

Background: Role of microRNAs in angiotensin II-mediated vascular smooth muscle cell (VSMC) dysfunction is unclear.
Results: Angiotensin II up-regulates miR-132 in VSMC. miR-132 induces MCP-1 partly via targeting PTEN, activates CREB, and regulates genes related to cell-cycle and motility.
Conclusion: miR-132/212 is a novel modulator of Ang II actions.
Significance: miRNAs may serve as new drug targets for Ang II-mediated cardiovascular diseases.
Keywords: Angiotensin II, Cardiovascular Disease, Inflammation, MicroRNA, Vascular Smooth Muscle Cells
Abstract
Angiotensin II (Ang II)-mediated vascular smooth muscle cell dysfunction plays a critical role in cardiovascular diseases. However, the role of microRNAs (miRNAs) in this process is unclear. We used small RNA deep sequencing to profile Ang II-regulated miRNAs in rat vascular smooth muscle cells (VSMC) and evaluated their role in VSMC dysfunction. Sequencing results revealed several Ang II-responsive miRNAs, and bioinformatics analysis showed that their predicted targets can modulate biological processes relevant to cardiovascular diseases. Further studies with the most highly induced miR-132 and miR-212 cluster (miR-132/212) showed time- and dose-dependent up-regulation of miR-132/212 by Ang II through the Ang II Type 1 receptor. We identified phosphatase and tensin homolog (PTEN) as a novel target of miR-132 and demonstrated that miR-132 induces monocyte chemoattractant protein-1 at least in part via PTEN repression in rat VSMC. Moreover, miR-132 overexpression enhanced cyclic AMP-response element-binding protein (CREB) phosphorylation via RASA1 (p120 Ras GTPase-activating protein 1) down-regulation, whereas miR-132 inhibition attenuated Ang II-induced CREB activation. Furthermore, miR-132 up-regulation by Ang II required CREB activation, demonstrating a positive feedback loop. Notably, aortas from Ang II-infused mice displayed similar up-regulation of miR-132/212 and monocyte chemoattractant protein-1, supporting in vivo relevance. In addition, microarray analysis and reverse transcriptase-quantitative PCR validation revealed additional novel miR-132 targets among Ang II-down-regulated genes implicated in cell cycle, motility, and cardiovascular functions. These results suggest that miR132/212 can serve as a novel cellular node to fine-tune and amplify Ang II actions in VSMC.
Introduction
Angiotensin II (Ang II)2 exerts various pathophysiological effects in the vessel wall, leading to not only vasoconstriction and hemodynamic effects but also proinflammatory, growth-promoting, and vascular remodeling events (1–3). Ang II activates vascular smooth muscle cells (VSMC) in the vessel wall and induces VSMC proliferation and hypertrophy (4, 5), key functions associated with hypertension, restenosis, and atherosclerosis (6). Moreover, Ang II induces the expression of cytokines such as interleukin-6 (IL-6) (7, 8), monocyte chemoattractant protein-1 (MCP-1) (9) in VSMC and osteopontin (10) in the arterial wall, key players involved in monocyte recruitment, neointimal formation, and atherosclerosis (2, 11).
The role of Ang II-mediated VSMC dysfunction in the pathogenesis of hypertension and atherosclerosis has been extensively studied in cultured VSMC and in vivo models. These diverse effects of Ang II are mostly mediated by the Ang II type 1 receptor (AT1R), leading to the activation of several key signaling pathways, including mitogen-activated protein kinases (MAPKs) (3) and transcription factors NF-κB (7) and cAMP response element-binding protein (CREB) (6). Ang II activates CREB through phosphorylation at the Ser-133 residue in a MAPK-dependent manner, and this is functionally associated with VSMC hypertrophy and inflammation (7, 8). However, the molecular mechanisms involved in Ang II-mediated VSMC dysfunction remain incompletely understood.
Growing evidence suggests that microRNAs (miRNAs) play critical roles in cardiovascular development and disorders (12). miRNAs are non-coding RNA molecules 20∼22 nucleotides in length that can negatively regulate gene expression and affect diverse biological processes. Typically, the seed sequences of mammalian miRNAs base pair with binding sites in the 3′-untranslated region (3′-UTR) of target mRNAs, leading to translational inhibition and/or mRNA degradation of these target genes (13). miRNA biogenesis and maturation begins with transcription of pri-miRNA by RNA polymerase II or III followed by pri-miRNA nuclear cleavage to generate pre-miRNA, which is transported to the cytoplasm and further processed by the RNase Dicer to form mature miRNA. Then these miRNAs are incorporated into the RNA-induced silencing complex and interact with target mRNAs to fine-tune gene regulation under diverse pathophysiological conditions (13). Recent studies with VSMC have identified functional roles for various miRNAs such as miR-143 and miR-145 which regulate VSMC differentiation (14), contractility, and Ang II-induced hypertension (15), miR-21 (16) and miR-31 (17) that regulate VSMC proliferation, and miR-125b that promotes proinflammatory responses in VSMC under diabetic conditions (18). However, the role of miRNAs in Ang II-mediated VSMC dysfunction has not yet been investigated.
In this study we utilized the recent technology of small RNA deep sequencing (smRNA-Seq) to profile Ang II-regulated miRNAs in rat VSMC (RVSMC) and also examined their functional relevance. We observed that Ang II increased the expression of miR-132 and miR-212 cluster (miR-132/212) in RVSMC in vitro and in aortas of Ang II-infused mice in vivo. We demonstrated that phosphatase and tensin homolog (PTEN) is a novel target of miR-132 and that miR-132/212 play key roles in Ang II-induced MCP-1 gene expression in RVSMC. Furthermore, we uncovered a positive feedback loop mechanism between Ang II-induced CREB activation and miR-132 expression in RVSMC. In addition, we observed that the predicted targets of miR-132 that were also down-regulated by Ang II have biological functions related to cell cycle, cell motility, and cardiovascular diseases.
EXPERIMENTAL PROCEDURES
Cell Culture
All animal studies were performed according to Institutional Animal Care and Use Committee-approved protocols. RVSMC were isolated from thoracic aortas of male Sprague-Dawley rats (Charles River) by enzymatic digestion (8) and cultured in M199 medium supplemented with 10% FBS and antibiotics. FACS analysis showed 99.2% cells stained positive for smooth muscle cell specific marker α-actin (data not shown). RVSMC were serum-depleted for 48 h in M199 medium supplemented with 0.2% BSA before Ang II stimulation unless mentioned otherwise.
smRNA-Seq
Total RNA was used for the construction of small RNA libraries, cluster generation, and then deep sequencing (at the Beckman Research Institute Sequencing Core) using the Alternative v1.5 Protocol (Illumina Inc., San Diego, CA) with minor optimization. Briefly, 0.5 μg of total RNA was ligated to sRNA 3′adapter (AUCUCGUAUGCCGUCUUCUGCUUG) using T4 RNA Ligase 2, truncated (New England BioLabs, Ipswich, MA) at 22 °C for 1 h, and subsequently ligated to the SRA 5′adapter (GUUCAGAGUUCUACAGUCCGACGAUC) with T4 RNA ligase (New England BioLabs, Ipswich, MA) at 20 °C for 1 h. The adaptor-linked RNA was converted to single-stranded cDNA using Superscript II reverse transcriptase (Invitrogen) and RT-Primer (5′-CAAGCAGAAGACGGCATACGA-3′) and then amplified with Phusion DNA Polymerase (Finnzymes Thermo Scientific, Pittsburgh, PA) for 12 cycles using primers (5′-CAAGCAGAAGACGGCATACGA-3′; 5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA-3′). PAGE purification was carried out to select small RNAs of 17–52 nucleotides in length. The purified library was quantified using quantitative PCR (qPCR), used for cluster generation on cBot, and sequenced using Genome Analyzer IIx (Illumina).
