

Abstract
The permanent cellular constituents of the heart include cardiac fibroblasts, myocytes, endothelial cells and vascular smooth muscle cells. Previous studies have demonstrated that there are undulating changes in cardiac cell populations during embryonic development, through neonatal development and into the adult. Transient cell populations include lymphocytes, mast cells and macrophages, which can interact with these permanent cell types to affect cardiac function. It has also been observed that there are marked differences in the makeup of the cardiac cell populations depending on the species, which may be important when examining myocardial remodeling. Current dogma states that the fibroblast makes up the largest cell population of the heart; however, this appears to vary for different species, especially mice. Cardiac fibroblasts play a critical role in maintaining normal cardiac function, as well as in cardiac remodeling during pathological conditions such as myocardial infarct and hypertension. These cells have numerous functions, including synthesis and deposition of extracellular matrix, cell-cell communication with myocytes, cell-cell signaling with other fibroblasts, as well as with endothelial cells. These contacts affect the electrophysiological properties, secretion of growth factors and cytokines, as well as potentiating blood vessel formation. While a plethora of information is known about several of these processes, relatively little is understood about fibroblasts and their role in angiogenesis during development or cardiac remodeling. In this review we provide insight into the various properties of cardiac fibroblasts that helps illustrate their importance in maintaining proper cardiac function, as well as their critical role in the remodeling heart.
Keywords: cardiac fibroblast, cardiac cell populations, cardiac remodeling, ECM, cytokines
Introduction
Studies in the 70’s and 80’s from Zak and Nag defined and quantified the cellular populations of the adult rat left ventricle based on morphological characteristics using transmission electron microscopy of rat left ventricular sections, as well as gradient centrifugation1, 2. These seminal studies on specific cardiac regions were further extended to the whole rat heart and suggested that the heart consists of approximately 70% non-myocytes and 30% cardiac myocytes1–5. In addition, various studies using cell-specific markers have demonstrated that the myocardium exhibits distinct regional differences that are influenced by the specific physiological nature of the region3–9. These data defining the cardiac cell populations in the rat have effectively been applied to all species. However, over the past decade the mouse model has become the standard organism to study the heart through the use of various transgenic, knockout and surgical models. There are specific physiological differences between the rat and mouse, such as heart rate and total collagen10, 11. Until recently, the cellular populations extrapolated from the early rat experiments had yet to be qualified in the mouse or any other species. During normal cardiac function, cellular components of the heart interact in a dynamic fashion to respond to changes in developmental, homeostatic and pathological stimuli. The main cellular constituents of the heart include cardiac fibroblasts, myocytes, endothelial cells and vascular smooth muscle cells, with the majority of cells consisting of fibroblasts and myocytes3–5. These cell types maintain the electrical, chemical and biomechanical responsive nature of the organ. Moreover, these cells help preserve the three-dimensional structure via autocrine and paracrine action of secreted factors, as well as via direct cell-cell interactions3–7, 9, 12. Alterations in these signals or biomechanical input can cause deleterious, adaptive and/or compensatory changes in the heart.
Gap junctions are essential in maintaining a normal heartbeat, and direct interactions between myocytes and cardiac fibroblasts occur via gap junctional connexins (Cx40, Cx43, and Cx45) to function in electrical conduction in the heart3, 4, 6, 7, 12. Indeed, Cx43 expression has been linked to arrhythmia and myocardial infarction size5, 8, 12–17. Moreover, abrogation of these interactions via remodeling can cause interference in this system leading to pathological conditions3, 7, 12, 18. Additionally, connexins have been shown to play an important role in endothelial cell interactions and may also be important for cardiac fibroblast-endothelial cell interactions, as discussed later in this review19–21. In addition to connexins, it has also been demonstrated that cadherins play a critical role in cardiac development and function. Previous studies have shown that N-cadherin is important for cell-cell interactions, as well as myofibril organization in the heart22, 23. It has also been demonstrated that cadherin-13, also known as T-cadherin, is important for vascular remodeling and may play a critical role in this process following myocardial infarct. Other studies have demonstrated that cytokines, such as interleukin-6 (IL-6) are critical in myocardial function and cardioprotection24–27. In vivo studies have shown that continuous activation of the gp130 receptor leads to myocardial hypertrophy25, 28. Additionally, studies in rats have demonstrated that constant IL-6Rα stimulation can reduce infarct size and protect against apoptosis29. Better understanding of these signaling pathways in cardiac fibroblasts may provide insight into potential therapeutic targets for the treatment of the failing heart. In this review, we summarize current studies involving the cardiac fibroblast and its importance in physiological and pathological cardiac remodeling.
The Cardiac Fibroblast
What is a fibroblast? Fibroblasts are widely distributed connective tissue cells that are found in all vertebrate organisms. They are usually defined as cells of mesenchymal origin that produce a variety of extracellular matrix (ECM) components, including multiple collagens, as well as fibronectin30, 31. In spite of this, synthesis and deposition of collagen are not often addressed in the identification of fibroblasts. This means that the definition of the fibroblast is solely based on morphological characteristics that can vary with location within the organism, as well as the overall activity level of the organism itself. Morphologically, fibroblasts are flat, spindle-shaped cells with multiple processes emanating from the main cell body. One characteristic of the fibroblast is that they lack a basement membrane, and it is one of the defining features that separate it from the other permanent cell types of the heart, all of which do contain a basement membrane.
Fibroblasts have typically been viewed as a uniform cell type with comparable functions regardless of whether they originate from the heart, skin or other tissue. This reductionist view has been challenged by data demonstrating extensive phenotypic heterogeneity among fibroblasts from different tissues and from a particular tissue under different physiological conditions. Indeed, lung fibroblasts have been shown to be heterogeneous in cell surface marker expression, as well as in their levels of collagen production32. Moreover, periodontal fibroblasts also show heterogeneity based on morphology, glycogen pools and collagen production32, 33. Furthermore, recent studies comparing the gene expression patterns of fifty fibroblast cultures from multiple sites showed that their gene expression patterns were highly diverse199. This phenotypic plasticity has created a unique challenge in attempts to better define the cardiac fibroblast. Further investigation and characterization of these different fibroblast subpopulations could lead to strategies to inhibit or reverse fibrosis.
As mentioned above, fibroblasts lack a basement membrane and tend to display a prominent Golgi apparatus and extensive rough endoplasmic reticulum, especially when active. While much research has been done examining the fibroblast, no truly definitive cell-specific marker has yet been defined. However, it has been recently shown that discoidin domain receptor 2 (DDR2) is expressed specifically by cardiac fibroblasts34, 35. DDR2 is a cell surface receptor that mediates a wide range of cellular functions, including growth, migration and differentiation. DDR2 is a collagen receptor that is expressed in cardiac fibroblasts, but not myocytes, endothelial cells or vascular smooth muscle cells34. DDR2 is also expressed on specific bone marrow-derived cells, termed fibrocytes, which will be discussed in further detail below36. Another marker that has been proposed to be a fibroblast-specific marker is fibroblast-specific protein-1 (FSP-1)37, 38; however, other papers in the literature have shown that FSP-1 is also expressed in a variety of other cell types, including leukocytes and a multitude of cancer cells39. It has also been shown that fibroblast-activation protein (FAP), a serine protease, is highly expressed on activated fibroblasts196, 197. Recent studies from our laboratory and others have shown that cadherin-11 is localized to fibroblasts40, 41. Moreover, work from Orlandini and Oliviero demonstrated that cell-cell interactions mediated by cadherin-11 lead to increased expression of vascular endothelial growth factor-D (VEGF-D) by fibroblasts42. Taken together, these studies suggest that cadherin-11 may be important for vascular remodeling following cardiac injury, such as myocardial infarction. In addition to these fibroblast markers, there are other genes that are highly expressed by cardiac fibroblasts including vimentin43, 44, β1-integrin45, 46, fibronectin47–49, connexins3, 4, 6, 7, 50 and the fasciclin gene, periostin51–53. Further identification of definitive fibroblast-specific markers should assist in continued efforts to understand this dynamic cell type.
Cardiac fibroblasts play numerous roles in cardiac development and remodeling. They also play a prominent role in defining cardiac structure and function. Below we discuss the numerous roles played by the cardiac fibroblast that help support and maintain proper cardiac function, as well as their role during cardiac pathology. Cardiac fibroblasts are both sources and targets of different stimuli, helping to coordinate chemical, mechanical and electrical signals between the cellular and acellular components of the heart. The global roles of cardiac fibroblast function, such as proliferation, migration, myofibroblast differentiation, matrix generation and degradation and secretion of cytokines and growth factors are illustrated in Figure 1.
Figure 1. Roles of the Cardiac Fibroblast.
