

Abstract
Type 2 diabetes (T2D) is a complex, polygenic disease affecting nearly 300 million people worldwide. T2D is primarily characterized by insulin resistance, and growing evidence has indicated the causative link between adipose tissue inflammation and the development of insulin resistance. Genetic association studies have successfully revealed a number of important genes consistently associated with T2D to date. However, these robust T2D-associated genes do not fully elucidate the mechanisms underlying the development and progression of the disease. Here, we report an alternative approach, gene expression-based genome-wide association study (eGWAS): searching for genes repeatedly implicated in functional microarray experiments (often publicly available). We performed an eGWAS across 130 independent experiments (totally 1,175 T2D case-control microarrays) to find additional genes implicated in the molecular pathogenesis of T2D and identified the immune-cell receptor CD44 as our top candidate (P = 8.5 × 10−20). We found CD44 deficiency in a diabetic mouse model ameliorates insulin resistance and adipose tissue inflammation and also found that anti-CD44 antibody treatment decreases blood glucose levels and adipose tissue macrophage accumulation in a high-fat, diet-fed mouse model. Further, in humans, we observed CD44 is expressed in inflammatory cells in obese adipose tissue and discovered serum CD44 levels were positively correlated with insulin resistance and glycemic control. CD44 likely plays a causative role in the development of adipose tissue inflammation and insulin resistance in rodents and humans. Genes repeatedly implicated in publicly available experimental data may have unique functionally important roles in T2D and other complex diseases.
Keywords: bioinformatics, meta-analysis, integration, obesity, hyperglycemia
Type 2 diabetes (T2D) is a common multifactorial disease characterized by hyperglycemia primarily resulting from peripheral insulin resistance, and growing functional evidence has indicated the causative link between adipose tissue inflammation and the development of insulin resistance (1, 2). In the past decade, a number of genetic genome-wide association studies (GWAS) have revealed 40 loci consistently associated with susceptibility to T2D and have rapidly expanded our knowledge of the genetic architecture of this disease (3–13). However, the genes located in or near these loci do not fully elucidate the tissue-specific molecular mechanisms underlying the development of T2D.
A large number of experiments using genome-wide gene-expression microarray measurements have been also performed over the past decade; however, there has been little success in fully identifying functionally important genes in the pathogenesis of T2D. Because a large number of genes are often detected as significant in each microarray experiment, it may be hard to subselect optimal candidates from individual studies for further verification. A combination of genome-wide data from two or more experiments has been performed for obesity (14–16), T2D (14), and other multifactorial disorders (17–20). Several of these methods have used microarray technology to focus on candidate genes already implicated in a region of a congenic or model animal (accelerated positional candidate identification). More recently, investigators have applied microarrays to genetics by considering gene expression levels as quantitative traits (expression quantitiative trait loci, eQTLs) and finding relations between gene variants and transcripts, with successful application to the identification of genes and targets for T2D (21, 22). The need for large numbers of simultaneously acquired genetic and gene-expression measurements within a single study makes this approach less scalable.
We suspected that the large number of molecular measurements from experimental results that are now publicly available, because of requirements from journals and funding agencies (23), could be used as an alternative scalable source of data. The strategy of finding commonly implicated genes across related—but deliberately varied—experimental conditions has been theorized to yield less overfit, potentially more generalizable causal factors (24), and publicly available data could be used as a source of these varied experimental vantage points for a condition. In this report, we propose the application of a gene expression-based genome-wide association study (eGWAS), a meta-analysis method for computing the likelihood of finding repeated differential expression for every gene across a large number of case and control microarray experiments, compared with expected. Our hypothesis is that those genes most repeatedly implicated across a large set of experimental representations of T2D can serve as data-driven causal T2D genes and candidates for validation. This approach is only feasible because many of these source raw experimental results are publicly available; here, we integrated 130 independent microarray experiments for T2D. In this case, our T2D candidates were found independent of any knowledge about insulin signaling, glucose, or lipid metabolism. For our top candidate gene, identification was followed by confirmatory functional studies using mouse models and samples from human subjects. (Fig. S1).
Results
eGWAS Identifies CD44 as a Functional Candidate Gene for T2D.
