;85:87–91. doi: 10.1136/heart.85.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi EY, Kwon HM, Ahn CW, Lee GT, Joung B, Hong BK, Yoon YW, Kim D, Byun KH, Kang TS, et al. Serum levels of advanced glycation end products are associated with in-stent restenosis in diabetic patients. Yonsei Med J. 2005;46:78–85. doi: 10.3349/ymj.2005.46.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meerwaldt R, Links T, Zeebregts C, Tio R, Hillebrands JL, Smit A. The clinical relevance of assessing advanced glycation endproducts accumulation in diabetes. Cardiovasc Diabetol. 2008;7:29. doi: 10.1186/1475-2840-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jakus V, Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol Res. 2004;53:131–142. [PubMed] [Google Scholar]
- 45.Nin JW, Jorsal A, Ferreira I, Schalkwijk CG, Prins MH, Parving HH, Tarnow L, Rossing P, Stehouwer CD. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: a 12-year follow-up study. Diabetes Care. 2011;34:442–447. doi: 10.2337/dc10-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- 47.Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T, Malmberg K, Sigurdsson G, Rydén L. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. 2005;28:612–616. doi: 10.2337/diacare.28.3.612. [DOI] [PubMed] [Google Scholar]
- 48.Gottdiener JS, Arnold AM, Aurigemma GP, Polak JF, Tracy RP, Kitzman DW, Gardin JM, Rutledge JE, Boineau RC. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol. 2000;35:1628–1637. doi: 10.1016/s0735-1097(00)00582-9. [DOI] [PubMed] [Google Scholar]
- 49.He CJ, Koschinsky T, Buenting C, Vlassara H. Presence of diabetic complications in type 1 diabetic patients correlates with low expression of mononuclear cell AGE-receptor-1 and elevated serum AGE. Mol Med. 2001;7:159–168. [PMC free article] [PubMed] [Google Scholar]
- 50.Kannel WB. The Framingham study. Br Med J. 1976;2:1255. doi: 10.1136/bmj.2.6046.1255-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zieman SJ, Kass DA. Advanced glycation endproduct crosslinking in the cardiovascular system: potential therapeutic target for cardiovascular disease. Drugs. 2004;64:459–470. doi: 10.2165/00003495-200464050-00001. [DOI] [PubMed] [Google Scholar]
- 52.Galderisi M. Diastolic dysfunction and diabetic cardiomyopathy: evaluation by Doppler echocardiography. J Am Coll Cardiol. 2006;48:1548–1551. doi: 10.1016/j.jacc.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 53.Steine K, Larsen JR, Stugaard M, Berg TJ, Brekke M, Dahl-Jørgensen K. LV systolic impairment in patients with asymptomatic coronary heart disease and type 1 diabetes is related to coronary atherosclerosis, glycaemic control and advanced glycation endproducts. Eur J Heart Fail. 2007;9:1044–1050. doi: 10.1016/j.ejheart.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 54.Willemsen S, Hartog JW, Hummel YM, van Ruijven MH, van der Horst IC, van Veldhuisen DJ, Voors AA. Tissue advanced glycation end products are associated with diastolic function and aerobic exercise capacity in diabetic heart failure patients. Eur J Heart Fail. 2011;13:76–82. doi: 10.1093/eurjhf/hfq168. [DOI] [PubMed] [Google Scholar]
- 55.Koyama Y, Takeishi Y, Arimoto T, Niizeki T, Shishido T, Takahashi H, Nozaki N, Hirono O, Tsunoda Y, Nitobe J, et al. High serum level of pentosidine, an advanced glycation end product (AGE), is a risk factor of patients with heart failure. J Card Fail. 2007;13:199–206. doi: 10.1016/j.cardfail.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 56.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 57.Koyama Y, Takeishi Y, Niizeki T, Suzuki S, Kitahara T, Sasaki T, Kubota I. Soluble Receptor for advanced glycation end products (RAGE) is a prognostic factor for heart failure. J Card Fail. 2008;14:133–139. doi: 10.1016/j.cardfail.