

Abstract
Ultrahigh-molecular-weight poly[(R)-3-hydroxybutyrate] [UHMW-P(3HB)] synthesized by genetically engineered Escherichia coli is an environmentally friendly bioplastic material which can be processed into strong films or fibers. An operon of three genes (organized as phaCAB) encodes the essential proteins for the production of P(3HB) in the native producer, Ralstonia eutropha. The three genes of the phaCAB operon are phaC, which encodes the polyhydroxyalkanoate (PHA) synthase, phaA, which encodes a 3-ketothiolase, and phaB, which encodes an acetoacetyl coenzyme A (acetoacetyl-CoA) reductase. In this study, the effect of gene order of the phaCAB operon (phaABC, phaACB, phaBAC, phaBCA, phaCAB, and phaCBA) on an expression plasmid in genetically engineered E. coli was examined in order to determine the best organization to produce UHMW-P(3HB). The results showed that P(3HB) molecular weights and accumulation levels were both dependent on the order of the pha genes relative to the promoter. The most balanced production result was achieved in the strain harboring the phaBCA expression plasmid. In addition, analysis of expression levels and activity for P(3HB) biosynthesis enzymes and of P(3HB) molecular weight revealed that the concentration of active PHA synthase had a negative correlation with P(3HB) molecular weight and a positive correlation with cellular P(3HB) content. This result suggests that the level of P(3HB) synthase activity is a limiting factor for producing UHMW-P(3HB) and has a significant impact on P(3HB) production.
INTRODUCTION
Polyhydroxyalkanoates (PHAs) are polyesters synthesized by more than 200 species of bacteria and are accumulated as intracellular carbon and energy storage materials (13, 16, 23, 24). PHAs have attracted much attention as environmentally friendly plastic materials because these plastics are derived from renewable biomass resources and are completely mineralized to CO2 and H2O by microorganisms in the environment after use (29).
A homopolymer of (R)-3-hydroxybutyrate [P(3HB)] is the most common type of PHA that bacteria accumulate in nature, and its production has been extensively studied. Ralstonia eutropha is the most widely studied native producer of P(3HB), and in this bacterium the polymer is synthesized from acetyl coenzyme A (acetyl-CoA) by the reaction of three enzymes: 3-ketothiolase (PhaA), NADPH-dependent acetoacetyl-CoA reductase (PhaB), and PHA synthase (PhaC). The weight-average molecular weight (Mw) of P(3HB) produced by native PHA-producing bacteria is usually in the range of 0.2 × 106 to 2 × 106 (24). However, this P(3HB) is a brittle and rigid material with low flexibility because of its high crystallinity. On the other hand, recombinant Escherichia coli harboring the R. eutropha pha operon (phaC-phaA-phaB) is capable of synthesizing ultrahigh-molecular-weight P(3HB) [UHMW-P(3HB)] with an Mw exceeding 3 × 106 from glucose (1, 2, 5, 8, 9, 10). The higher molecular weight gives UHMW-P(3HB) higher mechanical strength, and unlike lower-molecular-weight P(3HB) polymers, UHMW-P(3HB) can be processed into strong films or fibers by hot or cold drawing (7). It is expected that the improved material properties of UHMW-P(3HB) will expand the number of applications for this polymer. However, it is still difficult to accumulate high levels of UHMW-P(3HB) in bacterial cells, and thus the construction of a highly efficient UHMW-P(3HB) synthesis system is desirable to make the polymer commercially. In addition, the decrease of polymer molecular weight by pyrolysis during melt processing must be considered for industrial production of P(3HB) plastics. Therefore, synthesis of UHMW-P(3HB) will likely be necessary for the production of bulk commodity plastic products from this material.
In general, there is a trade-off between the molecular weight and yield of PHA polymers produced by microbial systems (21). The molecular weight and production of PHA polymers are generally determined by the ratio of phaC (PHA synthase gene) to phaAB (genes encoding the monomer-supplying enzymes) expression levels. In other words, the expression levels of these three genes could give rise to differences in PHA production. It is commonly known that for genes in operons, the gene that lies closer to the promoter will be more highly expressed (12). To data, the native pha operon (with a gene order of phaC-phaA-phaB) derived from R. eutropha has been used for UHMW-P(3HB) production by E. coli (1, 2, 5, 8, 9, 10). However, prior to our current study, rearranging the order of the phaA, phaB, and phaC genes within the pha operon to optimize the gene expression levels for UHMW-P(3HB) production had yet to be attempted.
In this study, we optimized the expression levels of the PHA synthesis genes for production of UHMW-P(3HB) with high accumulation levels by rearranging the pha gene order within the pha operon. To this end, the OGAB (ordered gene assembly in Bacillus subtilis) method (26), a single-step method for cloning multiple genes in a specific, desired order by using the transformation system of B. subtilis to arrange the gene order, was used to manipulate the pha gene order. The possible gene orders of the three genes (phaABC, phaACB, phaBAC, phaBCA, phaCAB, and phaCBA) were expressed in E. coli, and the effect of gene rearrangements on UHMW-P(3HB) production was investigated.
MATERIALS AND METHODS
Bacterial strains and plasmids.
