

Abstract
Detailed structural, mutational, and biochemical analyses of human FEN1/DNA complexes have revealed the mechanism for recognition of 5′ flaps formed during lagging strand replication and DNA repair. FEN1 processes 5′ flaps through a previously unknown, but structurally elegant double-stranded (ds) recognition/single stranded (ss) incision mechanism that both selects for 5′ flaps and selects against ss DNA or RNA, intact dsDNA, and 3′ flaps. Two major DNA binding interfaces, including a K+ bridge between the DNA and the H2TH motif, are spaced one helical turn apart and together select for substrates with dsDNA. A conserved helical gateway and a helical cap protects the two-metal active site and selects for ss flaps with free termini. Structures of substrate and product reveal an unusual step between binding substrate and incision that involves a double base unpairing with incision occurring in the resulting unpaired DNA or RNA. Ordering of the active site requires a disorder-to-order transition induced by binding of an unpaired 3′ flap, which ensures that the product is ligatable. Comparison with FEN superfamily members, including XPG, EXO1, and GEN1, identifies superfamily motifs such as the helical gateway that select for ss-dsDNA junctions and provides key biological insights into nuclease specificity and regulation.
Keywords: Structure-specific nuclease, two metal, DNA binding, replication, DNA repair, flap endonuclease
Flap endonuclease (FEN1) is an essential protein in DNA replication. It incises 5′ single stranded (ss) DNA or RNA flaps formed during lagging strand replication and during long patch base excision repair. In replication of human cells, it is responsible for incising an approximate 50 million Okazaki fragments. To achieve this, FEN1 is a highly efficient enzyme, enhancing the hydrolysis rate of phosphodiester bonds ~1017 (Finger et al., 2009). At the same time, incision must be precise, to enable productive ligation. FEN1 incises 5′ flaps, one nucleotide into the double strand (ds) DNA from the ssDNAdsDNA junction. Unpairing one nucleotide on the 3′ side, first observed in a FEN-DNA structure (Chapados et al., 2004), ensures that the product, with incision one nucleotide into the dsDNA, is immediately ligatable (Fig. 1A). In substrates where the 3′ side is paired, FEN1 will preferentially unpair and form a one nucleotide 3′ flap before incising the DNA (Finger et al., 2009).
Figure 1.

hFEN1 uses structural mechanisms to select for and incise 5′ flaps. (A) Schematic of hFEN1 substrate showing how an unpaired 3′ flap and incision 1 nt into the dsDNA generates a ligatable product. (B) Structure of hFEN1 bound to product DNA (3Q8K). The dsDNA is bound at a bent 100° angle. The inset shows the relative position of the 5′ flap and the 3′ flap. (C) Structure of the hFEN1-bound DNA with contact points 4 Å from the protein highlighted as spheres shows the biased distribution of protein-DNA contact points. Almost all interactions are to the complementary strand and terminal bases of the flaps. Interaction with the dsDNA is one helical turn apart (indicated approximately by brown lines), spacing that selects for 5′ flap and against 3′ flaps.
The major visual difference between normal intact dsDNA and the optimal FEN1 substrate with a 5′ flap is the single stranded 5′ flap. Thus, much of the biochemistry research has focused on a FEN1 mechanism with “ssDNA binding-dsDNA incision” (Bornarth et al., 1999; Ceska et al., 1996; Devos et al., 2007; Finger et al., 2009; Henricksen et al., 2000; Liu et al., 2006; Murante et al., 1995; Qiu et al., 2004). This ssDNA-initiated mechanism is based on FEN1 recognizing the ssDNA or RNA, sliding or clamping down to the ss-dsDNA junction, and incision in the dsDNA.
Previous structures of DNA-free FEN from us and others have shown that FEN protein is mixed α/β protein, with a central active site containing the seven invariantly conserved carboxylates (Ceska et al., 1996; Chapados et al., 2004; Dore et al., 2006; Feng et al., 2004; Sakurai et al., 2005). Intriguingly, an archway above the active site was observed in some of the FEN structures. It was conjectured that this arch was involved with ssDNA recognition, either by threading the ssDNA under the arch or by clamping the ssDNA with no threading.
Recent structural and biochemistry studies based on structures of FEN with product and substrate 5′ flap DNA support a different mechanism: “dsDNA binding–ssDNA incision” (Tsutakawa et al., 2011a). There are three structures of human FEN1 bound to product and substrate DNA. (1) WT hFEN1 with product DNA, K+, and Sm3+ at 2.2 Å. (2) WT hFEN1 with substrate 1 nucleotide (nt) flap DNA, K+, and Sm3+ at 2.3 Å. (3) D181A hFEN1 with substrate 1 nt flap DNA, and K+ at 2.6 Å. In the WT structures crystallized with Sm3+, two metals bound in the active site. In D181A, an inactive mutant with the mutation in one of the conserved carboxylates, we did not observe any metals in the active site although crystallization conditions included Ca2+. The bound DNA stretches across the length of hFEN1, with the 3′ flap bound unpaired and the 5′ flap product terminus centered over the two metals and the active site (Fig. 1B).