Data Analysis of smRNA-Seq
SmRNA-Seq data were analyzed as described earlier (18) with some modifications. Raw data in FASTQ format generated from the Illumina pipeline was aligned against Rat Nov. 2004 (rn4) assembly using Novoalign software. The 3′-adapter sequence of the raw reads was first trimmed by Novoalign, and reads with 16 or more bases were aligned to rat genome. For reads aligned to multiple locations on the genome, one aligned region was randomly selected for counting the number of reads (supplemental Table S1) as described (19). Genomic locus of each rat mature miRNA was generated by aligning rat mature miRNA sequences (miRBase v14) to rn4 assembly without allowing mismatches. Repetitive regions were not filtered out. For each sample, an aligned read was annotated to a specific genome feature in the order of mature miRNA, miRNA precursor, rRNA, tRNA, other non-coding RNA, Refseq gene, and intergenic region if it completely fell in the specified region with a 10-bp extension on both ends. Specifically, aligned reads were first annotated to rat mature miRNA (v14, miRbase) genomic loci. The reads that could not be annotated to mature miRNA regions were processed further to annotated pre-miRNA genomic regions (v14). Remaining reads were then annotated to non-coding RNAs other than miRNAs including rRNA and tRNA using a rat non-coding RNA annotation file downloaded from UCSC genome browser. Finally, the reads that were not annotated to any non-coding RNA regions were assigned to Refseq genes, and the remaining reads were defined as intergenic regions. For each sample, the reads corresponding to the mature miRNA genomic loci (including 10-base extension on both ends) were counted to obtain expression levels of total miRNAs (supplemental Table S2). The resulting miRNA expression dataset was further normalized by scaling the total mature miRNA counts in each sample to 8.5 million. Differentially expressed miRNAs induced by Ang II at the indicated time points were selected using the following criteria; (a) reads in at least one sample were above 256 and (b) showed at least a 1.5-fold change between Ang II-treated samples versus untreated.
Cluster Analysis of Differentially Expressed miRNAs
In each sample the raw read counts for each differentially expressed miRNA was scaled by the total aligned miRNA reads and log2-transformed. The transformed data were then mean-centered and subjected to unsupervised hierarchical clustering analysis using Euclidean distance as dissimilarity metric and average linkage.
Analysis of Potential Targets of Differentially Expressed miRNAs
Potential targets of differentially expressed miRNAs were identified using online bioinformatics tools TargetScan Human 5.1 (20), miRanda (21), and Diana-microT V3.0 (22). For each miRNA, only the targets that were predicted by at least two tools and conserved across rat, mouse, and human were selected. We pooled 2067 genes representing potential targets of all the differentially regulated miRNAs. This pooled target gene set was subsequently analyzed by DAVID v6.7 (david.abcc.ncifcrf.gov) to obtain enriched biological processes and Ingenuity pathway analysis (IPA) for potential pathways relevant to cardiovascular functions.
RNA Isolation and Gene Expression
RNA was extracted using miRNeasy columns (Qiagen, Inc., Valencia, CA). Gene expression analysis (miRNA and mRNA) was performed by reverse transcriptase-qPCR using the miScript Reverse Transcription kit and SYBR Green PCR kit (Qiagen) using primer sequences listed in supplemental Table S3. In this method, mature miRNAs present in RNA samples were first polyadenylated by poly(A) polymerase and then reverse-transcribed into cDNA using oligo-dT primer with a universal tag. Subsequently, miRNAs were amplified using specific mature miRNA sequences (designed by us or ordered from Qiagen) as forward primers and the universal primer provided in the kit as the reverse primer in the qPCR reaction. Expression levels of pri-miRNA of miR-132/212 were analyzed by Taqman pri-miRNA assay (Applied Biosystems, Carlsbad, CA).
Western Blotting
Preparation of protein lysates and Western blotting were performed as described earlier with some modifications (8, 18). Antibodies against Ser-133-phosphorylated CREB (pCREB), total CREB, and PTEN were purchased from Cell Signaling Technology (Beverly, MA). The RASA1 antibody was purchased from R&D systems (Minneapolis, MN).
VSMC Transfection
Transfection of VSMC was performed using Nucleofector II (Lonza, Basel, Switzerland), according to the manufacturer's instructions. RVSMC were trypsinized and resuspended in Basic Nucleofecton Solution for mammalian smooth muscle cells at 1 × 107 cells/ml. Next, 100 μl of cell suspension (1 × 106 cells) was mixed with miRNA mimic, 2′-O-methyl-modified hairpin inhibitor oligonucleotides, ON-TARGET plus siRNA (Dharmacon products, Thermo Fisher Scientific Inc., Waltham MA), or plasmids as indicated, and electroporated in a Lonza-certified cuvette with Nucleofector II device using the Nucleofector program D-33 optimized for RVSMC (optimization data not shown). In some experiments VSMC were transfected with plasmids expressing miR-132/212-resistant PTEN (plasmid 10750, Addgene, Cambridge, MA) (23) or RASA1 (24).
VSMC were also transfected using X-tremeGENE siRNA Transfection Reagent (Roche Applied Science) for luciferase assays according to the manufacturer's protocol. Transfected cells were harvested for RNA, protein, and luciferase assays as described (18) at the indicated times.
Transduction of VSMC with Lentiviruses
The lentiviral particles were generated in the Vector Laboratory at the Beckman Research Institute. Briefly, HEK 293T cells at a density of 4 × 106/10-cm culture dish were transfected with pCMV-G (containing the VSV-G gene), pCgp (containing the HIV-1 gag and pol genes), and pCMV-rev-2 (containing the rev gene) and precursor miRNA expression plasmid or miRNA inhibitor expression plasmid (Genecoepia, Rockville, MD) using calcium phosphate co-precipitation. Virus-containing supernatant was harvested at 24 and 36 h after transfection, and the titer was determined in HT1080 cells by flow cytometry analysis of respective fluorescence markers (25). To transduce RVSMC with lentiviral vectors expressing miRNA precursors and inhibitors, lentiviral particles were added to cells in the medium containing Polybrene (5 μg/ml), and medium was changed 24 h later.
Construction of 3′-UTR Reporter Plasmids
3′-UTRs of potential miR-132/212 targets were amplified from rat genomic DNA using specific primers and cloned downstream of firefly luciferase gene into ApaI and XbaI sites of pZeo/luc vector (26) or XhoI and NotI sites of psiCHECK-2 vector (Promega, Madison, WI). Cloned 3′-UTR fragments were verified by DNA sequencing. The PTEN 3′-UTR region was amplified using primers 5′-CTGCTCGAGCCTAGAGATTGATGATGCGTCCTCAG-3′ and 5′-GTAGCGGCCGCCAGGTAGGAGTCAACTCTGCCAAATAC-3′. RASA1 3′-UTR region was amplified using primers 5′-CATTCTAGAGCACACCTTTCCACATTCCAGTGATG-3′ and 5′-CACGGGCCCATACCTTCCTTTATACCTCCCTTGGTG-3′.
Ang II Infusion
Male C57BL/6J mice (from The Jackson Laboratory, Bar Harbor, ME) were implanted with Alzet osmotic mini-pumps (Model 2004) filled with either PBS or Ang II (2.5 μg/min/kg) for 2 weeks. At the end of the experiment, aortas were collected, and adventitia was carefully removed before subsequent RNA extraction and gene expression analysis by RT-qPCR.
Affymetrix Gene Array Experiments and Data Analysis
Microarray hybridization and data acquisition were carried out by the Functional Genomics and Bioinformatics Cores at the Beckman Research Institute of City of Hope. Briefly, biotinylated cDNA derived from total RNA was hybridized with Affymetrix Rat Gene 1.0 ST arrays representing 27,342 well characterized genes. Three independent biological replicates were performed for each culture condition. Raw intensity data in CEL file format were imported to Partek Genomics Suite (Partek Inc., St. Louis, MO) and then preprocessed and normalized by Robust Multichip Average method. Differentially expressed genes at different time points in RVSMC stimulated with Ang II relative to control (untreated) were subsequently identified using analysis of variance method in Partek.
