

Abstract
Risk of gastric infection with Vibrio parahaemolyticus increases with favorable environmental conditions and population shifts that increase prevalence of infective strains. Genetic analysis of New Hampshire strains revealed a unique population with some isolates similar to outbreak-causing strains and high-level diversity that increased as waters warmed.
TEXT
Vibrio parahaemolyticus, a prevalent estuarine resident, may cause acute inflammatory gastroenteritis and diarrheal disease following its consumption in raw or undercooked shellfish (5, 33). Food poisoning by V. parahaemolyticus is common in Asia, and incidence in the United States has increased not only where water temperatures are warm, but even in northern regions (2, 4, 5, 31). Typically, V. parahaemolyticus abundance is associated with increased temperature and moderate salinity (6, 8, 26, 29), and disease-causing strains often but not always harbor hemolysin genes (tdh and/or trh) (10). Outbreaks may relate to oceanic anomalies or climate variability, measured as increased temperature and changes in precipitation patterns (19, 23), which highlights the need to understand the ecology of endemic populations.
Previously, the diversity and the extent of recombination among clinical and environmental V. parahaemolyticus populations were examined in three studies using multilocus sequence analysis (MLSA) (3, 11, 32). These studies revealed highly diverse populations resulting from frequent recombination, and in one population, diversity was associated with geography (11). However, the relationship between specific environmental conditions (e.g., temperature and salinity) and strain diversity and the nature of endemic, cold-water populations remain unknown. Of particular interest is whether specific environmental variables may favor strains with greater infective risk. Therefore, we used MLSA to characterize the structure of a northeastern V. parahaemolyticus population in the Great Bay Estuary (GBE) of New Hampshire. Warm summertime water and unique environmental conditions make this site an ideal model for studying environmental change in northern waters and its effect on resident microbial ecology (16, 21).
Our analysis focused on 192 V. parahaemolyticus isolates collected from oysters, sediment, and water from the GBE between May and December from 2007 to 2009 (15, 18) (see Table S1 in the supplemental material). Collection and environmental sampling (to evaluate temperature, salinity, and dissolved oxygen content [25]) were conducted at two sites, one open to recreational shellfishing and another where shellfish fishing is prohibited due to proximity to a wastewater treatment facility. Bacteria were enumerated using a most-probable-number method, and Vibrio species were isolated on selective/differential media by standard methods (17, 20, 21, 25). Species identity and the absence of virulence markers tdh and trh were confirmed for all putative V. parahaemolyticus isolates by multiplex PCR (18, 22) (see Table S1 in the supplemental material).
We applied a seven-locus MLSA devised from two existing schemes to allow integration of our data into multiple MLSA databases and added three conserved virulence-associated genes to investigate whether these genes evolve differently and whether their inclusion in the analysis alters population structure. Target loci included recA, pyrH, and gyrB (24), used to determine genetic relationships more broadly among Vibrio spp., and dnaE, dtdS, pntA, and tnaA (11), from a V. parahaemolyticus scheme applied to both clinical and environmental isolates. Because standard virulence marker genes have no utility for MLSA if they are not present in all strains, we designed primers to amplify three conserved loci associated with the expression of virulence genes: gacA (forward, 5′ GCGTTTCAGCACATATTCATAG; reverse, 5′ TAAAGGGTCTCGGTGTCCA, toxR (forward, 5′ TGACTAACATCGGCACCAAA; reverse, 5′ GCTCAATAGAAGGCAACCAG), and vppC (forward, 5′ TATCAGTACCAACAACGGCGGC; reverse, 5′ CGAAGTCCCACAACACACTGACG). PCR amplicons were generated by standard protocols (25) and sequenced at the Hubbard Center for Genome Studies (Durham, NH) or by Functional Biosciences, Inc. (Madison, WI).