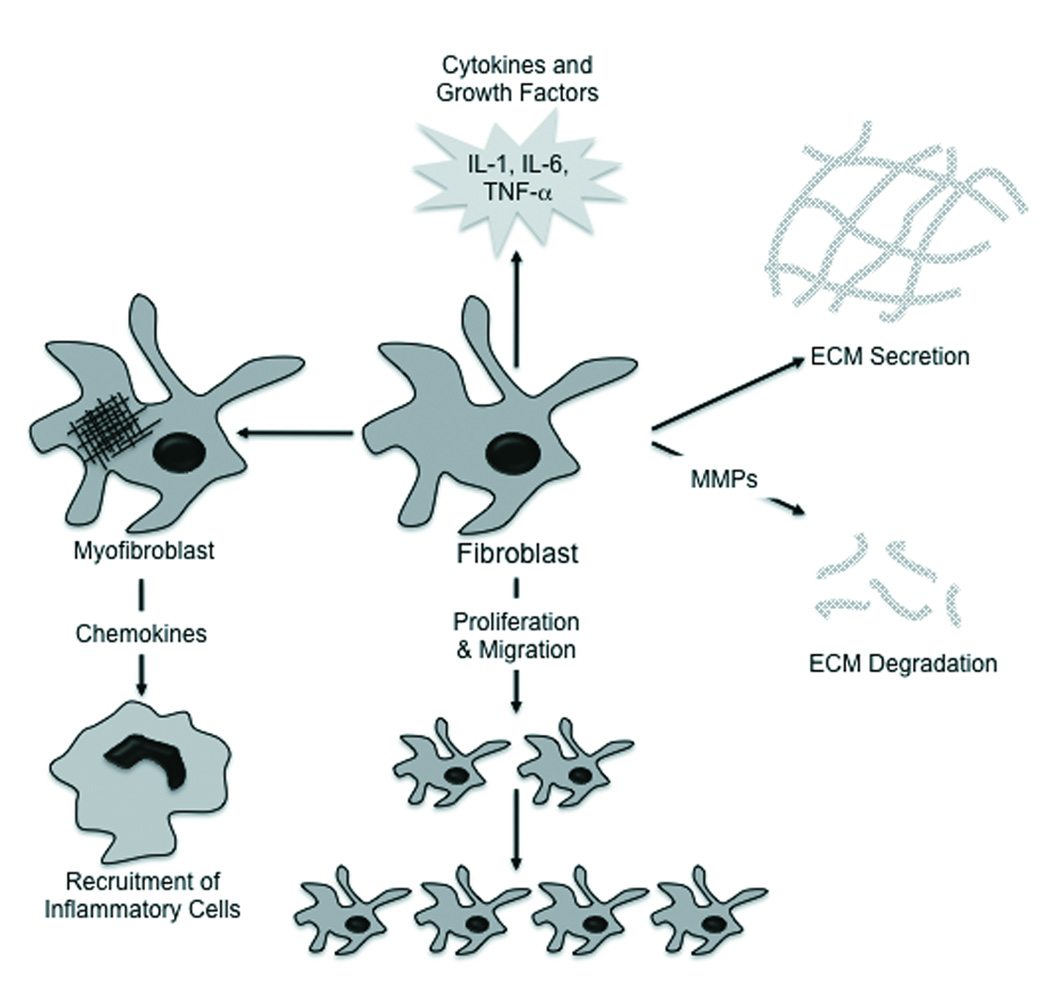
The cardiac fibroblast responds to stimuli in various ways, including secretion of cytokines and growth factors, differentiation into a myofibroblast, proliferation and migration and altering matrix generation and degradation.
The Myofibroblast
Research over the past twenty years has demonstrated that there is phenotypic heterogeneity among fibroblasts, and that some express features of smooth muscle differentiation. These smooth muscle-like cells were originally termed myofibroblasts by Gabbiani, who demonstrated that these cells take part in the growth, development and repair of various tissues54, 55. Under appropriate conditions, resting or quiescent fibroblasts can acquire an active, synthetic, contractile phenotype and express several smooth muscle cell markers that are not typically expressed in fibroblasts56. However, other smooth muscle cell markers, such as myosin heavy chain (MHC), are not expressed in these differentiated myofibroblasts. Additionally, myofibroblasts can be derived from bone marrow-derived cells or epithelial cells via epithelial-mesenchymal transformation (EMT). These cells express contractile proteins, are more mobile than “normal” fibroblasts, can contract collagen gels, and are thought to be important for wound closure and structural integrity of healing scars57. Indeed, research has demonstrated that myofibroblasts play a key role in reparative fibrosis in the infarcted heart58. In addition, myofibroblasts have been shown to be intimately associated with hypertrophic fibrotic scars in various injury models, and differentiation from fibroblast to myofibroblast is promoted by transforming growth factor-β (TGF-β), cytokines, the ECM and other growth factors59–62. Moreover, apoptosis of myofibroblasts has been linked in the progression of granulomatous tissue to a mature scar63. On the other hand, loss of apoptosis has been suggested to drive the progression towards fibrosis. With the exception of heart valve leaflets, myofibroblasts are not usually found in normal, healthy cardiac tissue. However, upon injury, myofibroblasts appear in the myocardium and seem to arise from interstitial and adventitial fibroblasts. It is also possible that myofibroblasts originate from resident progenitor stem cells in the heart or hematopoietic stem cells (HSCs) from the circulation. Whatever their origin, recent studies suggest that the expression and secretion of growth factors, cytokines, ECM and proteases by myofibroblasts is critical for tissue repair, fibrosis and organogenesis64. When myofibroblasts are not properly regulated, destructive tissue remodeling can occur65, 66. Other studies have demonstrated that fibroblasts cultured in vitro at low density will differentiate into myofibroblasts; however, it appears that these cells can be a transient phenotype as they can be reverted back to normal fibroblasts67. Indeed, it has been demonstrated that amniotic membrane stromal extract can reverse differentiated myofibroblasts back to fibroblasts in vitro68. Furthermore, it has also been observed that fibroblast growth factor (FGF) can block or reverse the myofibroblast phenotype69. As we will discuss, myofibroblasts play a critical role in cardiac pathology and remodeling.
Origin of Cardiac Fibroblasts
Considerable heterogeneity in both morphology and function occur in fibroblasts during development. For these reasons it is difficult to precisely define a fibroblast. To understand organogenesis, it is essential to know how, when and where the cell comes from, as well as how the cells migrate to their location in the tissue. Depending on the stage of development, fibroblasts in the heart can arise from various sources (Figure 2).
Figure 2. Sources of Fibroblasts and Myofibroblasts.

The pathways leading to fibrosis involve considerable cell plasticity, with the pro-fibrotic cells being derived from several sources. Epithelial cells can be transformed into mesenchymal cells and vice versa. Mesenchymal stem cells can differentiate into cardiac fibroblasts. Fibrocytes can also differentiate into fibroblasts or myofibroblasts. Finally, pericytes surrounding the vasculature can be differentiated into myofibroblasts.
During embryonic development, fibroblasts are mesenchymal in origin and appear to be closely involved in the formation of the heart. Fibroblasts are thought to arise from the differentiation of cells from the proepicardial organ. These epicardial-derived cells (EPDCs) are considered to be one of the major sources of cardiac fibroblasts70, 71. Additional studies have also demonstrated that fibroblasts can arise from mesoangioblasts. These multipotent progenitors have the ability to differentiate into either vascular (endothelial cells) or mesodermal (fibroblasts) tissues72. The origins of these progenitors in the bone marrow are hematopoietic stem cells. These cells express many lineage markers of vascular cells, such as CD34 and Flk-1 (VEGF-R2). These studies suggest that there is a progression from the mesoangioblast to endothelial cells and pericytes, which have recently been shown to have the potential to differentiate into myofibroblasts73, 74. Indeed, numerous studies in the adult animal suggest that cells, such as pericytes and mesenchymal stem cells (MSCs) of the bone marrow, can contribute to the fibroblast population75, 76.
In the neonatal and adult heart, fibroblasts arise from endogenous cell populations, via EMT and from bone marrow-derived cells. Rapid expansion of the heart occurs during fetal development and neonatal growth, with cardiac fibroblasts contributing ECM to several specific structures of the heart, including the valves, the atrial-ventricular node, the cardiac skeleton and the endomysial network. Fibroblastic cells derived through EMT form the cardiac valves77. This complex transformation process results in fibroblastic cells that contribute to the formation of cardiac cushions and ultimately formation of the collagenous valve leaflets, including the valve interstitium. Originally, these valve fibroblasts were believed to be long-lived cells; however, recent evidence has demonstrated that interstitial fibroblasts can be replaced by bone marrow-derived cells78. It has also been shown that myofibroblast precursors are present in peripheral blood79. In the adult heart, cardiac fibroblast turnover is low, with the sources of replacement being endogenous fibroblast populations and those derived through EMT. However, under pathophysiological conditions, such as cardiac hypertrophy or myocardial infarction, the fibroblasts can arise from bone marrow-derived cells. These latter cells are sometimes termed fibrocytes80–82.
Fibrocytes are typically defined by their growth characteristics and cell surface phenotype. These cells have many similarities to fibroblasts, in that they constitutively express collagen, but they also express cell surface markers indicative of leukocytes and hematopoietic progenitor cells83–88. These cells also express other defining cell surface markers, including chemokine receptors and adhesion molecules76, 89. Fibrocytes have been shown to deposit ECM during wound healing and fibrosis, as well as functioning in the immune response80–82. Taken together, these findings support the fact that HSCs and EPDCs are a major source of new fibroblasts in the adult cardiac valves, as well as other regions of the heart.
Organization of Cardiac Fibroblasts in the Heart
It has been stated that the adult heart consists of 30% myocytes and 70% non-myocytes1, 2. However, recent studies using flow cytometry have demonstrated that the adult murine heart consists of approximately 45% non-myocytes and 55% myocytes90. In addition, these studies confirmed that the adult rat heart consists of 30% myocytes and 70% non-myocytes as demonstrated by Zak and Nag90. These data suggest that the exact cellular makeup of the heart can vary dramatically from species to species. These differences in cardiac cell populations can lead to differences in ECM content as well.
The three-dimensional collagen network of the heart starts to form during fetal development and is primarily laid down during neonatal development. Within this connective tissue network lie the cardiac fibroblasts. During the formation of this three-dimensional network, myocyte-collagen attachments are made involving integrins91. Fibroblasts within the endomysial collagen network then surround the groups of myocytes in a lamellar function (Figure 3)34, 35, 92. The cardiac fibroblasts contain interconnected cellular processes that form a network of cells within the collagen network. This arrangement of fibroblasts within the network allows the fibroblasts to contract the endomysial collagen, exerting mechanical force on the myocytes. In addition, this organization allows for the fibroblasts to help maintain structural integrity of the heart through cell-cell and cell-ECM interactions, as well as through proliferation and ECM degradation and synthesis34, 63, 93, 94. In doing so, the fibroblasts are able to respond to a number of stimuli including chemical, mechanical and electrical signals. It is through these dynamic interactions that fibroblasts are able to maintain proper form and function of the heart95–98. Changes in any of these stimuli by the fibroblasts can also affect the function of other cardiac cell types, such as myocytes and endothelial cells.