Statistical Analysis
PRISM software (Graphpad, San Diego, CA) was used for data analysis. All data shown are the means ± S.E. p < 0.05 was regarded as statistically significant based on Student's t-tests (between two groups) or analysis of variance (between multiple groups).
RESULTS
Identification of Ang II-regulated miRNAs Using High Throughput smRNA-Seq
Evidence shows that various miRNAs play key roles in regulating VSMC differentiation and proliferation, but the involvement of miRNAs in Ang II-mediated effects in VSMC is still largely unknown. To identify differentially expressed miRNAs in response to Ang II in cultured RVSMC, we performed smRNA-Seq (22–30 nucleotide) with small RNA libraries generated using total RNA extracted from untreated (control) or Ang II-stimulated (100 nm) RVSMC at various time points. We identified several differentially expressed miRNAs using the stringent criteria that each miRNA has more than 256 reads in at least one sample, and the normalized reads show a more than 1.5-fold change after Ang II treatment compared with the control group (Fig. 1A). Ang II treatment triggered changes in some candidate miRNAs including up-regulation of miR-132, miR-212, miR-129, miR-21*, and miR-7a that were further validated by RT-qPCR (Fig. 1B). The levels of miR-145, known to be highly expressed in VSMC (14), were not significantly altered by Ang II (Fig. 1B).
FIGURE 1.

Identification of Ang II-regulated miRNAs in RVSMC using smRNA-Seq. A, unsupervised hierarchical clustering of differentially expressed miRNAs (>1.5-fold) in VSMC treated with Ang II (100 nm) at the indicated time points compared with Ctrl samples is shown. For each miRNA, red indicates high expression, and green indicates low expression relative to the average of all samples. RNA isolated from Ctrl and Ang II-treated samples was subjected to smRNA-Seq analysis as described under “Experimental Procedures.” B, RT-qPCR validation of some differentially expressed miRNAs in RVSMC treated with Ang II for 24 h is shown. Results were expressed as % of Ctrl (*, p < 0.05; **, p < 0.01; ***, p < 0.001, n = 6). C, a pie chart shows relative distribution of the Biological Processes highly enriched among predicted target genes of Ang II-regulated miRNAs identified by DAVID v6.7 (57) analysis (modified Fisher exact test, p < 0.05; actual values are shown). D, shown is IPA analysis of potential signaling pathways enriched among predicted targets of Ang II-regulated miRNAs.
Because each miRNA target prediction algorithm (TargetScan, miRanda, and Diana-microT V3.0) has its own advantages, targets theoretically predicted by multiple bioinformatics software and having conserved seed sequences are more likely to be true targets. We, therefore, used these bioinformatics tools and selected the targets that were predicted by at least two databases and conserved across rat, mouse, and human. These selected targets (2067 genes) were pooled and subjected to further bioinformatics analysis to identify biological processes and signaling networks regulated by them. Gene Ontology analysis by DAVID revealed significant enrichment of biological processes such as cell proliferation, cell migration, and cell adhesion, relevant to inflammatory and cardiovascular diseases (Fig. 1C). Furthermore, Ingenuity pathway analysis identified key signaling pathways including ERK/MAPK signaling and PTEN and CREB signaling, known to be involved in Ang II-mediated VSMC dysfunction (Fig. 1D). These results showed that Ang II can regulate several miRNAs in RVSMC and that their targets may have potential functional roles in Ang II-induced effects in VSMC.
Ang II Up-regulates miR-132 in RVSMC
Among the Ang II-induced miRNAs that were validated from the smRNA Seq data, miR-132 and miR-212 were the most highly up-regulated. These miRNAs are reported to be expressed as a highly conserved cluster in vertebrates and can modulate diverse functions in different cell systems. Therefore, we further investigated the functional roles of these miRNA in VSMC and Ang II actions. Time course experiments showed that miR-132/212 levels started to increase as early as 3 h after Ang II stimulation and remained elevated up to 24 h after stimulation compared with control (Ctrl) RVSMC (Fig. 2A). Dose-response experiments showed that Ang II potently induced both the miRNAs at as low as 1 nm that remained elevated up to 10,000 nm (Fig. 2B). In all subsequent experiments, RVSMC were stimulated with Ang II at a concentration of 100 nm unless otherwise indicated. Furthermore, pretreatment with AT1R blocker Losartan (10 μm) markedly blocked Ang II-induced miR-132/212 in RVSMC (Fig. 2C), demonstrating the role of AT1R activation.
FIGURE 2.

Up-regulation of miR-132 and miR-212 by Ang II in RVSMC. A, levels of miR-132 and miR-212 were analyzed by RT-qPCR in Ctrl or Ang II-treated (100 nm) RVSMC at the indicated time points, and results are expressed as % of Ctrl (**, p < 0.01; ***, p < 0.001, n = 8). B, expression of miR-132/212 in RVSMC treated with Ang II at the indicated concentrations for 6 h was determined by RT-qPCR. Results are shown as % of Ctrl (**, p < 0.01; ***, p < 0.001, n = 6). C, levels of miR-132 and miR-212 in RVSMC pretreated with vehicle or Losartan (10 μm) for 30 min and then treated with Ang II (100 nm) at indicated time points were determined by RT-qPCR. Data are from one experiment performed in duplicate.
We also examined whether the increased expression of these miRNAs in response to Ang II could be due to increases in primary transcript levels. We found that Pri-miR-132/212 levels were markedly up-regulated (by >20-fold) in response to Ang II. The levels increased by 1 h after Ang II stimulation and remained elevated up to 24 h, suggesting the involvement of transcriptional regulation in RVSMC (supplemental Fig. S1).
Because miR-132 and miR-212 share the same seed sequence (AACAGUC), indicating they have the same targets, and mature miR-132 is more abundant than miR-212 in RVSMC, we examined the functions and targets of this cluster mostly through modulating miR-132 levels in subsequent studies.
miR-132 Targets PTEN
TargetScan algorithm predicted four miR-132/212 binding sites in the 3′-UTR of PTEN in rat (Fig. 3A), and three of them were highly conserved in human and mouse, suggesting PTEN could be a potential target of miR-132. Because PTEN can have several protective effects in VSMC (27, 28), we examined whether Ang II and miR-132 could down-regulate PTEN in RVSMC. Results showed that Ang II significantly inhibits the expression of the putative miR-132 target PTEN in RVSMC (Fig. 3B). Next, we observed that transfection of RVSMC with miR-132 mimic (132-M) oligonucleotides also significantly down-regulated both PTEN mRNA (Fig. 3C) and protein (Fig. 3D) levels relative to those transfected with control mimic (NC-M) oligonucleotides. To verify if PTEN is a direct target of miR-132, we cloned the PTEN 3′-UTR containing four miR-132 binding sites (nucleotides 1119–2972) downstream of firefly luciferase reporter and co-transfected it along with 132-M or NC-M oligonucleotides into RVSMC. Luciferase assays were performed 48 h post-transfection. Results showed significantly reduced luciferase activity in 132-M-transfected cells compared with NC-M (Fig. 3E). In contrast, co-transfection with miR-132 hairpin inhibitor oligonucleotides (132-I) that inhibit miR-132 increased PTEN 3′-UTR activity relative to control inhibitor NC-I (Fig. 3F). These results clearly demonstrate that PTEN is a direct and novel target of miR-132 in RVSMC.
FIGURE 3.