B. subtilis BUSY9797 [RM125 proB::(cI spc)] (15), which can repress expression from the Pr promoter, was used for the OGAB assembly. E. coli DH5α [F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1] was used for general genetic manipulation and the production of P(3HB). The pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA) was used for the cloning of the phaA, phaB, and phaC genes. The pUC19 vector was used for making the 3-base cohesive ends required in OGAB method. The pGETS109 vector (26), a shuttle vector that can replicate in both B. subtilis and E. coli cells, was used as a final vector for phaABC expression plasmids.
Plasmid construction.
The protocol for the OGAB method was previously described (26). The phaA, phaB, and phaC genes, together with their ribosomal binding sites, were amplified from the R. eutropha H16 genomic DNA with Ex Taq DNA polymerase (Takara Bio, Otsu, Japan) using the following primers, where the underlined sequences show BspQI restriction sites: for the phaA gene, phaA-F (5′-TAGGCTCTTCAGTGATTGAAAGGACTACACAATGACTGAC-3′) and phaA-R (5′-TAGGCTCTTCATTATTTGCGCTCGACTGCC-3′); for the phaB gene, phaB-F (5′-TAGGCTCTTCAGTGCCAATCAAGGAGTGGACATG-3′) and phaB-R (5′-TAGGCTCTTCATTAGCCCATATGCAGGCC-3′); and for the phaC gene, phaC-F (5′-TAGGCTCTTCAGTGAGAGAGACAATCAAATCATGGCG-3′) and phaC-R (5′-TAGGCTCTTCATTATGCCTTGGCTTTGACGTATC-3′). The PCR products were subcloned into pCR2.1-TOPO (Invitrogen) and confirmed by DNA sequencing with an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). Each plasmid was digested with BspQI, and the resultant small fragments were inserted into the pUC19 destination vectors (pUC19V-1st-DraIII, pUC19V-2nd, and pUV19V-3rd10th-DraIII). The 9 plasmids (pUC19V-1st-phaA, pUC19V-1st-phaB, pUC19V-1st-phaC, pUC19V-2nd-phaA, pUC19V-2nd-phaB, pUC19V-2nd-phaC, pUC19V-3rd-phaA, pUC19V-3rd-phaB, and pUC19V-3rd-phaC) were digested with DraIII, which recognizes 5′-CACNNN/GTG-3′, to make the 3-base cohesive ends required to decide the gene order, and a final vector, pGETS109, was digested with SfiI, which recognizes 5′-GGCCNNNN/NGGCC-3′. These 10 fragments were gel purified. The vector and an appropriate combination of three insert genes were mixed (2 fmol/μl each) in ligation buffer (66 mM Tris-HCl [pH 7.6], 6.6 mM MgCl2, 10 mM dithiothreitol, 100 μM ATP, 150 mM NaCl, 10% polyethylene glycol 6000 [Wako, Osaka, Japan]) and incubated with 4 U of T4 DNA ligase (Toyobo, Osaka, Japan) at 37°C for 30 min. Each ligation reaction mixture was transformed into competent B. subtilis cells and incubated overnight on Luria-Bertani (LB) plates containing tetracycline at 37°C. The resultant plasmids, the pGES109-pha series (pGETS109-phaABC, pGETS109-phaABC, pGETS109-phaBAC, pGETS109-phaBCA, pGETS109-phaCAB, and pGETS109-phaCBA), were confirmed by restriction mapping. The DNA sequences of pha genes on the pGES109-pha series were also checked by DNA sequencing.
P(3HB) biosynthesis in E. coli.
E. coli DH5α was transformed with the pGETS109-pha series plasmids, and individual transformation events were isolated. To synthesize P(3HB), the DH5α transformants representing a transformation of each of the individual pGETS109-pha series plasmids were grown at 30°C for 72 h using a reciprocal shaker (130 strokes/min) in 500-ml flasks with 100 ml of LB medium (10 g/liter NaCl, 10 g/liter tryptone, and 5 g/liter yeast extract) supplemented with 20 g/liter of glucose. To maintain the plasmids within the cells, 100 μg/ml of ampicillin was added to the medium. The optical density at 600 nm (OD600) was measured after incubation for 6, 12, 24, 48, and 72 h. The collected cells were washed with double-distilled water (ddH2O), frozen, and lyophilized.
Characterization of P(3HB).
The P(3HB) content in the cells was determined by gas chromatography, using a GC14B instrument (Shimadzu, Kyoto, Japan), after methanolysis of approximately 15 mg lyophilized cells in the presence of 15% (vol/vol) sulfuric acid, as described previously (1). P(3HB) was extracted from cells by stirring with chloroform for 72 h at room temperature, and the cells were removed by filtration. The extracted P(3HB) was purified by precipitation with methanol. The number-average molecular weight (Mn) and Mw were determined by gel permeation chromatography (GPC) using a 10A GPC system (Shimadzu). Chloroform was used as the eluent at a flow rate of 0.8 ml/min, and each sample (concentration of 1 mg/ml) was applied. Polystyrene standards (peak molecular weight [Mp] = 1.3 × 103 to 7.5 × 106 g/mol) with a low polydispersity were used to make molecular weight calibration curves.
Analysis of gene expression levels by quantitative real-time PCR (qRT-PCR).