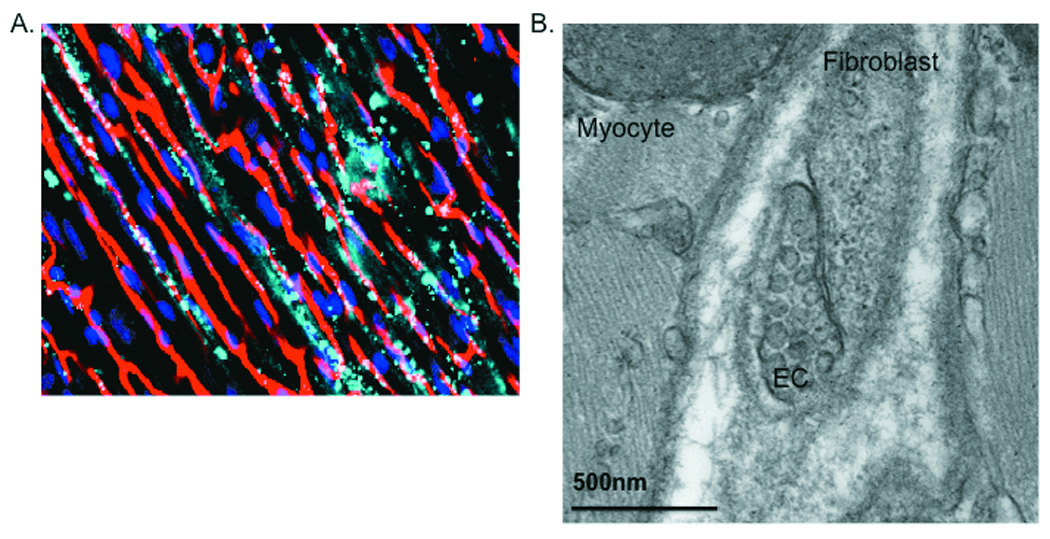
Figure 3. Organization and Interactions of Fibroblasts and Myocytes in the Heart.

A) Confocal micrograph of adult rat heart stained with phalloidin (red), DAPI (blue) and WGA (green). The fibroblasts lie within the endomysial collagen in the extracellular space as previously shown. B) Transmission electron micrograph showing cardiac myocytes in contact with a cardiac fibroblast. The cardiac fibroblasts are in contact with collagen, other fibroblasts, and myocytes forming a mechanosensitive three-dimensional arrangement.
Cardiac fibroblasts interact with the extracellular matrix through integrins and DDR2, while intercellular connections appear to be through two families of cell surface proteins, connexins and cadherins. Connexins 40, 43 and 45 have all been shown to be involved in these interactions and to provide electrical contact95–98. The connexin that connects fibroblasts to myocytes (heterotypic) is connexin 43 (Cx43) and fibroblasts to other fibroblasts (homotypic), connexin 45 (Cx45), although the distribution of these connexins can vary depending on species6, 34, 99. Besides connexins, other cell surface molecules that appear to play a role in forming cell-cell contacts are members of the cadherin family. Previous studies have demonstrated that cadherin-11 is highly expressed in fibroblasts and is associated with VEGF-D expression40, 42. In addition, western blot analyses have shown that cadherin-11 appears to be specifically expressed in cardiac fibroblasts in the mouse left ventricle (data not shown). Moreover, cadherin-13 has been shown to play a role in vascular remodeling, and may be important following myocardial infarction100. Furthermore, it has been demonstrated in vitro that N-cadherin is involved in myocyte-fibroblast and fibroblast-fibroblast interactions95. From these studies, it is clear that the fibroblast plays an essential role in chemical, mechanical and electrical signaling in the heart and disruption of these signaling pathways can lead to cardiac dysfunction.
Cardiac Fibroblasts and the ECM
Cardiac fibroblasts have been termed “sentinel” cells, as they can sense changes in chemical, mechanical and electrical signals in the heart and mount the appropriate response. However, one of the primary functions of the cardiac fibroblast is the synthesis and degradation of the ECM to provide a three-dimensional network for myocytes and other cells of the heart to ensure proper cardiac form and function. The ECM, which consists of the acellular components of the heart, includes interstitial collagens, proteoglycans, glycoproteins, cytokines, growth factors, matrikines and proteases101–104. The extracellular matrix serves multiple purposes, as it forms an organizational network that surrounds and interconnects cells, and provides a scaffold for cardiac cell populations. In addition, the ECM helps to distribute mechanical forces throughout the myocardium, convey mechanical signals to individual cells via cell surface ECM receptors and participate in fluid movement in the extracellular environment101, 102. The role of individual components present within the ECM is highly complicated and in many cases has been difficult to define. Proteoglycans and glycoproteins appear to play important roles in various functions of the ECM, including signaling and turnover of the ECM itself. Various growth factors and proteases are often bound as latent factors to the proteoglycans and glycoproteins105–107. Cytokines and growth factors are essentially short-range chemical signals that are critical for response to pathological stimuli. Extracellular proteases are part of a biochemical cascade within the ECM that is essential for turnover of ECM components, activation of latent factors and cardiac remodeling and are discussed in detail below.
Cardiac Fibroblasts and Remodeling
Remodeling is broadly defined as changes in the organization of the myocardium, and is a critical process that allows the heart to adapt to changes in mechanical, chemical and electrical signals108–110. Cardiac fibroblasts are key components of this process due to their ability to secrete and breakdown the ECM. Degradation of collagen requires the presence of matrix metalloproteinases (MMPs)111, 112. In the normal heart, MMP expression and function are tightly regulated; however, in pathological states MMP expression and activity are increased, leading to excessive ECM degradation, which can have profound effects on cardiac function. Following cardiac injury, fibroblast function can be influenced by chemical signals (i.e. cytokines, matrikines and growth factors) in a paracrine or autocrine manner. These factors can cause changes in fibroblast gene expression, as well as cell migration to the injured region to promote wound healing and scar formation.
Depending on the stage of heart failure, there can be considerable myocyte hypertrophy and cell death. Dilatation can also be observed in later stages; however, present at every stage are changes in the ECM, which are regulated by cardiac fibroblasts. There is also activation and differentiation of cardiac fibroblasts into myofibroblasts63, 93, 113. Upon maturation to myofibroblasts, an increase in the synthesis and secretion of fibronectin is observed114. As the heart undergoes remodeling associated with heart failure, an increase in cytokine and growth factor secretion is observed. In response to these various factors, myofibroblasts begin to proliferate, migrate and remodel the cardiac interstitium through increased secretion of MMPs and collagen63, 93, 115, 116. To further stimulate the remodeling process, cardiac fibroblasts secrete increased amounts of growth factors and cytokines, specifically IL-1β, IL-6 and tumor necrosis factor-α (TNF-α, which in turn activates MMPs leading to further cardiac remodeling63, 103, 117. Initially, all of these changes are critical to the reparative wound healing response; however, over time these changes become maladaptive leading to fibrosis and reduced cardiac function.
While not present in normal myocardium, myofibroblasts are highly localized to sites of injury where synthesis and deposition of collagen promotes scar formation and fibrosis118. In addition, these cells are also located near or associated with blood vessels. Since myofibroblasts express contractile proteins, such as smooth muscle actin (SMA), they are able to provide mechanical tension to the remodeling matrix helping to close the wound and reduce scarring57, 64, 114. As the scar matures, cells in the scar undergo apoptosis, leaving a scar that consists mainly of collagen and ECM proteins, but myofibroblasts are still present119. Indeed, myofibroblasts have been observed in mature scars in a rat model of myocardial infarct, as well as in scarred human tissue118, 120. Why myofibroblasts persist is unknown, but they are highly involved in regulating cardiac remodeling, cardiac dysfunction and ultimately cardiac failure.
It has been observed that valve fibroblasts can have an HSC origin78. It has also been demonstrated that fibrocytes can enter wounds, be detected in scar tissue and possibly participate with local fibroblasts in wound repair and pathological fibrosis. These studies help provide the foundation that different progenitor populations can possibly be harnessed and used as therapeutic agents in the treatment of cardiovascular disease.
In the normal heart, collagen and other ECM components help maintain heart structure and function. ECM is synthesized and degraded by cardiac fibroblasts in a coordinated fashion; however, during heart failure there is disruption of these regulatory pathways, leading to an imbalance of ECM synthesis and degradation that determines the level of cardiac remodeling. Increases in the extracellular matrix or fibrosis may be reparative, replacing areas of myocyte loss with a structural scar, or reactive, involving increases in ECM deposition at sites other than those of the primary injury. Fibrosis has significant consequences for cardiac function, as increases in ECM synthesis and deposition results in increased mechanical stiffness and contributes to diastolic dysfunction. Progressive increases in fibrosis can lead to systolic dysfunction and left ventricular hypertrophy. Moreover, increased levels of collagen can disrupt electrophysiological communication between myocytes. Furthermore, perivascular fibrosis around intracoronary vessels impairs oxygen and nutrient availability and intensifies myocyte ischemia. Heart failure is characterized by considerable differences in levels of disease severity and progression. This most likely reflects polygenic and environmental influences on the level and severity of heart disease in a patient-to-patient manner. Therefore, it is possible that cardiac fibroblasts and their role in cardiac remodeling act as disease modifiers and can potentially be used as predictive risk factors in heart failure.
Cardiac Fibroblasts and Chemical and Mechanical Signaling
Cardiac fibroblasts respond to a wide range of different stimuli during cardiac development and disease, including hypoxia, as well as changes in chemical and mechanical signals. Many of these stimuli function as activators of cardiac fibroblasts; however, chronic stimuli can lead to pathological remodeling and reduced cardiac output. During normal cardiac function, fibroblasts are constantly subject to mechanical stretch. Proper regulation of these mechanical signals is essential to maintaining normal cardiac function102, 121. Various studies have shown that mechanical stimulation of fibroblasts results in a marked upregulation of ECM components, ECM-specific receptors, as well as increased expression of various cytokines and growth factors122–127. In addition, mechanical stretch can induce MMP expression in cardiac fibroblasts leading to ECM degradation. Moreover, cytokine and growth factor expression has been shown to be upregulated in cardiac fibroblasts in response to mechanical stretch128–130. Furthermore, angiotensin II (Ang II), TGF-β and endothelin can regulate ECM synthesis and deposition by cardiac fibroblasts93, 131–133. These studies imply that mechanical load plays a critical role in the regulation of cardiac fibroblast gene expression. However, the response of cardiac fibroblasts to mechanical stimulation in tissue culture appears to be dependent on growth factor stimulation123, 134, 135. These observed differences between in vitro and in vivo studies may be due to improper cell-cell and/or cell-ECM contacts in vitro. Better in vitro models, as well as future in vivo studies, should help to define the interplay between mechanical and chemical signaling in cardiac fibroblasts.