In these hFEN1-DNA structures (Fig. 1B), DNA specificity is not simply from targeting the substrate but also preventing other substrates with similar characteristics from binding. The majority of the interaction of the substrate is to the dsDNA and most of that interaction is to the strand complementary to the flap strands (Fig. 1C and Table 1). Specificity for 5′ flap is first encoded in the 100° bend of the dsDNA. Only dsDNA with a break on one strand can bend to such a degree over a single phosphodiester linkage. Binding to the complementary strand and positioning the flaps using the complementary strand ensures that only substrates containing dsDNA will be in catalytic position and selects against inadvertent incision of ssDNA or RNA. Second, the dsDNA binding is non-contiguous and is spaced one helical turn apart from each other (Fig. 1C). One of these binding sites is mediated by a K+, bound by a H2TH motif, while the other side is at the 100° turn of the DNA. This periodicity again selects for dsDNA and in particular selects for 5′ flaps. The spacing between dsDNA binding sites selects against 3′ flaps that would require a more narrow spacing. The lack of significant interaction to the flap also enables FEN1 to incise 5′ flaps with either DNA or RNA in the ss region. Third, the single strand nature of the 5′ flap is selected for by a helical gateway that guards the active site (Fig. 2A and Table 1). The terminal base of the product is located between two helices, α2 and the N-terminal part of α4. The width between these two helices range between 13 to 15 Å, which could act as a gateway to the active site and to select for ssDNA or RNA. Fourth, the free terminus of the 5′ flap is selected by a helical cap that covers the gateway. The helical cap, formed by the rest of α4 and also α5, would impede structures without free ends, such as DNA bubbles or Holliday junctions, from going through the gateway to the active site. Use of this steric gateway and cap is similar to what we and others postulate for how XPD helicase selectively for a ssDNA region (Fan et al., 2008; Fuss and Tainer, 2011; Wolski et al., 2008; Wolski et al., 2010). The helical gateway contains three residues that directly interact with the terminal product nucleotide. Lys93 and Arg100 coordinate the phosphate, while Tyr40 stacks with the base. Mutation of the basic residues leads to ~400-fold decrease in activity, while mutation of Tyr40 reduced the reaction rate about 40-fold. It will be worth testing the roles of Tyr40 in catalysis and stability. For an analogous active site Tyr34 in superoxide dismutase, mutation of the Tyr increased stability but left the enzyme subject to product inhibition at high substrate concentrations (Guan et al., 1998).
Table 1.
Structural elements in hFEN1 select for and against a variety of DNA structures.
| FEN1 steric controls | Selection For | Selection Against |
|---|---|---|
| 100° bent dsDNA* | dsDNAwith break | Intact dsDNA |
| Binding complementary strand* | Substrates with dsDNA | ssDNA/RNAonly |
| Binding sites one helical turn apart* | 5′ flaps | 3′ flaps |
| Unpaired 3′ flap | Ligatable product | Gapped products |
| Helical Gateway over active site* | ssDNA or RNA | dsDNA |
| Helical Cap over gateway | Free End | DNA bubbles, Holliday junctions |
| Disorder->Order** | Presence of 3′ flap | DNA bubbles, dsDNA termini |
| Double base Unpairing occurring after ordering* | Checks for all elements (dsDNA, ss5′ flap, 3′ flap) before incision | ssDNA |
The asterisk highlights recognition motifs likely to be conserved in the FEN1 superfamily.
Figure 2.

The hFEN1 helical gateway and cap act to guard the active site and to sterically select for ssDNA and RNA with free termini. (A) In the product structure, the helical gateway and cap are located over the active site and catalytic metals. Tyr40 stacks against the terminal base of the product DNA. The 5′ phosphate of the product is coordinated to the two metals and to Arg90 and Lys100. (B) The terminal phosphate is coordinated to the two catalytic metals. These metals are coordinated by four of the seven superfamily-conserved carboxylates. (C) Model for a substrate flap going through the helical gateway. (D) Close-up view of the substrate model and the position of the scissile phosphate relative to the catalytic metals. We propose that the attacking water (red sphere) would be coordinated between the two metals.
The gateway covers the active site that is occupied by the two metals (Fig. 2B). The hFEN1 structures were crystallized with Sm3+ ions, whose addition to the crystallization enabled higher resolution of the crystallographic data. In the cell, these two ions are presumably Mg2+. In the DNA-free hFEN1 structures (Sakurai et al., 2005), two Mg2+ ions were identified in the electron density. The positions of the Sm3+ in the DNA-bound structures are similar to those of the Mg2+ ions. One phosphate oxygen of the terminal product nucleotide is coordinated by both metal ions. Based on a linear attack, that the DNA is walled in by the gateway helices, and that the position of the terminal phosphate did not shift after incision, we postulate that this phosphate oxygen was the attacking water. A similar positioning of the attacking water was observed in EndoIV, albeit through two zinc ions (Garcin et al., 2008b ; Ivanov et al., 2007). A model built on this assumption shows how the ss region of the 5′ flap could pass through the gateway (Fig. 2C and D).