Gene expression analysis was performed as described previously (30). Briefly, 0.5 ml of each recombinant E. coli strain was harvested after 12 h of growth. To each of these samples, 1 ml of RNA Protect Bacteria reagent (Qiagen, Hilden, Germany) was added for RNA stabilization. Cell pellets were stored at −80°C until RNA extraction. Total RNA was isolated from each sample using the RNeasy minikit (Qiagen). Remaining DNA in RNA samples was removed with RNase-free DNase I (NEB, Beverly, MA). The purity and concentration of RNA were checked spectrophotometrically using a UV-1700 PharmaSpec (Shimadzu). For each sample, cDNA synthesis was performed according to the manufacturer's recommendations using the iScript Select cDNA synthesis kit (Bio-Rad, Richmond, CA), and 100 ng total RNA was used as the template.
Gene expression levels were determined by real-time PCR using the cDNA samples as the templates. Primers for real-time PCR were designed using Primer 3 (http://frodo.wi.mit.edu/primer3/) and are listed in Table 1. The 16S rRNA gene was used as the housekeeping gene. The real-time PCRs were performed using the iQ SYBR green Supermix kit (Bio-Rad) with MxPro 3005P (Agilent Technology, Palo Alto, CA). The reaction conditions were as follows: 3 min at 95°C and then 40 cycles of 40 s at 95°C, 40 s at 60°C, and 40 s at 72°C. After the real-time PCR, melting curve analysis consisting of 76 cycles of 30 s each starting 60°C, increasing 0.5°C per cycle, and ending at 95°C was performed to check primer specificity. The relative gene expression levels of target genes (phaA, phaB, and phaC) were quantified using the following equation: gene expression level = 2[CT(16S rRNA gene) − CT(target gene)].
Table 1.
Primers used in real-time PCR
| Gene name | Oligonucleotides | Product length (bp) |
|---|---|---|
| phaA | 5′-GAGAACGTGGCCAAGGAATA-3′, 5′-GGGACGATCTCTTCGTCAAA-3′ | 119 |
| phaB | 5′-GATCGACACCAACCTGACCT-3′, 5′-TTCACCGACGAGATGTTGAC-3′ | 102 |
| phaC | 5′-GAACGACCTGGTGTGGAACT-3′, 5′-TCGTTCTGCAGGTAGGTGTG-3′ | 150 |
| 16S rRNA gene | 5′-AGAAGCTTGCTCTTTGCTGA-3′, 5′-CTTTGGTCTTGCGACGTTAT-3′ | 120 |
Enzyme activity assays.
Enzyme assays were performed as described previously (14). Recombinant E. coli strains were harvested from cells cultivated in LB medium for 12 h and were resuspended in 150 mM potassium phosphate (KPi) buffer (pH 7.0) with an appropriate volume corresponding to the OD600. The cells were disrupted by sonication for 2 min and then centrifuged at low speed (1,500 × g, 5 min, 4°C) to obtain a whole-cell extract containing PHA granules for PHA synthase (PhaC) analysis. The activities of the PHA synthase enzymes were determined spectrophotometrically by discontinuous assay. PHA synthase (PhaC) enzymes were assayed in 800 μl of 150 mM KPi buffer (pH 7.0) containing 0.1 mM (R)-3HB-CoA, 0.1 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (Wako) and 20 to 200 μg each whole-cell extract, and the thionitrobenzoic acid (TNB) increase corresponding to CoA release was detected at 420 nm. (R)-3HB-CoA was chemically synthesized by the mixed-anhydride method as described previously (22). One unit was defined as the activity for producing 1 μmol of TNB anion per minute (ε420 = 14.5 × 103 M−1 cm−1). The soluble cell extract was separated from the whole cells and disrupted cells by centrifugation of the crude extract (12,000 × g, 25 min, 4°C) for the 3-ketothiolase (PhaA) and acetoacetyl-CoA reductase (PhaB) assays. 3-Ketothiolase (PhaA) was assayed in 800 μl of 150 mM KPi buffer (pH 7.0) containing 0.3 mM acetoacetyl coenzyme A (Sigma-Aldrich, St. Louis, MO), 1 mM lithium coenzyme A (Sigma-Aldrich), 25 mM MgCl2 (Wako), and 2 to 10 μg each soluble cell extract, and the decrease in absorbance was recorded at 304 nm for the derived Mg2+-3-ketoacyl-CoA complex. One unit was defined as the activity for producing 1 μmol of Mg2+-3-ketoacyl-CoA complex per minute (ε304 = 19.5 × 103 M−1 cm−1). Acetoacetyl-CoA reductase (PhaB) was assayed in 800 μl of 150 mM KPi buffer (pH 7.0) containing 0.1 mM acetoacetyl coenzyme A, 0.1 mM NADPH (Sigma-Aldrich, MO), and 2.5 to 20 μg each soluble cell extract, and the decrease in absorbance of NADPH was measured at 340 nm. One unit was defined as the activity for consuming 1 μmol of NADPH per minute (ε340 = 6.22 × 103 M−1 cm−1). The concentration of total protein was determined with the Bradford assay using bovine serum albumin (BSA) as a standard.
Western blotting.