A major function of cardiac fibroblasts is to produce and secrete growth factors, cytokines and other signaling molecules. These signaling factors can have effects on all cardiac cell types. The specific signaling factors that are secreted by the cardiac fibroblasts largely depends on the stimuli. These stimuli may be chemical factors, such as pro- or anti-inflammatory cytokines and growth factors, electrical signals, hypoxia or mechanical stretch. In patients with heart failure, high levels of circulating pro-inflammatory (IL-1β, IL-6 and TNF-〈) and pro-fibrotic (TGF-β) cytokines are observed136. Due to the different expression levels and the complexity of the various factors involved, it is difficult to delineate the exact effects of individual factors on heart failure.
As mentioned above, cardiac fibroblasts are the main source of the pro-inflammatory cytokine, IL-1® in the heart following injury137. Expression of IL-1® can be induced via several different pathways, including hypoxia and TNF-α, while its expression can be inhibited by estrogen137, 138. IL-1®, acting through its receptor (IL-1R) inhibits cardiac fibroblast proliferation, while promoting cell migration139–143. Moreover, it also promotes turnover of the ECM through reduced collagen type I and III synthesis and increased secretion of MMPs144, 145. Furthermore, IL-1® can also induce IL-6 expression, as well as angiotensin type 1 receptor (AT1R) expression in neonatal rat cardiac fibroblasts146.
Previous studies have demonstrated a tight connection between plasma TNF-〈 levels and the progression of left ventricular remodeling and heart failure147. Additional studies in animals demonstrated that infusion of TNF-α resulted in cardiac dysfunction and heart failure148. Moreover, it has been shown that TNF-α increases expression of IL-1β and IL-6 in cardiac fibroblasts in vitro149. These data further demonstrate the multiple levels of gene regulation that occur in the cardiac fibroblast and how this regulation can affect cardiac function.
IL-6 is a pleiotropic cytokine that is responsible for numerous processes, such as regulation of cell growth, apoptosis, differentiation and survival in various cell types and organs, including the heart. Recent studies have indicated that IL-6 is a critical component in cell-cell interactions that occur between myocytes and cardiac fibroblasts150. The biological activity of IL-6 is regulated by binding to the IL-6Rα/gp130 signal transduction complex and subsequent activation of several signal transduction pathways151, 152. Data from several studies suggests that IL-6 and the gp130-JAK-STAT signaling pathway are important in cardiac function and cardioprotection24–27. Indeed, cardiac hypertrophy results in a dramatic increase in the expression of IL-6153–155. Other studies have shown that continuous activation of gp130 leads to myocardial hypertrophy28. Furthermore, constant IL-6/sIL-6Rα stimulation can reduce infarct size and protect against apoptosis29. One mechanism through which IL-6 acts is by inducing proliferation of cardiac fibroblasts and altering ECM turnover144. IL-6 can be produced by both cardiac fibroblasts and myocytes156; however, the majority of IL-6 appears to be secreted by cardiac fibroblasts, but both cell types can respond to IL-6 stimulation150. As mentioned above, IL-1® can induce IL-6 expression, but it can also be regulated by other factors including TNF-α and Ang II138, 157.
Angiotensin II is the effector molecule of the renin-angiotensin system (RAS) that is important for regulating blood pressure and volume. In the circulatory RAS, renin cleaves angiotensinogen into Ang I, which is further processed into Ang II by angiotensin-converting enzyme (ACE-1). Recently, it has been demonstrated that a local RAS exists in the heart and plays an key role in cardiac function168, 169. Cardiac fibroblasts can express angiotensinogen, renin and ACE-1, allowing them to effectively produce Ang II170. Moreover, it has been shown that Ang II can increase collagen production and secretion from cardiac fibroblasts, which occurs through the AT1R171, 172. These studies suggest that the intracellular RAS is a critical component of the heart and normal cardiac function and open up alternative avenues for therapeutic intervention.
Another cytokine that is typically upregulated during heart failure is TGF-β, which acts through interactions with its cell surface receptors, with cardiac fibroblasts being the primary producers158. TGF-β is involved in cell proliferation, differentiation, migration, apoptosis and ECM production. Stimulation of cardiac fibroblasts by TGF-β results in increased synthesis of fibrillar collagen, fibronectin, proteoglycans and expression of contractile genes leading to differentiation into myofibroblasts159–163. Additionally, there are conflicting reports of whether TGF-β acts in pro- or anti-proliferative manner, suggesting multiple roles for TGF-β in the heart164–167. Better understanding of cytokine and growth factor regulation and signaling in the future may provide potential therapeutic targets in heart failure patients.
Cardiac Fibroblasts and Electrical Signaling
Recent studies have demonstrated that fibroblasts have other critical functions other than ECM synthesis, deposition and remodeling173. Cardiac fibroblasts have a high membrane resistance, which makes them good conductors. Cell junctions, such as through connexins, are important for cellular communications in other organ systems and likely play similar roles in physical communication between fibroblasts and other cells within the myocardium175. Indeed, it has been demonstrated in vitro through Cx43, that electrical coupling of myocytes and cardiac fibroblasts can occur8, 95–97. In addition, it has also been shown that fibroblast-myocyte coupling can occur, via Cx45, in the sinoatrial node95–97. Studies from Louault and colleagues also demonstrated function coupling of cardiac fibroblasts through Cx40 and Cx4398. These studies suggest that cardiac fibroblasts could act as bridges that connect different regions of myocytes that would normally be electrically isolated by connective tissue. Moreover, in vitro cell-cell interaction assays have demonstrated that cardiac fibroblasts and myocytes communicate through the formation of tight cell–cell junctions176 (Figure 4). Additionally, ion channels also play an intriguing and important method of signaling because abnormalities in these channels can lead to cardiac dysfunction177. It has been demonstrated that K+ channels are activated by contact between cardiac fibroblasts and myocytes178. Activation of K+ channels has also been linked to Ang II sensitivity179. Furthermore, recent studies have shown that cardiac fibroblasts express several different voltage-gated K+ channels180. Both of these types of signals can have profound implications on cardiac repair and remodeling.
Figure 4. Cell Communication Between Cardiac Fibroblasts and Myocytes.

Z-section of a cell aggregate containing cardiac fibroblasts that were dual loaded with Lucifer Yellow and CMRA and myocytes that were unloaded. Cardiac fibroblasts appear as yellow or orange cells. The orange cells are fibroblasts that have transferred their green dye to an adjacent cell (orange arrow). Note the green cells, which are myocytes that have received green dye from an adjacent fibroblast (white arrow).
Cardiac Fibroblasts and Angiogenesis
Formation of blood vessels depends upon environmental cues that modulate endothelial cell function181, 182. During wound healing, various angiogenic factors cooperate to assemble and stabilize endothelial cells into blood vessels. It has been demonstrated by several laboratories that fibroblasts play a critical role in the wound healing process183–185. In addition, we have recently observed a tight association between cardiac fibroblasts and endothelial cells both in vitro and in vivo, suggesting that fibroblasts are important for blood vessel formation during development and potentially disease (Figure 5). Therefore, understanding the interactions between endothelial cells and fibroblasts is essential for understanding how angiogenesis is regulated.
Figure 5. Cardiac Fibroblasts and Endothelial Cells are Intimately Associated with One Another.
A) Confocal micrograph demonstrating tight association of fibroblasts and endothelial cells (ECs) in the coronary vasculature. Mice were perfused with fluorescent microspheres (red), hearts collected and stained with DAPI (blue) to visualize nuclei and DDR2 to visualize fibroblasts (teal). Note the tight association between the fibroblasts and the vasculature. White spots are co-staining of microspheres and 1611. B) TEM imaging of adult mouse hearts illustrates the close relationship between fibroblasts and ECs. Note the pinocytotic vesicles present in the ECs. Cells in such close proximity have several avenues by which they communicate, including gap junctions and the ECM.
As mentioned, fibroblasts play a role in the development, growth, and remodeling of tissues. They do so through synthesis and deposition of ECM, cytokines and growth factors, which allow them to modulate their environment through autocrine and paracrine signaling. Fibroblast involvement in blood vessel formation was first reported several years ago186, 187; however, their exact role in the angiogenic process remains unresolved. Fibroblast growth factors are potent inducers of angiogenesis. In addition to the cytokines listed above, fibroblasts can also secrete FGF and vascular endothelial growth factor (VEGF)188. Both FGF and VEGF act on vascular endothelial cells and are important for stimulating angiogenesis and coronary collateral formation for restoring the blood supply to injured myocardium. It is possible that chemokines regulate the fibrotic process through recruitment and activation of fibroblast progenitors, fibrocytes, by exerting direct effects on resident cardiac fibroblasts and regulating angiogenesis. In addition, it has been demonstrated that pigment epithelium-derived growth factor (PEDF) can be expressed by both cardiac fibroblasts and myocytes. PEDF was shown to inhibit VEGF-induced endothelial cultures in vitro189. Taken together, these studies indicate that cardiac fibroblasts can express both pro-and anti-angiogenic factors, and that proper regulation of these factors is critical to vascular development and remodeling. To date, few studies have directly investigated how fibroblasts contribute to blood vessel formation.
Other factors that have been shown to play a role in angiogenesis are also produced by fibroblasts, such as MMPs and tissue inhibitors of metalloproteinases (TIMPs)65. MMPs can regulate endothelial cell proliferation, adhesion and survival leading to activation or inhibition of angiogenesis. MMPs act by degrading the ECM, which can promote endothelial cell migration and sprouting, or MMPs can cleave and release anti-angiogenic factors that inhibit these events190, 191. On the other hand, TIMPs have been shown to inhibit angiogenesis192, 193, as well as promote vessel formation194, 195. Thus, like MMPs, it appears that the function of TIMPs is context-dependent. Indeed, recent studies from the Lilly group demonstrated that TIMP-1 is secreted by fibroblasts and increases vessel formation174. In these studies, they observed that the activities of TIMP-1 were MMP-dependent. In addition, they observed that direct interaction between fibroblasts and endothelial cells is necessary for optimal vessel formation174. These studies further underline the significance of fibroblast-derived factors and cell-cell interactions in regulating angiogenesis. Future studies should be aimed at defining the cell surface molecules that play a role in these cell-cell interactions.
Conclusions
Recent studies have helped identify, quantify and map the various cell lineages that are involved in cardiac development, maintenance and disease. Dynamic interactions between the various cell types, the ECM and the biochemical factors that are present in the heart are essential for proper form and function of the heart. Despite recent progress, our current understanding of the how and where cardiac fibroblasts arise from is still relatively unclear. Identification of better cellular markers with absolute specificity for fibroblasts will help in understanding these lingering lineage issues. In addition, identification of cell-specific markers defining cardiac fibroblast progenitor cells (i.e. MSCs, fibrocytes, pericytes) would prove invaluable for the isolation and further characterization of these cells for potential therapeutic applications.