There are seven absolutely conserved carboxylate residues in FEN1 and its 5′ nuclease superfamily (Harrington and Lieber, 1994). Four of these carboxylates are directly coordinating the metals, while the remainder are involved in water-mediated coordination. It is intriguing that seven carboxylates are invariably conserved in the FEN superfamily, but only four are directly coordinating the metal. Although the other three are involved in water-mediated coordination of the metal, it is possible that they are playing additional roles in the mechanism that we do not currently understand. For example, the metals may act as a negative repulsive cushion as the 5′ flap is pushed through the gateway and under the cap and thereby prevent incidental incision of the ss region of the 5′ flap before the substrate is fully bound. Another possibility is that the metals are shifting their coordination, as suggested from the variable positions of different metal atoms in the superfamily-related EXO1 structures (Orans et al., 2011).
Specificity for an unpaired 3′ flap is encoded by a hydrophobic wedge that encloses the 3′ flap backbone and stacks against the base-pair next to the 3′ flap. It is notable that this region as well as the helical gateway and cap are disordered in DNA-free hFEN1 (Sakurai et al., 2005) (Fig. 3). We postulate that binding of the 3′ flap region orders the hydrophobic wedge, which in turn orders the helical gateway and cap, including catalytically important residues in α2 and α4. Tyr40 in α2 and Lys93 and Arg100 in α4 are disordered in the DNA-free hFEN1 but are ordered in the substrate and product-bound hFEN1. Mutation of Arg47 that connects the hydrophobic wedge to the helical cap and gateway reduces activity 30-fold (Tsutakawa et al., 2011a), comparable to the decrease in reaction rate for Tyr40 mutation at the active site.
Figure 3.

hFEN1 disorder-to-order transition induced by DNA. Comparison of the three DNA-free structures (1ULI.pdb) and the DNA-bound (3Q8K.pdb) structure reveals a disorder-to-order transition of the gateway (blue), cap (pink), and hydrophobic wedge (green). The hydrophobic wedge forms part of the 3′ flap binding pocket. The regions not modeled in the DNA-free structures are drawn in as dashed lines.
The mechanism for ssDNA incision is suggested from the comparison of the substrate and product complexes (Fig. 4). The 1 nt flap of the substrate flaps (+1) is basepaired, but in the product complex, the terminal product base (−1) is unpaired and is coordinated to the active site metals. The “+” refers to nucleotides 5′ to the scissile phosphate, while “−” refers to nucleotides 3′ to the scissile phosphate. If we assume that the scissile phosphate must be within catalytic distance to the active site metals, then both the +1 and −1 nucleotides must be unpaired for the scissile phosphate to be close to the metals and the DNA was single stranded during the incision. We thus propose an unusual endonuclease mechanism: double base unpairing followed by single-stranded incision. The reason why the incision takes place on ssDNA is not from physical constraints of incising dsDNA. Restriction endonucleases cleave the easily accessible backbone in dsDNA. We postulate that the double base unpairing provides an additional regulation. Incision can occur only when multiple criteria are met. Unpairing requires an ordered gateway, and an ordered gateway requires a disorder-to-order transition. The disorder-to-order transition is induced by binding an unpaired 3′ flap and binding dsDNA between the hydrophobic wedge/active site and the K+/H2TH. Based on FEN1 substrate structures, this ordering is not dependent on unpairing. We found that when the 1 nt flap has a 5′ hydroxyl, the substrate DNA was basepaired and not incised under crystallographic conditions. Importantly, the gateway and arch are already ordered. The same substrate with a 5′ phosphate was incised in the crystals. Biochemically, we measured an 8.2 fold increase in the normalized initial catalytic rate with a 5′ phosphate compared to a 5′ hydroxyl. The 5′ phosphate at the +1 position is apparently critical for catalytic activity, and we postulate that the 5′ phosphate is involved in the unpairing step. The 5′ phosphate of the +1 nucleotide would be pulled by the metals and basic residues Lys93 and Arg100, with the +1 nucleotide becoming unpaired. The scissile phosphate would still be too far for incision to occur, and the −1 nucleotide would be pulled in a similar fashion and unpaired. Once the scissile phosphate has moved closer to the active site metals, incision can take place. Thus, although FEN1 is incising one nucleotide into the dsDNA, it is in fact unpairing the dsDNA and incising ssDNA. Presumably, if there is dsDNA in the helical gateway as might be found for a three-way junction, the gateway cannot form properly. Lys93 and Arg100 cannot position themselves to pull on the phosphates of the +1 and −1 nucleotides on either side of the scissile phosphate. The unpairing and thus the incision would not occur.