Separation of proteins in whole-cell extracts and soluble cell extracts was done by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 5 μg of the whole-cell extract for PHA synthase (PhaC) and 1 μg of the soluble cell extract for 3-ketothiolase (PhaA) and acetoacetyl-CoA reductase (PhaB). Separated proteins were transferred to a polyvinylidene fluoride membrane using a Trans-Blot SD semidry transfer cell (Bio-Rad) at 40 mA for 600 min. Western blot analysis of PHA synthase (PhaC) was carried out using a specific rabbit antiserum raised against the C terminus of PHA synthase (PhaC). Rabbit antisera to 3-ketothiolase (PhaA) and acetoacetyl-CoA reductase (PhaB) provided by Seiichi Taguchi (Hokkaido University, Japan) were used. Proteins were detected using goat anti-rabbit antibodies with horseradish peroxidase (HRP) as a secondary antibody. Phosphate-buffered saline (PBS)-Tween (0.8 g/liter NaCl, 0.02 g/liter KCl, 0.29 g/liter Na2HPO4 · 12H2O, 0.02 g/liter KH2PO4, 0.1 g/liter Tween 20) was used for membrane washing and dilution of primary and secondary antibodies. Proteins that cross-reacted with the antibodies were visualized using the ECL Plus Western blotting detection reagent (GE Healthcare, Little Chalfont, United Kingdom), and data were recorded with a charge-coupled device (CCD) camera.
RESULTS
Assembly of pha genes by the OGAB method.
Figure 1 shows maps of the six expression plasmids (pGETS109-pha series) carrying individual pha operons consisting of phaA, phaB, and phaC from R. eutropha with various gene orders constructed in this study by the OGAB method. The pha genes were inserted in the same orientation to ensure colinear expression. These genes were under the transcriptional control of the Pr promoter of the λ phage, which is constitutively expressed in E. coli. The ribosomal binding sites for each gene were derived from the native Shine-Dalgarno sequences for each corresponding gene in R. eutropha.
Fig 1.

Plasmids (pGETS109-pha series) used in this study. The phaA, phaB, and phaC genes are derived from R. eutropha. pha genes were inserted into the B. subtilis-E. coli shuttle vector pGETS109. Plasmids were constructed by the OGAB method (see Materials and Methods) with various pha gene orders (1 to 6).
Effect of gene order on P(3HB) production.
In order to assess the effect of gene order on P(3HB) production, recombinant E. coli DH5α harboring pGETS109-pha series plasmids (Fig. 1) were cultured at 30°C in LB medium containing 20 g/liter glucose and 100 mg/liter ampicillin for 72 h. The growth curves of the six recombinant E. coli strains are shown in Fig. 2. The growth behavior of each strain was dependent on the order of the pha genes on the pGETS109-pha plasmid that each strain harbored. All recombinant strains reached stationary phase at 24 h. After 72 h of cultivation, the highest OD600 was 50 in strains 6 (gene order, phaC-phaB-phaA [CBA]) and 5 (CAB) and the lowest OD600 was 30 in strain 1 (ABC).
Fig 2.
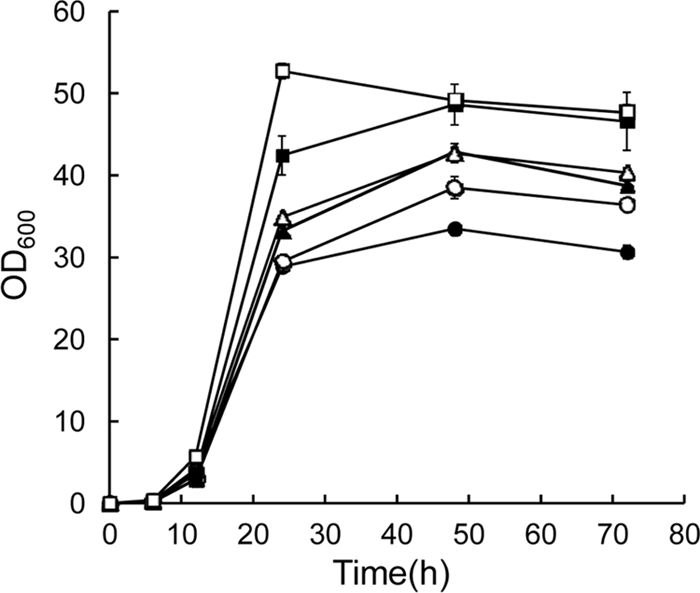
Growth curves of recombinant E. coli DH5α harboring the pGETS109-pha series plasmids. Cells were cultivated in LB medium containing glucose (20 g/liter) at 30°C for 72 h with shaking at 130 rpm. Cultivation for each strain was carried out in triplicate, and the average value for samples taken at each time point is shown. The error was less than 8% per sample. Strains: ●, 1 (ABC); ○, 2 (ACB); ▲, 3 (BAC); △, 4 (BCA); ■, 5 (CAB); □, 6 (CBA).
The results for P(3HB) production after 12 h and 72 h of cultivation of recombinant E. coli strains are summarized in Table 2. The ranges of dry cell weights were 1.3 to 2.6 g/liter at 12 h and 8.0 to 11.2 g/liter at 72 h. At both cultivation times (12 h and 72 h), the highest dry cell weight was obtained in strain 6 (CBA).
Table 2.