miR-132 directly targets PTEN in RVSMC. A, shown is sequence alignment of four potential miR-132 binding sites in rat PTEN 3′-UTR predicted by TargetScan. Nucleotide locations of the four binding sites are shown. B, PTEN mRNA levels were determined by RT-qPCR in Ctrl and Ang II-treated RVSMC at the indicated time points. Results were expressed as % of Ctrl (**, p < 0.01; ***, p < 0.001, n = 6). C, PTEN mRNA levels in RVSMC transfected with NC-M or 132-M. Data were expressed as % of NC-M (**, p < 0.01, n = 6). D, shown are representative immunoblots of PTEN protein in RVSMC transfected with NC-M or 132-M in duplicate samples. β-Actin was used as the internal control. The bar graph (right panel) shows quantification of PTEN protein levels (**, p < 0.01, n = 6). E and F, shown is relative activity of luciferase (luc) reporter with PTEN 3′-UTR containing miR-132 binding sites co-transfected with the indicated mimic (E) or inhibitor (F) oligonucleotides in RVSMC. Results were shown as % of respective NC oligonucleotides (***, p < 0.001, n = 8).
miR-132 Enhances MCP-1 Gene Expression in Part through PTEN Inhibition
Next, we tested the functional relevance of PTEN down-regulation. Previous studies in mouse VSMC showed that PTEN knockdown increased the expression of MCP-1 (29), which is also known to be induced by Ang II in VSMC. In addition, PTEN overexpression was reported to suppress Ang II-induced MCP-1 mRNA expression (30). Therefore, we next examined the effect of miR-132 overexpression on MCP-1 gene expression. Results showed that MCP-1 expression was significantly increased in RVSMC transfected with 132-M oligonucleotides relative to cells transfected with NC-M (Fig. 4A). Our data also showed increased levels of MCP-1 mRNA in RVSMC transduced with lentiviral vectors expressing precursor miR-132 (LV-132-M) relative to the control vector (LV-Scr-M) (Fig. 4B). In contrast, inhibition of endogenous miR-132 by lentiviral-expressed anti-miR-132 (LV-132-I) significantly blocked both basal and Ang II-induced MCP-1 mRNA levels relative to control vector (LV-Scr-I) (Fig. 4C), further confirming the role of miR-132 in MCP-1 expression. Moreover, PTEN gene silencing by specific siRNAs (siPTEN) (Fig. 4D) (mimicking miR-132 actions) indeed significantly increased MCP-1 expression relative to control non-targeting oligonucleotide (siNTC) in RVSMC (Fig. 4E). In contrast, the overexpression of PTEN without its 3′-UTR (Fig. 4F) significantly suppressed 132-M-induced MCP-1 mRNA expression (Fig. 4G), further supporting the role of PTEN in miR-132-induced regulation of MCP-1. Together, these data suggest that the down-regulation of PTEN by miR-132 could be one of the mechanisms involved in augmenting MCP-1 expression in Ang II-treated RVSMC.
FIGURE 4.

miR-132 regulates MCP-1 mRNA, partly through PTEN down-regulation. A, MCP-1 mRNA levels in RVSMC transfected with control (NC-M) and miR-132 mimic (132-M) were determined by RT-qPCR 48 h post-transfection. Results were expressed as % of NC-M (***, p < 0.001, n = 6). B, MCP-1 mRNA levels in RVSMC transduced with lentiviral vector expressing pre-miR-132 (LV-132-M) or control scramble sequence (LV-Scr-M) were determined by RT-qPCR, and results are shown as % of LV-Scr-M (***, p < 0.001, n = 6). C, inhibition of MCP-1 mRNA expression in basal and Ang II-stimulated (12 h) RVSMC transduced with lentiviral vector expressing anti-miR-132 (LV-132-I) relative to those transduced with scrambled sequence (LV-Scr-I). *, p < 0.05; **, p < 0.01, n = 6). D and E, PTEN knockdown increases MCP-1 expression. RVSMC transfected with siRNAs targeting PTEN (siPTEN) or non-targeting control (siNTC) were cultured in complete medium for 24 h and serum-depleted for 48 h. Cell lysates were immunoblotted with indicated antibodies (D), and total RNA was used to analyze MCP-1 mRNA expression by RT-qPCR (E). Results were expressed as % of siNTC (**, p < 0.01, n = 6). F and G, PTEN overexpression reduces MCP-1 levels. RVSMC were transfected with PTEN expression plasmid or control plasmid pSG5L with or without 132-M as indicated. 48 h later cell lysates were immunoblotted with indicated antibodies (F), whereas total RNA was extracted to determine MCP-1 mRNA levels by RT-qPCR (G). Results were expressed as % of NC-M+ pSG5L (***, p < 0.001, n = 6).
miR-132 and CREB Form Positive Regulatory Feedback Loop
Activation of CREB through MAPK-mediated Ser-133 phosphorylation plays an important role in Ang II-induced VSMC dysfunction, including hypertrophy, extracellular matrix gene expression, IL-6 up-regulation, and vascular remodeling (8, 31–33). Evidence shows that miR-132 could increase CREB phosphorylation by targeting inhibitors of upstream Ras-Raf1-ERK signaling, including ras GTPase-activating protein 1 (RASA1 or p120RasGAP), in rat neuronal cells and HEK293 cells (34). RASA1 was also validated as a miR-132 target in endothelial cells, and this was associated with vascular endothelial growth factor-induced angiogenesis (24). Therefore, we tested if miR-132 can also regulate CREB activity in RVSMC. We first observed that transfection with 132-M significantly increased phospho-CREB (pCREB) levels without changes in the internal controls including total CREB (tCREB) and β-actin (Fig. 5A, left panel). Next, we examined the effect of miR-132 inhibition on pCREB levels. Results showed a significant reduction in Ang II-induced pCREB levels in RVSMC transfected with a mixture of 132-I and 212-I relative to RVSMC transfected with NC-I (Fig. 5B, left panel). The ratio of pCREB to β-actin determined by a calibrated densitometer is shown in the right panels (Fig. 5, A and B). Fig. 5C shows the conserved miR-132 and miR-212 binding sites in the rat RASA1 3′-UTR. Interestingly, both RASA1 mRNA and luciferase activity of the reporters containing rat RASA1 3′-UTR (nucleotides 117–1308) were also significantly down-regulated by 132-M (Fig. 5, D and E, respectively), supporting RASA1 as a target of miR-132 also in RVSMC. To further verify the role of RASA1 in CREB phosphorylation, we next examined whether expression of RASA1 without the miR-132/212 binding sites can attenuate Ang II effects on CREB activation. Results showed that overexpression of a miR-resistant RASA1 construct lacking its 3′-UTR (24) decreased Ang II-induced pCREB levels without affecting the internal controls (Fig. 5F). Together, these data demonstrate that miR-132 can amplify CREB activation and enhance Ang II functions in RVSMC.
FIGURE 5.

miR-132 and CREB form a positive feedback loop. A, shown are representative immunoblots of total cell lysates from RVSMC transfected with NC-M or 132-M for 18 h probed with indicated antibodies. The bar graph on the right shows quantification of relative pCREB levels expressed as % of NC-M (*, p < 0.05, n = 6). B, shown are representative immunoblots of cell lysates from RVSMC transfected with 132-I + 212-I or NC-I for 18 h, serum-depleted for 24 h, and treated with Ang II (15 min), probed with the indicated antibodies. The bar graph on the right shows quantification of relative pCREB levels expressed as % of NC-I basal (**, p < 0.01, n = 4). C, shown is alignment of Rat RASA1 3′-UTR with miR-132 and miR-212 as predicted by TargetScan. D, RASA1 mRNA levels in RVSMC transfected with NC-M or 132-M are shown. Data are expressed as % of NC-M (***, p < 0.001, n = 6). E, relative luciferase activity of rat RASA1 3′-UTR in RVSMC co-transfected with NC-M or 132-M (**, p < 0.01, n = 8). F, shown are representative immunoblots of total cell lysates from RVSMC transfected with miR-resistant RASA1 constructs or EGFP as control plasmid for 24 h, serum-depleted for 24 h, treated with Ang II (15 min), and probed with indicated antibodies. G, CREB gene silencing in RVSMC is shown. RVSMC were transfected with control non-targeting (siNTC) and CREB siRNA (siCREB) oligonucleotides and 72 h later stimulated with Ang II for 15 min. Cell lysates were immunoblotted with indicated antibodies. Data are representative of three independent experiments. H, shown is expression of miR-132 and miR-212 in RVSMC transfected with the indicated oligonucleotides under basal conditions or after stimulation with Ang II (100 nm) for 6 h. Results were expressed as % of siNTC basal (**, p < 0.01; ***, p < 0.001, n = 6).