We carried out an eGWAS for T2D by using 130 independent microarray experiments, totaling 1,175 samples collected from public repositories (Fig. 1, Tables S1 and S2, and SI Materials and Methods). We ranked all 24,898 genes by the likelihood that repeated differential expression for that gene was due to chance, then controlled for multiple-hypothesis testing. To overview which molecular functions are most shared in the highest-ranked genes in our T2D eGWAS, we took the top 127 genes (Table S3; Bonferroni threshold, P < 2.0 × 10−6) from our eGWAS and then estimated the enrichment of Gene Ontology (GO) terms. Interestingly, “receptor activity” and “receptor binding” functions were the most implicated of the top-ranked genes (receptor activity, 38%; receptor binding, 19%) (Fig. S2). These activities suggested that a number of top-ranked genes on our list are involved in intra- and intertissue signaling cascades in the development of T2D (1).
Fig. 1.

eGWAS for T2D using a χ2 analysis. Plot of -log10 (P value) (y axis) by chromosomal position (x axis). P values for each gene were calculated from our eGWAS across 130 microarray experiments with 1,175 T2D case-control microarray samples (591 T2D cases and 584 controls) as the likelihood of finding repeated differential expression compared with expected using a χ2 analysis, or a Fisher’s exact test (Fig. S3). Our top gene, CD44, showed a significant differential expression in 78 experiments (P = 8.5 × 10−20). The red line indicates the Bonferroni threshold (P = 2.0 × 10−6). The green dots indicate several well known T2D-susceptibility genes.
Our top-most T2D candidate gene was CD44 (Fig. 1: χ2 analysis, P = 8.5 × 10−20; Fig. S3: Fisher’s exact test, P = 6.1 × 10−17; Fig. S4: weighted Z-method), markedly differentially expressed in experiments studying diabetes in adipose tissue compared with other tissues (Fig. S5). CD44 is located on chromosome 11p13 and codes for a cell-surface glycoprotein, an immunological cell (macrophage/T-cell) receptor, involved in inflammatory cell migration and activation. Interestingly, one of the known ligands for CD44, secreted phosphoprotein 1 [SPP1; also known as osteopontin (OPN)], a Th1 cytokine secreted by immunological cells (macrophages), was also included in the top-ranked genes (Fig. 1: χ2 analysis, P = 1.3 × 10−11; Fig. S3: Fisher’s exact test, P = 3.8 × 10−10). Recent studies have indicated that obese adipose tissue is hallmarked with chronic, low-grade inflammation, and that inflammation plays a central role in the development of insulin resistance (1, 2). Although the contributions of the CD44 encoded protein to the molecular pathogenesis of T2D have not yet been reported, SPP1 was reported as a link between adipose tissue inflammation (stromal infiltration by inflammatory cells) and the development of insulin resistance in a murine model of diet-induced obesity (25). Furthermore, the expression profile of CD44 and SPP1 are coordinately dysregulated, especially in adipose tissue (Fig. S6; coordinate dysregulation rate = 0.90). These findings suggest that CD44 might have a key role in mediating obesity-induced adipose tissue inflammation and insulin resistance.
CD44 Expression Increases in Obese Adipose Tissue.
High-fat feeding in C57BL/6J mice leads to the development of obesity, adipose inflammation, and insulin resistance (25, 26). To examine whether the CD44 mRNA transcript is expressed in adipose tissue and modulated by obesity, C57BL/6J mice were maintained either on a normal-fat diet (NFD; 12% of total calories from fat) or high-fat diet (HFD; 60% of total calories from fat) for 16 wk (n = 8 per group). Compared with the NFD group, mice fed a HFD gained 37% more weight after a 16-wk feeding period (29.9 ± 0.5 g versus 40.9 ± 1.6 g; P = 5.8 × 10−8). Epididymal white adipose tissue (EWAT) was removed from these mice to analyze CD44 mRNA expression levels. Feeding a HFD resulted in a significant 11.3-fold increase of CD44 mRNA levels in adipose tissue compared with NFD (Fig. 2A). To establish the presence of CD44 protein in adipose tissue, an immunohistochemical localization of CD44 was performed on EWAT isolated from HFD mice. We found that CD44 was abundantly expressed in inflammatory cells within adipose tissue (Fig. 2B). These results clearly indicate that CD44+ cells accumulated in EWAT of diet-induced obese mice. In addition, we confirmed that the expression levels of SPP1 mRNA in adipose tissue in the HFD group was significantly higher than that in the NFD mice (0.005 ± 0.003 versus 0.06 ± 0.02; P < 0.05), similar to previous reports (25). Interestingly, we also found that CD44 mRNA expression level was positively correlated with the SPP1 mRNA expression level in the HFD mice (Fig. 2C; r = 0.78, P = 0.02), suggesting that CD44 and SPP1 may be closely related in obese adipose tissue.