Biosynthesis of P(3HB) in recombinant E. coli DH5α harboring pGETS109-pha series plasmids and cultivated for 12 h or 72 ha
| Strain | Gene order | Culture time (h) | Dry cell wt (g/liter) | P(3HB) content (wt %) | Mol wt |
||
|---|---|---|---|---|---|---|---|
| Mn (×106) | Mw (×106) | Mw/Mn | |||||
| 1 | ABC | 12 | 1.6 ± 0.1 | 4.9 ± 0.4 | 3.3 ± 0.3 | 6.2 ± 0.4 | 1.9 |
| 72 | 8.0 ± 0.2 | 31 ± 1.0 | 3.8 ± 0.2 | 6.2 ± 0.1 | 1.7 | ||
| 2 | ACB | 12 | 1.3 ± 0.1 | 5.3 ± 0.7 | 1.5 ± 0.1 | 4.4 ± 0.3 | 2.9 |
| 72 | 8.6 ± 0.2 | 36 ± 2.4 | 1.4 ± 0.1 | 3.8 ± 0.4 | 2.7 | ||
| 3 | BAC | 12 | 1.4 ± 0.1 | 4.9 ± 0.2 | 2.2 ± 0.2 | 5.4 ± 0.2 | 2.5 |
| 72 | 9.4 ± 0.2 | 43 ± 0.9 | 1.6 ± 0.2 | 4.1 ± 0.4 | 2.7 | ||
| 4 | BCA | 12 | 1.5 ± 0.1 | 7.0 ± 0.9 | 2.8 ± 0.3 | 6.2 ± 0.1 | 2.5 |
| 72 | 9.3 ± 0.1 | 41 ± 1.4 | 2.3 ± 0.1 | 5.0 ± 0.1 | 2.2 | ||
| 5 | CAB | 12 | 1.7 ± 0.1 | 8.2 ± 0.9 | 1.6 ± 0.1 | 3.4 ± 0.3 | 3.1 |
| 72 | 10.8 ± 0.4 | 52 ± 3.3 | 0.8 ± 0.2 | 2.5 ± 0.4 | 3.3 | ||
| 6 | CBA | 12 | 2.6 ± 0.1 | 8.5 ± 1.0 | 1.5 ± 0.2 | 4.2 ± 0.2 | 2.8 |
| 72 | 11.2 ± 0.1 | 57 ± 1.6 | 0.5 ± 0.1 | 2.0 ± 0.1 | 3.8 | ||
Cells were cultivated in LB medium containing glucose (20 g/liter) at 30°C. The results are the averages from three independent experiments ± standard errors.
P(3HB) content was also dependent on the pha gene order within the operon. Recombinant E. coli strains produced P(3HB) at levels ranging from 31 to 57% (wt/wt) of the dry cell weight after 72 h of cultivation. The highest P(3HB) content was observed in strain 6 (CBA).
Effect of the gene order on P(3HB) molecular weight.
As listed in Table 2, the four strains produced UHMW-P(3HB) with Mws exceeding 3 × 106 after 72 h of cultivation. The highest-molecular-weight P(3HB) was observed in strain 1 (ABC), and the P(3HB) with the next highest molecular weight was accumulated in strain 4 (BCA). In contrast, strains 5 (CAB) and 6 (CBA) produced relatively low-molecular-weight P(3HB).
The molecular weight distributions of the P(3HB)s synthesized by the six strains after 12 h and 72 h of cultivation are shown in Fig. 3. The most narrow molecular weight distribution was observed for the P(3HB) synthesized by strain 1 (ABC) at 72 h. The polymers produced in this strain had a polydispersity (Mw/Mn) of 1.7, as shown in Table 2. The P(3HB) synthesized by strain 4 (BCA) also had a narrow polydispersity of 2.2. On the other hand, the P(3HB)s synthesized by strains 5 (CAB) and 6 (CBA) had broad molecular weight distributions, and the polydispersities (Mw/Mn) were 3.3 and 3.8, respectively. Comparison of the P(3HB) molecular weight distributions produced by the strains at 12 h and 72 h revealed that molecular weight of P(3HB) collected from cells at 12 h tended to be higher than that of P(3HB) collected from cells at 72 h.
Fig 3.

Molecular weight distributions of P(3HB) synthesized by recombinant E. coli DH5α harboring pGET109-pha series plasmids. The cultivation time was 12 h (dashed lines) or 72 h (solid lines). Determination of molecular weight was carried out in triplicate, and typical distributions for each polymer are shown.
Expression levels of pha genes.
The relationship between pha gene expression level and gene order is shown in Fig. 4. The x axis displays the gene order; that is, the gene closest to the promoter is represented by 1, whereas the gene furthest from the promoter is represented by 3. The y axis shows the relevant mRNA abundances of pha genes compared to 16S rRNA gene abundance. These three graphs clearly indicate that the gene closest to the promoter showed higher levels of gene expression. In addition, when ihfB (integration host factor subunit β gene) (3, 4, 6) was used as a housekeeping gene, the same expression pattern of pha genes was obtained (data not shown). The expression level of phaC was relatively lower than those of phaA and phaB. One reason for this difference may be the difference in the efficiency of reverse transcription among target genes. This phenomenon was also observed in a previous transcriptional analysis of R. eutropha (11).
Fig 4.

pha gene expression levels with respect to gene order in recombinant E. coli DH5α harboring pGETS109-pha series plasmids. The x axis displays the gene order, with the gene closest to the promoter represented by 1 and the gene farthest from the promoter represented by 3. The y axis shows the relevant mRNA abundances of pha genes compared to 16S rRNA gene abundance determined by real-time PCR analysis. Experiments were carried out in triplicate, and the average and standard deviation for each point is shown.
Levels of production of P(3HB) synthesis enzymes.