The ECM is also a crucial component in trying to understand the dynamic nature of the heart. The role of the extracellular matrix in mechanical signaling is well-known, but how the ECM functions in chemical signaling is relatively unknown. Future studies will involve more quantitative approaches to examine interactions between the fibroblasts and other cardiac cell types (myocytes and endothelial cells), as well as the interactions of these cells with the ECM.
The unique features of cardiac fibroblasts make it an appealing target for reducing pathological remodeling. Recent studies demonstrating the importance of the fibroblast to blood vessel formation opens up additional avenues for cardiac therapies. Moreover, studies have demonstrated that the heart does have some intrinsic reparative responses ranging from recruitment of progenitor cells from the bone marrow to the injury myocardium, as well as fibroblast and pericyte differentiation into myofibroblasts at the site of injury. A better understanding of the underlying mechanisms of these processes holds the key to successful therapy of the injured heart.
Acknowledgements
The authors thank Dr. Thomas Borg for technical support, helpful suggestions and critical reading of the manuscript.
Sources of Funding
This work was supported by National Heart, Lung and Blood Institute Grant 1RO1-HL-85847.
Non-standard Abbreviations and Acronyms
- ACE-1
Angiotensin-converting enzyme
- Ang II
Angiotensin II
- AT1R
Angiotensin type I receptor
- CMRA
Chloromethyl rhodamine-acetylated
- Cx
Connexin
- DAPI
4’,6-diamidino-2-phenylindole
- DDR2
Discoidin domain receptor 2
- EC
Endothelial cell
- ECM
Extracellular matrix
- EMT
Epithelial-mesenchymal transformation
- EPDC
Epicardial-derived cells
- FAP
Fibroblast activation protein
- FGF
Fibroblast growth factor
- FSP-1
Fibroblast-specific protein 1
- HSC
Hematopoietic stem cells
- IL-1β
Interleukin-1beta
- IL-1R
Interleukin-1 receptor
- IL-6
Interleukin-6
- IL-6Rα
Interleukin-6 receptor-alpha
- JAK-STAT
Janus kinases-signal transducers and activators of transcription
- K+
Potassium
- MHC
Myosin heavy chain
- MMP
Matrix metalloproteinases
- MSC
Mesenchymal stem cells
- PEDF
Pigment epithelium-derived growth factor
- RAS
Renin-angiotensin system
- SMA
Smooth muscle actin
- TEM
Transmission electron microscopy
- TGF-β
Transforming growth factor-beta
- TIMP
Tissue inhibitors of metalloproteinases
- TNF-α
Tumor necrosis factor-alpha
- VEGF
Vascular endothelial growth factor
- WGA
Wheat germ agglutinin
Footnotes
Disclosures
None
References
- 1.Nag A. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 2.Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res. 1974;32(Suppl 2):17–26. [PubMed] [Google Scholar]
- 3.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 4.Camelliti P, Borg TK, Kohl P. Structural and functional characterization of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 5.Harvey PR, Rosenthal N. Heart Development. vol. 1. New York: Academic; 1999. [Google Scholar]
- 6.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004;94:828–835. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 7.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 8.Kohl P. Cardiac cellular heterogeneity and remodeling. Cardiovasc Res. 2004;64:195–197. doi: 10.1016/j.cardiores.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 9.Sussman MA, McCulloch A, Borg TK. Dance band on the Titanic: biomechanical signaling in cardiac hypertrophy. Circ Res. 2002;91:888–898. doi: 10.1161/01.res.0000041680.43270.f8. [DOI] [PubMed] [Google Scholar]
- 10.Bregagnollo EA, Zornoff LA, Okoshi K, Sugizaki M, Mestrinel MA, Padovani CR, Cicogna AC. Myocardial contractile dysfunction contributes to the development of heart failure in rats with aortic stenosis. Int J Cardiol. 2007;117:109–114. doi: 10.1016/j.ijcard.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Wong AK, Osborn TG, Miller JG, Wickline SA. Quantification of ventricular remodeling in the tight-skin mouse cardiomyopathy with acoustic microscopy. Ultrasound Med Biol. 1993;19:356–374. doi: 10.1016/0301-5629(93)90055-s. [DOI] [PubMed] [Google Scholar]
- 12.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 13.Jugdutt BI, Joljart MJ, Khan MI. Rate of collagen deposition during healing and ventricular remodeling after myocardial infarction in rat and dog models. Circulation. 1996;94:94–101. doi: 10.1161/01.cir.94.1.94. [DOI] [PubMed] [Google Scholar]
- 14.Jurko A., Jr Echocardiographic evaluation of left ventricle postnatal growth in newborns and infants. Bratisl Lek Listy. 2004;105:78–85. [PubMed] [Google Scholar]
- 15.Li F, Wang X, Capasso JM, Gerdes AM. Transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 16.Mercier I, Pham-Dang M, Clement R, Gosselin H, Colombo F, Rouleau J, Calderone A. Elevated mean arterial pressure in ovariectomized rat was normalized by ETA receptor antagonist therapy: absence of cardiac hypertrophy and fibrosis. Br J Pharmacol. 2002;136:685–692. doi: 10.1038/sj.bjp.0704765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molkentin JD, Jobe SM, Markham BE. α-Myosin heavy chain gene regulation: delineation and characterization of the cardiac muscle-specific enhancer and muscle-specific promoter. J Mol Cell Cardiol. 1996;28:1211–1225. doi: 10.1006/jmcc.1996.0112. [DOI] [PubMed] [Google Scholar]
- 18.van Empel VP, De Windt LJ. Myocyte hypertrophy and apoptosis: a balancing act. Cardiovasc Res. 2004;63:487–499. doi: 10.1016/j.cardiores.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid Endothelial Cell-Selective Loading of Connexin 40 Antibody Blocks Endothelium-Derived Hyperpolarizing Factor Dilation in Rat Small Mesenteric Arteries. Circ Res. 2005;97:399–408. doi: 10.1161/01.RES.0000178008.46759.d0. [DOI] [PubMed] [Google Scholar]
- 20.Pepper MS, Montesano R, el Aoumari A, Gros D, Orci L, Meda P. Coupling and connexin 43 expression in microvascular and large vessel endothelial cells. Am J Physiol. 1992;262:C1246–C1257. doi: 10.1152/ajpcell.1992.262.5.C1246. [DOI] [PubMed] [Google Scholar]
- 21.Johnson T, Nerem R. Endothelial Connexin 37, Connexin 40 and Connexin 43 Respond Uniquely to Substrate and Shear Stress. Endothelium. 2007;14:215–226. doi: 10.1080/10623320701617233. [DOI] [PubMed] [Google Scholar]
- 22.Goncharova EJ, Kam Z, Geiger B. The involvement of adherens junction components in myofibrillogenesis in cultured cardiac myocytes. Development. 1992;114:173–183. doi: 10.1242/dev.114.1.173. [DOI] [PubMed] [Google Scholar]
- 23.Soler AP, Knudsen KA. N-cadherin involvement in cardiac myocyte interaction and myofibrillogenesis. Dev Biol. 1994;162:9–17. doi: 10.1006/dbio.1994.1062. [DOI] [PubMed] [Google Scholar]
- 24.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr, Mueller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 25.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92:4862–4866. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivey BL. Microvascular plasticity and experimental heart failure. Hypertension. 2006;47:1–3. doi: 10.1161/01.HYP.0000215283.53943.39. [DOI] [PubMed] [Google Scholar]
- 27.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;203:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 28.Kunisada K, Tone E, Fujio Y, Matsui H, Yamauchi-Takihara K, Kishimoto T. Activation of gp130 transduces hypertrophic signals via STAT3 in cardiac myocytes. Circulation. 1998;98:346–352. doi: 10.1161/01.cir.98.4.346. [DOI] [PubMed] [Google Scholar]
- 29.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 30.Eghbali-Webb M. Molecular Biology Intelligence Unit Molecular Biology of Collagen Matrix in the Heart. Austin, TX: Landes; 1994. [Google Scholar]
- 31.Kanekar S, Hirozanne T, Terracio L, Borg TK. Cardiac fibroblasts: form and function. Cardiovasc Pathol. 1998;7:127–133. doi: 10.1016/s1054-8807(97)00119-1. [DOI] [PubMed] [Google Scholar]
- 32.Fries KM, Blieden T, Looney RJ, Sempowski GD, Silvera MR, Willis RA, Phipps RP. Evidence of Fibroblast Heterogeneity and the Role of Fibroblast Subpopulations in Fibrosis. Clin Immunol Immunopathol. 1994;72:283–292. doi: 10.1006/clin.1994.1144. [DOI] [PubMed] [Google Scholar]
- 33.Lekic PC, Pender N, McCulloch CAG. Is Fibroblast Heterogeneity Relevant to the Health, Diseases and Treatments of Periodontal Tissues? Crit Rev Oral Biol Med. 1997;8:253–268. doi: 10.1177/10454411970080030201. [DOI] [PubMed] [Google Scholar]
- 34.Camelliti P, Borg TK, Kohl P. Structural and functional characterization of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 35.Goldsmith EC, Hoffman A, Morales MO, Potts JD, Price RL, McFadden A, Rice M, Borg TK. Organization of fibroblasts in the heart. Dev Dyn. 2004;230:787–794. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 36.Ebihara Y, Masuya M, Larue AC, Fleming PA, Visconti RP, Minamiguchi H, Drake CJ, Ogawa M. Hematopoietic origins of fibroblasts: II. In vitro studies of fibroblasts, CFU-F and fibrocytes. Exp Hematol. 2006;34:219–229. doi: 10.1016/j.exphem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossini M, Cheunsuchon B, Donnert E, Ma LJ, Thomas JW, Neilson EG, Fogo AB. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 2005;68:2621–2628. doi: 10.1111/j.1523-1755.2005.00734.x. [DOI] [PubMed] [Google Scholar]
- 39.Mazzucchelli L. Protein S100A4: Too long overlooked by pathologists? Am J Pathol. 2002;160:7–13. doi: 10.1016/S0002-9440(10)64342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valencia X, Higgins JMG, Kiener HP, Lee DM, Podrebarac TA, Dascher CC, Watts GFM, Mizguchi E, Simmons B, Patel DD, Bhan AK, Brenner MB. Cadherin-11 Provides Specific Cellular Adhesion between Fibroblast-like Synoviocytes. J Exp Med. 2004;12:1673–1679. doi: 10.1084/jem.20041545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simoneau L, Kitagawa M, Suzuki S, Thiery JP. Cadherin-11 expression marks the mesenchymal phenotype: towards new functions for cadherins? Cell Adhes Commun. 1995;3:115–130. doi: 10.3109/15419069509081281. [DOI] [PubMed] [Google Scholar]