Fig. 2.

CD44 expression in adipose tissue of obese mice. (A) CD44 mRNA expression levels in epididymal adipose tissues in C57BL/6J mice fed either a NFD or HFD (n = 8 per group). (B) CD44 immunoreactivity (arrows, DAB chromogen; brown) in epididymal adipose tissues from C57BL/6J mice fed a HFD. Sections were counterstained with hematoxylin (blue). (C) CD44 and SPP1 gene expression profiles in the HFD (n = 8; circles) and NFD (n = 8; triangles) groups. Correlation between CD44 and SPP1 mRNA expression in the HFD group (circles) was analyzed by using the Pearson’s correlation test. Gene expression was monitored by using real-time RT-PCR and normalized to expression of GAPDH mRNA.
CD44 Deficiency Ameliorates Adipose Tissue Inflammation and Insulin Resistance.
To next determine the contribution of CD44 to the development of adipose tissue inflammation and insulin resistance, we fed male CD44−/− and diabetes-prone C57BL/6J (CD44+/+) mice with either a HFD (n = 16 per group) or a NFD (n = 10 per group) for 12 wk and performed immunohistochemical analysis and metabolic measurements on these mice. There were no significant differences in body weights between CD44−/− and CD44+/+ mice after feeding a NFD or a HFD (NFD: CD44−/− 28.5 ± 0.5 g versus CD44+/+ 29.5 ± 0.5 g; HFD: 38.1 ± 1.5 g versus 39.5 ± 1.2 g). In CD44+/+ mice that were fed a HFD, we frequently observed the accumulation of inflammatory cells (macrophages) forming crown-like structures (CLSs) surrounding adipocytes in obese visceral adipose tissue. However, CD44−/− mice fed a HFD exhibited strikingly less macrophage infiltration into the stroma of adipose tissue compared with CD44+/+ mice fed a HFD (Fig. 3A). Fasting blood glucose levels were significantly lower in CD44−/− mice fed a HFD compared with the diabetes-prone CD44+/+ mice fed a HFD (Fig. 3B). Glucose tolerance tests also indicated that CD44−/− mice fed a HFD were significantly more efficient in their ability to clear intraperitoneally injected glucose than CD44+/+ mice fed a HFD (Fig. 3C, solid lines) despite similar insulin secretory responses after the injection of glucose. Furthermore, insulin sensitivity, as measured by insulin tolerance test, showed that CD44−/− mice fed a HFD were significantly more efficient at insulin-mediated suppression of blood glucose than CD44+/+ mice fed a HFD (Fig. 3D, solid lines). Interestingly, even in the mice fed a NFD, the ameliorative effects of CD44 deficiency on insulin sensitivity was observed in both glucose and insulin tolerance tests (Fig. 3 C and D, dashed lines). These results that we obtained with CD44-deficient mice confirm that CD44 molecules are essential for macrophage recruitment and inflammation in adipose tissue and the development of insulin resistance in diet-induced obese mice.
Fig. 3.

