

Summary
Differential methylation of the two parental genomes in placental mammals is essential for genomic imprinting and embryogenesis. To systematically study this epigenetic process, we have generated a base-resolution, allele specific DNA methylation (ASM) map in the mouse genome. We find parent-of-origin dependent (imprinted) ASM at 1,952 CG dinucleotides. These imprinted CGs form 55 discrete clusters including virtually all known germline differentially methylated regions (DMRs) and 24 previously unknown DMRs, with some occurring at microRNA genes. We also identify sequence dependent ASM at 131,765 CGs. Interestingly, methylation at these sites exhibits a strong dependence on the immediate adjacent bases, allowing us to define a conserved sequence preference for the mammalian DNA methylation machinery. Finally, we report a surprising presence of non-CG methylation in the adult mouse brain, with some showing evidence of imprinting. Our results provide a resource for understanding the mechanisms of imprinting and allele-specific gene expression in mammalian cells.
Introduction
In mammals, DNA methylation plays a critical role in genomic imprinting, X chromosome inactivation, cellular differentiation and development (Bird, 2002). Occurring primarily on cytosine within a CG dinucleotide, DNA methylation is considered a major epigenetic mark responsible for silencing of cell fate regulators during development (Reik et al., 2001). DNA methylation is established by the de novo DNA methyltransferases DNMT3a and DNMT3b, and maintained by the DNA methyltransferase DNMT1 (Chen and Li, 2004). Mutations that compromise the DNA methylation machinery result in early embryonic lethality (Li et al., 1992; Okano et al., 1999). Cytosine methylation can also occur in non-CG contexts including CHH and CHG (where H = A, C or T) as shown in embryonic stem cells (Lister et al., 2009; Ramsahoye et al., 2000; Ziller et al., 2011), oocytes, and pre-implantation embryos (Haines et al., 2001; Imamura et al., 2005; Tomizawa et al., 2011). Non-CG methylation is largely depleted from adult somatic cells previously examined (Lister et al., 2011; Ramsahoye et al., 2000; Ziller et al., 2011) with a few exceptions (Dyachenko et al., 2010).
A subset of mammalian genes are only transcribed from one parental allele leading to parent-of-origin specific expression or genomic imprinting (Bartolomei and Ferguson-Smith, 2011; Reik and Walter, 2001). Such genomic imprinting is crucial for embryonic development as mouse embryos containing only maternal or paternal genomes failed to develop normally (Surani et al., 1990). In humans, loss of imprinting contributes to the development of a number of diseases including Prader-Willi Syndrome, Angelman Syndrome, Beckwith-Wiedemann Syndrome and cancer (Lalande, 1996). Many imprinted genes are known to be expressed in the brain and are involved in neurodevelopment (Wilkinson et al., 2007). Imprinted expression is often directly controlled by the differentially methylated regions (DMRs) harboring parent-of-origin dependent allele specific DNA methylation (ASM). Some DMRs acquire their allelic methylation status during gametogenesis (germline DMRs, or gDMRs), which is then maintained throughout development (Reik and Walter, 2001). Other DMRs become allelicly methylated only later in development (somatic DMRs, or sDMRs), often in a tissue specific manner. In mice, several large-scale efforts have been carried out to identify imprinted DMRs (Hayashizaki et al., 1994; Hiura et al., 2010; Kelsey et al., 1999; Peters et al., 1999; Plass et al., 1996; Singh et al., 2011; Smith et al., 2003). Yet, currently the number of known imprinted DMRs is still very limited. Less than 30 well-validated germline DMRs have been reported in mice or humans (Chotalia et al., 2009; Hiura et al., 2010; Schulz et al., 2008).
Allelic DNA methylation can also arise in a way dependent on the sequence context (Tycko, 2010). Such ASM has been found in humans and mice, with some linked to allele specific gene expression (Chen et al., 2011; Gertz et al., 2011; Hellman and Chess, 2010; Kerkel et al., 2008; Schalkwyk et al., 2010; Schilling et al., 2009; Shoemaker et al., 2010; Zhang et al., 2009). Currently, it is not entirely clear what sequence determinants are important for such allelic DNA methylation.
Previous large-scale approaches identifying ASM primarily relied upon methylation-sensitive restriction enzyme or immunoprecipitation of methylated DNA (Cooper and Constancia, 2010; Tycko, 2010). These methods suffered from a low resolution constrained by the limited number of restriction sites, or the size of fragmented DNA. A novel microarray based approach allowed the investigation of over 27,000 CG sites in human promoter regions for possible imprinted ASM sites at single-nucleotide resolution (Avila et al., 2010; Choufani et al., 2011). Recently, next generation sequencing based tools such as MethylC-Seq, BS-Seq, and RRBS (Reduced Representation Bisulfite Sequencing) enabled efficient base-resolution mapping of DNA methylation (Cokus et al., 2008; Lister et al., 2008; Meissner et al., 2008). Their application to mammalian cells has led to the identification of ASM at thousands of CG sites in the human genome (Chen et al., 2011; Gertz et al., 2011; Shoemaker et al., 2010).
Here we present a genome-wide, base-resolution ASM map in mice, generated by applying MethylC-Seq to the mouse frontal cortex from reciprocal crosses between two distantly related inbred strains. Taking advantage of ∼20 million single nucleotide polymorphisms (SNPs) present in these two strains, we were able to identify virtually all known imprinted germline DMRs and 24 candidate imprinted DMRs. Further, we demonstrated the presence of non-CG methylation in the adult mouse brain and showed that it could also occur in an allele specific manner. Finally, we investigated the determinants underlying sequence dependent ASM at 131,765 CG sites, and revealed a conserved sequence preference of DNA methylation machinery.
Results
Identification of parent-of-origin and sequence dependent ASM at base resolution in the mouse frontal cortex
To investigate allele specific DNA methylation genome wide, we performed reciprocal crosses between two inbred mouse strains 129X1/SvJ (129) and Cast/EiJ (Cast), and conducted MethylC-Seq (Lister et al., 2009) using frontal cortex DNA from adult F1 progenies of the initial cross 129 (mother) × Cast (father) (denoted hereafter as F1i) and the reciprocal cross Cast (mother) × 129 (father) (denoted hereafter as F1r). We generated 1.54 billion (25.4 × per strand) and 1.33 billion (22.1 × per strand) uniquely mapped reads, respectively, from F1i and F1r (Figure 1A and Figure S1A). The bisulfite conversion rates were 99.50% for F1i and 99.51% for F1r (Supplementary Methods). To distinguish parental origins for alleles in the progeny strains, we first identified 20.4 million SNPs between the 129 genome (sequenced in this study, 14.7 × coverage) and the Cast genome (Keane et al., 2011). Due to these genetic polymorphisms, 9.7% of CGs, 1.8% of CHGs and 1.2% CHHs in the 129 genome are disrupted in the Cast genome. In subsequent analyses, we focused primarily on the CGs, CHGs and CHHs common to both the 129 and Cast strains. In F1i, 1.15 billion cytosine methylation events were found in all mapped reads (Figure 1B). Surprisingly, a significant fraction of these events correspond to cytosines in non-CG contexts (8% from CHG and 27% from CHH). The average non-CG methylation levels found here are comparable to those observed in human embryonic stem cells (hESCs) (Lister et al., 2011) (Figure S1B). Similar observations were also made for F1r (Figure 1B and Figure S1B), suggesting that non-CG methylation is also present in the mouse frontal cortex (discussed in detail later).
Figure 1. Genome-wide base-resolution identification of ASM in the mouse frontal cortex.