Figure 4.
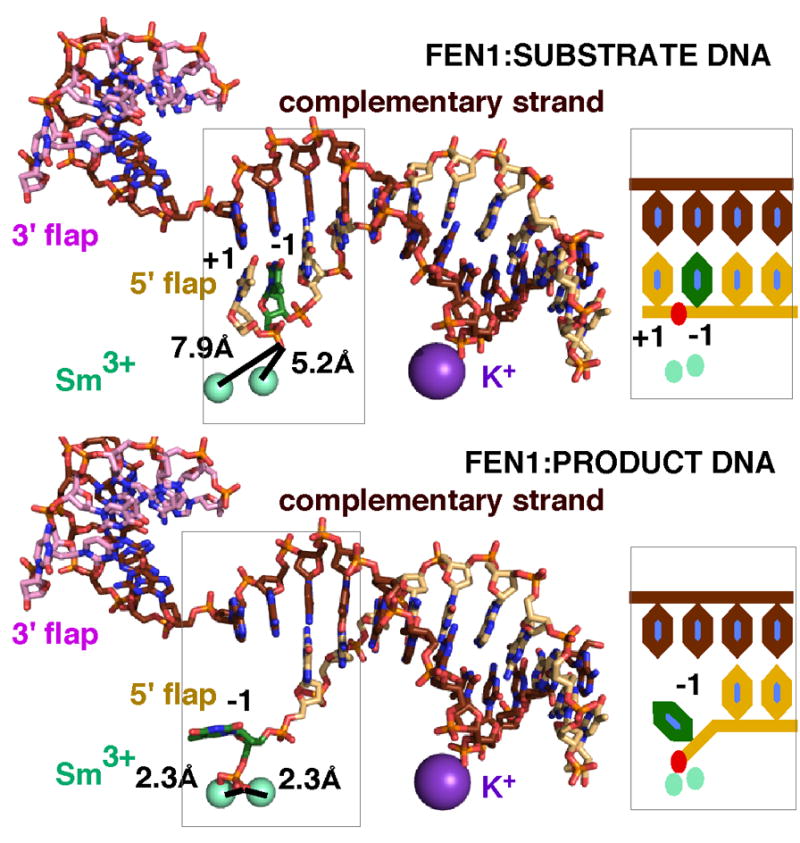
hFEN1 incises ssDNA formed by double base unpairing around the scissile phosphate. Comparison of the substrate (3Q8L.pdb, upper panel) and product (3Q8K.pdb, lower panel) DNA complexes with hFEN1 reveals a double base unpairing. In the substrate complex, the scissile phosphate is too distant from the metals for catalysis. In the product complex where the scissile phosphate is coordinated to the metals, the −1 base (green) of the product DNA is unpaired. This difference in the substrate and product DNA suggests that a double base unpairing of the +1 and −1 nucleotides is required to move the scissile phosphate into catalytic distance of the metals. On the right is shown a schematic of the unpairing.
Thus FEN1 uses a number of structural mechanisms to identify its substrate and exclude other substrates (ssDNA, RNA, dsDNA, 3′ flaps, bubble DNA, Holliday junctions) (Table 1). Interestingly, most of these FEN1 structural elements appear to be conserved in the 5′ nuclease superfamily that includes Exo1, XPG, and GEN. Such a conservation of functional elements and motifs have proven to provide key insights among other superfamily, as seen for the DNA repair photolyases and clock cryptochromes (Hitomi et al., 2009). The structure of EXO1 with different DNA substrates and metals have been determined (Orans et al., 2011) and hFEN1 structures can be directly compared. Although the structures for XPG and GEN have not been determined, secondary structure predictions and conservation of key residues indicate conservation of structure (Tsutakawa et al., 2011a). The helical gateway is conserved in all members, consistent with these nucleases all acting on ssDNA-dsDNA junctions. The seven carboxylates in the active site and the residues in the α4 gateway helix, Lys93 and Arg100 in FEN, are all absolutely conserved. A potential base stacking residue is conserved at Tyr40. We believe that the non-contiguous DNA binding spaced one helical turn apart is also conserved, with one side being bound by the H2TH/K+ and the other at a sharp bend in the DNA. We also believe that all family members unpair the basepairs around the scissile phosphate and in fact incise ssDNA after unpairing. These elements are observed in the Exo1-DNA structure (Orans et al., 2011). What is not conserved in the helical cap that is found only in Exo1 and FEN1 sequences. We postulate that there is no cap in XPG or GEN, enabling their substrates, bubble DNA and Holliday junctions respectively, access to the helical gateway and the active site. Such differential binding specificity can help control pathway selection and progression as seen for alkyl-guanine transferase, which flips and binds alkylated damaged guanine in DNA and remains bound to promote nucleotide excision repair (NER) for this base damage by handoff to the NER pathway enzyme XPG (Tubbs et al., 2009). How FEN1 superfamily enzymes such as XPG, which acts at different times during the process of NER and during replication fork rescue (Sarker et al., 2005; Trego et al., 2011), control pathway selection by binding and conformational change will be of substantial interest to elucidate. Another interesting facet of binding and regulation concerns the ability of protein domains to regulate processes by DNA mimicry, as was first structurally defined by crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with uracil-DNA glycosylase (Putnam et al., 1999; Shroyer et al., 1999): it will be important to see if FEN1 superfamily enzymes, such as XPG an EXOI, also employ DNA mimicry in regulating their interactions with DNA substrates.