Histological and metabolic analyses of wild-type CD44+/+ and CD44−/− mice. (A) Inflammatory cell (macrophage) content determined by immunohistochemical staining for Mac-2 (DAB, brown; hematoxylin, blue) in epididymal adipose tissues from CD44−/− and CD44+/+ mice fed a HFD. (B–D) Metabolic measurements on CD44+/+ (open bars and symbols; diabetes-prone) and CD44−/− (filled bars and symbols) mice fed either a HFD (n = 16 per group; solid lines) or a NFD (n = 10 per group; dashed lines). (B) Fasting blood glucose. (C) Glucose tolerance tests [i.p. glucose (2 g/kg body weight)] after a 14-h overnight fast. Venous blood was obtained for measurement of blood glucose at 0, 15, 30, 60, 90, and 120 min after the injection. (D) Insulin tolerance tests [i.p. insulin (1.0 unit/kg body weight)] after a 4-h fast. Venous blood was obtained for measurement of blood glucose at 0, 30, and 45 min after the injection.
CD44 Blockade Decreases Blood Glucose Levels and Adipose Macrophage Infiltration.
Several in vivo studies have shown that anti-CD44 monoclonal antibody (CD44 mAb) treatment exhibits robust antiinflammatory effects in animal models of immune-mediated diseases (27–30). We therefore sought to investigate whether CD44 blockade might demonstrate a therapeutic effect on T2D. We performed daily i.p. injections of CD44 mAb in diabetic model mice for 1 wk and found that blood glucose levels and adipose macrophage accumulation were significantly reduced in CD44 mAb treated mice compared with isotype-control treated mice, despite continuing on the HFD and similar body weight increase during the treatment (Fig. 4 A and B). When adipose inflammation was quantified as the average number of CLSs per low power field, EWAT from CD44 mAb treated mice contained significantly fewer inflammatory cells compared with control treated mice (2.4 ± 0.5 versus 5.4 ± 0.7; P = 0.0005). Collectively, these effects of CD44 mAb clearly show that CD44 molecules are required for the recruitment of macrophages into obese adipose tissue and the maintenance of inflammatory reactions there, and the CD44 receptor may be useful as a therapeutic target for T2D.
Fig. 4.

Anti-CD44 antibody treatment for diabetic mice. (A) HFD-fed C57BL/6J mice were injected intraperitoneally with purified rat anti-mouse CD44 (IM7; 553131, BD Pharmingen) (n = 6; filled circles) or purified rat IgG2b, κ isotype control (A95-1; 559478, BD Pharmingen) (n = 8; open circles) for 1 wk (100 μg at day 0 and 50 μg at day 1–7). Morning blood glucose was measured at day 0, 1, 3, 5, and 7 during the treatment. The effect of anti-CD44 treatment on blood glucose levels was evaluated with two-way repeated measures ANOVA (*P; treatment × time). Comparisons between two groups were performed by using the two-tailed Welch's t test. Data are represented as mean ± SE. (B) Epididymal adipose tissues from control and anti-CD44 antibody-treated mice were analyzed for inflammatory cell (macrophage) content by using a Mac-2 antibody (magnified as indicated). Arrows indicate crown-like structures (CLSs) surrounding individual adipocytes.
To gain additional insight into the clinical importance of CD44 in obese fat, we performed an immunohistochemical analysis of CD44 in omental adipose tissue in human obesity. Consistent with our mouse model observation, we discovered that CD44+ cells infiltrated into the stroma of adipose tissue in human obese subjects, suggesting that CD44 molecules may mediate macrophage migration into obese adipose tissue in humans (Fig. 5A).
Fig. 5.

CD44 functional experiments in human subjects. (A) Paraffin-embedded omental adipose tissue from an obese woman [age (yr); 57, BMI (kg/m2); 36.9] was analyzed for CD44 immunoreactivity. (B and C) The correlation between serum levels of standard soluble CD44 (sCD44std) and either an index of glycemic control, HbA1c (B), or an index of insulin resistance, HOMA-IR (C), determined by using a linear regression model estimated with minimal square method in human subjects [n = 55: mean ± SD age (yr), 60.3 ± 15; BMI (kg/m2), 23.2 ± 4.3; fasting plasma glucose (mg/dL), 109 ± 13; fasting plasma insulin (μU/mL), 6.22 ± 3.84; HbA1c (%), 5.9 ± 0.34].