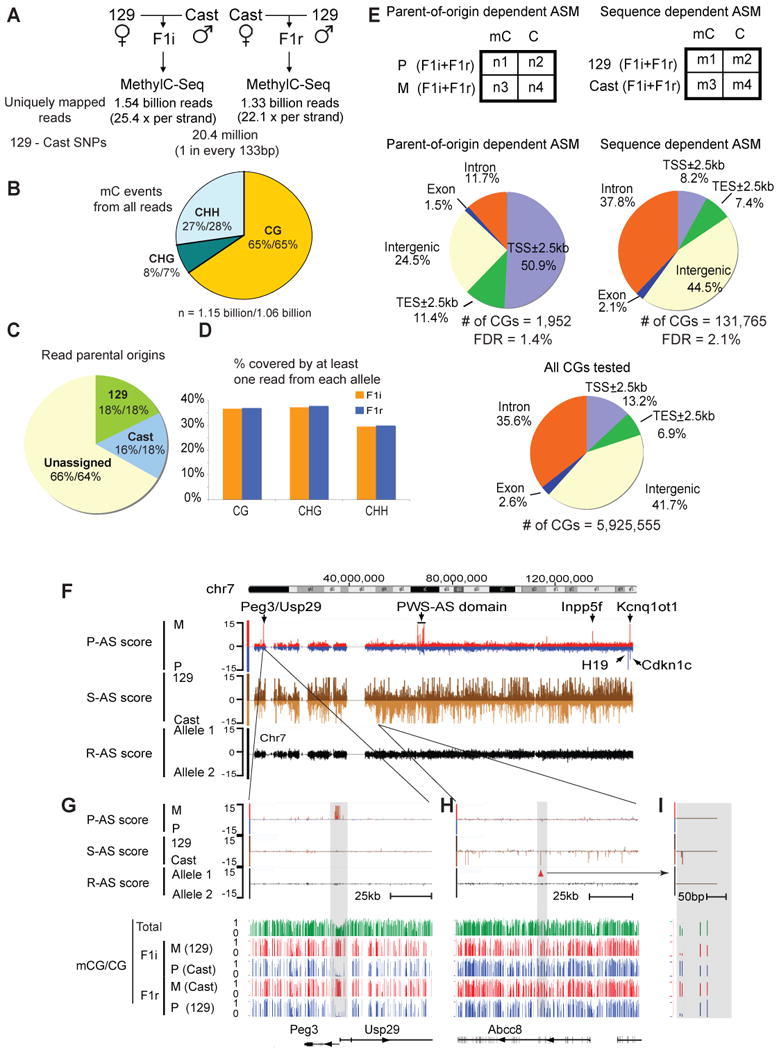
(A) The methylome sequencing depths for F1i and F1r, and the number of SNPs between the parental 129 and Cast genomes are shown. (B) A pie-chart showing the percentages of total methylcytosine events that occur in the contexts of CG, CHG and CHH for F1i (the first numbers) and F1r (the second numbers). Numbers of methylcytosine events resulting from bisulfite conversion failure (based on the conversion rate) were subtracted from the total numbers of methylcytosine events for CGs, CHGs and CHHs. (C) A pie-chart showing the percentages of MethylC-Seq reads assigned to their parental origins for F1i (the first numbers) and F1r (the second numbers). (D) The percentages of cytosines in the mouse genome covered by at least one read from both alleles are shown as bar graphs for F1i (orange) and F1r (blue). (E) The Fisher's exact test was used to identify parent-of-origin and sequence dependent ASM (top). The genomic distributions are shown for the identified ASM events (middle) or all CGs subjected to the Fisher's exact test (bottom). TSS, transcription start site; TES, transcription end site. (F) A chromosome-wide (chr7) view of AS scores for parent-of-origin dependent ASM (P-AS score; red, maternally methylated (M); blue, paternally methylated (P)) and sequence dependent ASM (S-AS score; dark brown, the 129 allele methylated (129); light brown, the Cast allele methylated (Cast)). A control track shows AS scores by assigning reads randomly to two arbitrary alleles (R-AS score; black). (G) A zoomed-in view of (F) for a region near imprinted genes Peg3 and Usp29 (top). The CG methylation levels (data from both strands combined; green, total; red, maternal; blue, paternal) in each strain are also shown (bottom). (H) A zoomed-in view of (F) for a region near Abcc8 showing sequence dependent ASM. A further enlarged region with two sequence dependent ASM sites is shown in (I). See also Figure S1.
We next determined parent-of-origin dependent (imprinted) and sequence dependent ASM. Using the above SNP table, we assigned 527 million MethylC-Seq reads to their parental origins in F1i (34% of total reads, Figure 1C). Throughout the genome, 36.7%, 37.3% and 29.5% of CG, CHG and CHH sites, respectively, are covered by at least one read from each parental allele (Figure 1D). Similar observations were made for F1r. We first focused our studies on CGs, and investigated only those that had at least 5x coverage of each allele in each strain (n = 5,925,555). We selected CGs that showed consistent allele bias (parent-of-origin or sequence dependent) for DNA methylation in both strains. The significance of such bias at each CG site was assessed by the Fisher's exact test using allelic reads pooled from both strains (Figure 1E, top). We then used the p-values from the test and computed an “allele specific score” (AS score, -log10(p-value)) to reflect DNA methylation bias for the parent of origin (P-AS score, with positive and negative values assigned for the maternal and paternal preferences, respectively) or the strain background (sequence) (S-AS score, with positive and negative values assigned for the 129 and the Cast preferences, respectively). To estimate the false discovery rate (FDR), we randomly permuted the allele-assignment of each read and computed the AS scores in parallel (R-AS score, Supplementary Methods). As shown in Figure 1F, clusters of parent-of-origin dependent ASM can be readily revealed by the AS scores at known imprinted loci on chromosome 7, including Peg3/Usp29 (with a zoomed-in view shown in Figure 1G), the PWS-AS domain, Inpp5f, H19, Kcnq1ot1 and Cdkn1c. Furthermore, sequence dependent ASM sites were also identified, which appear to be much more abundant (Figure 1F). The majority of these ASM sites exist in isolation and scatter along the chromosome (Figure 1H and I, discussed in detail later).
Using a cutoff of AS score 3 (absolute value, corresponding to p-value = 0.001), we identified sequence dependent ASM at 131,765 CGs, compared to 2,737 ASM sites in random datasets (FDR = 2.1%, Figure S1C). The same criterion however yielded 8,335 imprinted ASM sites with a high FDR of 32.8% (2,737/8,335). By further applying more stringent criteria on these 8,335 CGs, we selected those that show either higher AS scores (AS score ≥ 5, absolute value, Figure S1C) or clustering with other imprinted CGs (Figure S1D), resulting in a total of 1,952 imprinted ASM sites with a FDR of 1.4% (Figure S1E). Compared to the total CGs that we analyzed (Figure 1E, bottom), imprinted ASM sites preferentially occur in the proximal promoters (Figure 1E, middle left). By contrast, sequence dependent ASM sites are typically found in intergenic and intronic regions, and are relatively depleted from the proximal promoters (Figure 1E, middle right). Therefore these results suggest distinct molecular basis underlying these two types of ASM.
Identification and analyses of imprinted DMRs
As noted above, imprinted CGs are frequently found in clusters. In fact, we found that the 1,952 imprinted ASM sites can be grouped into 55 discrete genomic regions (Supplementary Methods), including 31 known DMRs (Table 1; see Table S2 for full references). We expect to identify most of the germline DMRs. Indeed, among 22 germline DMRs previously reported in mice (Chotalia et al., 2009; Hiura et al., 2010; Schulz et al., 2008), 21 (95%) are found in our list (Table 1, marked by “*”). A germline DMR near Nnat was not identified due to poor SNP coverage of the locus. Further examination of MethylC-Seq reads covering this region showed that CGs in these reads are either fully methylated or not methylated at all, supporting the presence of ASM events (Figure S2A). For the majority of the known DMRs, their sizes we identified are consistent with those reported previously (Figure S2B). Certain variations of DMR boundaries identified in this and prior studies may reflect incomplete coverage of SNPs, different assays, or the dynamic changes of DMRs in various cell types or developmental stages (Tomizawa et al., 2011).
Table1.
Imprinted DMRs identified in this study.
| Known imprinted DMRs (n=31) | Novel DMRs (n = 24) | |||||
|---|---|---|---|---|---|---|
| Novel DMRs within or near known imprinted loci (n=13) | ||||||
| Chr | Locus | Chr | Locus | Imprinting gene activity nearby | CGI | GC content |
| chr1 | Gpr1/Zdbf2* | chr2 | H13 DMR2 (3′ end) | Known imprinting locus, K4me3 | Yes | 0.60 |
| chr2 | Mcts2/H13* | chr7 | Snrpn U exon | Known imprinting locus, RNA | No | 0.52 |
| chr2 | Nesp | chr7 | AK086712 promoter | Known imprinting locus, K4me3, RNA | No | 0.47 |
| chr2 | Nespas/Gnasxl* | chr7 | U80893 upstream | Known imprinting locus, K4me3 | No | 0.39 |
| chr2 | Gnas1a* | chr7 | mir344b | Known imprinting locus, K4me3, K27ac | No | 0.42 |
| chr6 | Peg10/Sgce* | chr7 | mir344c | Known imprinting locus, K27ac | No | 0.41 |
| chr6 | Mest (Peg1)* | chr7 | mir344 | Known imprinting locus, K27ac | No | 0.45 |
| chr6 | Herc3/Nap1l5* | chr7 | mir344-2 | Known imprinting locus, K4me3, K27ac | No | 0.44 |
| chr7 | Peg3/Usp29* | chr7 | mir344g | Known imprinting locus, K4me3, K27ac | No | 0.37 |
| chr7 | Snurf/Snrpn* | chr7 | Magel2 promoter (known in humans) | Known imprinting locus, K4me3 | Yes | 0.52 |
| chr7 | Ndn | chr7 | Magel2-Mrkn3 intergenic | Known imprinting locus, K4me3 | No | 0.51 |
| chr7 | Mkrn3 | chr11 | Grb10 DMR2 (intragenic) | Known imprinting locus, RNA | No | 0.55 |
| chr7 | Peg12 | chr11 | Commd1 DMR2 (intragenic) | Known imprinting locus, RNA | No | 0.5 |
| chr7 | Inpp5f* | Novel diffuse DMRs within or near known imprinted loci (n=2) | ||||
| chr7 | H19 promoter | |||||
| chr7 | H19 ICR* | |||||
| chr7 | Kcnq1ot1* | Chr | Locus | Imprinting gene activity nearby | CGI | GC content |
| chr7 | Cdkn1c | chr12 | Gtl2-Mirg diffuse DMR | Known imprinting locus, RNA | NA | 0.45 |
| chr7 | Cdkn1c upstream | chr15 | Eif2c2 diffuse DMR | Known imprinting locus, RNA | NA | 0.49 |
| chr9 | Rasgrf1* | Novel DMRs outside of known imprinted loci (n=9) | ||||
| chr10 | Plagl1* | |||||
| chr11 | Grb10* | Chr | Locus | Imprinting gene activity nearby | CGI | GC content |
| chr11 | Zrsr1/Commd1* | chr6 | Casc1 intragenic | K4me3 | No | 0.51 |
| chr12 | Dlk1 | chr7 | 6330408a02Rik 3′ end | K4me3 | Yes | 0.62 |
| chr12 | Dlk1-Gtl2 IG* | chr11 | FR149454 promoter | K4me3 | No | 0.53 |
| chr12 | Gtl2 | chr12 | FR085584 promoter | K4me3 | Yes | 0.52 |
| chr15 | Peg13* | chr15 | Myo10 intragenic | K4me3 | Yes | 0.61 |
| chr15 | Slc38a4* | chr6 | Vwde promoter | No | No | 0.61 |
| chr17 | Airn* | chr10 | Neurog3 upstream | No | No | 0.46 |
| chr17 | Igf2r | chr13 | Nhlrc1 downstream | No | Yes | 0.54 |
| chr18 | Impact* | chr15 | Pvt1 promoter | No | No | 0.47 |
Known germline DMRs are marked by “*”. CGI, DMRs overlapping with CpG islands; GC content, the GC content of DMRs (or a 200bp window around the DMR if a DMR is less than 100bp); known imprinting locus, DMRs within or near known imprinted loci; K4me3, allele specific H3K4me3 enrichment observed on the opposite allele of DNA methylation in the DMR; K27ac, allele specific H3K27ac enrichment observed on the opposite allele of DNA methylation in the DMR; RNA, allele specific RNA transcripts observed on the opposite allele of DNA methylation in nearby regions; NA, not available.