Both miR-132 and miR-212 are transcribed from an intergenic region on rat chromosome 10 and regulated by CREB in neurons (35). However, it is not known whether CREB activation plays a role in Ang II-induced miR-132 up-regulation in VSMC. To examine this, we knocked down CREB protein levels by transfection with siRNAs targeting CREB (siCREB) relative to non-targeting control oligonucleotides (siNTC) (Fig. 5G, tCREB). Levels of pCREB were also reduced (Fig. 5G, pCREB). Furthermore, CREB down-regulation by siCREB led to significant inhibition of both basal and Ang II-induced expression of mature miR-132 and miR-212 (Fig. 5H). Some minimal residual Ang II effects may due to either the incomplete knockdown of CREB or that other transcription factors are also involved in up-regulating miR-132 expression (36). These results demonstrate that CREB activation is required for miR-132/212 up-regulation by Ang II and also the operation of a positive feedback loop that can amplify miR-132 expression and CREB activation in RVSMC.
Ang II Infusion Increases miR-132/212 in Vivo in Mice Aorta
We next examined the in vivo relevance using aortas from Ang II-infused mice. Alzet miniosmotic pumps were implanted subcutaneously in male C57BL/6J mice to deliver Ang II or the vehicle PBS for a 2-week period according to reported protocols (37). At the end of the infusion period, aortas were collected from both PBS- and Ang-II infused mice, adventitia were removed, and gene expression was examined by RT-qPCR. Results showed that the expression of miR-132 (Fig. 6A) and miR-212 (Fig. 6B) as well as the downstream inflammatory marker MCP-1 (Fig. 6C) were significantly increased in aortas from Ang II-infused mice compared with PBS treated mice. Furthermore, expression of the miR-132 target PTEN showed a decreased trend that was, however, not statistically significant (Fig. 6D). Overall, these results suggest that Ang II infusion can significantly induce miR-132/212, enhance MCP-1, and attenuate PTEN gene expression, supporting the in vivo relevance.
FIGURE 6.

Regulation of miR-132 and miR-212 in the aortas of Ang II-infused mice. A–D, expression of indicated genes in aortas derived from PBS (Ctrl, n = 7) or Ang II-infused mice (n = 10) was analyzed by RT-qPCR and expressed as % of Ctrl (data are from two independent infusion experiments; ***, p < 0.001). Male C57BL/6J mice were implanted with miniosmotic pumps to deliver vehicle (PBS) or Ang II (2.5 μg/min/kg) and 2 weeks later aortas were collected, adventitia was removed, and gene expression was analyzed by RT-qPCR.
Unbiased Microarray Screening Reveals That Subset of Genes Down-regulated by Ang II in RVSMC Are Predicted Targets of miR-132
To determine if Ang II induced up-regulation of miR-132 can affect its target genes related to VSMC functions or cardiovascular disease, we next adopted a more unbiased profiling approach. Because mammalian miRNAs can function through repression of mRNA levels (38), we performed Affymetrix transcriptome profiling of RNA obtained from RVSMC treated with Ang II for various time periods at which miR-132 was also up-regulated. Then we selected putative miR-132 targets predicted by TargetScan (context score <−0.20, pCT > 0.20) from genes down-regulated by Ang II (p < 0.05, −fold change >1.2) (Fig. 7A) as they are likely to be directly repressed by miR-132 in response to Ang II. This approach revealed that 49 potential miR-132 target genes were down-regulated in Ang II-treated RVSMC (Fig. 7B). RASA1 is not represented in this heatmap because it was decreased by 17% in the original microarray, whereas for this comparison our selection criteria was at least 20% downregulation by Ang II. IPA was next used to uncover potential biological processes mediated by these 49 down-regulated genes. Results showed that these miR-132 targets could be associated with processes such as regulation of cell cycle, cell morphology, and cellular movement as well as cardiovascular system development, function, and disease (Fig. 7C). We next tested five genes enriched in these biological functions for further validation by RT-qPCR in both RVSMC treated with Ang II and those transfected with 132-M. Results demonstrated that all these five potential miR-132 targets were significantly down-regulated by Ang II at the indicated time points relative to control cells (Fig. 7D). Furthermore, transfection of 132-M oligonucleotides also attenuated their expression in RVSMC compared with those transfected with control NC-M (Fig. 7E). Together these data suggest that, by targeting multiple genes with key relevant functions, miR-132 may have multiple roles in Ang II-mediated VSMC dysfunction.
FIGURE 7.

A subset of genes down-regulated by Ang II are putative miR-132 targets. A, a flowchart shows the strategy used to identify multiple miR-132 targets in Ang II-treated RVSMC by overlapping TargetScan-predicted miR-132 targets (context score <-0.2, pCT >0.2) with Ang II-down-regulated genes. Expression profiling experiments with Affymetrix arrays were performed in triplicate using RVSMC treated with or without Ang II at the indicated time points (p < 0.05, -fold change >1.2). B, a Heatmap was generated based on the strategy shown in A, listing Ang II-down-regulated genes that are also potential miR-132 targets in RVSMC. The log2 intensity of gene expression levels was subtracted by the mean expression of the corresponding gene in control samples to directly reflect gene expression changes after Ang II treatment. Unsupervised hierarchical clustering was then applied to cluster genes down-regulated by Ang II at different time points. C, IPA analysis of potential Ang II relevant biological functions enriched in genes listed in B is shown. D, shown is validation of down-regulation of a set of novel miR-132 targets by Ang II. RT-qPCR analysis of potential miR-132 targets were identified by microarray in RVSMC stimulated with Ang II at the indicated time points. Results are expressed as -fold over untreated (Ctrl) cells (**, p < 0.01; ***, p < 0.001, n = 6). E, shown is RT-qPCR validation of the repression of biological function-related key Ang II down-regulated genes/miR-132 targets in RVSMC transfected with 132-M. *, p < 0.05; **, p < 0.01; ***, p < 0.001 132-M versus NC-M, n = 6).
DISCUSSION
In this study we used the smRNA-Seq method for the first time to profile Ang II-regulated miRNAs in VSMC and also observed that some of these miRNAs can act as novel mediators and modulators of Ang II actions in VSMC. Importantly, we demonstrated the up-regulation of miR-132/212 cluster by Ang II both in vitro in RVSMC and in vivo in Ang II-infused mice. We also showed that miR-132 could modulate Ang II-mediated MCP-1 gene expression in VSMC at least in part through its novel target PTEN. Moreover, our results demonstrated the role of AT1R and CREB in miR-132/212 regulation and established a positive feedback loop between CREB activation and miR-132/212 expression in RVSMC, potentially through down-regulation of the miR-132 target RASA1 (Fig. 8). Furthermore, we used a more unbiased approach to demonstrate that other potential targets of miR-132 could modulate processes associated with cardiovascular disease functions such as cell cycle and cell motility.
FIGURE 8.