Soluble CD44 shed from cell surfaces exists in normal human serum. To estimate the relevance between CD44 protein and glucose homeostasis in human subjects, we evaluated the relationship between serum levels of CD44 and metabolic traits in human and found that serum CD44 was positively correlated with glycemic control and insulin resistance as estimated through HbA1c (n = 55, r = 0.49, P < 0.001) and HOMA-IR (n = 55, r = 0.29, P = 0.03) (Fig. 5 B and C). We then classified the 55 subjects into two groups according to the WHO criteria (31): hyperglycemia (n = 21: “diabetes mellitus” + “impaired glucose regulation”) and normoglycemia (n = 34: “Normal Glucose Tolerance”), and found that the serum levels of CD44 were significantly higher in the hyperglycemic group than the normoglycemic group (246.9 ± 15.0 versus 209.5 ± 9.8 ng/mL; P = 0.02). These results suggest that CD44 protein may be released from insulin-resistant and diabetic tissues into circulation in humans.
Discussion
Adipose tissue inflammation is thought to be a pivotal event leading to the metabolic syndrome, insulin resistance, and T2D. Genome-wide experimental methods to identify disease genes, such as association studies (GWAS), linkage studies (GWL), and eQTL analyses, have been performed for T2D by many researchers to date, and these methods have revealed a number of loci to be linked with T2D (3–14, 21, 32–35). However, these genetically mapped loci do not fully account for the tissue-specific mechanisms underlying the development of T2D. In this report, we proposed an alternative methodology, eGWAS, computing the likelihood of finding repeated differential expression of a gene in disease-related tissues by using thousands of case-control microarray samples. To detect additional genes functionally implicated in the molecular pathogenesis of T2D, we successfully performed an eGWAS to identify T2D candidate genes and verified our top candidate gene, CD44, an immunological cell receptor, plays a significant role in the development of adipose tissue inflammation and insulin resistance in mouse models and human subjects.
In our T2D eGWAS, we identified in total 127 genes as significantly repeatedly dysregulated with P values <2.0 × 10−6 (under the Bonferroni-corrected threshold) and rediscovered several genes that have been shown to be important in T2D pathogenesis, including TCF7L2, PPARG, KCNQ1, IDE, CD36, GLUT4, and LEPR, supporting the validity of our methodology. Of the 127 genes, we found that more than one-half were implicated in the “receptor” or “ligand” activity by using the GO term enrichment analysis (Fig. S2). Interestingly, SPP1, encoding a ligand for the CD44 receptor, was shown to be included also in our top-ranked gene list (Fig. 1 and Table S3). Furthermore, our eGWAS provided the prediction of tissue specificity of gene expression by calculating a distribution of scores for each gene across tissues, indicating that CD44 mRNA expression was more highly up-regulated in adipose tissue in diabetes than other tissues (Fig. S5). These analysis results led us to the speculation that our top-most candidate gene, CD44, encoding an immune-cell surface receptor, may be implicated in adipose tissue inflammation causing insulin resistance in obesity, given the fact that the CD44 receptor is known to regulate immune-cell migration and activation and its ligand SPP1 has been reported to mediate macrophage infiltration into obese adipose tissue (25). When interpreting this hypothesis, however, we need to consider the possibility that the dysregulation of CD44 expression in relevant tissues can result secondarily from hyperglycemia and diabetes. We therefore performed verification experiments to see whether the dysregulated expression of CD44 gene in obese adipose tissue can be accepted as the cause of insulin resistance.
In our functional tests for the CD44 gene products, we showed that knocking out the receptor CD44 leads to striking reduction in immune-cell infiltration into visceral adipose tissue and improvements in insulin sensitivity in mouse models, and anti-CD44 monoclonal antibody treatment decrease the blood glucose levels and visceral adipose tissue macrophages in diabetic obese mice. In addition, we showed that higher serum levels of the soluble form of CD44 correlate with increasing hyperglycemia and insulin resistance in humans. Although the expression of CD44 in macrophages and T cells in obese adipose tissue has been reported (36, 37), this study directly addresses the functional role of CD44 in adipose tissue inflammation. The findings that systemic glucose intolerance is ameliorated by CD44 depletion and blockade by using CD44 mAb strongly suggest that CD44-dependent adipose inflammation has an impact on systemic metabolism. However, further studies are needed to determine which immune-cell population primarily expresses CD44 receptor (macrophages or T cells?) and how the CD44 molecules initiate and maintain immune-cell infiltration into adipose tissue (through the SPP1 signals?). Even so, these results indicate the significance of CD44 immune-receptor as a possible therapeutic target for T2D and a unique biomarker for insulin resistance.