- 42.Orlandini M, Oliviero S. In fibroblasts Vegf-D expression is induced by cell-cell contact mediated by cadherin-11. J Biol Chem. 2001;276:6576–6581. doi: 10.1074/jbc.M009573200. [DOI] [PubMed] [Google Scholar]
- 43.Lane EB, Hogan BL, Kurkinen M, Garrels JI. Co-expression of vimentin and cytokeratins in parietal endoderm cells of early mouse embryo. Nature. 1983;303:701–704. doi: 10.1038/303701a0. [DOI] [PubMed] [Google Scholar]
- 44.Duprey P, Paulin D. What can be learned from intermediate filament gene regulation in the mouse embryo? Int J Dev Biol. 1995;39:443–457. [PubMed] [Google Scholar]
- 45.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 46.Sutherland AE, Calarco PG, Damsky CH. Developmental regulation of integrin expression at the time of implantation in the mouse embryo. Development. 1993;119:1175–1186. doi: 10.1242/dev.119.4.1175. [DOI] [PubMed] [Google Scholar]
- 47.MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in modulating cardiac fibroblasts function and extracellular matrix synthesis. Cardiovasc Res. 2000;46:257–263. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- 48.Perkinson RA, Norton PA. Expression of the mouse fibronectin gene and fibronectin-lacZ transgenes during somitogenesis. Dev Dyn. 1997;208:244–254. doi: 10.1002/(SICI)1097-0177(199702)208:2<244::AID-AJA11>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Manso AM, Kang SM, Plotnikov SV, Thievessen I, Oh J, Beggs HE, Ross RS. Cardiac fibroblasts require focal adhesion kinase for normal proliferation and migration. Am J Physiol Heart Circ Physiol. 2009;296:H627–H638. doi: 10.1152/ajpheart.00444.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohl P, Camelliti P, Burton FL, Smith GL. Electrical coupling of fibroblasts and myocytes: relevance for cardiac propagation. J Electrocardiol. 2005;38:45–50. doi: 10.1016/j.jelectrocard.2005.06.096. [DOI] [PubMed] [Google Scholar]
- 51.Norris RA, Potts JD, Yost MJ, Junor L, Brooks T, Tan H, Hoffman S, Hart MM, Kern MJ, Damon B, Markwald RR, Goodwin RL. Periostin promotes a fibroblastic lineage pathway in atrioventricular valve progenitor cells. Dev Dyn. 2009;238:1052–1063. doi: 10.1002/dvdy.21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of Postn expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 54.Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–550. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- 55.Gabbiani G. The cellular derivation and the life span of the myofibroblast. Pathol Res Pract. 1996;192:708–711. doi: 10.1016/S0344-0338(96)80092-6. [DOI] [PubMed] [Google Scholar]
- 56.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodeling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 57.Sun Y, Weber KT. RAS and connective tissue in the heart. Int J Biochem Cell Biol. 2003;35:919–931. doi: 10.1016/s1357-2725(02)00276-5. [DOI] [PubMed] [Google Scholar]
- 58.Calderone A, Bel-Hadj S, Drapeau J, El-Helou V, Gosselin H, Clement R, Villeneuve L. Scar myofibroblasts of the infarcted rat heart express natriuretic peptides. J Cell Physiol. 2006;207:165–173. doi: 10.1002/jcp.20548. [DOI] [PubMed] [Google Scholar]
- 59.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 60.Kaden JJ, Kiliç R, Sarikoç A, Hagl S, Lang S, Hoffmann U, Brueckmann M, Borggrefe M. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med. 2005;16:869–872. [PubMed] [Google Scholar]
- 61.Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta 1. J Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 64.Brown E, Dejana E. Cell-to-cell contract and the extracellular matrix. Curr Opin Cell Biol. 2003;15:505–508. [Google Scholar]
- 65.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol Cell Physiol. 1999;277:C1–C19. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 66.Adler KB, Low RB, Leslie KO, Mitchel J, Evans JN. Contractile cells in normal and fibrotic lung. Lab Invest. 1989;60:473–485. [PubMed] [Google Scholar]
- 67.Masur SK, Dewal HS, Dinh TT, Erenburg I, Petridou S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc Natl Acad Sci USA. 1996;93:4219–4223. doi: 10.1073/pnas.93.9.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor β-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–192. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 69.Cushing MC, Mariner PD, Liao JT, Sims EA, Anseth KS. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008;22:1769–1177. doi: 10.1096/fj.07-087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–1267. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- 71.Norris RA, Borg TK, Butcher JT, Baudino TA, Banerjee I, Markwald RR. Neonatal and adult cardiovascular pathophysiological remodeling and repair: developmental role of periostin. Ann N Y Acad Sci. 2008;1123:30–40. doi: 10.1196/annals.1420.005. [DOI] [PubMed] [Google Scholar]
- 72.Cossu G, Bianco P. Mesoangioblasts-vascular progenitors for extravascular mesodermal tissues. Curr Opin Genet Dev. 2003;13:537–542. doi: 10.1016/j.gde.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 73.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and Perivascular Fibroblasts Are the Primary Source of Collagen-Producing Cells in Obstructive Fibrosis of the Kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo, Diaz Flores, Pericytes L. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Hisolo Histopathol. 2009;24:909–969. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- 75.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling. Circ Res. 2001;89:1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 76.Sundberg C, Ivarsson M, Gerdin B, Rubin K. Pericytes as collagen-producing cells in excessive dermal scarring. Lab Invest. 1996;74:452–466. [PubMed] [Google Scholar]
- 77.Potts JD, Runyan RB. Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta. Dev Biol. 1989;134:392–401. doi: 10.1016/0012-1606(89)90111-5. [DOI] [PubMed] [Google Scholar]
- 78.Visconti RP, Ebihara Y, Larue AC, Fleming PA, McQuinn TC, Mauya M, Minamiguchi H, Markwald RR, Ogawa M, Drake CJ. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 79.Wilcox JN, Okamoto EI, Nakahara KI, Vinten-Johansen J. Perivascular responses after angioplasty which may contribute to postangioplasty restenosis: a role for circulating myofibroblast precursors? Ann NY Acad Sci. 2001;947:68–90. doi: 10.1111/j.1749-6632.2001.tb03931.x. [DOI] [PubMed] [Google Scholar]
- 80.Wu Y, Wang JF, Scott PG, Tredget EE. Bone marrow-derived stem cells in wound healing: a review. Wound Rep Reg. 2007;15:S18–S26. doi: 10.1111/j.1524-475X.2007.00221.x. [DOI] [PubMed] [Google Scholar]
- 81.Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukocyte Biol. 2009;86:1–8. doi: 10.1189/jlb.0309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87:858–870. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- 83.Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–425. [PubMed] [Google Scholar]
- 84.Chesney J, Bucala R. Peripheral blood fibrocytes: novel fibroblast-like cells that present antigen and mediate tissue repair. Biochem Soc Trans. 1997;25:520–524. doi: 10.1042/bst0250520. [DOI] [PubMed] [Google Scholar]
- 85.Grab DJ, Lanners H, Martin LN, Chesney J, Cai C, Adkisson HD, Bucala R. Interaction of Borrelia burgdorferi with peripheral blood fibrocytes, antigen-presenting cells with the potential for connective tissue targeting. Mol Med. 1999;5:46–54. [PMC free article] [PubMed] [Google Scholar]
- 86.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 87.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 88.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 89.Metz CN. Fibrocytes: a unique cell population implicated in wound healing. Cell Mol Life Sci. 2003;60:1342–1350. doi: 10.1007/s00018-003-2328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–H1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 91.Ross RS. The extracellular connections: The role of integrins in myocardial remodeling. J Cardiac Failure. 2002;8:S326–S331. doi: 10.1054/jcaf.2002.129263. [DOI] [PubMed] [Google Scholar]
- 92.Borg TK, Ranson WF, Moslehy FA, Caulfield JB. Structural basis of ventricular stiffness. Lab Invest. 1981;44:49–54. [PubMed] [Google Scholar]
- 93.Weber KT. Fibrosis in hypertensive heart disease: focus on cardiac fibroblasts. J Hypertens. 2004;22:47–50. doi: 10.1097/00004872-200401000-00011. [DOI] [PubMed] [Google Scholar]
- 94.Baxter SC, Morales MO, Goldsmith EC. Adaptive changes in cardiac fibroblast morphology and collagen organization as a result of mechanical environment. Cell Biochem Biophys. 2008;51:33–44. doi: 10.1007/s12013-008-9013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic Interactions between myocytes, fibroblasts and extracellular matrix. Ann NY Acad Sci. 2006;1080:76–84. doi: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- 96.Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007;583:225–236. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kohl P. Heterogeneous cell coupling in the heart: an electrophysiological role for fibroblasts. Circ Res. 2003;93:381–383. doi: 10.1161/01.RES.0000091364.90121.0C. [DOI] [PubMed] [Google Scholar]
- 98.Louault C, Benamer N, Faivre JF, Potreau D, Bescond J. Implication of connexins 40 and 43 in functional coupling between mouse cardiac fibroblasts in primary culture. Biochim Biophys Acta. 2008;1778:2097–2104. doi: 10.1016/j.bbamem.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 99.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc Res. 2004;62:415–425. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 100.Ivanov D, Philippova M, Tkachuk V, Erne P, Resnik T. Cell adhesion molecule T-cadherin regulates vascular cell adhesion, phenotype and motility. Exp Cell Res. 2004;293:207–218. doi: 10.1016/j.yexcr.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 101.Bowers SL, Banerjee I, Baudino TA. The extracellular matrix: At the center of it all. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.08.024. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–253. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- 103.Corda S, Samuel JL, Rappaport L. Extracellular matrix and growth factors during heart growth. Heart Fail Rev. 2000;5:119–130. doi: 10.1023/A:1009806403194. [DOI] [PubMed] [Google Scholar]