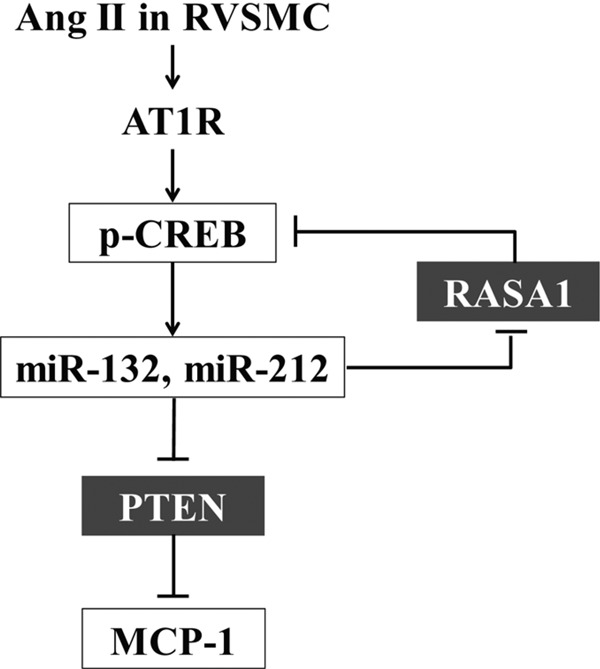
Schematic diagram shows the role of miR-132/212 in Ang II-induced VSMC dysfunction. Ang II signaling through AT1R activates CREB (Ser-133 phosphorylation) and increases levels of miR-132 and miR-212 cluster. Increased levels of miR-132 inhibit the expression of PTEN via targeting its 3′-UTRs. PTEN inhibition increases key proinflammatory gene MCP-1. On the other hand, down-regulation of RASA1, an inhibitor of Ras/Raf/MAPK pathway, leads to activation of CREB, a key regulator of VSMC dysfunction. CREB activation can also promote a positive feedback mechanism by further increasing miR-132 and miR-212 levels.
Recent advances in next generation sequencing technologies have led to high throughput and robust approaches such as smRNA-Seq that can quantify miRNA expression genome-wide and also lead to the discovery of new miRNAs. These approaches have provided highly useful and novel information with human and mouse cells. However, similar analysis in rat cells has been difficult because the rat genome annotation is still relatively incomplete. Our studies provide the first smRNA-Seq data base catalogue of RVSMC. We observed that Ang II can differentially regulate several miRNAs in RVSMC, with miR-132/212 being the most highly induced. Our smRNA-Seq data also showed that miR-143/145 cluster, miR-21, and let-7 family members are the most abundant miRNAs in RVSMC, similar to that reported in mouse and human VSMC (14–16, 39), but Ang II treatment in vitro did not affect their expression significantly (<1.5-fold changes). However, this does not rule out the possibility that in vivo these miRNAs may contribute to Ang II-mediated vascular dysfunction by fine-tuning target gene expression (12).
Bioinformatics approaches indicated that the predicted targets of differentially expressed miRNAs could be enriched in several well known Ang II-regulated biological processes and signaling pathways. DAVID analysis suggested that these predicted targets of Ang II-regulated miRNAs are likely to be involved in cell proliferation, migration, and translation regulation processes (Fig. 1C) relevant to VSMC hyperplasia and hypertrophy associated with hypertension, restenosis, and atherosclerosis. Several signaling pathways activated by Ang II contribute to VSMC proliferation, hypertrophy, and migration, including tyrosine kinases (6), MAPKs, Akt, and CREB (6, 32). IPA analysis of Ang II-responsive miRNA targets revealed enrichment of several of these pathways including ERK/MAPK, PI3K/Akt, PTEN, and CREB signaling (p < 1 × 10−7), supporting our hypothesis that these miRNAs can regulate VSMC dysfunction. This was further supported by experimental evidence that miR-132 could activate CREB and down-regulate RASA1 and PTEN, key signaling molecules associated with aberrant VSMC gene expression and signaling. Further experimental studies are needed to confirm the functional roles of other Ang II-regulated miRNAs and their predicted targets.
In this study we focused on the conserved miR-132/212 cluster as they were highly induced by Ang II in vitro and in vivo. Moreover, although the functions of these miRNAs have been demonstrated in different biological contexts such as nervous system, endocrine system, innate immunity, and vascular endothelium, their role in VSMC is not known. These miRNAs are transcribed from an intergenic region on rat chromosome 10 and have been shown to be regulated by upstream CREB binding sites in neuronal cells (35). Our studies demonstrated that CREB knockdown decreased Ang II-induced miR-132/212 expression, suggesting a direct role for CREB in mature miR-132/212 cluster regulation by Ang II. Furthermore, miR-132 in turn could activate CREB, demonstrating a positive feedback loop between CREB and miR-132/212 in RVSMC (Figs. 5 and 8). A similar regulatory mechanism was also recently implicated in cocaine-stimulated neurons (34). Ang II-induced CREB activation plays a key role in inflammatory and extracellular matrix gene expression and hypertrophy (8, 31–33). Thus, miR-132/212 could play an important role in these processes by amplifying CREB signaling. Evidence also shows a key role for CREB in VSMC phenotypic modulation (40); however, the role of miR-132/212-induced CREB activation in these events awaits further investigation.
Reports show that miR-132 has diverse effects on inflammatory responses in different cell types. These include inhibition of inflammatory gene expression by targeting proinflammatory acetylcholinesterase in neurons (41) and coactivator histone acetyl transferase p300 in monocytes (42). In contrast, miR-132 enhanced inflammatory gene expression through targeting Sirtuin1, a histone deacetylase that inhibits NF-κB activation in preadipocytes and adipocytes (43). We observed that miR-132 mimic increased MCP-1 expression in VSMC, whereas its inhibitor blocked Ang II-induced MCP-1 expression. However, miR-132 did not have a significant effect on another proinflammatory cytokine IL-6, suggesting some level of specificity. MCP-1 is a major inflammatory mediator and chemoattractant (11) that also promotes VSMC migration and proliferation (44, 45). Ang II-induced MCP-1 promotes the accumulation of macrophages and lymphocytes in the atherosclerotic lesions of animal models (46, 47). We also identified PTEN as a direct target of miR-132. Furthermore, PTEN gene silencing by specific siRNAs increased MCP-1 expression, whereas its overexpression inhibited both Ang II- and miR-132-induced MCP-1, suggesting PTEN down-regulation as one of the mechanisms involved in miR-132-induced MCP-1 expression. However, increases in MCP-1 expression by PTEN gene silencing was much lower than that induced by miR-132 transfection, suggesting that other pathways such as NF-κB activation through targeting Sirtuin 1 (43) and RASA1 might also be involved. Previous studies showed that PTEN gene silencing or VSMC-specific PTEN knockdown increased inflammatory gene expression, neointima formation, growth, migration, and vascular remodeling (27, 29, 30, 48). Thus, miR-132 may augment Ang II-mediated VSMC dysfunction by modulating some of these processes. Hydrogen peroxide (H2O2), a downstream effector of Ang II actions (49), plays a key role in Ang II-induced MCP-1 expression in VSMC (9). Furthermore, H2O2 directly stimulates CREB activation (50) and increases miR-132 (51) in VSMC. Based on our data that CREB is required for Ang II-induced miR-132 transcription, it is possible that Ang II-generated H2O2 might also promote MCP-1 expression through increasing miR-132 in RVSMC.
In vivo relevance was supported by our observations that Ang II infusion could significantly induce miR-132/212 and enhance MCP-1 expression in mice aortas. Under the same conditions, the miR-132/212 target PTEN was also attenuated, although not statistically significant. This may be due to the fact that, besides smooth muscle cells, aortas also contain other cell types. Because miRNA effects are known to be cell type-specific, it is possible that PTEN was not efficiently targeted by miR-132/212 in cell types other than VSMC.
We also identified additional novel miR-132 targets by microarray profiling of Ang II down-regulated genes in RVSMC coupled with bioinformatics analysis. This method is advantageous because it can identify multiple putative VSMC-specific miR-132 targets among the down-regulated genes in an unbiased manner. However, we might have also missed some true miR-132 targets due to the stringent criteria used as well as other mechanisms such as translation inhibition of target mRNAs. Interestingly, IPA analysis of the miR-132 targets identified in this comparative analysis indicated their potential regulatory roles in cell growth, morphology, and movement (functions related to Ang II effects in VSMC and cardiovascular disease). Similar to PTEN, Foxo3 (52) was reported to inhibit VSMC cell cycle progression and, hence, neointimal hyperplasia. Moreover, Zeb2 (53), Arhgef11 (54), Sox4 (55), and Ssh2 (56) have been shown in other systems to modulate cell cycle, cellular apoptosis, or morphological changes, which are again Ang II-related biological processes. Therefore, biological functions mediated by these potential miR-132 targets are worthy of further investigations in the future.