In conclusion, we discovered that an immune-cell receptor gene, CD44, is pathogenetically implicated in the development of adipose tissue inflammation and insulin resistance, using a data-driven candidate gene approach by using an eGWAS method of integrating >1,000 publicly available genome-wide functional microarrays related to T2D. Application of the eGWAS methodology to publicly available data can yield promising candidate genes that are differentially expressed in T2D-relevant tissues, independently of knowledge about insulin signaling, glucose, or lipid metabolism. We suggest that a data-driven approach can enable investigators to consider glucose homeostasis phenotypes from a different point of view and notice new pathways that could be involved in the development of T2D. Although GWAS and other genetic analyses will continue as the method of choice for the next few years, an eGWAS approach could complement these studies to yield additional pathogenetically important genes for many other complex diseases by using this integrated data-driven approach.
Materials and Methods
See SI Materials and Methods for further descriptions.
eGWAS.
All T2D-related genome-wide microarray experiments used for this study were collected from three public data sources: the NCBI Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo), the Diabetes Genome Anatomy Project (DGAP; www.diabetesgenome.org), and the Nuclear Receptor Signaling Atlas (NURSA; www.nursa.org). There were a total of 1,175 samples (591 T2D cases and 584 controls) in 130 independent datasets. To estimate differences between groups of samples from diabetic subjects and groups representing control, raw postquantitation microarray data were reanalyzed by using Significance Analysis of Microarrays software (SAM) (38). For each gene in every microarray experiment with three or more samples in each group, we calculated a d score (di), which denotes the standardized change in gene expression:
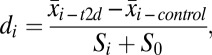 |
where 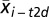 is the mean expression level of gene i in group T2D,
is the mean expression level of gene i in group T2D, 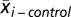 is the mean expression level of gene i in group control, Si is the SD for the numerator calculation, and S0 is a small positive constant. We considered genes to be significantly dysregulated with either an absolute value of the d score ≥2 or a fold change ≥2 between controls and cases. We then converted all probe identifiers across the various microarray platforms for mouse, rat, and human to the latest human Entrez Gene identifiers by using our published AILUN system (39). Gene expression profiles were assigned in our eGWAS database according to the standardized (human) Entrez Gene ID. There were 24,898 genes in the database in total. For every one of the 24,898 genes, we counted the observed number of microarray experiments in which each gene was significantly dysregulated. We then calculated P values from the number of positive/negative experiments for every one of genes and sum of the number of positive/negative experiments for all other genes, using a 2 × 2 χ2 analysis (Fig. 1) or a Fisher’s exact test as an alternative (Fig. S3), and ranked all of the genes according to their P values [-log10(P)]. A third method, Liptak–Stouffer’s weighted Z-method (40), provided additional support for CD44 (Fig. S4).
is the mean expression level of gene i in group control, Si is the SD for the numerator calculation, and S0 is a small positive constant. We considered genes to be significantly dysregulated with either an absolute value of the d score ≥2 or a fold change ≥2 between controls and cases. We then converted all probe identifiers across the various microarray platforms for mouse, rat, and human to the latest human Entrez Gene identifiers by using our published AILUN system (39). Gene expression profiles were assigned in our eGWAS database according to the standardized (human) Entrez Gene ID. There were 24,898 genes in the database in total. For every one of the 24,898 genes, we counted the observed number of microarray experiments in which each gene was significantly dysregulated. We then calculated P values from the number of positive/negative experiments for every one of genes and sum of the number of positive/negative experiments for all other genes, using a 2 × 2 χ2 analysis (Fig. 1) or a Fisher’s exact test as an alternative (Fig. S3), and ranked all of the genes according to their P values [-log10(P)]. A third method, Liptak–Stouffer’s weighted Z-method (40), provided additional support for CD44 (Fig. S4).
Animal Experiments.