In order to assess the 3-ketothiolase (PhaA), acetoacetyl-CoA reductase (PhaB), and PHA synthase (PhaC) expression levels and activity levels in each recombinant E. coli strain, enzyme activity assays and Western blotting were performed using cells collected at 12 h (in exponential growth phase). Figure 5 shows the 3-ketothiolase (PhaA), acetoacetyl-CoA reductase (PhaB), and PHA synthase (PhaC) proteins detected by Western blotting and the specific activity of each enzyme determined as described in Materials and Methods. Enzyme expression levels were different for each strain (Fig. 5). The specific activities of the enzymes were positively correlated with the expression of each specific enzyme as determined by Western blotting. This result suggests that expressed enzymes from pGETS109 plasmids were properly folded and enzymatically active in the cells. Additionally, there was a tendency for protein expression levels of the PHA synthesis enzymes to be positively correlated with mRNA levels of the pha genes.
Fig 5.

Western blot analysis and enzyme activities of PHA synthase (PhaCRe) and the monomer-supplying enzymes (PhaARe and PhaBRe) in recombinant E. coli DH5α harboring pGETS109-pha series plasmids. A crude extract (1 μg for PhaARe and PhaBRe, 5 μg for PhaCRe) was prepared from each recombinant strain and was used for Western blot analysis. Enzyme assays were performed spectrophotometrically using appropriate amounts of crude extract and calculated specific activities (see Materials and Methods). Each enzyme assay was carried out in triplicate, and results are shown as averages with standard deviations.
Relationship of PhaC activity level to P(3HB) accumulation level and P(3HB) molecular weight.
In order to understand the contribution of each enzyme activity level to P(3HB) production, the relationship between enzyme activity level and synthesized P(3HB) was investigated. As shown in Fig. 6, there is a negative correlation between the number-average molecular weight (Mn) and PHA synthase (PhaC) activity. That is, the lower the enzymatic activity detected for PHA synthase (PhaC), the higher the molecular weight of the P(3HB) polymer that is produced. In addition, the P(3HB) contents accumulated by the cells were positively correlated with PhaC activity; the higher the PHA synthase (PhaC) activity, the higher the P(3HB) content produced relative to the dry cell weight. On the other hand, 3-ketothiolase (PhaA) and acetoacetyl-CoA reductase (PhaB) activity levels did not correlate with P(3HB) molecular weight or content. This phenomenon suggested that ample 3-ketothiolase (PhaA) and acetoacetyl-CoA reductase (PhaB) are expressed and supply 3HB precursors in excess, followed by the polymerization of these intracellular precursors depending on the PHA synthase (PhaC) expression level. Therefore, PHA synthase (PhaC) would be the limiting factor for PHA synthesis in this study.
Fig 6.

Effect of PHA synthase (PhaCRe) specific activity on the molecular weight (Mn) and content of P(3HB) synthesized in recombinant E. coli strains. P(3HB) characterization and P(3HB) synthesis enzyme assays revealed a negative correlation between Mn and PHA synthase (PhaCRe) activity and a positive correlation between P(3HB) content and PHA synthase (PhaCRe) activity. ▲, number-average molecular weight (Mn); □, P(3HB) content.
DISCUSSION
In this study, we focused on the relationship of gene order to various aspects of PHA production. Our results demonstrated that genes positioned closer to the promoter were more highly expressed than genes positioned further away from the promoter. In addition, the gene expression levels were positively correlated to the protein expression levels. The ubiquity of this relationship between gene expression, protein expression, and promoter proximity has been previously demonstrated by using synthetic operons consisting of fluorescent protein genes (12). However, the order of genes in the operon had not been considered to be important for PHA production. Specific placement and ordering of genes in the operon constitute an effective approach for regulating gene expression and differ from conventional methods of controlling gene expression such as promoter switching or varying the concentrations of inducers.
This study used the OGAB method to specify the order of genes in synthetic operons. In previous studies, application of the OGAB method led to improved production of bioproducts such as plipastatin (27) and carotenoids (15). Our study demonstrates that the OGAB method can be successfully applied to PHA production as well. For the production of PHA, one must consider not only yield but also physical properties, such as the mechanical strength of the polymers produced. The optimization of these two parameters (PHA yield and molecular weight) was successfully achieved by switching the gene order within the pha operon. As a result, the synthetic phaBCA operon delivered an increased molecular weight of P(3HB) in addition to a relatively high P(3HB) yield (Table 2).
Additionally, we have shown that PHA synthase (PhaC) activity is the critical factor for PHA yield and molecular weight (Fig. 6). Early studies (2, 17, 20, 21) reported correlations of PhaC activity to the PHA yield and the molecular weight of the PHA polymers produced. The results of our current study support the findings of Sim et al., who clearly demonstrated that PhaC activity has a negative correlation with P(3HB) molecular weight and a positive correlation with cellular P(3HB) content (21). On the other hand, some reports suggest that the limiting factor for PHA production and a main limitation of PHA synthesis flux is a shortage of NAPDH, which is required as a cofactor for the acetoacetyl-CoA reductase (PhaB) reaction (18, 19), or the low expression levels of phaB (28). In most cases, the native pha operon (phaC-phaA-phaB) from R. eutropha has been used for PHA production, and the phaB expression levels are lower since phaB is the last gene in the native operon. Therefore, by increasing concentrations of intracellular NADPH (18, 19) and phaB overexpression (28), increased levels of PHA accumulation could be achieved. In this study, PHA synthase (PhaC) activity was highly correlated with the production and molecular weight of PHA. However, unlike in previous studies (18, 19, 28), no correlation between acetoacetyl-CoA reductase (PhaB) and PHA productivity was observed based on transcriptional, protein expression, and PHA accumulation data (Table 2). It is probable that sufficient acetoacetyl-CoA reductase (PhaB) was expressed despite the rearrangement of gene order in this study. The NADPH supply might be intrinsically sufficient because intracellular NADPH concentrations are much higher than the Michaelis constant (Km) for NADPH of acetoacetyl-CoA reductase (PhaB) (28).