In addition to reported imprinted DMRs, we also found 24 novel DMRs, among which 15 are either near or within the known imprinted domains (Table 1). Interestingly, ten of these fifteen DMRs (those on chromosome 7) reside in the PWS-AS domain, mutations in which are responsible for Prader-Willi Syndrome and Angelman Syndrome (Nicholls and Knepper, 2001). We also found two large DNA domains (the Gtl2-Mirg and the Eif2c2 diffuse DMRs) that contain lower density of imprinted CGs than that of other DMRs (Figure 4A and Figure S5A, discussed later). Lastly, nine novel DMRs (Casc1 intragenic, 6330408a02Rik 3′ end, FR149454 promoter, FR085584 promoter, Myo10 intragenic, Vwde promoter, Neurog3 upstream, Nhlrc1 downstream and Pvt1 promoter) are distant from any known imprinted domains (>5 megabase pairs). Of these nine DMRs, four co-localize with CpG islands and seven are in GC-rich regions (GC content >0.5, compared to 0.42 for the genome average) (Table 1).
Figure 4. Parent-of-origin dependent non-CG methylation in the mouse frontal cortex.
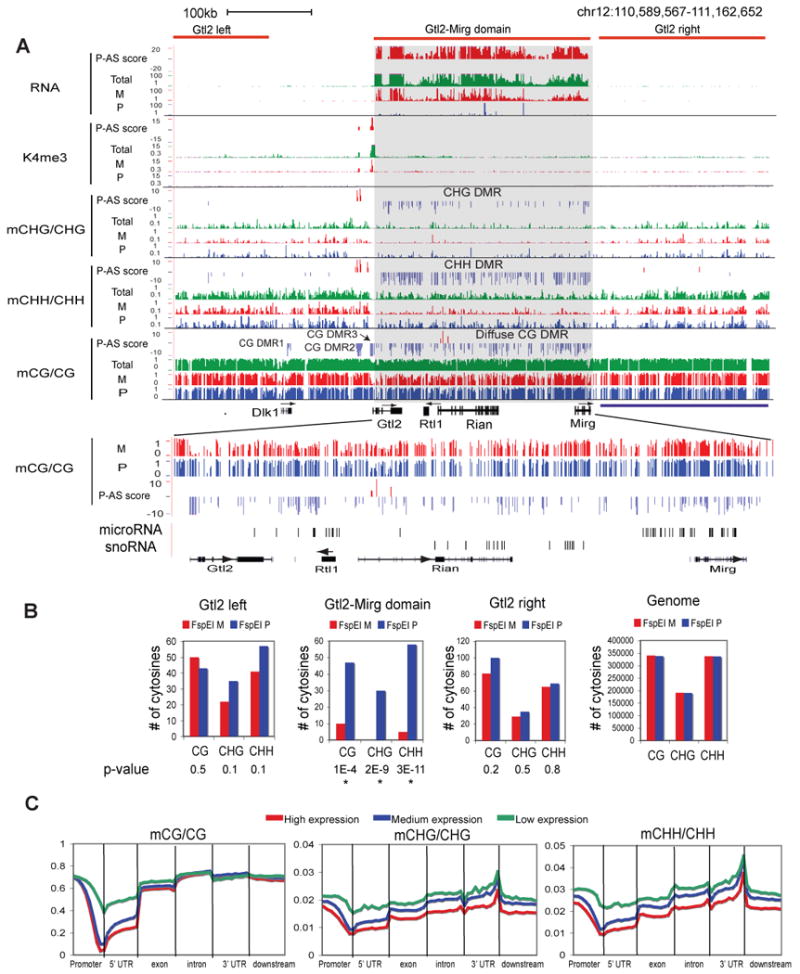
(A) Total and/or allelic levels of RNA, K4me3, CHG methylation, CHH methylation and CG methylation, together with their P-AS scores, are shown for the Dlk1-Gtl2-Mirg domain (top). CG DMRs (DMR1-3 and the diffuse CG DMR) and non-CG DMRs are indicated. A zoomed-in region for the Gtl2-Mirg domain is shown with the locations of microRNA and snoRNA genes indicated (bottom). (B) The numbers of CGs, CHGs and CHHs corresponding to FspEI cut sites on the maternal and paternal alleles in the Gtl2 domain, two regions nearby (“Gtl2-left” and “Gtl2-right”) or the entire genome are shown as bar graphs. The p-values for the allelic bias (binomial distribution) are also shown (“*”, p-value < 0.01). (C) The average methylation levels for CG, CHG and CHH are shown along the promoter (2.5kb upstream of TSSs), 5′UTR, exon, intron, 3′UTR and downstream regions (2.5kb downstream of TESs), for all RefSeq genes with high (top 1/3, red), medium (middle 1/3, blue) and low (bottom 1/3, green) levels of expression (FPKM values, average of F1i and F1r). See also Figure S5.
To search for potential imprinted transcriptional activities near these novel DMRs, we performed RNA-Seq in the mouse frontal cortex. In the same tissue we also carried out ChIP-Seq assays for two histone modifications associated with gene activities: H3K4me3 (K4me3) and H3K27ac (K27ac). Parent-of-origin AS scores were computed for each data type to assess their allelic bias (Supplementary Methods). As shown in Figure 2A, AS scores for RNA and histone modifications accurately reflect preferential paternal enrichment of K4me3, K27ac and RNA transcripts at Peg3 and Usp29, two genes known to be paternally expressed. In sum, for 20 out of 24 novel DMRs reported in this study, we have found evidence in nearby regions (∼ 135kb for the Snrpn U exon DMR and < 20kb for the rest 19 DMRs) for parent-of-origin dependent transcription and/or active histone mark enrichment (Table 1 and described below). For the remaining 4 DMRs (Vwde promoter, Neurog3 upstream, Nhlrc1 downstream and Pvt1 promoter), we did not find imprinted gene activity within five megabase pairs.
Figure 2. Identification of known and novel imprinted DMRs in the mouse genome.