Mice for breeding, C57BL/6J wild-type (diabetes-prone) and CD44-deficient mice backcrossed to C57BL/6J for at least 10 generations (B6.Cg-Cd44tm1Hbg/J), were obtained from The Jackson Laboratory. Mice were housed in a barrier facility under specific pathogen-free conditions. The Animal Care and Use Committee of Kitasato University approved all animal experiments.
Human Studies.
Venous peripheral blood samples were collected from human subjects who went through a 75 g oral glucose tolerance test after an overnight fast [n = 55: sex (M/F); 36/19]. HbA1c was measured in Japan Diabetes Society (JDS)-HbA1c units and then converted to National Glycohemoglobin Standardization Program (NGSP) levels by the formula HbA1c (%) (NGSP) = HbA1c (JDS) (%) + 0.4% (41). We then calculated homeostasis model assessment as an index of insulin resistance [HOMA-IR = fasting plasma insulin (μU/mL) × fasting plasma glucose (mg/dL)/405] as described (42). Serum sCD44std (standard soluble CD44) and SPP1 concentrations were determined by using a quantitative ELISA technique (sCD44std ELISA, Bender MedSystems; Human Osteopontin Quantikine ELISA, R&D Systems).
Informed consent was obtained from all of the subjects enrolled in this study, and the protocol was approved by the ethics committee of the University of Tokyo.
Immunohistochemistry.
For histological analysis of CD44 expression in adipose tissue, EWAT was removed from mouse models, and omental adipose tissue obtained from consented donors undergoing elective gastric bypass surgery (lot no. OM020304B) was purchased from Zen-Bio. Formalin-fixed paraffin-embedded sections were stained with mouse monoclonal antibody against CD44 at 1:50 dilution (DF1485/sc-7297; Santa Cruz Biotechnology), followed by reactions with anti-mouse immunoglobulins-HRP, and anti-fluorescein-HRP. 3,3′-Diaminobenzidine (DAB) was used as a chromogen.
Analysis of inflammatory cell (macrophage) content in EWAT was performed on tissue pads isolated from model mice. Formalin-fixed paraffin-embedded sections were incubated overnight with primary antibody: Purified Anti Mouse MAC-2 Monoclonal Antibody (CL8942AP, 1:100; Cedarlane Laboratories) and stained by using Histofine Simple Stain Mouse MAX-PO secondary antibody (Nichirei Biosciences) with a DAB solution.
Statistics.
For verification studies in mouse models and human subjects, comparisons between two groups were performed by using the two-tailed Welch’s t test. Two-way repeated measures ANOVA was used to examine the treatment (antibodies type) × time interaction on blood glucose changes from baseline. P values of <0.05 were considered significant. All experimental data are represented as mean ± SE unless otherwise noted.
Supplementary Material
Acknowledgments
We thank Saori Ohta of Kitasato University School of Pharmacy for her support in animal experiments; Dr. Shojiro Morinaga of Department of Pathology, Kitasato Research Institute Hospital, for his assistance in histological analysis; and Dr. Damon Tojjar (Department of Clinical Sciences, Lund University, Scania University Hospital) and Dr. Purvesh Khatri (Division of Systems Medicine, Stanford University School of Medicine) for their suggestions in preparing the manuscript. This work was supported in part by the Howard Hughes Medical Institute, National Library of Medicine Grants (R01 LM009719 and K22 LM008261), and by the Lucile Packard Foundation for Children's Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.D.A. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1114513109/-/DCSupplemental.