- 104.Visconti RP, Markwald RR. Recruitment of new cells into the postnatal heart: potential modification of phenotype by periostin. Ann N Y Acad Sci. 2006;1080:19–33. doi: 10.1196/annals.1380.003. [DOI] [PubMed] [Google Scholar]
- 105.Lopez B, Gonzalez A, Diez J. Role of matrix metalloproteinases in hypertension-associated cardiac fibrosis. Curr Opin Nephrol Hypertens. 2004;13:197–204. doi: 10.1097/00041552-200403000-00008. [DOI] [PubMed] [Google Scholar]
- 106.Maquart FX, Bellon G, Pasco S, Monboisse JC. Matrikines in the regulation of extracellular matrix degradation. Biochimie. 2005;87:353–360. doi: 10.1016/j.biochi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 107.Schor SL, Schor AM. Tumor-stroma interactions phenotypic and genetic alterations in mammary stroma: implications for tumour progression. Breast Cancer Res. 2001;3:373–379. doi: 10.1186/bcr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol. 2002;283:H518–H525. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 109.Chancey AL, Brower GL, Janicki JS. Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am J Physiol Heart Circ Physiol. 2002;282:H2152–H2158. doi: 10.1152/ajpheart.00777.2001. [DOI] [PubMed] [Google Scholar]
- 110.Stewart JA, Jr, Wei CC, Brower GL, Rynders PE, Hankes GH, Dillon AR, Lucchesi PA, Janicki JS, Dell’Italia LJ. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35:311–319. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 111.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–359. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 113.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in repurfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain. Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 114.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 115.Lindsey ML, Escobar GP, Mukherjee R, Goshorn DK, Sheats NJ, Bruce JA, Mains IM, Hendrick JK, Hewett KW, Gourdie RG, Matrisian LM, Spinale FG. Matrix metalloproteinase-7 affects connexin-43 levels, electrical conduction, and survival after myocardial infarction. Circulation. 2006;113:2919–2928. doi: 10.1161/CIRCULATIONAHA.106.612960. [DOI] [PubMed] [Google Scholar]
- 116.Raizman JE, Komijenovic J, Chang R, Deng C, Bedosky KM, Rattan SG, Cunnington RH, Freed DH, Dixon IM. The participation of the Na+-Ca2+ exchanger in primary cardiac myofibroblast migration, contraction and proliferation. J Cell Physiol. 2007;213:540–551. doi: 10.1002/jcp.21134. [DOI] [PubMed] [Google Scholar]
- 117.Brown RD, Mitchell MD, Long CS. Proinflammatory cytokines and cardiac extracellular matrix: regulation of fibroblast phenotype. In: Villarreal FJ, editor. Interstitial Fibrosis in Heart Disease. New York: Springer; 2004. pp. 57–81. [Google Scholar]
- 118.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 119.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 120.Willems IE, Havenith MG, DeMey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 121.Catalucci D, Latronico MV, Ellingsen O, Condorelli G. Physiological myocardial hypertrophy: how and why? Front Biosci. 2008;13:312–324. doi: 10.2741/2681. [DOI] [PubMed] [Google Scholar]
- 122.Chapman D, Weber KT, Eghbali M. Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res. 1990;67:787–794. doi: 10.1161/01.res.67.4.787. [DOI] [PubMed] [Google Scholar]
- 123.Butt RP, Bishop JE. Mechanical load enhances the stimulatory effect of serum growth factors on cardiac fibroblast procollagen synthesis. J Mol Cell Cardiol. 1997;29:1141–1151. doi: 10.1006/jmcc.1996.0347. [DOI] [PubMed] [Google Scholar]
- 124.Carver W, Nagpal ML, Nachtigal M, Borg TK, Terracio L. Collagen expression in mechanically stimulated cardiac fibroblasts. Circ Res. 1991;69:116–122. doi: 10.1161/01.res.69.1.116. [DOI] [PubMed] [Google Scholar]
- 125.Hsieh AH, Tsai CM, Ma OJ, Lin T, Banes AJ, Villarreal FJ, Akeson WH, Sung KL. Time-dependent increases in type-III collagen gene expression in medical collateral ligament fibroblasts under cyclic strains. J Orthop Res. 2000;18:220–227. doi: 10.1002/jor.1100180209. [DOI] [PubMed] [Google Scholar]
- 126.Lee AA, Delhaas T, Waldman LK, MacKenna DA, Villarreal FJ, McCulloch AD. An equibiaxial strain system for cultured cells. Am J Physiol Cell Physiol. 1996;271:C1400–C1408. doi: 10.1152/ajpcell.1996.271.4.C1400. [DOI] [PubMed] [Google Scholar]
- 127.Carver W, Terracio L, Borg TK. Expression and accumulation of interstitial collagen in the neonatal rat heart. Anat Rec. 1993;236:511–520. doi: 10.1002/ar.1092360311. [DOI] [PubMed] [Google Scholar]
- 128.van Wamel AJ, Ruwhof C, van der Valk-Kokshoom LE, Schrier PE, van der Laarse A. The role of angiotensin. II. endothelin-1 and transforming growth factor-β as autocrine/paracrine mediators of stretch-induced cardiomyocyte hypertrophy. Mol Cell Biochem. 2001;218:113–124. doi: 10.1023/a:1007279700705. [DOI] [PubMed] [Google Scholar]
- 129.Yokoyama T, Sekiguchi K, Tanaka T, Tomaru K, Arai M, Suzuki T, Nagai R. Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am J Physiol. 1999;276:H1968–H1976. doi: 10.1152/ajpheart.1999.276.6.H1968. [DOI] [PubMed] [Google Scholar]
- 130.Ruwhof C, van Wamel AE, Egas JM, van der Laarse A. Cyclic stretch induces the release of growth promoting factors from cultured neonatal cardiomyocytes and cardiac fibroblasts. Mol Cell Biochem. 2000;208:89–98. doi: 10.1023/a:1007046105745. [DOI] [PubMed] [Google Scholar]
- 131.Brilla CG, Reams GP, Maisch B, Weber KT. Renin-angiotensin system and myocardial fibrosis in hypertension: regulation of the myocardial collagen matrix. Eur Heart J. 1993;14:57–61. [PubMed] [Google Scholar]
- 132.Elias JA, Freundlich B, Adams S, Rosenbloom J. Regulation of human lung fibroblast collagen production by recombinant interleukin-1, tumor necrosis factor, and interferon gamma. Ann NY Acad Sci. 1990;580:233–244. doi: 10.1111/j.1749-6632.1990.tb17932.x. [DOI] [PubMed] [Google Scholar]
- 133.Goldstein RH, Poliks CF, Pilch PF, Smith BD, Fine A. Stimulation of collagen formation by insulin and insulin-like growth factor I in cultures of human lung fibroblasts. Endocrinology. 1989;124:964–970. doi: 10.1210/endo-124-2-964. [DOI] [PubMed] [Google Scholar]
- 134.Atance J, Yost MJ, Carver W. Influence of the extracellular matrix on the regulation of cardiac fibroblast behavior by mechanical stretch. J Cell Physiol. 2004;200:377–386. doi: 10.1002/jcp.20034. [DOI] [PubMed] [Google Scholar]
- 135.Bishop JE. Regulation of cardiovascular collagen deposition by mechanical forces. Mol Med Today. 1998;4:69–75. doi: 10.1016/S1357-4310(97)01193-3. [DOI] [PubMed] [Google Scholar]
- 136.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 137.Long CS. The role of interleukin-1 in the failing heart. Heart Fail Rev. 2001;6:81–94. doi: 10.1023/a:1011428824771. [DOI] [PubMed] [Google Scholar]
- 138.Turner NA, Aley PK, Hall KT, Warburton P, Galloway S, Midgley L, O'Regan DJ, Wood IC, Ball SG, Porter KE. Simvastatin inhibits TNFα-induced invasion of human cardiac myofibroblasts via both MMP-9-dependent and -independent mechanisms. J Mol Cell Cardiol. 2007;43:168–176. doi: 10.1016/j.yjmcc.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 139.Mitchell MD, Laird RE, Brown RD, Long CS. IL-1β stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol Heart Circ Physiol. 2007;292:H1139–H1147. doi: 10.1152/ajpheart.00881.2005. [DOI] [PubMed] [Google Scholar]
- 140.Mughal RS, Warburton P, O'Regan DJ, Ball SG, Turner NA, Porter KE. Peroxisome proliferator-activated receptor γ-independent effects of thiazolidinediones on human cardiac myofibroblast function. Clin Exp Pharmacol Physiol. 2009;36:478–486. doi: 10.1111/j.1440-1681.2008.05088.x. [DOI] [PubMed] [Google Scholar]
- 141.Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1β induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest. 1995;95:2555–2564. doi: 10.1172/JCI117956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Koudssi F, Lopez JE, Villegas S, Long CS. Cardiac fibroblasts arrest at the G1/S restriction point in response to interleukin (IL)-1β. Evidence for IL-1β-induced hypophosphorylation of the retinoblastoma protein. J Biol Chem. 1998;273:25796–25803. doi: 10.1074/jbc.273.40.25796. [DOI] [PubMed] [Google Scholar]
- 143.Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M, Karliner JS. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol. 2000;32:565–576. doi: 10.1006/jmcc.2000.1109. [DOI] [PubMed] [Google Scholar]
- 144.Siwik DA, Chang DL, Colucci WS. Interleukin-1β and tumor necrosis factor-α decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 145.Xie Z, Singh M, Singh K. Differential regulation of matrix metalloproteinase-2 and -9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1β. J Biol Chem. 2004;279:39513–39519. doi: 10.1074/jbc.M405844200. [DOI] [PubMed] [Google Scholar]
- 146.Cowling RT, Zhang X, Reese VC, Iwata M, Gurantz M, Dillmann WH, Greenberg BH. Effects of cytokine treatment on angiotensin II type 1A receptor transcription and splicing in rat cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2005;289:H1176–H1183. doi: 10.1152/ajpheart.00088.2005. [DOI] [PubMed] [Google Scholar]