UHMW-P(3HB) is expected to be useful as an actual plastic material due to its high mechanical strength (7). However, previous attempts at producing UHMW-P(3HB) resulted in relatively low yields; thus, effective production of UHMW-P(3HB) has been desired. Previously, improved UHMW-P(3HB) production has been achieved by optimizing the plasmid vector (2, 8), host bacterial strain (1), cultivation conditions (1, 5, 9), PHA synthase (PhaC) (1), and point mutation insertions to PHA synthase (PhaC) (1). Previous studies revealed that specific culture conditions such as slightly acidic growth media (pH 6.0) (5, 9) and higher temperature (1) increased the Mw up to 20 × 106. Other studies demonstrated that the use of a low-copy-number expression plasmid as opposed to a high-copy-number plasmid is effective in increasing both the production and molecular weight of P(3HB) (2, 8). Additionally, it was confirmed that the PHA synthase from Delftia acidovorans (PhaCDa) was more effective than the PHA synthase from R. eutropha (PhaCRe) for higher-molecular-weight P(3HB) production in E. coli JM109 (1). A previous in vitro evolutionary study demonstrated that a point mutation (F420S) in the PhaCRe (25) enzyme led to increases in both P(3HB) content per cell and P(3HB) molecular weight (1). Despite the advances of these studies, the levels of production of UHMW-P(3HB) are still not high enough for commercial production.
In this study, in order to increase both the production and molecular weight of P(3HB), we investigated the optimal balance between monomer supply and PHA synthase abundance by switching the gene order of the phaCAB operon relative to the promoter. As a result, we have shown that the optimization of gene order has great influence on the production of P(3HB) in recombinant E. coli. This knowledge can be effectively applied to control both the yield and molecular weight of recombinantly produced PHA polymers. Additionally, by combining the knowledge gained from previous studies aimed at improving P(3HB) production with the optimization of gene order within the pha operon, the development of a superior UHMW-P(3HB) production system can be expected.
ACKNOWLEDGMENTS
We thank S. Taguchi (Hokkaido University, Sapporo, Japan) for providing antibodies for the PhaARe, PhaBRe, and PhaCRe proteins.
This work was supported by funding from the New Energy and Industrial Technology Development Organization (NEDO) to T. Tsuge and by NSF grant DMR 0907085 to C. T. Nomura.
Footnotes
Published ahead of print 17 February 2012
REFERENCES
- 1. Agus J, Kahar P, Abe H, Doi Y, Tsuge T. 2006. Molecular weight characterization of poly[(R)-3-hydroxybutyrate] synthesized by genetically engineered strains of Escherichia coli. Polym. Degrad. Stab. 91:1138–1146 [Google Scholar]
- 2. Agus J, Kahar P, Abe H, Doi Y, Tsuge T. 2006. Altered expression of polyhydroxyalkanoate synthase gene and its effect on poly[(R)-3-hydroxybutyrate] synthesis in recombinant Escherichia coli. Polym. Degrad. Stab. 91:1645–1650 [Google Scholar]
- 3. Baez-Viveros LJ, et al. 2007. Metabolic transcription analysis of engineered Escherichia coli strains that overproduce l-phenylalanine. Microb. Cell. Fact. 6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caspeta L, Flores N, Perez ON, Bolivar F, Ramirez TO. 2009. The effect of heating rate on Escherichia coli metabolism, physiological stress, transcriptional response, and production of temperature-induced recombinant protein: a scale-down study. Biotechnol. Bioeng. 102:468–482 [DOI] [PubMed] [Google Scholar]
- 5. Choi J, Lee SY. 2004. High level production of supra molecular weight poly((R)-3-hydroxybutyrate) by metabolically engineered Escherichia coli. Biotechnol. Bioprocess. Eng. 9:196–200 [Google Scholar]
- 6. Flores N, et al. 2005. Adaptation for fast growth on glucose by differential expression of central carbon metabolism and gal regulon genes in an Escherichia coli strain lacking the phosphoenolpyruvate: carbohydrate phosphotransferase system. Metab. Eng. 7:70–87 [DOI] [PubMed] [Google Scholar]
- 7. Iwata T. 2005. Strong fibers and films of microbial polyesters. Macromol. Biosci. 5:689–701 [DOI] [PubMed] [Google Scholar]
- 8. Kahar P, et al. 2004. Effective production and kinetic characterization of ultra-high-molecular-weight poly[(R)-3-hydroxybutyrate] in recombinant Escherichia coli. Polym. Degrad. Stab. 87:161–169 [Google Scholar]
- 9. Kusaka S, Abe H, Lee Y, Doi Y. 1997. Molecular mass of poly[(R)-3-hydroxybutyric acid] produced in a recombinant Escherichia coli. Appl. Microbiol. Biotechnol. 47:140–143 [DOI] [PubMed] [Google Scholar]