Handoff of DNA intermediates to FEN1 and from FEN1 is likely to be highly coordinated, since DNA intermediates are potentially toxic (Parikh et al., 1999). DNA polymerases such as Pol δ and Pol β displace the preceding DNA to form a flap. FEN1 incises the flap, passing on the nicked DNA to Ligase 1 (LIG1). Examination of the dsDNA in a Pol β structure that includes both upstream and downstream DNA structures shows an uncanny similarity to the dsDNA bound by FEN1 (Krahn et al., 2003) (Fig. 5A). Both FEN1 and Pol β bind the DNA at an approximately 90–100° bend. The 3′ OH region that will form a 3′ flap for FEN1 is well covered by the polymerase, and it is unlikely that there is direct handoff of this region. Instead, we postulate that FEN1 is waiting on the downstream DNA through its K+/H2TH dsDNA binding motif. Pol β is known for its tendency to fall off its substrate, and FEN1 can then move in to incise the 5′ flap. The DNA bend has already been formed, so there is no energetic loss from disrupting the base stacking and bending the DNA for FEN1. For FEN1 and LIG1, it is more likely that the handoff is directly handled. The DNA binding domain (DBD) of LIG1 binds within the minor groove of the downstream DNA (Pascal et al., 2004) while hFEN1 binds on the major groove side. Overlaying the downstream DNA using the 5′ terminal phosphate to register the overlay, the DBD of LIG1 and FEN1 can bind the same downstream DNA from opposite sides (Fig. 5B). There is a slight overlap of the cap region, suggesting that the DBD might promote a order-to-disorder transition in FEN1, resulting in release of FEN1 from the DNA. This in turn would then allow the DNA to straighten and LIG1 to wrap all of its domains around the nicked DNA.
Figure 5.

Structural basis of coordination in hFEN1:DNA complexes. Handoff to and from FEN1. (A) The structures of FEN1-bound DNA (blue) and Pol β–DNA (magenta) are aligned showing how similarly the DNA is distorted. FEN1-DNA interactions are mapped onto the hFEN1-bound DNA (blue sphere, left). A cartoon of Pol β shows how Pol β binding overlaps hFEN1 binding on the DNA (blue spheres). (B) hFEN1 and the DNA binding domain (DBD) of Ligase 1 bind opposite sides of the downstream DNA. For Ligase 1, the downstream DNA of hFEN1 (3Q8K) and of LIG1 were superimposed. Shown are the hFEN1:DNA structure with hFEN1:DNA interactions mapped onto the DNA (blue sphere) and the Lig1 DBD (1X9N.pdb) with its interactions mapped onto its DNA substrate (green spheres). For ligation, FEN1 must come off the DNA, the upstream DNA must straighten, and the remaining Ligase 1 domains must wrap around the straightened DNA. (C) Model for FEN1 interaction with PCNA. Superimposition of hFEN1:Product DNA (3Q8K) with the Y chain of hFEN1:PCNA (1ULI.pdb) and extension of the duplex DNA is consistent with the dsDNA on the 3′ flap-side going through the PCNA ring.
In the cell, these handoffs are likely functioning with the polymerase, FEN1, and LIG1 all simultaneously bound to PCNA (Chapados et al., 2004; Tsutakawa et al., 2011a). Based on the PCNA bound to a FEN1 peptide (Chapados et al., 2004) and DNA-free hFEN1 structures bound to PCNA (Sakurai et al., 2005), overlay of one of these PCNA-bound hFEN1 structures with the hFEN1-DNA structures provides a feasible model where the DNA on the 3′ flap side would feed directly to the PCNA center hole (Fig. 5C). Interestingly the PCNA-bound hFEN1 structures suggest a flexible linkage between the PCNA and DNA binding regions that may nonetheless influence each other. How such flexible tethering might still allow allosteric coordination between binding sites is an exciting area of research and was recently implicated to occur for both Nbs1-Mre11 interactions (Williams et al., 2009) and for Mre11-Rad50 interactions (Williams et al., 2011) during DNA break repair. Techniques such as small angle x-ray scattering (SAXS) and deuterium-hydrogen exchange will help to test these models and aid in defining protein-protein interfaces during handoff exchanges (Hammel et al., 2011; Hammel et al., 2010a; Hammel et al., 2010b; Pascal et al., 2006).