Our analyses indicated that the GBE population is highly diverse in comparison to other previously characterized populations and nearly as diverse as the current worldwide database (see Table S2 in the supplemental material) (11). Neighbor-joining phylogenetic trees were constructed for the entire GBE collection by alignment and concatenation of sequences from sets of both 7 loci (Fig. 1) and 10 loci and from genome sequences available for V. parahaemolyticus outbreak strains (see Table S3 in the supplemental material) using MEGA version 5.0 and ClustalW (25, 28). The mean nucleotide distance (calculated using the Nei-Gojobori method and the Jukes-Cantor correction with 1,000 bootstrap replications) for 7 housekeeping loci indicated a high level of diversity that did not significantly change with the addition of virulence-associated genes (results for the 7- and 10-locus analyses were 0.0131 and 0.0129 nucleotide differences per site, respectively). Analyses of each housekeeping locus by the codon-based Z test of purifying selection reflected purifying selection as expected, but this was also the case for the three virulence-associated genes. Given that ToxR and GacA are global regulators of genes not exclusive to virulence, they too likely experienced purifying selection (27). Of the isolates, 74% had unique genotypes that formed only 20 clonal groups of no more than five isolates each (Fig. 1). However, certain clones and several strain groups with greater than 70% bootstrap support were found in multiple collection years, indicating that the population is endemic and persists in the estuary (Fig. 1; also see Table S1 in the supplemental material). The lack of population structure is consistent with the findings from other MLSA studies of V. parahaemolyticus (11, 30, 32). More notably, isolates from cold water (<11°C) tended to cluster into certain clades (Fig. 1, bold labels), and another clade with ambiguous resolution included pandemic and nonpandemic strains AQ4037 and AQ3810, which we included in our analysis for context (Fig. 1, bootstrap value of 53%).
Fig 1.

Population structure of GBE V. parahaemolyticus isolates and several clinical isolates. A consensus neighbor-joining tree was constructed from seven concatenated housekeeping gene loci including dnaE, dtdS, gyrB, pntA, recA, and tnaA sequences (2,988 bp) by using a Jukes-Cantor model for 192 GBE isolates collected from 2007 to 2009. Clinical V. parahaemolyticus strains RIMD 2210633, AQ3810, AN-5034, and AQ4037 were included for comparison. Cold-water strains (from water at <11°C) are highlighted in larger type. The bar indicates 0.2% divergence, and branches with 70% or greater bootstrap support are labeled.
To further explore effects of seasonality on strain diversity, a linear regression between genotype diversity and water temperature (for strain groups corresponding to similar temperatures) was conducted (Fig. 2), revealing a strong positive relationship (r2 = 0.91). In general, strains cultured from colder waters (1 to 11°C) were less diverse than the overall collection (mean nucleotide distance per site ± standard deviation, 0.0122 ± 0.0019 versus 0.0131 ± 0.0011), suggesting that a cold-tolerant subpopulation was replaced by more diverse strains as waters warmed. Genetic diversity did not correlate with any other environmental factor (i.e., salinity, sampling site, substrate, or year) as determined by linear regression (see Table S4 in the supplemental material). Moreover, neither the clade identified by splits decomposition analysis with SplitsTree version 4.0 (13) (Fig. 3) nor the small GBE clade that was related to outbreak strains (Fig. 1) was associated with a particular habitat or condition. These results suggest that most genotypes in the GBE are ecological generalists and capable of persistence in a wide variety of habitats.
Fig 2.
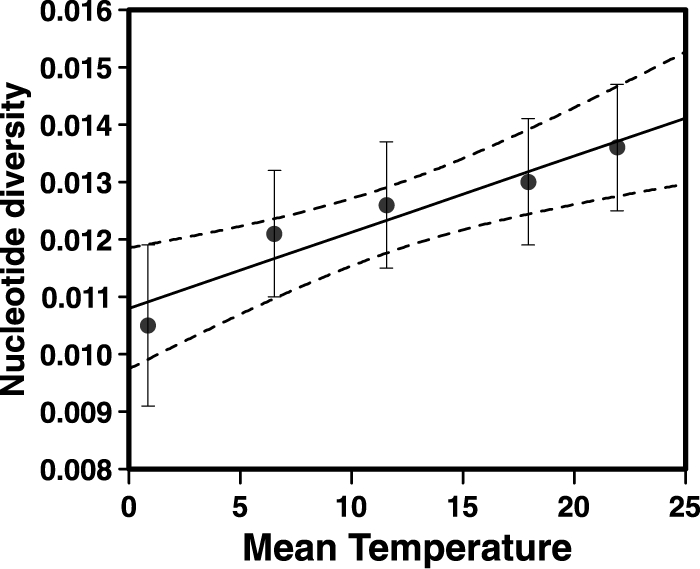
Linear regression of nucleotide diversity of V. parahaemolyticus isolates with temperature. Isolates are grouped by temperature windows of −1 to 4.9°C (n = 4), 5 to 9.9°C (n = 31), 10 to 14.9°C (n = 39), 15 to 19.9°C (n = 50), and 20 to 24.9°C (n = 59), with mean temperature of isolation. Dashed lines reflect the 95% confidence interval of the linear model (F = 32.12; P = 0.011; r2 = 0.91). Error bars indicate standard errors.
Fig 3.