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 58.Raposeiras-Roubín S, Rodiño-Janeiro BK, Grigorian-Shamagian L, Moure-González M, Seoane-Blanco A, Varela-Román A, Almenar-Bonet L, Alvarez E, González-Juanatey JR. Relation of soluble receptor for advanced glycation end products to predict mortality in patients with chronic heart failure independently of Seattle Heart Failure Score. Am J Cardiol. 2011;107:938–944. doi: 10.1016/j.amjcard.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 59.Raposeiras-Roubin S, Rodiño-Janeiro BK, Grigorian-Shamagian L, Moure-González M, Seoane-Blanco A, Varela-Román A, Alvarez E, González-Juanatey JR. Soluble receptor of advanced glycation end products levels are related to ischaemic aetiology and extent of coronary disease in chronic heart failure patients, independent of advanced glycation end products levels: New Roles for Soluble RAGE. Eur J Heart Fail. 2010;12:1092–1100. doi: 10.1093/eurjhf/hfq117. [DOI] [PubMed] [Google Scholar]
- 60.Campbell DJ, Somaratne JB, Jenkins AJ, Prior DL, Yii M, Kenny JF, Newcomb AE, Schalkwijk CG, Black MJ, Kelly DJ. Impact of type 2 diabetes and the metabolic syndrome on myocardial structure and microvasculature of men with coronary artery disease. Cardiovasc Diabetol. 2011;10:80. doi: 10.1186/1475-2840-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katakami N, Matsuhisa M, Kaneto H, Matsuoka TA, Sakamoto K, Nakatani Y, Ohtoshi K, Hayaishi-Okano R, Kosugi K, Hori M, et al. Decreased endogenous secretory advanced glycation end product receptor in type 1 diabetic patients: its possible association with diabetic vascular complications. Diabetes Care. 2005;28:2716–2721. doi: 10.2337/diacare.28.11.2716. [DOI] [PubMed] [Google Scholar]
- 62.Koyama H, Shoji T, Yokoyama H, Motoyama K, Mori K, Fukumoto S, Emoto M, Shoji T, Tamei H, Matsuki H, et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:2587–2593. doi: 10.1161/01.ATV.0000190660.32863.cd. [DOI] [PubMed] [Google Scholar]
- 63.Cipollone F, Iezzi A, Fazia M, Zucchelli M, Pini B, Cuccurullo C, De Cesare D, De Blasis G, Muraro R, Bei R, et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation. 2003;108:1070–1077. doi: 10.1161/01.CIR.0000086014.80477.0D. [DOI] [PubMed] [Google Scholar]
- 64.Schram MT, Schalkwijk CG, Bootsma AH, Fuller JH, Chaturvedi N, Stehouwer CD. Advanced glycation end products are associated with pulse pressure in type 1 diabetes: the EURODIAB Prospective Complications Study. Hypertension. 2005;46:232–237. doi: 10.1161/01.HYP.0000164574.60279.ba. [DOI] [PubMed] [Google Scholar]
- 65.Genuth S, Sun W, Cleary P, Sell DR, Dahms W, Malone J, Sivitz W, Monnier VM. Glycation and carboxymethyllysine levels in skin collagen predict the risk of future 10-year progression of diabetic retinopathy and nephropathy in the diabetes control and complications trial and epidemiology of diabetes interventions and complications participants with type 1 diabetes. Diabetes. 2005;54:3103–3111. doi: 10.2337/diabetes.54.11.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerrits EG, Lutgers HL, Kleefstra N, Graaff R, Groenier KH, Smit AJ, Gans RO, Bilo HJ. Skin autofluorescence: a tool to identify type 2 diabetic patients at risk for developing microvascular complications. Diabetes Care. 2008;31:517–521. doi: 10.2337/dc07-1755. [DOI] [PubMed] [Google Scholar]
- 67.Miyata T, Ueda Y, Horie K, Nangaku M, Tanaka S, van Ypersele de Strihou C, Kurokawa K. Renal catabolism of advanced glycation end products: the fate of pentosidine. Kidney Int. 1998;53:416–422. doi: 10.1046/j.1523-1755.1998.00756.x. [DOI] [PubMed] [Google Scholar]
- 68.Soulis-Liparota T, Cooper ME, Dunlop M, Jerums G. The relative roles of advanced glycation, oxidation and aldose reductase inhibition in the development of experimental diabetic nephropathy in the Sprague-Dawley rat. Diabetologia. 1995;38:387–394. doi: 10.1007/BF00410275. [DOI] [PubMed] [Google Scholar]