The hFEN1 structural and mutational analyses have revealed how hFEN1 as a structure-specific endonuclease identifies and double checks all the properties of its substrate-helical properties of duplex DNA, bendability, presence of a unpaired 3′ flap, and ssDNA nature of the 5′ flap, before incision. This type of detailed knowledge is fundamental to aiding the redesign of proteins for new functions, which can span from covalently linked assemblies, such as done for superoxide dismutase (Hallewell et al., 1989) to the development of sensitive detection assays, as has been done with FEN1 (Allawi et al., 2006). Furthermore, analyses of metalloprotein active sites provide the basis to understand how toxic metal ions may block essential DNA repair and replication proteins, as seen for mismatch repair inhibition by cadmium (McMurray and Tainer, 2003). Significantly, we are discovering that metalloproteins are largely uncharacterized even in microbial metalloproteomes and that the same reactions can be accomplished by different metal ions in related enzymes, so the structural characterization of metal ion binding sites is valuable (Cvetkovic et al., 2010; Perry et al., 2010b). Besides the active site metal ions, the arch and gateway disorder-to-order transitions involved in specific end recognition by FEN1 and in bubble interactions by XPG and GEN1 merit added study such as might be accomplished by biochemical experiments, by advanced SAXS on enzyme-DNA complexes in solution, and by NMR (Hura et al., 2009; Perry et al., 2010a; Putnam et al., 2007; Rambo and Tainer, 2010, 2011). All these approaches to characterize flexible complexes will be aided by these detailed structures of hFEN1 in complex with DNA substrate and product, as seen for Mre11-Rad50 complexes (Williams et al., 2011), XRCC4-XLF complexes for doublestrand break repair (Hammel et al., 2011), and ubiquitin-PCNA complexes for translesion bypass (Tsutakawa et al., 2011b). Another question to be addressed is how flexible and unstructured regions avoid toxic mis-assemblies and instabilities that can occur when mutations disrupt the framework stability of enzymes, such as seen for superoxide dismutase and XPD (DiDonato et al., 2003; Fan et al., 2008; Shin et al., 2009). Also how important is protein flexibility in different types of enzyme-DNA interactions and structure-based specificity? For example, the interaction sites for ssDNA at the flap junction may involve more flexibility than those for dsDNA, based for example upon the flexible interactions observed by SAXS on RPA bound to ssDNA (Pretto et al., 2010). One key practical implication of flexibility may be to employ active site plasticity to create superior specific chemical inhibitors by structure-based design, as successfully achieved for nitric oxide synthase isozymes (Garcin et al., 2008a).
Overall, the combined results on FEN1-DNA complexes adds to our understanding of how DNA sequence-independent damage recognition and exquisite catalytic specificity for DNA junctions are both accomplished, which are areas of great interest (Hitomi et al., 2007; Huffman et al., 2005; Min and Pavletich, 2007). Collectively, FEN1 structures, mutational analyses, and biochemistry support a new model for its FEN1 mechanism of “dsDNA binding, ssDNA incision” that we propose is likely to be a prototype for the 5′ nuclease superfamily. Tight regulation of nuclease activity either by its substrate or regulatory proteins may be a recurring theme for other nucleases, as seen as well for MRE11, XPG, and EXO1 plus WRN (Orans et al., 2011; Rahal et al., 2010; Schlacher et al., 2011; Staresincic et al., 2009; Trego et al., 2011; Williams et al., 2010; Williams et al., 2008). On the flip side, some nucleases such as endonuclease V, which acts in base repair, seem to be able to initiate a deaminated adenine repair pathway relatively independently (Dalhus et al., 2009), raising questions about how DNA integrity is maintained so well in the context of so many different nucleases. Going forward, an important structural challenge will be to characterize the roles for flexibility, conformational change, and structural dynamics in the exquisite specificity and regulation of nucleases in cell biology, as these enzymes control critical decision points at the interfaces for replication, recombination, and repair.
Highlights.
FEN1: DNA structures and mutations reveal 5′-flap recognition mechanism and incision.
FEN1 recognizes and incises 5′ flaps through dsDNA binding – ssDNA incision.
FEN1 binds 100° bent dsDNA and unpaired 3′-flap to thread 5′-end near the active site for incision.
Binding of unpaired 3′ flap and dsDNA induces disorder-to-order transition to align residues for catalysis.
Two nucleotides of 5′-flap must unpair to productively position phosphodiester near the two catalytic metals.