Recombination network revealed by splits decomposition analysis of genotypes of GBE V. parahaemolyticus isolates. The analysis is based on 10 concatenated gene loci including dnaE, dtdS, gyrB, pntA, pyrH, recA, tnaA, toxR, gacA, and vppC sequences (4,805 bp) from 192 isolates. Removal of any one locus did not alter topology.
This population also showed a history of recombination both within and between major clades (Fig. 3), which only partly explains its diversity. Nonredundant allelic profiles were built for both the 7-locus and 10-locus analyses using NRDB Align (14), and these profiles were used to determine the extent of recombination using LIAN 3.5 (12). For loci shared with prior studies (dnaE, pntA, tnaA, recA, and dtdS; reported at http://pubMLST.org/), very few alleles or sequence types (STs) were reported previously. Of tnaA sequences, 48% were identical to database sequences, yet at other loci fewer than 20% of sequences were reported previously. In total, 94% of GBE isolates defined unique STs. Analysis of STs by eBURST (data not shown) revealed only 27 related groups from 192 isolates (9). For both the 7-locus and 10-locus allelic profiles, LIAN tests of recombination revealed that alleles were essentially in linkage equilibrium (index of association [IA] = 0.1721 [P < 0.001] and IA = 0.1659 [P < 0.001], respectively). We found equivalent levels of recombination at all loci, with no significant difference between housekeeping and virulence-associated genes. The reticulate nature of the phylogeny (Fig. 3) produced by SplitsTree (13) reflects this history of recombination. Frequent recombination has been detected previously among environmental V. parahaemolyticus strains but not among strains associated with disease (30), yet some isolates within this collection surprisingly cluster with pathogenic strains. Nonetheless, the amount of recombination (r) among these GBE isolates was smaller than the amount of mutation (m; r/m = 0.872 [95% confidence range, 0.648 to 1.101]) (7). Together, these analyses suggest that this population has evolved in situ from a broad sample of the global species diversity.
In summary, the V. parahaemolyticus population from the GBE is highly diverse and genotypically unique compared to other reported populations. Even with the absence of trh and tdh, which are associated with outbreak strains, some GBE strains are genetically similar to pandemic and nonpandemic isolates from infections. In light of the amount of recombination within the GBE population and among V. parahaemolyticus strains in general and the increased abundance and diversity associated with warming waters, new pathogenic lineages may emerge with climate change and warming water (1).
Supplementary Material
ACKNOWLEDGMENTS
We thank Anna Tyzik, Rachel Donner, Jong Yu, Jenny Mahoney, Matthew Gerding, Colin Edwards, and Eliot Jones for assistance with sampling, oyster processing, strain isolation, and PCR amplification and Victoria Seetaran and Victoria Sicard for development of virulence-specific MLSA primers.
This research was supported by Sea Grant R/CE-137 and NIH 1R03AI081102-01.
Footnotes
Published ahead of print 9 March 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Baker-Austin C, Stockley L, Rangdale R, Martinez-Urtaza J. 2010. Environmental occurrence and clinical impact of Vibrio vulnificus and Vibrio parahaemolyticus: a European perspective. Environ. Microbiol. Rep. 2:7–18 [DOI] [PubMed] [Google Scholar]
- 2. Chiou C, Hsu S, Chiu S, Wang T, Chao C. 2000. Vibrio parahaemolyticus serovar O3:K6 as cause of unusually high incidence of food-borne disease outbreaks in Taiwan from 1996 to 1999. J. Clin. Microbiol. 38:4621–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chowdhury NR, Stine OC, Morris JG, Nair GB. 2004. Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J. Clin. Microbiol. 42:1280–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colwell RR, Kaper J, Joseph SW. 1977. Vibrio cholerae, Vibrio parahaemolyticus, and other vibrios: occurrence and distribution in Chesapeake Bay. Science 198:394–396 [PubMed] [Google Scholar]
- 5. Daniels NA, et al. 2000. Vibrio parahaemolyticus infections in the United States, 1973–1998. J. Infect. Dis. 181:1661–1666 [DOI] [PubMed] [Google Scholar]