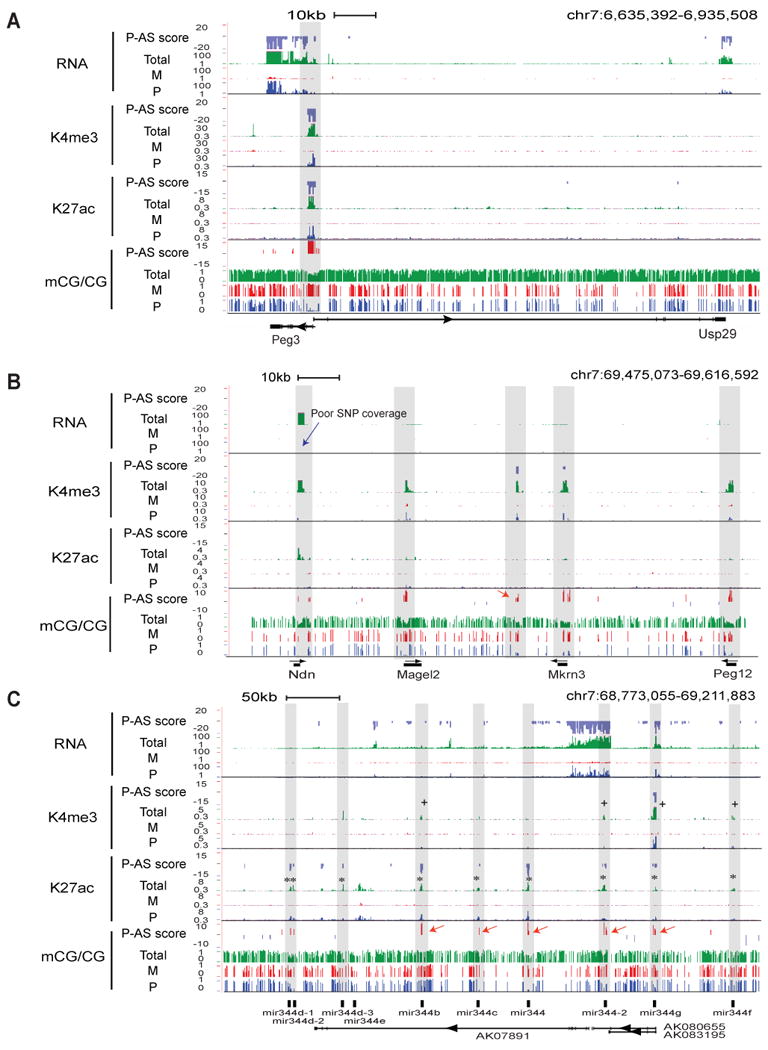
(A) Total (green) and allelic (red, maternal; blue, paternal) levels of RNA, K4me3, K27ac (RPKM values) and CG methylation, together with their P-AS scores, are shown for a region containing Peg3 and Usp29. The ChIP-Seq data were input-normalized. The shade denotes the approximate area harboring the identified DMR in this study. (B) A similar graph as (A) is shown for a region containing Ndn, Magel2, Mkrn3 and Peg12 with DMRs shaded. A novel DMR (red arrow) and a region with poor SNP coverage (blue arrow) are indicated. (C) A similar graph as (A) is shown for a region containing a microRNA gene cluster (mir344). DMRs, K27ac and K4me3 peaks that co-localize with microRNA genes (shaded) are indicated by red arrows, “*” and “+”, respectively. See also Figure S3.
Figure 2B shows an example of newly identified DMRs in the PWS-AS domain including a cluster of paternally expressed genes: Ndn, Magel2, Mkrn3 and Peg12 (in the mouse frontal cortex, Magel2, Mkrn3 and Peg12 are not expressed based on our RNA-Seq data). In mice, evidence of DMRs was reported for Mkrn3, Peg12, Ndn, and a DMR was found in humans for Magel2 (see Table S2 for full references). Consistently, we found maternally methylated DMRs at the promoters of all four genes. Further we found a novel DMR in the intergenic region between Magel2 and Mkrn3 (Figure 2B, red arrow). This DMR is marked by a paternal K4me3 peak, suggesting the existence of an un-annotated gene that is potentially imprinted.
Notably, we also found maternally methylated DMRs, each containing 1-5 CGs, at 5 microRNA genes (mir344b, mir344c, mir344, mir344-2 and mir344g) in the PWS-AS domain (Figure 2C, red arrows). These genes are part of the mir344 gene cluster that includes 5 other microRNA genes (Figure 2C). It is currently unknown if genes in the mir344 cluster are imprinted (Royo and Cavaille, 2008). The lack of SNPs in the mature microRNA sequences has prevented us from directly assessing the imprinting status of these microRNA genes. Our RNA-Seq analysis, which only assayed RNA molecules greater than 50bp and therefore cannot capture microRNA expression, did reveal a paternal transcript that appears to initiate from the promoter of an upstream gene AK086712 (data not shown) and extend into the mir344 cluster (Figure 2C, track “RNA Total”). Interestingly, we found strong paternal enrichment of K4me3 at mir344g (which shares a promoter region with AK080655 and AK083195) and weak paternal peaks of K4me3 at mir344b, mir344-2 and mir344f (Figure 2C, marked by “+”). Paternal enrichment of K27ac at these microRNA genes is even more evident, appearing at 9 out of the 10 microRNA genes (Figure 2C, marked by “*”). Therefore, the presence of imprinted DMRs and active histone marks at the mir344 gene cluster not only strongly supports their imprinted status, but also suggests an autonomous transcription mechanism for these microRNA genes by utilizing their own promoters. The remaining novel DMRs are included in Figure S3.
Identification of non-CG methylation in the mouse frontal cortex
As described above, a large fraction of methylcytosines occur in the non-CG context in the adult mouse frontal cortex (Figure 1B and Figure 3A). While the methylation level for most non-CG sites is low in the frontal cortex genome, a significant number of non-CG sites are highly methylated (Figure 3B). We detected over 3.1 million and 2.6 million non-CG sites with methylation levels greater than 0.4 (coverage ≥ 10) in F1i and F1r, respectively. These are comparable to the number of methylated non-CG sites using the same threshold (0.4) in hESCs (∼ 2.3 million, calculated from Lister et al., 2009). To validate the presence of non-CG methylation, we took three experimental approaches. First, we showed that the MethylC-Seq data were well reproduced using bisulfite-PCR coupled with Sanger sequencing at three genomic loci (Figure S4A). Second, we determined DNA methylation genome wide using a DNA methylation-dependent enzyme FspEI. FspEI recognizes the CmC motif, in which the second cytosine is methylated and can be in the context of CG, CHG or CHH (Zheng et al., 2010). We sequenced the FspEI digested genomic DNA from the mouse frontal cortex (F1i and F1r) and control cells IMR90 and MEF (Figure S4B-C). In IMR90 and MEF, methylcytosines corresponding to the FspEI cut sites are predominantly CGs (Figure 3C). By contrast, we found a large fraction of non-CG methylation at the FspEI cut sites in the frontal cortex genome. Importantly, the average number of FspEI cuts per cytosine positively correlates with cytosine methylation levels obtained from MethylC-Seq for CGs, CHGs and CHHs (Figure 3D). This is not the case when BstNI, a methylation independent restriction enzyme, was used in DNA digestion before subsequent sequencing (Figure 3D). Finally, abundant non-CG methylation is also observed in the parental strains 129 and Cast when we sequenced their methylomes (12.5× and 12.8× per strand, respectively)(data not shown). Therefore, we conclude that non-CG methylation is indeed present in the adult mouse brain.
Figure 3. Non-CG methylation is present in the mouse frontal cortex.
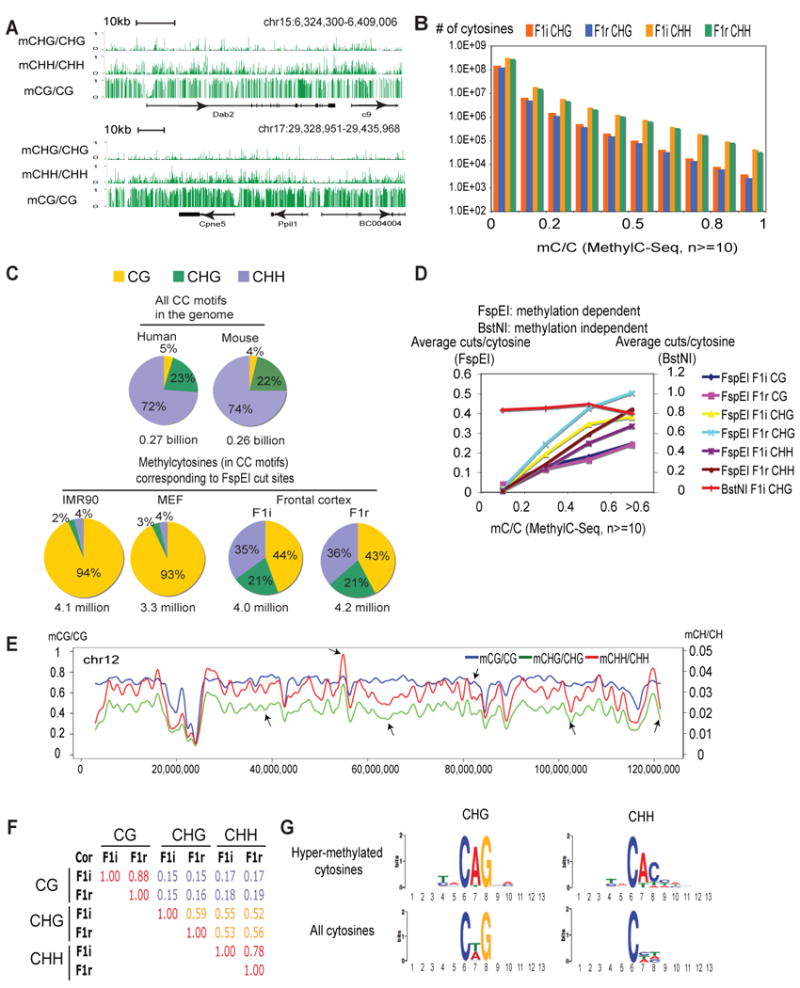
(A) The methylation levels for CHG, CHH and CG are shown near two examples of genomic loci (pooled data from F1i and F1r, coverage ≥10). For simplicity, only data from the forward strand are shown for non-CG methylation. (B) The numbers of cytosines (coverage ≥10) at various methylation levels are shown as bar graphs for CHG and CHH in F1i and F1r. (C) The percentages of CGs, CHGs and CHHs corresponding to FspEI cut sites in IMR90, MEF, F1i and F1r are plotted as pie-charts. The percentages of CGs, CHGs and CHHs in the CC motif (the second cytosine) in the mouse and human genomes are also shown. (D) The average numbers of FspEI or BstNI cuts per recognized cytosine are plotted against the cytosine methylation levels determined by MethylC-Seq. BstNI recognizes CCWGG (W=A or T) where the second cytosine in the CHG context shows abundant methylation in the mouse cortex (data not shown). (E) A chromosome view (chr12) of CG (blue), CHG (green) and CHH (red) methylation levels (10-kb window). Arrows indicate regions where CG and non-CG methylation show different distributions. (F) Pearson correlation coefficients are shown for pairwise comparison of CG, CHG and CHH methylation levels in F1i and F1r genome wide. (G) Sequence logos are shown for bases proximal to hyper-methylated CHGs (mCHG/CHG ≥ 0.3, coverage ≥ 10) and CHHs (mCHH/CHH ≥ 0.5, coverage ≥ 10). See also Figure S4.