- 69.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murata T, Nagai R, Ishibashi T, Inomuta H, Ikeda K, Horiuchi S. The relationship between accumulation of advanced glycation end products and expression of vascular endothelial growth factor in human diabetic retinas. Diabetologia. 1997;40:764–769. doi: 10.1007/s001250050747. [DOI] [PubMed] [Google Scholar]
- 71.Stitt AW. Advanced glycation: an important pathological event in diabetic and age related ocular disease. Br J Ophthalmol. 2001;85:746–753. doi: 10.1136/bjo.85.6.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stitt AW, Li YM, Gardiner TA, Bucala R, Archer DB, Vlassara H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am J Pathol. 1997;150:523–531. [PMC free article] [PubMed] [Google Scholar]
- 73.Stitt AW, Bhaduri T, McMullen CB, Gardiner TA, Archer DB. Advanced glycation end products induce blood-retinal barrier dysfunction in normoglycemic rats. Mol Cell Biol Res Commun. 2000;3:380–388. doi: 10.1006/mcbr.2000.0243. [DOI] [PubMed] [Google Scholar]
- 74.Lu M, Kuroki M, Amano S, Tolentino M, Keough K, Kim I, Bucala R, Adamis AP. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–1224. doi: 10.1172/JCI1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meerwaldt R, Links TP, Graaff R, Hoogenberg K, Lefrandt JD, Baynes JW, Gans RO, Smit AJ. Increased accumulation of skin advanced glycation end-products precedes and correlates with clinical manifestation of diabetic neuropathy. Diabetologia. 2005;48:1637–1644. doi: 10.1007/s00125-005-1828-x. [DOI] [PubMed] [Google Scholar]
- 76.Veves A, Akbari CM, Primavera J, Donaghue VM, Zacharoulis D, Chrzan JS, DeGirolami U, LoGerfo FW, Freeman R. Endothelial dysfunction and the expression of endothelial nitric oxide synthetase in diabetic neuropathy, vascular disease, and foot ulceration. Diabetes. 1998;47:457–463. doi: 10.2337/diabetes.47.3.457. [DOI] [PubMed] [Google Scholar]
- 77.Chen AS, Taguchi T, Sugiura M, Wakasugi Y, Kamei A, Wang MW, Miwa I. Pyridoxal-aminoguanidine adduct is more effective than aminoguanidine in preventing neuropathy and cataract in diabetic rats. Horm Metab Res. 2004;36:183–187. doi: 10.1055/s-2004-814344. [DOI] [PubMed] [Google Scholar]
- 78.Paul RG, Bailey AJ. The effect of advanced glycation end-product formation upon cell-matrix interactions. Int J Biochem Cell Biol. 1999;31:653–660. doi: 10.1016/s1357-2725(99)00023-0. [DOI] [PubMed] [Google Scholar]
- 79.Corman B, Duriez M, Poitevin P, Heudes D, Bruneval P, Tedgui A, Levy BI. Aminoguanidine prevents age-related arterial stiffening and cardiac hypertrophy. Proc Natl Acad Sci USA. 1998;95:1301–1306. doi: 10.1073/pnas.95.3.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. 2003;92:785–792. doi: 10.1161/01.RES.0000065620.39919.20. [DOI] [PubMed] [Google Scholar]
- 81.Cai W, He JC, Zhu L, Peppa M, Lu C, Uribarri J, Vlassara H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation. 2004;110:285–291. doi: 10.1161/01.CIR.0000135587.92455.0D. [DOI] [PubMed] [Google Scholar]
- 82.Sobal G, Menzel EJ, Sinzinger H. Calcium antagonists as inhibitors of in vitro low density lipoprotein oxidation and glycation. Biochem Pharmacol. 2001;61:373–379. doi: 10.1016/s0006-2952(00)00548-7. [DOI] [PubMed] [Google Scholar]
- 83.Lagadic-Gossmann D, Buckler KJ, Le Prigent K, Feuvray D. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am J Physiol. 1996;270:H1529–H1537. doi: 10.1152/ajpheart.1996.270.5.H1529. [DOI] [PubMed] [Google Scholar]
- 84.Bidasee KR, Nallani K, Henry B, Dincer UD, Besch HR. Chronic diabetes alters function and expression of ryanodine receptor calcium-release channels in rat hearts. Mol Cell Biochem. 2003;249:113–123. [PubMed] [Google Scholar]