- 6. DePaola A, Kaysner CA, Bowers J, Cook DW. 2000. Environmental investigation of Vibrio parahaemolyticus in oysters after outbreaks in Washington, Texas, and New York (1997 and 1998). Appl. Environ. Microbiol. 66:4649–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eiler A, Johansson M, Bertilsson S. 2006. Environmental influences on Vibrio populations in northern temperate and boreal coastal waters (Baltic and Skagerrak Seas). Appl. Environ. Microbiol. 72:6004–6011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia KR, et al. 2009. Dynamics of clinical and environmental Vibrio parahaemolyticus strains during seafood-related summer diarrhea outbreaks in southern Chile. Appl. Environ. Microbiol. 75:7482–7487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gonzalez-Escalona N, et al. 2008. Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190:2831–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haubold B, Hudson RR. 2000. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16:847–849 [DOI] [PubMed] [Google Scholar]
- 13. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267 [DOI] [PubMed] [Google Scholar]
- 14. Jolley KA, Feil EJ, Chan M-S, Maiden MCJ. 2001. Sequence type analysis and recombinational tests (START). Bioinformatics 17:1230–1231 [DOI] [PubMed] [Google Scholar]
- 15. Jones SH, Striplin MJ, Mahoney JC, Cooper VS, Whistler CA. 2010. Incidence and abundance of pathogenic Vibrio species in the Great Bay Estuary, New Hampshire, p 127–134 In Lassus P. (ed), Proceedings of the Seventh International Conference on Molluscan Shellfish Safety. Quae Publishing, Nantes, France [Google Scholar]
- 16. Jones SH, Summer-Brason BW. 1998. Incidence and detection of pathogenic Vibrio sp. in a northern New England estuary, USA. J. Shellfish Res. 17:1665–1669. [Google Scholar]
- 17. Kaysner CA, DePaola A., Jr May 2004, revision date. Chapter 9, Vibrio. In Hammack T, et al. (ed), Bacterial analytical manual. Food and Drug Administration, Washington, DC: http://www.fda.gov/Food/ScienceResearch/LaboratoryMethods/BacteriologicalAnalyticalManualBAM/ucm070830.htm [Google Scholar]
- 18. Mahoney JC, Gerding MJ, Jones SH, Whistler CA. 2010. Comparison of the pathogenic potentials of environmental and clinical Vibrio parahaemolyticus strains indicates a role for temperature regulation in virulence. Appl. Environ. Microbiol. 76:7459–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martinez-Urtaza J, Bowers JC, Trinanes J, DePaola A. 2010. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res. Int. 43:1780–1790 [Google Scholar]
- 20. Massad G, Oliver JD. 1987. New selective and differential plating medium for Vibrio vulnificus and Vibrio cholerae. Appl. Environ. Microbiol. 53:2262–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Neill KR, Jones SH, Grimes DJ. 1992. Seasonal incidence of Vibrio vulnificus in the Great Bay estuary of New Hampshire and Maine. Appl. Environ. Microbiol. 58:3257–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Panicker G, Douglas R, Call M, Krug MJ, Bej AK. 2004. Detection of pathogenic Vibrio spp. in shellfish by using multiplex PCR and DNA microarrays. Appl. Environ. Microbiol. 70:7436–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rose JB, et al. 2001. Climate variability and change in the United States: potential impacts on water- and foodborne diseases caused by microbiologic agents. Environ. Health Perspect. 109:211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sawabe T, Kita-Tsukamoto K, Thompson FL. 2007. Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 189:7932–7936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuster BM, et al. 2011. Ecology and genetic structure of a northern temperate Vibrio cholerae population related to toxigenic isolates. Appl. Environ. Microbiol. 77:7568–7575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singleton FL, Attwell RW, Jangi MS, Colwell RR. 1982. Influence of salinity and organic nutrient concentration on survival and growth of Vibrio cholerae in aquatic microcosms. Appl. Environ. Microbiol. 43:1080–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Souza JT, de Mazzola M, Raaijmakers JM. 2003. Conservation of the response regulator gene gacA in Pseudomonas species. Environ. Microbiol. 5:1328–1340 [DOI] [PubMed] [Google Scholar]
- 28. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson FL, Iida T, Swings J. 2004. Biodiversity of vibrios. Microbiol. Mol. Biol. Rev. 68:403–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vos M, Didelot X. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3:199–208 [DOI] [PubMed] [Google Scholar]
- 31. Wong H, et al. 2000. Characteristics of Vibrio parahaemolyticus O3:K6 from Asia. Appl. Environ. Microbiol. 66:3981–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan Y, et al. 2011. Extended MLST-based population genetics and phylogeny of Vibrio parahaemolyticus with high levels of recombination. Int. J. Food Microbiol. 145:106–112 [DOI] [PubMed] [Google Scholar]
- 33. Yeung PS, Boor KJ. 2004. Epidemiology, pathogenesis, and prevention of foodborne Vibrio parahaemolyticus infections. Foodborne Pathog. Dis. 1:74–88 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.