In summary, we have demonstrated a new role for miRNAs in Ang II actions and in particular the up-regulation of miR-132 and miR-212 by Ang II in vitro and in vivo as well as the role of miR-132 in key Ang II responses including CREB activation and inflammatory gene expression. We also demonstrated a positive feedback loop between CREB signaling and miR-132, suggesting that this circuitry may fine-tune and amplify Ang II-mediated gene expression (Fig. 8). Because miRNAs generally regulate multiple genes, miR-132 and miR-212 up-regulation could have profound effects on several genes involved in VSMC dysfunction and thus serve as potential therapeutic targets for Ang II-mediated cardiovascular diseases.
Supplementary Material
Acknowledgments
We thank Dr. Ali Ehsani and Dr. Amy Leung for helpful discussions and Mei Wang for technical assistance. We are extremely grateful to Dr. Harry Gao, Charles Warden, Dr. Zheng Liu, and Dr. Yate-Ching Yuan (Beckman Research Institute Sequencing and Functional Genomics Cores) for tremendous help with the smRNA-Seq and microarray work. We also appreciate the help from Dr. Jiing-Kuan Yee and the Vector Laboratory for the lentiviral vector preparation. We thank Merck & Co., Inc. (Whitehouse Station, NJ) for the gift of Losartan, Dr. David Cheresh and Dr. Anand Sudarshan (Uinversity of California, San Diego) for the gift of miRNA-resistant RASA1 plasmid, Dr. William Sellers' help for providing the PTEN plasmid and its control plasmid through Addgene, Debra Rateri (University of Kentucky, Lexington) for advice on the Ang II infusions in mice, and Dr. Mary Weiser-Evans (University of Colorado, Denver, CO) for help with the PTEN vector.
This work was supported, in whole or in part, by National Institutes of Health Grants NIH RO1 HL087864 and NIH RO1 HL106089 (NHLBI; to R. N.).

This article contains supplemental Fig. S1 and Tables S1–S3.
- Ang II
- angiotensin II
- MCP-1
- monocyte chemoattractant protein-1
- CREB
- cAMP responsive element-binding protein
- pCREB
- phosphorylated CREB
- RASA1
- p120 Ras GTPase-activating protein 1 (p120RasGAP)
- PTEN
- phosphatase and tensin homolog
- VSMC
- vascular smooth muscle cell(s)
- RVSMC
- rat VSMC(s)
- miRNA
- microRNA
- AT1R
- Ang II type 1 receptor
- qPCR
- quantitative PCR
- IPA
- ingenuity pathway analysis
- smRNA-Seq
- small RNA deep sequencing
- 132-M
- miR-132 mimic
- NC-M
- control mimic
- 132-I
- miR-132 inhibitor
- Ctrl
- control.
REFERENCES
- 1. Daugherty A., Cassis L. (2004) Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc. Med. 14, 117–120 [DOI] [PubMed] [Google Scholar]
- 2. Marchesi C., Paradis P., Schiffrin E. L. (2008) Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 29, 367–374 [DOI] [PubMed] [Google Scholar]
- 3. Duff J. L., Marrero M. B., Paxton W. G., Schieffer B., Bernstein K. E., Berk B. C. (1995) Angiotensin II signal transduction and the mitogen-activated protein kinase pathway. Cardiovasc. Res. 30, 511–517 [PubMed] [Google Scholar]
- 4. Gibbons G. H., Pratt R. E., Dzau V. J. (1992) Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-β1 expression determines growth response to angiotensin II. J. Clin. Invest. 90, 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Natarajan R., Gonzales N., Lanting L., Nadler J. (1994) Role of the lipoxygenase pathway in angiotensin II-induced vascular smooth muscle cell hypertrophy. Hypertension 23, I142–I147 [DOI] [PubMed] [Google Scholar]
- 6. Mehta P. K., Griendling K. K. (2007) Angiotensin II cell signaling. Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol 292, C82–C97 [DOI] [PubMed] [Google Scholar]
- 7. Han Y., Runge M. S., Brasier A. R. (1999) Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-κB transcription factors. Circ. Res. 84, 695–703 [DOI] [PubMed] [Google Scholar]
- 8. Sahar S., Reddy M. A., Wong C., Meng L., Wang M., Natarajan R. (2007) Cooperation of SRC-1 and p300 with NF-κB and CREB in angiotensin II-induced IL-6 expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 27, 1528–1534 [DOI] [PubMed] [Google Scholar]
- 9. Chen X. L., Tummala P. E., Olbrych M. T., Alexander R. W., Medford R. M. (1998) Angiotensin II induces monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells. Circ. Res. 83, 952–959 [DOI] [PubMed] [Google Scholar]
- 10. Bruemmer D., Collins A. R., Noh G., Wang W., Territo M., Arias-Magallona S., Fishbein M. C., Blaschke F., Kintscher U., Graf K., Law R. E., Hsueh W. A. (2003) Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J. Clin. Invest. 112, 1318–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charo I. F., Taubman M. B. (2004) Chemokines in the pathogenesis of vascular disease. Circ. Res. 95, 858–866 [DOI] [PubMed] [Google Scholar]
- 12. Small E. M., Olson E. N. (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469, 336–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartel D. P. (2009) MicroRNAs. Target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cordes K. R., Sheehy N. T., White M. P., Berry E. C., Morton S. U., Muth A. N., Lee T. H., Miano J. M., Ivey K. N., Srivastava D. (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boettger T., Beetz N., Kostin S., Schneider J., Krüger M., Hein L., Braun T. (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Invest. 119, 2634–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ji R., Cheng Y., Yue J., Yang J., Liu X., Chen H., Dean D. B., Zhang C. (2007) MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ. Res. 100, 1579–1588 [DOI] [PubMed] [Google Scholar]
- 17. Liu X., Cheng Y., Chen X., Yang J., Xu L., Zhang C. (2011) MicroRNA-31 regulated by the extracellular-regulated kinase is involved in vascular smooth muscle cell growth via large tumor suppressor homolog 2. J. Biol. Chem. 286, 42371–42380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Villeneuve L. M., Kato M., Reddy M. A., Wang M., Lanting L., Natarajan R. (2010) Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 59, 2904–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weng L., Wu X., Gao H., Mu B., Li X., Wang J. H., Guo C., Jin J. M., Chen Z., Covarrubias M., Yuan Y. C., Weiss L. M., Wu H. (2010) MicroRNA profiling of clear cell renal cell carcinoma by whole-genome small RNA deep sequencing of paired frozen and formalin-fixed, paraffin-embedded tissue specimens. J. Pathol. 222, 41–51 [DOI] [PubMed] [Google Scholar]
- 20. Friedman R. C., Farh K. K., Burge C. B., Bartel D. P. (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. John B., Enright A. J., Aravin A., Tuschl T., Sander C., Marks D. S. (2004) Human microRNA targets. PLoS Biol. 2, e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maragkakis M., Reczko M., Simossis V. A., Alexiou P., Papadopoulos G. L., Dalamagas T., Giannopoulos G., Goumas G., Koukis E., Kourtis K., Vergoulis T., Koziris N., Sellis T., Tsanakas P., Hatzigeorgiou A. G. (2009) DIANA-microT web server. Elucidating microRNA functions through target prediction. Nucleic Acids Res. 37, W273–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramaswamy S., Nakamura N., Vazquez F., Batt D. B., Perera S., Roberts T. M., Sellers W. R. (1999) Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 2110–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anand S., Majeti B. K., Acevedo L. M., Murphy E. A., Mukthavaram R., Scheppke L., Huang M., Shields D. J., Lindquist J. N., Lapinski P. E., King P. D., Weis S. M., Cheresh D. A. (2010) MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat. Med. 16, 909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yam P. Y., Li S., Wu J., Hu J., Zaia J. A., Yee J. K. (2002) Design of HIV vectors for efficient gene delivery into human hematopoietic cells. Mol. Ther. 5, 479–484 [DOI] [PubMed] [Google Scholar]