We next investigated the genomic distribution of non-CG methylation. A chromosome-wide view of CG and non-CG methylation revealed that, while CHG and CHH methylation correlate fairly well, CG and non-CG methylation show both similarities and differences (Figure 3E). This is also true genome wide as non-CG methylation only moderately correlates with CG methylation (Figure 3F), suggesting that non-CG methylation is not simply a side product of CG methylation. An analysis of DNA sequences around hyper-methylated CHGs and CHHs revealed strong enrichment of motifs that largely resemble those found in hESCs (Figure 3G) (Lister et al., 2009). In summary, non-CG methylation has distinct distributions compared to that of CG methylation in the frontal cortex.
Allele specific non-CG methylation at imprinted loci in the mouse frontal cortex
We then asked if non-CG methylation might also occur in a parent-of-origin dependent manner. We computed the parent-of-origin AS scores for non-CG methylation (Supplementary Methods) and examined the methylation allele bias at known imprinted loci. Indeed, parent-of-origin dependent non-CG methylation is evident at 8 imprinted loci (the Gtl2-Mirg domain, the PWS-AS domain, Kcnqot1, Trappc9/Peg13, Gpr1, Sgce, Rasgrf1 and Grb10), including most imprinted regions of large size (see below and data not shown). One such locus, the Gtl2-Mirg domain, is located in the Dlk1-Dio3 imprinting cluster, which is known to be essential for embryonic development (da Rocha et al., 2008). The Dlk1-Dio3 domain contains at least three paternally expressed genes Dlk1, Rtl1 and Dio3 (which all appear to be silenced in the mouse frontal cortex, Figure 4A and data not shown), and multiple maternally expressed non-coding RNA genes including Gtl2, Rian and Mirg. We observed a single H3K4me3 peak at the Gtl2 promoter, followed by a region of continuous maternal transcription that appears to span the entire Gtl2-Mirg domain (Figure 4A, shaded), supporting the existence of a single non-coding transcript initiating from Gtl2 (Tierling et al., 2006). In the same region we observed paternal enrichment of non-CG methylation. This is true for both CHG and CHH methylation (Figure 4A) and in both F1i and F1r (data not shown), thus strongly arguing that the presence of non-CG methylation is not due to the failure of bisulfite conversion, in which case both parental alleles would be affected equally.
Interestingly, in addition to the non-CG methylation DMRs present in the Gtl2-Mirg domain, we also found evidence of a large CG DMR (206 kbp) in the same region that contains at least 205 paternally methylated CGs. These imprinted CGs in this DMR are relatively scattered (the median number of neighboring imprinted CGs in a 5kb window is 8, compared to 31 for all other imprinted CGs, t test p-value = 4E-205). This is in contrast to other CG DMRs including those previously identified in the Dlk1-Dio3 cluster (DMR1-DMR3, Figure 4A)(Takada et al., 2002). These imprinted CGs do not appear to co-localize with the promoters of annotated genes in this region including microRNA genes and snoRNA genes (Figure 4A, bottom). Therefore, we considered it as a special “diffuse DMR”. A similar diffuse DMR is observed at the Eif2c2 locus just outside of the Trapp9/Peg13 imprinted domain (Figure S5A). In summary, parent-of-origin dependent DMRs are present for both CG and non-CG methylation in the Gtl2-Mirg domain.
Notably, the non-CG methylation in the Gtl2-Mirg domain is present on a silenced allele (Figure 4A). This is also confirmed by the FspEI digestion assay, which shows preferential cut of the paternal allele in the Gtl2 domain, but not in two regions nearby (“Gtl2 left” and “Gtl2 right”) (Figure 4B). Similarly we found non-CG methylation occurring on the repressed allele of the imprinted Kcnq1ot1 (Figure S5B-C), and four other imprinted genes (Peg13, Sgce, Grb10 and Rasgrf1, data not shown). Further, while CG DMRs (except for diffuse CG DMRs) in these imprinted loci are preferentially located at the promoters/upstream regions, non-CG DMRs often extend into gene bodies (Figure S5B and data not shown). We then examined the relationship of non-CG methylation and gene activity in the entire genome. Consistent with previous findings(Lister et al., 2009), we found that at promoters, both CHG and CHH methylation inversely correlate with gene expression (Figure 4C). However, in gene bodies, in striking contrast to the reported positive correlation between non-CG methylation and gene activity in hESCs (Lister et al., 2009), both CHG and CHH methylation negatively correlate with gene expression in the mouse frontal cortex (see Discussion). Taken together, these data not only demonstrate that non-CG methylation in the mouse frontal cortex correlate with gene activity, but also suggest that it may be regulated differently from that in hESCs.
Characterization of genomic regions associated with sequence dependent ASM
Compared to parent-of-origin dependent ASM, sequence dependent ASM sites are very abundant in the mouse genome (Figure 1F). We confirmed that sequence dependent ASM was not due to mapping bias between the two alleles (Figure S6A). Such methylation bias is not only present between the 129 and the Cast alleles in F1i and F1r, but is also apparent between the parental 129 and Cast strains (Figure 5A and Figure S6B), indicating that it is likely inherited from parental strains in a sequence dependent manner. A genomic distribution analysis revealed that the level of sequence dependent ASM (S-AS score) is largely uniform in regions near genes with the exception of the proximal promoters, where ASM is depleted at genes with high or medium expression levels (Figure 5B and Figure S6C). This phenomenon may be partly due to low levels of CG methylation, SNP density and high level of conservation associated with active genes (Figure S6D-H). Nevertheless, genes depleted of ASM are strongly enriched in those coding for homeobox proteins, transcription factors, development regulators, as well as histones and ribosome proteins (Figure S6I). We did not find any gene ontology enrichment for genes that show the most abundant sequence dependent ASM. These results suggest that DNA methylation at the promoters of some key developmental regulators and housekeeping genes is subject to stringent regulation.
Figure 5. Genome-wide localization of sequence dependent ASM.
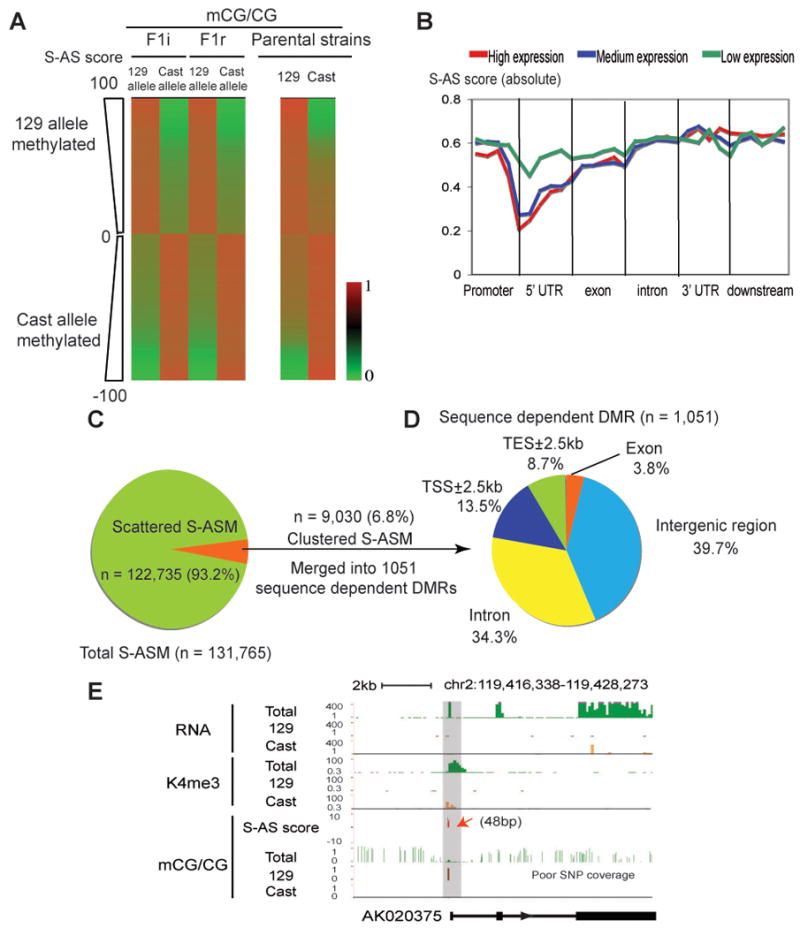
(A) The CG methylation levels of the sequence dependent ASM sites (ranked by the S-AS scores) are shown for the 129 allele and Cast allele for F1i and F1r (left). The methylation levels for the same CG sites in the parental 129 and Cast strains (coverage ≥10) are also shown (right). (B) The average S-AS scores (absolute value) are shown along the promoter (2.5kb upstream of TSSs), 5′UTR, exon, intron, 3′UTR and downstream regions (2.5kb downstream of TESs) for all RefSeq genes with high (top 1/3, red), medium (middle 1/3, blue) and low (bottom 1/3, green) levels of expression. (C) The percentages of scattered and clustered sequence dependent ASM sites are shown in a pie chart. (D) Genomic distribution of sequence dependent DMRs (median length = 1,010bp) is shown in a pie chart. (E) An example gene AK020375 shows the 129 allele specific promoter CG methylation (red arrow) and the Cast allele specific K4me3 enrichment and transcription. A region with poor SNP coverage is indicated. See also Figure S6.