- 85.Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, Inoko H, Ikemura T. Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics. 1994;23:408–419. doi: 10.1006/geno.1994.1517. [DOI] [PubMed] [Google Scholar]
- 86.Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 87.Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta. 2009;1793:993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 88.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 89.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 90.Sturchler E, Galichet A, Weibel M, Leclerc E, Heizmann CW. Site-specific blockade of RAGE-Vd prevents amyloid-beta oligomer neurotoxicity. J Neurosci. 2008;28:5149–5158. doi: 10.1523/JNEUROSCI.4878-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McDonald DR, Bamberger ME, Combs CK, Landreth GE. beta-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 93.Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med. 2000;6:643–651. doi: 10.1038/76216. [DOI] [PubMed] [Google Scholar]
- 94.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–31749. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 95.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198:1507–1515. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lin L. RAGE on the Toll Road? Cell Mol Immunol. 2006;3:351–358. [PubMed] [Google Scholar]
- 100.Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci. 2011;36:625–632. doi: 10.1016/j.tibs.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 101.Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE) J Biol Chem. 2008;283:27255–27269. doi: 10.1074/jbc.M801622200. [DOI] [PubMed] [Google Scholar]
- 102.Xue J, Rai V, Singer D, Chabierski S, Xie J, Reverdatto S, Burz DS, Schmidt AM, Hoffmann R, Shekhtman A. Advanced glycation end product recognition by the receptor for AGEs. Structure. 2011;19:722–732. doi: 10.1016/j.str.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem. 2007;282:4218–4231. doi: 10.1074/jbc.M608888200. [DOI] [PubMed] [Google Scholar]
- 104.Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130:323–334. doi: 10.1016/j.cell.2007.05.055. [DOI] [PubMed] [Google Scholar]
- 105.Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22:1572–1580. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- 106.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 107.Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler Thromb. 1994;14:1521–1528. doi: 10.1161/01.atv.14.10.1521. [DOI] [PubMed] [Google Scholar]
- 108.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–9897. [PubMed] [Google Scholar]
- 109.Penfold SA, Coughlan MT, Patel SK, Srivastava PM, Sourris KC, Steer D, Webster DE, Thomas MC, MacIsaac RJ, Jerums G, et al. Circulating high-molecular-weight RAGE ligands activate pathways implicated in the development of diabetic nephropathy. Kidney Int. 2010;78:287–295. doi: 10.1038/ki.2010.134. [DOI] [PubMed] [Google Scholar]
- 110.Riehl A, Bauer T, Brors B, Busch H, Mark R, Németh J, Gebhardt C, Bierhaus A, Nawroth P, Eils R, et al. Identification of the Rage-dependent gene regulatory network in a mouse model of skin inflammation. BMC Genomics. 2010;11:537. doi: 10.1186/1471-2164-11-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 112.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 113.Neumann A, Schinzel R, Palm D, Riederer P, Münch G. High molecular weight hyaluronic acid inhibits advanced glycation endproduct-induced NF-kappaB activation and cytokine expression. FEBS Lett. 1999;453:283–287. doi: 10.1016/s0014-5793(99)00731-0. [DOI] [PubMed] [Google Scholar]