Acknowledgments
We thank Jane Grasby, Dave Finger, Binghui Shen, Priscilla Cooper, Altaf Sarker, Brian Chapados, Andy Arvai, Scott Classen, Tom Ellenberger, Ivaylo Ivanov, Gareth Williams, Jill Fuss, Sophia Hartung, Peter Burgers, Jerzy Majka, and Joanne Sweasy for many fruitful scientific discussions. Work on hFEN1 is supported by the National Cancer Institute through RO1CA081967 and P01 CA092584 (JAT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allawi HT, Li H, Sander T, Aslanukov A, Lyamichev VI, Blackman A, Elagin S, Tang YW. Invader plus method detects herpes simplex virus in cerebrospinal fluid and simultaneously differentiates types 1 and 2. J Clin Microbiol. 2006;44:3443–3447. doi: 10.1128/JCM.01175-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornarth CJ, Ranalli TA, Henricksen LA, Wahl AF, Bambara RA. Effect of flap modifications on human FEN1 cleavage. Biochemistry. 1999;38:13347–13354. doi: 10.1021/bi991321u. [DOI] [PubMed] [Google Scholar]
- Ceska TA, Sayers JR, Stier G, Suck D. A helical arch allowing singlestranded DNA to thread through T5 5′-exonuclease. Nature. 1996;382:90–93. doi: 10.1038/382090a0. [DOI] [PubMed] [Google Scholar]
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL, 2nd, Jenney FE, Jr, Lancaster WA, Praissman JL, Shanmukh S, Vaccaro BJ, Trauger SA, Kalisiak E, Apon JV, Siuzdak G, Yannone SM, Tainer JA, Adams MW. Microbial metalloproteomes are largely uncharacterized. Nature. 2010;466:779–782. doi: 10.1038/nature09265. [DOI] [PubMed] [Google Scholar]
- Dalhus B, Arvai AS, Rosnes I, Olsen OE, Backe PH, Alseth I, Gao H, Cao W, Tainer JA, Bjoras M. Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat Struct Mol Biol. 2009;16:138–143. doi: 10.1038/nsmb.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos JM, Tomanicek SJ, Jones CE, Nossal NG, Mueser TC. Crystal structure of bacteriophage T4 5′ nuclease in complex with a branched DNA reveals how flap endonuclease-1 family nucleases bind their substrates. J Biol Chem. 2007;282:31713–31724. doi: 10.1074/jbc.M703209200. [DOI] [PubMed] [Google Scholar]
- DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, Kassmann CJ, Lo TP, Bruns CK, Powers ET, Kelly JW, Getzoff ED, Tainer JA. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–615. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- Dore AS, Kilkenny ML, Jones SA, Oliver AW, Roe SM, Bell SD, Pearl LH. Structure of an archaeal PCNA1-PCNA2-FEN1 complex: elucidating PCNA subunit and client enzyme specificity. Nucleic Acids Res. 2006;34:4515–4526. doi: 10.1093/nar/gkl623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M, Patel D, Dervan JJ, Ceska T, Suck D, Haq I, Sayers JR. Roles of divalent metal ions in flap endonuclease-substrate interactions. Nat Struct Mol Biol. 2004;11:450–456. doi: 10.1038/nsmb754. [DOI] [PubMed] [Google Scholar]
- Finger LD, Blanchard MS, Theimer CA, Sengerova B, Singh P, Chavez V, Liu F, Grasby JA, Shen B. The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J Biol Chem. 2009;284:22184–22194. doi: 10.1074/jbc.M109.015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat Chem Biol. 2008a;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcin ED, Hosfield DJ, Desai SA, Haas BJ, Bjoras M, Cunningham RP, Tainer JA. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat Struct Mol Biol. 2008b;15:515–522. doi: 10.1038/nsmb.1414. [DOI] [PubMed] [Google Scholar]
- Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- Hallewell RA, Laria I, Tabrizi A, Carlin G, Getzoff ED, Tainer JA, Cousens LS, Mullenbach GT. Genetically engineered polymers of human CuZn superoxide dismutase. Biochemistry and serum half-lives. J Biol Chem. 1989;264:5260–5268. [PubMed] [Google Scholar]
- Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, Schriemer DC, Lees-Miller SP, Tainer JA. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–32650. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010a;18:1431–1442. doi: 10.1016/j.str.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010b;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington JJ, Lieber MR. Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev. 1994;8:1344–1355. doi: 10.1101/gad.8.11.1344. [DOI] [PubMed] [Google Scholar]
- Henricksen LA, Tom S, Liu Y, Bambara RA. Inhibition of flap endonuclease 1 by flap secondary structure and relevance to repeat sequence expansion. J Biol Chem. 2000;275:16420–16427. doi: 10.1074/jbc.M909635199. [DOI] [PubMed] [Google Scholar]
- Hitomi K, DiTacchio L, Arvai AS, Yamamoto J, Kim ST, Todo T, Tainer JA, Iwai S, Panda S, Getzoff ED. Functional motifs in the (6-4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc Natl Acad Sci U S A. 2009;106:6962–6967. doi: 10.1073/pnas.0809180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I, Tainer JA, McCammon JA. Unraveling the three-metal-ion catalytic mechanism of the DNA repair enzyme endonuclease IV. Proc Natl Acad Sci U S A. 2007;104:1465–1470. doi: 10.1073/pnas.0603468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn JM, Beard WA, Miller H, Grollman AP, Wilson SH. Structure of DNA polymerase beta with the mutagenic DNA lesion 8-oxodeoxyguanine reveals structural insights into its coding potential. Structure. 2003;11:121–127. doi: 10.1016/s0969-2126(02)00930-9. [DOI] [PubMed] [Google Scholar]