- 26. Kato M., Zhang J., Wang M., Lanting L., Yuan H., Rossi J. J., Natarajan R. (2007) MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-β-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. U.S.A. 104, 3432–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang J., Kontos C. D. (2002) Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler. Thromb. Vasc. Biol. 22, 745–751 [DOI] [PubMed] [Google Scholar]
- 28. Chen W. J., Lin K. H., Lai Y. J., Yang S. H., Pang J. H. (2004) Protective effect of propylthiouracil independent of its hypothyroid effect on atherogenesis in cholesterol-fed rabbits. PTEN induction and inhibition of vascular smooth muscle cell proliferation and migration. Circulation 110, 1313–1319 [DOI] [PubMed] [Google Scholar]
- 29. Furgeson S. B., Simpson P. A., Park I., Vanputten V., Horita H., Kontos C. D., Nemenoff R. A., Weiser-Evans M. C. (2010) Inactivation of the tumorsuppressor, PTEN, in smooth muscle promotes a proinflammatory phenotype and enhances neointima formation. Cardiovasc. Res. 86, 274–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koide S., Okazaki M., Tamura M., Ozumi K., Takatsu H., Kamezaki F., Tanimoto A., Tasaki H., Sasaguri Y., Nakashima Y., Otsuji Y. (2007) PTEN reduces cuff-induced neointima formation and proinflammatory cytokines. Am. J. Physiol. Heart Circ. Physiol. 292, H2824–H2831 [DOI] [PubMed] [Google Scholar]
- 31. Funakoshi Y., Ichiki T., Takeda K., Tokuno T., Iino N., Takeshita A. (2002) Critical role of cAMP-response element-binding protein for angiotensin II-induced hypertrophy of vascular smooth muscle cells. J. Biol. Chem. 277, 18710–18717 [DOI] [PubMed] [Google Scholar]
- 32. Ichiki T. (2006) Role of cAMP response element binding protein in cardiovascular remodeling. Good, bad, or both? Arterioscler. Thromb. Vasc. Biol. 26, 449–455 [DOI] [PubMed] [Google Scholar]
- 33. Reddy M. A., Thimmalapura P. R., Lanting L., Nadler J. L., Fatima S., Natarajan R. (2002) The oxidized lipid and lipoxygenase product 12(S)-hydroxyeicosatetraenoic acid induces hypertrophy and fibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMP response element-binding protein activation. Mediation of angiotensin II effects. J. Biol. Chem. 277, 9920–9928 [DOI] [PubMed] [Google Scholar]
- 34. Hollander J. A., Im H. I., Amelio A. L., Kocerha J., Bali P., Lu Q., Willoughby D., Wahlestedt C., Conkright M. D., Kenny P. J. (2010) Striatal microRNA controls cocaine intake through CREB signalling. Nature 466, 197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vo N., Klein M. E., Varlamova O., Keller D. M., Yamamoto T., Goodman R. H., Impey S. (2005) A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 16426–16431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee J., Li Z., Brower-Sinning R., John B. (2007) Regulatory circuit of human microRNA biogenesis. PLoS Comput. Biol. 3, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daugherty A., Cassis L. (1999) Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor −/− mice. Ann. N.Y. Acad. Sci. 892, 108–118 [DOI] [PubMed] [Google Scholar]
- 38. Guo H., Ingolia N. T., Weissman J. S., Bartel D. P. (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng Y., Liu X., Yang J., Lin Y., Xu D. Z., Lu Q., Deitch E. A., Huo Y., Delphin E. S., Zhang C. (2009) MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res. 105, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reusch J. E. (2011) Beyond phosphorylation. Nuclear export in vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 31, 1955–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shaked I., Meerson A., Wolf Y., Avni R., Greenberg D., Gilboa-Geffen A., Soreq H. (2009) MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 31, 965–973 [DOI] [PubMed] [Google Scholar]
- 42. Lagos D., Pollara G., Henderson S., Gratrix F., Fabani M., Milne R. S., Gotch F., Boshoff C. (2010) miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 12, 513–519 [DOI] [PubMed] [Google Scholar]
- 43. Strum J. C., Johnson J. H., Ward J., Xie H., Feild J., Hester A., Alford A., Waters K. M. (2009) MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol. Endocrinol. 23, 1876–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Viedt C., Vogel J., Athanasiou T., Shen W., Orth S. R., Kübler W., Kreuzer J. (2002) Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-κB and activator protein-1. Arterioscler. Thromb. Vasc. Biol. 22, 914–920 [DOI] [PubMed] [Google Scholar]
- 45. Ma J., Wang Q., Fei T., Han J. D., Chen Y. G. (2007) MCP-1 mediates TGF-β-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 109, 987–994 [DOI] [PubMed] [Google Scholar]
- 46. Daugherty A., Manning M. W., Cassis L. A. (2000) Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 105, 1605–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hernández-Presa M., Bustos C., Ortego M., Tuñon J., Renedo G., Ruiz-Ortega M., Egido J. (1997) Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-κB activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 95, 1532–1541 [DOI] [PubMed] [Google Scholar]
- 48. Nemenoff R. A., Simpson P. A., Furgeson S. B., Kaplan-Albuquerque N., Crossno J., Garl P. J., Cooper J., Weiser-Evans M. C. (2008) Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1α. Circ. Res. 102, 1036–1045 [DOI] [PubMed] [Google Scholar]
- 49. Griendling K. K., Minieri C. A., Ollerenshaw J. D., Alexander R. W. (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74, 1141–1148 [DOI] [PubMed] [Google Scholar]
- 50. Ichiki T., Tokunou T., Fukuyama K., Iino N., Masuda S., Takeshita A. (2003) Cyclic AMP response element-binding protein mediates reactive oxygen species-induced c-fos expression. Hypertension 42, 177–183 [DOI] [PubMed] [Google Scholar]
- 51. Lin Y., Liu X., Cheng Y., Yang J., Huo Y., Zhang C. (2009) Involvement of microRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J. Biol. Chem. 284, 7903–7913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Littlewood T. D., Bennett M. R. (2007) Foxing smooth muscle cells. FOXO3a-CYR61 connection. Circ. Res. 100, 302–304 [DOI] [PubMed] [Google Scholar]
- 53. Mejlvang J., Kriajevska M., Vandewalle C., Chernova T., Sayan A. E., Berx G., Mellon J. K., Tulchinsky E. (2007) Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol. Biol. Cell 18, 4615–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Panizzi J. R., Jessen J. R., Drummond I. A., Solnica-Krezel L. (2007) New functions for a vertebrate Rho guanine nucleotide exchange factor in ciliated epithelia. Development 134, 921–931 [DOI] [PubMed] [Google Scholar]
- 55. Hur W., Rhim H., Jung C. K., Kim J. D., Bae S. H., Jang J. W., Yang J. M., Oh S. T., Kim D. G., Wang H. J., Lee S. B., Yoon S. K. (2010) SOX4 overexpression regulates the p53-mediated apoptosis in hepatocellular carcinoma. Clinical implication and functional analysis in vitro. Carcinogenesis 31, 1298–1307 [DOI] [PubMed] [Google Scholar]
- 56. Kligys K., Claiborne J. N., DeBiase P. J., Hopkinson S. B., Wu Y., Mizuno K., Jones J. C. (2007) The slingshot family of phosphatases mediates Rac1 regulation of cofilin phosphorylation, laminin-332 organization, and motility behavior of keratinocytes. J. Biol. Chem. 282, 32520–32528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.