- 114.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–592. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 115.Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, Kislinger T, Stern DM, Schmidt AM, De Caterina R. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation. 2002;105:816–822. doi: 10.1161/hc0702.104183. [DOI] [PubMed] [Google Scholar]
- 116.Mizutani K, Ikeda K, Tsuda K, Yamori Y. Inhibitor for advanced glycation end products formation attenuates hypertension and oxidative damage in genetic hypertensive rats. J Hypertens. 2002;20:1607–1614. doi: 10.1097/00004872-200208000-00024. [DOI] [PubMed] [Google Scholar]
- 117.Norton GR, Candy G, Woodiwiss AJ. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation. 1996;93:1905–1912. doi: 10.1161/01.cir.93.10.1905. [DOI] [PubMed] [Google Scholar]
- 118.Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004;24:32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- 119.Wilkinson-Berka JL, Kelly DJ, Koerner SM, Jaworski K, Davis B, Thallas V, Cooper ME. ALT-946 and aminoguanidine, inhibitors of advanced glycation, improve severe nephropathy in the diabetic transgenic (mREN-2)27 rat. Diabetes. 2002;51:3283–3289. doi: 10.2337/diabetes.51.11.3283. [DOI] [PubMed] [Google Scholar]
- 120.Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27:605–614. doi: 10.1159/000108104. [DOI] [PubMed] [Google Scholar]
- 121.Stirban A, Negrean M, Stratmann B, Gawlowski T, Horstmann T, Götting C, Kleesiek K, Mueller-Roesel M, Koschinsky T, Uribarri J, et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care. 2006;29:2064–2071. doi: 10.2337/dc06-0531. [DOI] [PubMed] [Google Scholar]
- 122.Du X, Edelstein D, Brownlee M. Oral benfotiamine plus alpha-lipoic acid normalises complication-causing pathways in type 1 diabetes. Diabetologia. 2008;51:1930–1932. doi: 10.1007/s00125-008-1100-2. [DOI] [PubMed] [Google Scholar]
- 123.Asif M, Egan J, Vasan S, Jyothirmayi GN, Masurekar MR, Lopez S, Williams C, Torres RL, Wagle D, Ulrich P, et al. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc Natl Acad Sci USA. 2000;97:2809–2813. doi: 10.1073/pnas.040558497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, deGroof RC, Lakatta EG. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104:1464–1470. doi: 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- 125.Vasan S, Foiles P, Founds H. Therapeutic potential of breakers of advanced glycation end product-protein crosslinks. Arch Biochem Biophys. 2003;419:89–96. doi: 10.1016/j.abb.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 126.Little WC, Zile MR, Kitzman DW, Hundley WG, O’Brien TX, Degroof RC. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J Card Fail. 2005;11:191–195. doi: 10.1016/j.cardfail.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 127.Thohan V, Koerner MM, Pratt CM, Torre GA. Improvements in diastolic function among patients with advanced systolic heart failure utilizing alagebrium (an oral advanced glycation endproduct cross-link breaker) Circulation. 2005;112(Suppl 2):2647. [Google Scholar]
- 128.Hartog JW, Willemsen S, van Veldhuisen DJ, Posma JL, van Wijk LM, Hummel YM, Hillege HL, Voors AA. Effects of alagebrium, an advanced glycation endproduct breaker, on exercise tolerance and cardiac function in patients with chronic heart failure. Eur J Heart Fail. 2011;13:899–908. doi: 10.1093/eurjhf/hfr067. [DOI] [PubMed] [Google Scholar]
- 129.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 130.Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 131.Kaji Y, Usui T, Ishida S, Yamashiro K, Moore TC, Moore J, Yamamoto Y, Yamamoto H, Adamis AP. Inhibition of diabetic leukostasis and blood-retinal barrier breakdown with a soluble form of a receptor for advanced glycation end products. Invest Ophthalmol Vis Sci. 2007;48:858–865. doi: 10.1167/iovs.06-0495. [DOI] [PubMed] [Google Scholar]
- 132.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]