We then examined the relationship between sequence dependent ASM and allele specific gene expression (ASE). Unlike imprinted ASM, most sequence dependent ASM sites (93.2%) are present in isolation (Figure 5C). Such ASM does not appear to correlate with ASE genome wide (data not shown). A small fraction of sequence dependent ASM sites (6.8%, n=9030) do show clustering and can be grouped into 1,051 DMRs (Figure 5D). Of these sequence dependent DMRs, the majority fall into intergenic regions (39.7%) and introns (34.3%), yet 141 (13.5%) are present at gene promoters. We examined the downstream genes that are likely to be regulated by promoter-associated sequence dependent DMRs. Among the 94 genes for which allelic expression or K4me3 state could be ascertained, 20 (21.3%) show allele specific transcription or K4me3 enrichment that inversely correlates with the DNA methylation status (see Figure 5E for an example). The rest display no significant allelic bias in gene activity. These data are consistent with a study in humans (Gertz et al., 2011), suggesting that a small fraction of sequence dependent ASM sites are clustered and may influence allele specific gene expression.
Sequence dependent ASM reveals a sequence preference for DNA methylation
To determine what genetic variations may contribute to sequence dependent ASM, we next examined the SNP frequency near sequence dependent ASM sites. Indeed, an elevated SNP density is associated with these allelicly methylated cytocines (Figure 6A). Interestingly, the SNPs at the -1 and +1 position show a strong bias in base composition (Figure 6B). On the hyper-methylated allele, there is a strong enrichment of G and C at the -1 and +1 positions, respectively. By contrast, on the hypo-methylated allele A and T/A are preferentially present at the -1 and +1 positions, respectively. Importantly, this is not observed for a random set of CGs (Figure 6B). Togther, these results revealed the over-representation of GCG/CGC and ACG/CGT motifs on the hyper- and hypo-methylated alleles, respectively (Figure 6C). We next hypothesized that such sequence preference for DNA methylation may exist in the entire genome. To test this, we examined methylation levels of various 4-mer CG motifs (CG plus -1 and +1 bases, or NCGN) throughout the genome using combined F1i and F1r methylome data. We excluded CpG islands (CGIs) and promoters in our analysis, as these regions are generally depleted of DNA methylation in part due to the presence of antagonistic H3K4me3 (Jia et al., 2007; Ooi et al., 2007; Thomson et al., 2010). Indeed, GCGC exhibits the highest level of methylation among all 4-mer motifs (Figure 6D), and it is followed by motifs that contain either a GCG or CGC signatures. Those containing an ACG or CGT motif are ranked lowest in DNA methylation. This is not simply related to GC content, as motifs with similar GC contents (Figure 6D, marked by “*” or “#”) demonstrate distinct methylation levels. The hyper- and hypo-methylated motifs also do not show significant differences in their locations in relation to genes (excluding the promoters, Figure S7A) or repetitive sequences (Figure S7B). We conclude that the CG methylation dependence on the -1 and +1 flanking positions is observed both at the sequence dependent ASM sites and on a genome-wide scale.
Figure 6. Sequence dependent ASM reveals sequence determinants of DNA methylation.
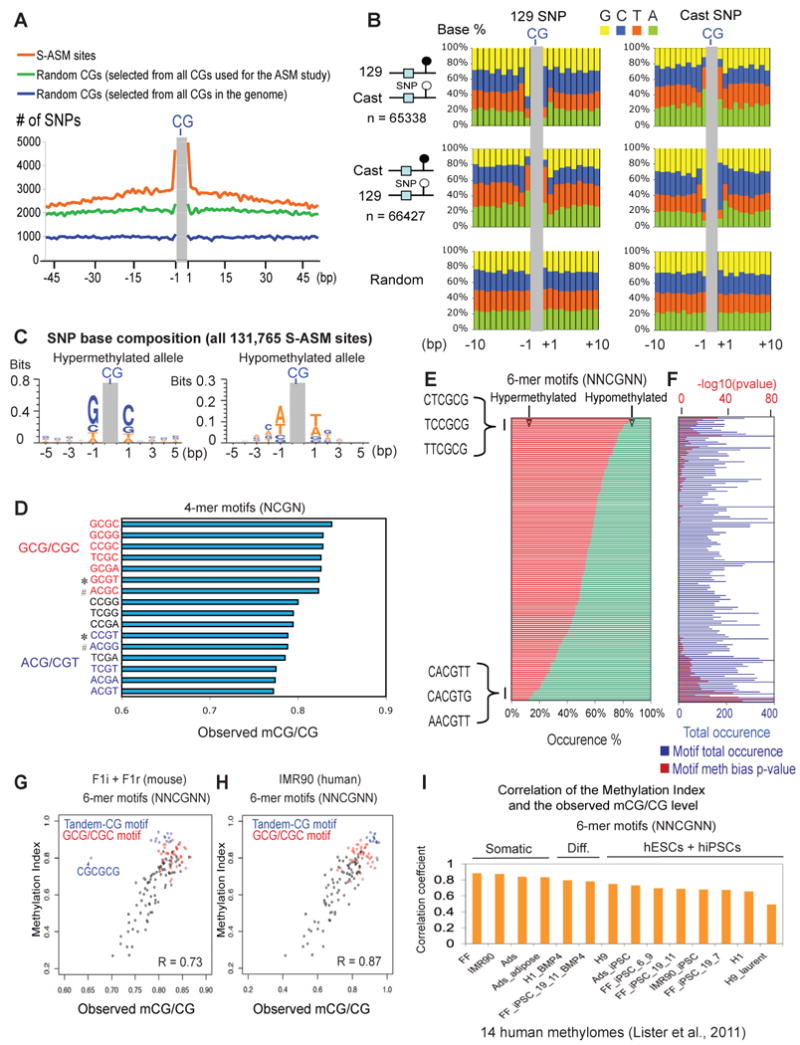
(A) The total number of SNPs at each base within +/- 50bp of 131,765 sequence dependent ASM sites (orange) is shown. A similar plot is shown for 131,765 random CG sites drawn from either all CGs selected for the ASM study (green) or all CGs in the whole genome (blue). (B) The SNP base composition on the 129 (left) or the Cast (right) allele is shown for those near ASM sites that are preferentially methylated on the 129 allele (top) or the Cast allele (middle). A similar analysis was done for a control set of random CGs of equal size drawn from all CGs selected for the ASM study (bottom). (C) The base composition for SNPs on the hyper- (left) or hypo- (right) methylated alleles near all sequence-dependent ASM sites is shown as a sequence logo. (D) The median methylation levels of various 4-mer CG motifs (“Observed mCG/CG”) across the frontal cortex genome (excluding the promoters and CGIs) are shown. Motifs that contain GCG/CGC or ACG/CGT signatures are in red or blue, respectively. Examples of motif pairs (marked by “*” or “#”) with similar GC content but with distinct methylation levels are indicated. (E) The percentages of occurrences on hyper- (red) and hypo- (green) methylated alleles are shown for each 6-mer CG motif. (F) The number of total occurrences on both alleles (blue bars, with the scale at the bottom) and the p-value (binomial test, after Bonferroni multiple test correction) reflecting allele occurrence bias (red bars, with the scale at the top) for each motif in (E) are shown. (G) A scatter plot is shown for various 6-mer CG motifs comparing the Methylation Indexes to the median methylation levels across the frontal cortex genome. Motifs with tandem CGs (blue) or GCG/CGC signatures (red) are indicated. R, the Pearson correlation coefficient. (H) A similar plot as (G) is shown for various 6-mer CG motifs comparing the Methylation Indexes derived from mice to the median methylation levels across the genome of IMR90 (Lister et al., 2011). (I) The Pearson correlation coefficients are shown comparing the Methylation Indexes derived from mice to the median methylation levels across the genomes of 14 human lines (Lister et al., 2011) for various 6-mer CG motifs. See also Figure S7.