References
- 1.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 2.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saxena R, et al. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 6.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 7.Zeggini E, et al. Wellcome Trust Case Control Consortium Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Unoki H, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda K, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008;40:1092–1097. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- 10.Dupuis J, et al. DIAGRAM Consortium GIANT Consortium Global BPgen Consortium Anders Hamsten on behalf of Procardis Consortium MAGIC investigators New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saxena R, et al. GIANT consortium MAGIC investigators Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42:142–148. doi: 10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voight BF, et al. MAGIC investigators GIANT Consortium Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamauchi T, et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet. 2010;42:864–868. doi: 10.1038/ng.660. [DOI] [PubMed] [Google Scholar]
- 14.Aitman TJ, et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- 15.Schadt EE, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37:710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karp CL, et al. Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat Immunol. 2000;1:221–226. doi: 10.1038/79759. [DOI] [PubMed] [Google Scholar]
- 18.Klein RF, et al. Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science. 2004;303:229–232. doi: 10.1126/science.1090985. [DOI] [PubMed] [Google Scholar]
- 19.Yagil C, et al. Identification of hypertension-related genes through an integrated genomic-transcriptomic approach. Circ Res. 2005;96:617–625. doi: 10.1161/01.RES.0000160556.52369.61. [DOI] [PubMed] [Google Scholar]
- 20.Hsu YH, et al. An integration of genome-wide association study and gene expression profiling to prioritize the discovery of novel susceptibility Loci for osteoporosis-related traits. PLoS Genet. 2010;6:e1000977. doi: 10.1371/journal.pgen.1000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong H, et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derry JM, et al. Identification of genes and networks driving cardiovascular and metabolic phenotypes in a mouse F2 intercross. PLoS ONE. 2010;5:e14319. doi: 10.1371/journal.pone.0014319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perou CM. Show me the data! Nat Genet. 2001;29:373. doi: 10.1038/ng1201-373. [DOI] [PubMed] [Google Scholar]
- 24.Paylor R. Questioning standardization in science. Nat Methods. 2009;6:253–254. doi: 10.1038/nmeth0409-253. [DOI] [PubMed] [Google Scholar]
- 25.Nomiyama T, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. 2007;117:2877–2888. doi: 10.1172/JCI31986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 27.Hutás G, et al. CD44-specific antibody treatment and CD44 deficiency exert distinct effects on leukocyte recruitment in experimental arthritis. Blood. 2008;112:4999–5006. doi: 10.1182/blood-2008-04-150383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikecz K, Brennan FR, Kim JH, Glant TT. Anti-CD44 treatment abrogates tissue oedema and leukocyte infiltration in murine arthritis. Nat Med. 1995;1:558–563. doi: 10.1038/nm0695-558. [DOI] [PubMed] [Google Scholar]
- 29.Katoh S, et al. A role for CD44 in an antigen-induced murine model of pulmonary eosinophilia. J Clin Invest. 2003;111:1563–1570. doi: 10.1172/JCI16583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brocke S, Piercy C, Steinman L, Weissman IL, Veromaa T. Antibodies to CD44 and integrin alpha4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc Natl Acad Sci USA. 1999;96:6896–6901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 32.Hanis CL, et al. A genome-wide search for human non-insulin-dependent (type 2) diabetes genes reveals a major susceptibility locus on chromosome 2. Nat Genet. 1996;13:161–166. doi: 10.1038/ng0696-161. [DOI] [PubMed] [Google Scholar]
- 33.Mahtani MM, et al. Mapping of a gene for type 2 diabetes associated with an insulin secretion defect by a genome scan in Finnish families. Nat Genet. 1996;14:90–94. doi: 10.1038/ng0996-90. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh S, et al. Type 2 diabetes: Evidence for linkage on chromosome 20 in 716 Finnish affected sib pairs. Proc Natl Acad Sci USA. 1999;96:2198–2203. doi: 10.1073/pnas.96.5.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe RM, et al. The Finland-United States investigation of non-insulin-dependent diabetes mellitus genetics (FUSION) study. II. An autosomal genome scan for diabetes-related quantitative-trait loci. Am J Hum Genet. 2000;67:1186–1200. [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 37.Zeyda M, et al. Osteopontin is an activator of human adipose tissue macrophages and directly affects adipocyte function. Endocrinology. 2011;152:2219–2227. doi: 10.1210/en.2010-1328. [DOI] [PubMed] [Google Scholar]
- 38.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen R, Li L, Butte AJ. AILUN: Reannotating gene expression data automatically. Nat Methods. 2007;4:879. doi: 10.1038/nmeth1107-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitlock MC. Combining probability from independent tests: The weighted Z-method is superior to Fisher’s approach. J Evol Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- 41.Seino Y, et al. Report of the Committee on the Classification and Diagnostic Criteria of Diabetes Mellitus. J Diabetes Invest. 2010;1:212–228. doi: 10.1111/j.2040-1124.2010.00074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matthews DR, et al. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.