- 10. Kusaka S, Iwata T, Doi Y. 1999. Properties and biodegradability of ultra-high-molecular-weight poly[(R)-3-hydroxybutyrate] produced by a recombinant Escherichia coli. Int. J. Biol. Macromol. 25:87–94 [DOI] [PubMed] [Google Scholar]
- 11. Lawrence AG, et al. 2005. Transcriptional analysis of Ralstonia eutropha genes related to poly-(R)-3-hydrozybutyrate homeostasis during batch fermentation. Appl. Microbiol. Biotechnol. 68:663–672 [DOI] [PubMed] [Google Scholar]
- 12. Lim HN, Lee Y, Hussein R. 2011. Fundamental relationship between operon organization and gene expression. Proc. Natl. Acad. Sci. U. S. A. 108:10626–10631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu J, Tappel RC, Nomura CT. 2009. Biosynthesis of poly(hydroxyalkanoates). J. Macromol. Sci. Polym. Rev. 49:226–248 [Google Scholar]
- 14. Mifune J, Nakamura S, Fukui T. 2010. Engineering of pha operon on Cupriavidus necator chromosome for efficient biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) from vegetable oil. Polym. Degrad. Stab. 95:1305–1312 [Google Scholar]
- 15. Nishizaki T, Tsuge K, Itaya M, Doi N, Yanagawa H. 2007. Metabolic engineering of carotenoid biosynthesis in Escherichia coli by ordered gene assembly in Bacillus subtilis. Appl. Environ. Micobiol. 73:1355–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rehm BHA. 2003. Polyester synthases: natural catalysts for plastics. Biochem. J. 376:15–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ren Q, et al. 2005. Poly(3-hydroxyalkanoate) polymerase synthesis and in vitro activity in recombinant Escherichia coli and Pseudomonas putita. Appl. Microbiol. Biotechnol. 69:286–292 [DOI] [PubMed] [Google Scholar]
- 18. Sanchez MA, Andrews J, Hussein I, Bennett NH, San YK. 2006. Effect of overexpression of a soluble pyridine nucleotide transhydrogenase (UdhA) on the production of poly(3-hydroxybutyrate) in Escherichia coli. Biotechnol. Prog. 22:420–425 [DOI] [PubMed] [Google Scholar]
- 19. Shi H, Nikawa J, Shimizu K. 1999. Effect of modifying metabolic network on poly-3-hydroxybutyrate biosynthesis in recombinant Escherichia coli. J. Biosci. Bioeng. 87:666–677 [DOI] [PubMed] [Google Scholar]
- 20. Sim SJ, et al. 1997. PHA synthase activity controls the molecular weight and polydispersity of polyhydroxybutyrate in vivo. Nat. Biotechnol. 15:63–67 [DOI] [PubMed] [Google Scholar]
- 21. Sim SJ, Sneel KD, Kim BW, Rha KC, Sinskey AJ. 2001. Increased poy-β-hydroxybutyrate (PHB) chain length by the modulation of PHA synthase activity in recombinant Escherichia coli. Biotechnol. Lett. 23:2057–2061 [Google Scholar]
- 22. Stadtman RE. 1957. Preparation and assay of acyl coenzyme A and other thiol esters; use of hydroxylamine. Methods Enzymol. 3:931–941 [Google Scholar]
- 23. Steinbuchel A. 2001. Perspectives for biotechnological production and utilization of biopolymers: metabolic engineering of polyhydroxyalkanoate biosynthesis pathway as a successful example. Macromol. Biosci. 1:1–24 [Google Scholar]
- 24. Sudesh K, Abe H, Doi Y. 2000. Synthesis structure and properties of polyhydroxyalkanoates: biological polyesters. Prog. Polym. Sci. 25:1503–1555 [Google Scholar]
- 25. Taguchi S, Nakamura H, Hiraishi T, Yamato I, Doi Y. 2002. In vitro evolution of a polyhydroxybutyrate synthase by intragenic suppression-type mutagenesis. J. Biochem. 131:801–806 [DOI] [PubMed] [Google Scholar]
- 26. Tsuge K, Matsui K, Itaya M. 2003. One step assembly of multiple DNA fragments with a designed order and orientation in Bacillus subtilis plasmid. Nucleic Acids Res. 31:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsuge K, Matsui K, Itaya M. 2007. Production of the non-ribosomal peptide plipastatin in Bacillus subtilis regulated by three relevant gene blocks assembled in a single movable DNA segment. J. Biotechnol. 129:592–603 [DOI] [PubMed] [Google Scholar]
- 28. Tyo KE, Fischer CR, Simeon F, Stephanopoulos G. 2010. Analysis of polyhydroxybutyrate flux limitations by systematic genetic and metabolic perturbations. Metab. Eng. 12:187–195 [DOI] [PubMed] [Google Scholar]
- 29. Volova TG, Belyaeva OG, Plotnikov VF, Puzyr AP. 1998. Studies of biodegradation of microbial polyhydroxyalkanoates. Appl. Biochem. Microbiol. 34:488–492 [Google Scholar]
- 30. Wang Q, Nomura CT. 2010. Monitoring differences in gene expression levels and polyhydrozyalkanoate (PHA) production in Pseudomonas putida KT2440 grown on different carbon sources. J. Biosci. Bioeng. 110:653–659 [DOI] [PubMed] [Google Scholar]