- 147.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD) J Am Coll Cardiol. 1996;27:1201–1206. doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- 148.Bozkurt B, Kribbs SB, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor-promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 149.Turner NA, Mughal RS, Warburton P, O’Regan DJ, Ball SG, Porter KE. Mechanism of TNFα-induced IL-1α, IL-1β and IL-6 expression in human cardiac fibroblasts: Effects of statins and thiazolidediones. Cardiovas Res. 2007;76:81–90. doi: 10.1016/j.cardiores.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 150.Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694–H1704. doi: 10.1152/ajpheart.00908.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O'Donnell VB. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol. 2007;171:315–325. doi: 10.2353/ajpath.2007.061078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tsuruda T, Costello-Boerrigter LC, Burnett JC., Jr Matrix metalloproteinases: pathways of induction by bioactive molecules. Heart Fail Rev. 2004;9:53–61. doi: 10.1023/B:HREV.0000011394.34355.bb. [DOI] [PubMed] [Google Scholar]
- 153.Diaz JA, Booth AJ, Lu G, Wood SC, Pinsky DJ, Bishop DK. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant. 2009;9:1773–1783. doi: 10.1111/j.1600-6143.2009.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol. 2005;202:891–899. doi: 10.1002/jcp.20197. [DOI] [PubMed] [Google Scholar]
- 155.Camelliti P, Green CR, Kohl P. Structural and functional coupling of cardiac myocytes and fibroblasts. Adv Cardiol. 2006;42:132–149. doi: 10.1159/000092566. [DOI] [PubMed] [Google Scholar]
- 156.Ancey C, Corbi P, Froger J, Delwail A, Wijdenes J, Gascan H, Potreau D, Lecron JC. Secretion of IL-6, IL-11 and LIF by human cardiomyocytes in primary culture. Cytokine. 2002;18:199–205. doi: 10.1006/cyto.2002.1033. [DOI] [PubMed] [Google Scholar]
- 157.Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M, Saito Y, Ogawa S. ERK and p38 MAPK, but not NF-κB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res. 2001;89:661–669. doi: 10.1161/hh2001.098873. [DOI] [PubMed] [Google Scholar]
- 158.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor β receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004;95:1167–1173. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 159.Eghbali M, Tomek R, Woods C, Bhambi B. Cardiac fibroblasts are predisposed to convert into myocyte phenotype: specific effect of transforming growth factor β. Proc Natl Acad Sci USA. 1991;88:795–799. doi: 10.1073/pnas.88.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Heimer R, Bashey RI, Kyle J, Jimenez SA. TGF-beta modulates the synthesis of proteoglycans by myocardial fibroblasts in culture. J Mol Cell Cardiol. 1995;27:2191–2198. doi: 10.1016/s0022-2828(95)91479-x. [DOI] [PubMed] [Google Scholar]
- 161.Villarreal FJ, Lee AA, Dillmann WH, Giordano FJ. Adenovirus-mediated overexpression of human transforming growth factor-beta 1 in rat cardiac fibroblasts, myocytes and smooth muscle cells. J Mol Cell Cardiol. 1996;28:735–742. doi: 10.1006/jmcc.1996.0068. [DOI] [PubMed] [Google Scholar]
- 162.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.07.015. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Leask A. TGFβ, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 164.Drobic V, Cunnington RH, Bedosky KM, Raizman JE, Elimban VV, Rattan SG, Dixon IM. Differential and combined effects of cardiotrophin-1 and TGF-β1 on cardiac myofibroblast proliferation and contraction. Am J Physiol Heart Circ Physiol. 2007;293:H1053–H1064. doi: 10.1152/ajpheart.00935.2006. [DOI] [PubMed] [Google Scholar]
- 165.Sigel AV, Centrella M, Eghbali-Webb M. Regulation of proliferative response of cardiac fibroblasts by transforming growth factor-β1. J Mol Cell Cardiol. 1996;28:1921–1929. doi: 10.1006/jmcc.1996.0185. [DOI] [PubMed] [Google Scholar]
- 166.Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, Mains IM, Mingoia JT, Flack EC, Lindsey ML. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39:699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 167.Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L, Renfrow MB, Chen YF. Atrial natriuretic peptide inhibits transforming growth factor β-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–192. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 168.Kumar R, Singh VP, Baker KM. The intracellular rennin-angiotensin system: implications in cardiovascular remodeling. Curr Opin Nephrol Hypertens. 2008;17:168–173. doi: 10.1097/MNH.0b013e3282f521a8. [DOI] [PubMed] [Google Scholar]
- 169.Dostal DE, Baker KM. The cardiac rennin-angiotensin system: conceptual, or a regulator of cardiac function? Circ Res. 1999;85:643–655. doi: 10.1161/01.res.85.7.643. [DOI] [PubMed] [Google Scholar]
- 170.Sanghi S, Kumar R, Smith M, Baker KM, Dostal DE. Activation of protein kinase A by atrial natriuretic peptide in neonatal rat cardiac fibroblasts: role in regulation of the local renin-angiotensin system. Regul Pept. 2005;132:1–8. doi: 10.1016/j.regpep.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 171.Lijnen PJ, Petrov VV, Fagard RH. Angiotensin II-induced stimulation of collagen secretion and production in cardiac fibroblasts is mediated via angiotensin II subtype 1 receptors. J Renin Angiotensin Aldosterone Syst. 2001;2:117–122. doi: 10.3317/jraas.2001.012. [DOI] [PubMed] [Google Scholar]
- 172.Autelitano DJ, Ridings R, Pipolo L, Thomas WG. Adrenomedullin inhibits angiotensin AT1A receptor expression and function in cardiac fibroblasts. Regul Pept. 2003;112:131–137. doi: 10.1016/s0167-0115(03)00031-4. [DOI] [PubMed] [Google Scholar]
- 173.Deschamps AM, Spinale FG. Disruptions and detours in the myocardial matrix highway and heart failure. Curr Heart Fail Rep. 2005;2:10–17. doi: 10.1007/s11897-005-0002-6. [DOI] [PubMed] [Google Scholar]
- 174.Liu H, Chen B, Lilly B. Fibroblasts potentiate blood vessel formation partially through secreted factor TIMP-1. Angiogenesis. 2008;11:223–234. doi: 10.1007/s10456-008-9102-8. [DOI] [PubMed] [Google Scholar]
- 175.Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI, Gourdie RG. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- 176.Baudino TA, McFadden A, Fix C, Hastings J, Price R, Borg TK. Cell patterning: interaction of cardiac myocytes and fibroblasts in three-dimensional culture. Microsc Micoranal. 2008;14:117–125. doi: 10.1017/S1431927608080021. [DOI] [PubMed] [Google Scholar]
- 177.Shibukawa Y, Chilton EL, Maccannell KA, Clark RB, Giles WR. K+ currents activated by depolarization in cardiac fibroblasts. Biophys J. 2005;88:3924–3935. doi: 10.1529/biophysj.104.054429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chilton L, Ohya S, Freed D, George E, Drobic V, Shibukawa Y, Maccannell KA, Imaizumi Y, Clark RB, Dixon IM, Giles WR. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am J Physiol Heart Circ Physiol. 2005;288:H2931–H2939. doi: 10.1152/ajpheart.01220.2004. [DOI] [PubMed] [Google Scholar]
- 179.Delpon E, Caballero R, Gomez R, Nunez L, Tamargo J. Angiotensin II, angiotensin II antagonists and spironolactone and their modulation of cardiac repolarization. Trends Pharmacol Sci. 2005;26:155–161. doi: 10.1016/j.tips.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 180.Walsh KB, Zhang J. Neonatal rat cardiac fibroblasts express three types of voltage-gated K+ channels: regulation of a transient outward current by protein kinase C. Am J Physiol Heart Circ Physiol. 2008;294:H1010–H1017. doi: 10.1152/ajpheart.01195.2007. [DOI] [PubMed] [Google Scholar]
- 181.Darland DC, D’Amore PA. Cell–cell interactions in vascular development. Curr Top Dev Biol. 2001;52:107–149. doi: 10.1016/s0070-2153(01)52010-4. [DOI] [PubMed] [Google Scholar]
- 182.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 183.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 184.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 185.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 186.Montesano R, Pepper MS, Orci L. Paracrine induction of angiogenesis in vitro by Swiss 3T3 fibroblasts. J Cell Sci. 1993;105:1013–1024. doi: 10.1242/jcs.105.4.1013. [DOI] [PubMed] [Google Scholar]
- 187.Villaschi S, Nicosia RF. Paracrine interactions between fibroblasts and endothelial cells in a serum-free coculture model. Modulation of angiogenesis and collagen gel contraction. Lab Invest. 1994;71:291–299. [PubMed] [Google Scholar]
- 188.Murakami M, Simons M. Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol. 2008;15:215–220. doi: 10.1097/MOH.0b013e3282f97d98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rychli K, Kaun C, Hohensinner P, Dorfner A, Pfaffenberger S, Niessner A, Bauer M, Dietl W, Podesser B, Maurer G, Huber K, Wojta J. The antiangiogenic factor PEDF is present in the human heart and is regulated by anoxia in cardiac myocytes and fibroblasts. J Cell Mol Med. 2009 Feb 27; doi: 10.1111/j.1582-4934.2009.00731.x. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bix G, Iozzo RV. Matrix revolutions: “tails” of basement membrane components with angiostatic functions. Trends Cell Biol. 2005;15:52–60. doi: 10.1016/j.tcb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 191.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chirco R, Liu XW, Jung KK, Kim HR. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006;25:99–113. doi: 10.1007/s10555-006-7893-x. [DOI] [PubMed] [Google Scholar]
- 193.Lambert E, Dasse E, Haye B, Petitfrere E. TIMPs as multifacial proteins. Crit Rev Oncol Hematol. 2004;49:187–198. doi: 10.1016/j.critrevonc.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 194.Yamada E, Tobe T, Yamada H, Okamoto N, Zack DJ, Werb Z, Soloway PD, Campochiaro PA. TIMP-1 promotes VEGF induced neovascularization in the retina. Histol Histopathol. 2001;16:87–97. doi: 10.14670/HH-16.87. [DOI] [PubMed] [Google Scholar]
- 195.Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, Davis GE. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J Cell Biol. 2006;175:179–191. doi: 10.1083/jcb.200603176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rettig WJ, Garin-Chesa P, Healey JH, Su SL, Ozer HL, Schwab M, Albino AP, Old LJ. Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer Res. 1993;53:3327–3335. [PubMed] [Google Scholar]
- 197.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]