- Liu R, Qiu J, Finger LD, Zheng L, Shen B. The DNA-protein interaction modes of FEN-1 with gap substrates and their implication in preventing duplication mutations. Nucleic Acids Res. 2006;34:1772–1784. doi: 10.1093/nar/gkl106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT, Tainer JA. Cancer, cadmium and genome integrity. Nat Genet. 2003;34:239–241. doi: 10.1038/ng0703-239. [DOI] [PubMed] [Google Scholar]
- Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- Murante RS, Rust L, Bambara RA. Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J Biol Chem. 1995;270:30377–30383. doi: 10.1074/jbc.270.51.30377. [DOI] [PubMed] [Google Scholar]
- Orans J, McSweeney EA, Iyer RR, Hast MA, Hellinga HW, Modrich P, Beese LS. Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell. 2011;145:212–223. doi: 10.1016/j.cell.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Hosfield DJ, Tainer JA. Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- Pascal JM, Tsodikov OV, Hura GL, Song W, Cotner EA, Classen S, Tomkinson AE, Tainer JA, Ellenberger T. A flexible interface between DNA ligase and PCNA supports conformational switching and efficient ligation of DNA. Mol Cell. 2006;24:279–291. doi: 10.1016/j.molcel.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Perry JJ, Cotner-Gohara E, Ellenberger T, Tainer JA. Structural dynamics in DNA damage signaling and repair. Curr Opin Struct Biol. 2010a;20:283–294. doi: 10.1016/j.sbi.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JJ, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta. 2010b;1804:245–262. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretto DI, Tsutakawa S, Brosey CA, Castillo A, Chagot ME, Smith JA, Tainer JA, Chazin WJ. Structural dynamics and single-stranded DNA binding activity of the three N-terminal domains of the large subunit of replication protein A from small angle X-ray scattering. Biochemistry. 2010;49:2880–2889. doi: 10.1021/bi9019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Shroyer MJ, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, Tainer JA. Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase. J Mol Biol. 1999;287:331–346. doi: 10.1006/jmbi.1999.2605. [DOI] [PubMed] [Google Scholar]
- Qiu J, Liu R, Chapados BR, Sherman M, Tainer JA, Shen B. Interaction interface of human flap endonuclease-1 with its DNA substrates. J Biol Chem. 2004;279:24394–24402. doi: 10.1074/jbc.M401464200. [DOI] [PubMed] [Google Scholar]
- Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. Cell Cycle. 2010;9:2866–2877. doi: 10.4161/cc.9.14.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–137. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24:683–693. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell. 2005;20:187–198. doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Doublestrand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Didonato M, Barondeau DP, Hura GL, Hitomi C, Berglund JA, Getzoff ED, Cary SC, Tainer JA. Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. J Mol Biol. 2009;385:1534–1555. doi: 10.1016/j.jmb.2008.11.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroyer MJ, Bennett SE, Putnam CD, Tainer JA, Mosbaugh DW. Mutation of an active site residue in Escherichia coli uracil-DNA glycosylase: effect on DNA binding, uracil inhibition and catalysis. Biochemistry. 1999;38:4834–4845. doi: 10.1021/bi982986j. [DOI] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trego KS, Chernikova SB, Davalos AR, Perry JJ, Finger LD, Ng C, Tsai MS, Yannone SM, Tainer JA, Campisi J, Cooper PK. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle. 2011;10:1998–2007. doi: 10.4161/cc.10.12.15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, Cooper PK, Grasby JA, Tainer JA. Human Flap Endonuclease Structures, DNA Double-Base Flipping, and a Unified Understanding of the FEN1 Superfamily. Cell. 2011a;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Van Wynsberghe AW, Freudenthal BD, Weinacht CP, Gakhar L, Washington MT, Zhuang Z, Tainer JA, Ivanov I. Solution X-ray scattering combined with computational modeling reveals multiple conformations of covalently bound ubiquitin on PCNA. Proc Natl Acad Sci U S A. 2011b;108:17672–17677. doi: 10.1073/pnas.1110480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, Guenther G, SilDas S, Hammel M, Russell P, Tainer JA. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol. 2011;18:423–431. doi: 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, Tainer JA. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolski SC, Kuper J, Hanzelmann P, Truglio JJ, Croteau DL, Van Houten B, Kisker C. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolski SC, Kuper J, Kisker C. The XPD helicase: XPanDing archaeal XPD structures to get a grip on human DNA repair. Biol Chem. 2010;391:761–765. doi: 10.1515/BC.2010.076. [DOI] [PubMed] [Google Scholar]