We further asked if any bases beyond the -1 and +1 positions may also influence CG methylation, particularly those at the -2 and +2 positions, where SNPs show the highest A+T percentages on the hyper-methylated allele and the lowest A+T percentages on the hypo-methylated allele (Figure 6B). We therefore examined 13,584 sequence dependent ASM sites that contain SNPs at the -1, -2, +1 or +2 positions. At these sites, various 6-mer motifs (NNCGNN) demonstrated distinct frequencies on the hyper- and hypo-methylated alleles (Figure 6E and Table S3), many of which are of high statistical significance (Figure 6F). For example, CTCGCG is observed 235 times (86%) on hyper-methylated alleles but only 39 times (14%) on hypo-methylated alleles (p-value = 2E-33, binomial test). To quantify such methylation preference for each motif, we computed a “Methylation Index” based on its relative occurrence on hyper- and hypo-methylated alleles using a Bayesian model (Supplementary Methods). Similar as for the 4-mer motifs, we asked if such DNA methylation preference for the 6-mer motifs also holds true in the genome. Indeed, we observed a positive correlation (R = 0.73) between the median methylation level in the genome and the Methylation Index for each motif (Figure 6G). Interestingly, the correlation is higher (R = 0.85) when excluding motifs containing tandem CGs (such as CGCGCG or CGCGGT). Further, we also examined 14 recently published human methylomes (Lister et al., 2011). Again, we observed strong correlation for various 6-mer motifs between their Methylation Indexes derived from mice and their methylation levels in humans (see Figure 6H for an example in IMR90). Interestingly, the correlations are lower for hESCs and hiPSCs than those for human somatic cells (Figure 6I), possibly due to the high levels of DNA methylation in hESCs and hiPSCs which likely diminish the differences of methylation levels among various motifs (Figure S7C). In summary, we found that CG methylation is significantly influenced by the immediate flanking bases, a feature appearing to be conserved from mice to humans.
Discussion
A genome-wide, base-resolution survey of imprinted ASM in the mouse genome
Differentially methylated regions between two alleles are critical for the genomic imprinting and proper embryogenesis (Bartolomei and Ferguson-Smith, 2011). In this study, we have performed a comprehensive survey of ASM in the mouse genome, uncovering virtually all known imprinted germline DMRs, as well as 24 new imprinted DMRs. These novel DMRs should help identify new regulatory regions for known imprinted genes, or discover new imprinted loci. Among them, of particular interest are two atypical DMRs (the Gtl2-Mirg and the Eif2c2 diffuse DMRs) containing relatively scattered imprinted CGs. Currently, it is not clear whether the diffuse DMRs are a cause or a result of the allele specific transcription. Therefore, a novel imprinting mechanism may exist in this DMD that calls for future study. In addition, such DMRs allowed the identification of novel imprinted genes whose imprinting status is difficult to determine, including those that show mono-allelic expression only in certain tissues, and microRNA genes which have short mature transcripts. We also compared DMRs identified in this study to a recent genome-wide survey of imprinted genes in the mouse (Gregg et al., 2010), which reported over a thousand imprinted genes in the embryonic brain, adult cortex and hypothalamus. Surprisingly, we found that most of the novel imprinted genes found by Gregg et al. are far away from the DMRs identified in the present study (93% are at least 1 megabase pairs away from any DMRs, compared to 2% for known imprinted genes). Similar to two previous studies (Babak et al., 2008; Wang et al., 2008), our own RNA-Seq data also failed to reveal the imprinting status of most novel genes reported in Gregg et al. (data not shown). It is possible that DNA methylation independent imprinting mechanisms may be responsible for the large number of imprinted genes reported by Gregg and colleagues. Alternatively, the discrepancy may also arise from differences in the strains or methods of data analyses used in each study. Nevertheless, results from our study reveal significant epigenetic differences between the two parental genomes that will help elucidate the mechanisms of genomic imprinting.
Evidence for non-CG methylation in the mouse frontal cortex
The discovery of abundant non-CG methylation events in the adult mouse frontal cortex is surprising. In contrast to non-CG methylation in hESCs (Lister et al., 2009), our data suggest that non-CG methylation in the mouse frontal cortex is negatively correlated with gene activity in transcribed regions. In addition, we found CHHs are more likely to be methylated than CHGs in the mouse brain, while an opposite observation was made in hESCs (Lister et al., 2009). It is currently unclear why non-CG methylation displays distinct distribution patterns in these two types of cells. Interestingly, it has been shown that Dnmt3a, which has been implicated in methylation at non-CG sites (Ramsahoye et al., 2000), is expressed in different isoforms in ESCs and the brain. The major isoform expressed in ESCs Dnmt3a2 is preferentially enriched at euchromatin, while the mouse brain only expresses Dnmt3a1 which selectively targets heterochromatin (Chen et al., 2002), suggesting that different DNA methylation machinery may exist in ESCs and the frontal cortex. Recently, 5-hydroxymethylcytosine (5hmC) has been found in the mouse brain cells (Kriaucionis and Heintz, 2009). The lack of a base-resolution approach to measure 5hmC prevents us from quantitatively distinguishing it from methylcytosine in the MethylC-seq data. However, in the mouse brain, 5hmC appears to be detected only at CG sites, but not at non-CG (CA) sites (or below the detection limit) (Kriaucionis and Heintz, 2009). It shows positive correlation with gene activity over transcribed regions (Song et al., 2011), where non-CG methylation shows negative correlation, suggesting that non-CG methylation is unlikely to be a simple result of 5hmC. In conclusion, these findings suggest that non-CG methylation is not limited to pluripotent cells and may be subject to regulations by different mechanisms in hESCs and the mouse brain.
Sequence dependent ASM reveals a sequence code for DNA methylation
Although imprinted ASM is critical for development, our genome-wide data suggest that the vast majority of differences in DNA methylation between two parental genomes are sequence dependent. In this study, we have focused on ASM that does not involve the change of CG identities. We showed that while most of such ASM events are isolated and appear to have little effect on gene expression, they provide a unique opportunity for us to determine the sequence determinants of DNA methylation. We demonstrate that DNA methylation at CGs is strongly influenced by defined sequences in the immediate neighborhood. Such sequence preference is not unique to mouse frontal cortex, but is also observed in multiple human cell types, suggesting a conserved mechanism for regulation of DNA methylation by adjacent sequences. These findings are consistent with previous studies showing that Dnmt3a and Dnmt3b, or the Dnmt3a interacting protein Dmnt3L, may be affected by the sequence context of their substrates (Chedin et al., 2002; Jia et al., 2007; Wienholz et al., 2010). The hyper- and hypo-methylated motifs found here appear to be different from those derived from DNA methylation patterns at several CpG islands using the episomal methylation assay in a recent study (Wienholz et al., 2010), but agree with DNA methylation motifs discovered in Arabidopsis (Cokus et al., 2008; Lister et al., 2008). Taken together, these data suggest the existence of an evolutionarily conserved sequence code for DNA methylation. Given that CpG islands and promoter regions are actively maintained in a hypo-methylated state by H3K4me3 or other factors (Jia et al., 2007; Ooi et al., 2007; Thomson et al., 2010), the DNA methylation pattern is likely a result of methyltransferase (or demethylase) actions influenced by transcription factors, local sequence context and chromatin environment. Our findings set the stage for further investigation of how these factors work together to establish the global DNA methylation landscape in mammalian genomes.
Experimental Procedures
Strain crosses
The crosses of the two mouse strains were performed at Jackson Laboratories. The male parental strains and the F1 offspring were shipped at 8 to 9 weeks of age.
MethylC-Seq library generation and sequencing
Genomic DNA was extracted from the frontal cortex of the F1 crosses or the parental strains, and was spiked in with unmethylated lambda DNA (Promega). The DNA was fragmented by sonication. Purified DNA fragments were end-repaired and ligated to paired-end cytosine- methylated adapters provided by Illumina. Size-selected adapter-ligated DNA was treated with sodium bisulfite using the EZ DNA methylation-Gold Kit (Zymo Research). The resulting DNA molecules were enriched by PCR, purified and sequenced following standard protocols from Illumina.
ChIP-Seq library generation and sequencing
Frozen frontal cortex from the F1 crosses was thawed on ice and processed with a razor blade into small pieces. The tissue was then crosslinked with formaldehyde, washed, homogenized, and proceeded following a ChIP protocol as described in the Supplemental Information. ChIP libraries were prepared and sequenced following standard protocols from Illumina.
RNA-Seq library generation and sequencing
The frontal cortex from the F1 crosses was dissected and RNA was isolated followed by DNAseI treatment. RNA was treated with RiboMinus (Invitrogen) to remove the ribosomal RNA. Libraries were prepared according to the SOLiD sequencing protocol and sequenced at EdgeBio.
Data analyses
Details of bioinformatic analyses can be found in the Supplemental Information.
Supplementary Material
Highlights.
A base-resolution genome-wide allelic methylation map for CG and non-CG in mice
Novel imprinted CG DMRs include those at microRNA genes and two large diffuse DMRs
Abundant non-CG methylation is present in the mouse brain and can be allele specific
A sequence preference for CG methylation is likely evolutionarily conserved
Acknowledgments
We thank Drs. Ryan Lister and Joseph Ecker for sharing the MethylC-Seq protocol and for valuable input on the experimental design. We are grateful to Dr. Paul Soloway for comments on the manuscript, Dr. Wei Wang for discussions, Ms. Lee Edsall and Samantha Kuan for technical support in deep sequencing, Richard Logan, Yu Feng and Lissette Gomez for technical support in dissection of frontal cortex and extraction of DNA/RNA, Karen Wigg for initial RNA-Seq bioinformatics support, and members of the Ren laboratory for discussions. This study was funded in part by grants from the Krembil Seed Development Fund (CB), an Applied Biosystems (Life Technologies) 10K Genome Award (CB), and by funding from the Ludwig Institute for Cancer Research (BR) and the National Human Genome Research Institute R01 HG003991 (BR).
Footnotes
Data Accession: All sequencing data were deposited to GEO under the accession number GSE33722.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avila L, Yuen RK, Diego-Alvarez D, Penaherrera MS, Jiang R, Robinson WP. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta. 2010;31:1070–1077. doi: 10.1016/j.placenta.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Babak T, Deveale B, Armour C, Raymond C, Cleary MA, van der Kooy D, Johnson JM, Lim LP. Global survey of genomic imprinting by transcriptome sequencing. Curr Biol. 2008;18:1735–1741. doi: 10.1016/j.cub.2008.09.044. [DOI] [PubMed] [Google Scholar]
- Bartolomei MS, Ferguson-Smith AC. Mammalian Genomic Imprinting. Cold Spring Harb Perspect Biol. 2011 doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002;99:16916–16921. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Feng S, Joo JW, Jacobsen SE, Pellegrini M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011;12:R62. doi: 10.1186/gb-2011-12-7-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Li E. Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol. 2004;60:55–89. doi: 10.1016/S0070-2153(04)60003-2. [DOI] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Xie S, Li E. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J Biol Chem. 2002;277:38746–38754. doi: 10.1074/jbc.M205312200. [DOI] [PubMed] [Google Scholar]
- Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105–117. doi: 10.1101/gad.495809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Shapiro JS, Susiarjo M, Butcher DT, Grafodatskaya D, Lou Y, Ferreira JC, Pinto D, Scherer SW, Shaffer LG, et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome research. 2011;21:465–476. doi: 10.1101/gr.111922.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper WN, Constancia M. How genome-wide approaches can be used to unravel the remaining secrets of the imprintome. Brief Funct Genomics. 2010;9:315–328. doi: 10.1093/bfgp/elq018. [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–316. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Dyachenko OV, Schevchuk TV, Kretzner L, Buryanov YI, Smith SS. Human non-CG methylation: are human stem cells plant-like? Epigenetics. 2010;5:569–572. doi: 10.4161/epi.5.7.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, Kucera KS, Willard HF, Myers RM. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7:e1002228. doi: 10.1371/journal.pgen.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines TR, Rodenhiser DI, Ainsworth PJ. Allele-specific non-CpG methylation of the Nf1 gene during early mouse development. Dev Biol. 2001;240:585–598. doi: 10.1006/dbio.2001.0504. [DOI] [PubMed] [Google Scholar]
- Hayashizaki Y, Shibata H, Hirotsune S, Sugino H, Okazaki Y, Sasaki N, Hirose K, Imoto H, Okuizumi H, Muramatsu M, et al. Identification of an imprinted U2af binding protein related sequence on mouse chromosome 11 using the RLGS method. Nature genetics. 1994;6:33–40. doi: 10.1038/ng0194-33. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin. 2010;3:11. doi: 10.1186/1756-8935-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura H, Sugawara A, Ogawa H, John RM, Miyauchi N, Miyanari Y, Horiike T, Li Y, Yaegashi N, Sasaki H, et al. A tripartite paternally methylated region within the Gpr1-Zdbf2 imprinted domain on mouse chromosome 1 identified by meDIP-on-chip. Nucleic Acids Res. 2010;38:4929–4945. doi: 10.1093/nar/gkq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Kerjean A, Heams T, Kupiec JJ, Thenevin C, Paldi A. Dynamic CpG and non-CpG methylation of the Peg1/Mest gene in the mouse oocyte and preimplantation embryo. J Biol Chem. 2005;280:20171–20175. doi: 10.1074/jbc.M501749200. [DOI] [PubMed] [Google Scholar]
- Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–251. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey G, Bodle D, Miller HJ, Beechey CV, Coombes C, Peters J, Williamson CM. Identification of imprinted loci by methylation-sensitive representational difference analysis: application to mouse distal chromosome 2. Genomics. 1999;62:129–138. doi: 10.1006/geno.1999.6022. [DOI] [PubMed] [Google Scholar]
- Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalande M. Parental imprinting and human disease. Annu Rev Genet. 1996;30:173–195. doi: 10.1146/annurev.genet.30.1.173. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Wroe SF, Wells CA, Miller HJ, Bodle D, Beechey CV, Williamson CM, Kelsey G. A cluster of oppositely imprinted transcripts at the Gnas locus in the distal imprinting region of mouse chromosome 2. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3830–3835. doi: 10.1073/pnas.96.7.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass C, Shibata H, Kalcheva I, Mullins L, Kotelevtseva N, Mullins J, Kato R, Sasaki H, Hirotsune S, Okazaki Y, et al. Identification of Grf1 on mouse chromosome 9 as an imprinted gene by RLGS-M. Nature genetics. 1996;14:106–109. doi: 10.1038/ng0996-106. [DOI] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nature reviews Genetics. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- Royo H, Cavaille J. Non-coding RNAs in imprinted gene clusters. Biol Cell. 2008;100:149–166. doi: 10.1042/BC20070126. [DOI] [PubMed] [Google Scholar]
- Schalkwyk LC, Meaburn EL, Smith R, Dempster EL, Jeffries AR, Davies MN, Plomin R, Mill J. Allelic skewing of DNA methylation is widespread across the genome. Am J Hum Genet. 2010;86:196–212. doi: 10.1016/j.ajhg.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling E, El Chartouni C, Rehli M. Allele-specific DNA methylation in mouse strains is mainly determined by cis-acting sequences. Genome research. 2009;19:2028–2035. doi: 10.1101/gr.095562.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R, Woodfine K, Menheniott TR, Bourc'his D, Bestor T, Oakey RJ. WAMIDEX: a web atlas of murine genomic imprinting and differential expression. Epigenetics. 2008;3:89–96. doi: 10.4161/epi.3.2.5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20:883–889. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Wu X, Lee DH, Li AX, Rauch TA, Pfeifer GP, Mann JR, Szabo PE. Chromosome-wide analysis of parental allele-specific chromatin and DNA methylation. Mol Cell Biol. 2011;31:1757–1770. doi: 10.1128/MCB.00961-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Dean W, Konfortova G, Kelsey G. Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome research. 2003;13:558–569. doi: 10.1101/gr.781503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani MA, Kothary R, Allen ND, Singh PB, Fundele R, Ferguson-Smith AC, Barton SC. Genome imprinting and development in the mouse. Dev Suppl. 1990:89–98. [PubMed] [Google Scholar]
- Takada S, Paulsen M, Tevendale M, Tsai CE, Kelsey G, Cattanach BM, Ferguson-Smith AC. Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with Igf2-H19. Hum Mol Genet. 2002;11:77–86. doi: 10.1093/hmg/11.1.77. [DOI] [PubMed] [Google Scholar]
- Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr AR, Deaton A, Andrews R, James KD, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierling S, Dalbert S, Schoppenhorst S, Tsai CE, Oliger S, Ferguson-Smith AC, Paulsen M, Walter J. High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics. 2006;87:225–235. doi: 10.1016/j.ygeno.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, Sasaki H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development. 2011;138:811–820. doi: 10.1242/dev.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko B. Allele-specific DNA methylation: beyond imprinting. Hum Mol Genet. 2010;19:R210–220. doi: 10.1093/hmg/ddq376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sun Q, McGrath SD, Mardis ER, Soloway PD, Clark AG. Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One. 2008;3:e3839. doi: 10.1371/journal.pone.0003839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholz BL, Kareta MS, Moarefi AH, Gordon CA, Ginno PA, Chedin F. DNMT3L modulates significant and distinct flanking sequence preference for DNA methylation by DNMT3A and DNMT3B in vivo. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson LS, Davies W, Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci. 2007;8:832–843. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Rohde C, Reinhardt R, Voelcker-Rehage C, Jeltsch A. Non-imprinted allele-specific DNA methylation on human autosomes. Genome Biol. 2009;10:R138. doi: 10.1186/gb-2009-10-12-r138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Cohen-Karni D, Xu D, Chin HG, Wilson G, Pradhan S, Roberts RJ. A unique family of Mrr-like modification-dependent restriction endonucleases. Nucleic Acids Res. 2010;38:5527–5534. doi: 10.1093/nar/gkq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. Genomic Distribution and Inter-Sample Variation of Non-CpG Methylation across Human Cell Types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.