

Abstract
Autotransporters have been employed as the anchoring scaffold for cell surface display by replacing their passenger domains with heterologous proteins to be displayed. We adopted an autotransporter (YfaL) of Escherichia coli for the cell surface display system. The critical regions in YfaL for surface display were identified for the construction of a ligation-independent cloning (LIC)-based display system. The designed system showed no detrimental effect on either the growth of the host cell or overexpressing heterologous proteins on the cell surface. We functionally displayed monomeric red fluorescent protein (mRFP1) as a reporter protein and diverse agarolytic enzymes from Saccharophagus degradans 2-40, including Aga86C and Aga86E, which previously had failed to be functional expressed. The system could display different sizes of proteins ranging from 25.3 to 143 kDa. We also attempted controlled release of the displayed proteins by incorporating a tobacco etch virus protease cleavage site into the C termini of the displayed proteins. The maximum level of the displayed protein was 6.1 × 104 molecules per a single cell, which corresponds to 5.6% of the entire cell surface of actively growing E. coli.
INTRODUCTION
Displaying heterologous peptides or proteins of interest (POI) on the surface of bacterial cells has been developed for many biotechnological applications (10). Although many proteins exposed to the outside of cells can be potential templates for a surface display system, only some genes, such as those for outer membrane proteins, lipoproteins, subunits of surface appendages, S-layer proteins, and autotransporters (ATs), have been used (29).
ATs have recently emerged as a popular target for bacterial surface display in Gram-negative bacteria because they have several advantages over other scaffold proteins (28). ATs are known to transport passenger domains if various sizes, ranging from short peptides to full-size proteins having over 3,000 residues (11), whereas other scaffold proteins often have a size limitation for surface display (28). ATs are capable of high-level functional expression of a target protein on the cell surfaces, avoiding toxic effects of overexpression (28). In addition, a relatively large number of AT proteins (about 50,000 to 100,000 copies per cell) can be presented on the cell surface (20, 28).
An AT is typically composed of three distinct functional regions: (i) a signal peptide region, (ii) an N-terminal passenger domain that is displayed outside the cell, and (iii) a C-terminal translocator (β-barrel) domain that delivers the passenger domain across the outer membrane (11). The C-terminal translocator domain shares a common secondary and tertiary structure in the AT family, but the passenger domain is sequentially or functionally diverse (e.g., adhesins, enzymes, or cytotoxins) (11). Because of this diversity of the passenger domain, the region can be substituted for various target proteins to be displayed.
In Escherichia coli, the yfaL gene was initially annotated as encoding a hypothetical protein, but later its product was predicted to be an AT (in the AIDA-I family) by its domain organization and a sequence similarity (34). Even though the exact cellular function of the yfaL gene is still unknown, a few studies on YfaL showed that (i) its overexpression induced aggregation of cells without a detrimental effect (27) and (ii) the passenger domain was released by an unknown mechanism (23). Based on these observations, we adopted YfaL as a potential scaffold protein for a novel E. coli surface display system that can possibly overexpress and release a displayed protein under appropriate conditions.
In this study, we present a surface display system using the autotransporter YfaL of E. coli. In order to determine the optimal regions in YfaL for displaying heterologous proteins, we performed sequence and domain analysis because the correct recognition of the passenger domain is critical for successful surface display. A ligation-independent cloning (LIC)-based display system was constructed for rapid and reliable cloning. We evaluated the system for functionality of displayed proteins and enumerated the total number of displayed proteins in a single cell. The controlled release of displayed proteins was also attempted by incorporating the designed cleavage site into the scaffold protein.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
E. coli DH5α was used as a general cloning host. E. coli BW25113 was used as a host strain for the surface display system. The E. coli strains were grown aerobically in Luria-Bertani (LB) broth (BD Difco) at 37°C. Saccharophagus degradans 2-40T (ATCC 43961) was grown in a sea salt minimal medium as described by Ekborg et al. (13). The bacterial strains, plasmids, and primers for PCR are listed in Table 1.
Table 1.
Bacterial strains, plasmids, and oligonucleotide primers used in this study
| Strain, plasmid, or primer | Relevant characteristic(s), description, or sequencea | Source or referenceb |
|---|---|---|
| Strains | ||
| E. coli DH5α | F−endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 hsdR17 (rK− mK+) λ− | Invitrogen |
| E. coli BW25113 | F− Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ−rph-1 Δ(rhaD-rhaB)568 hsdR514 | CGSC |
| S. degradans 2-40 | Source of agarase genes and NABH | ATCC |
| Plasmids | ||
| pBAD vector | araBAD promoter (PBAD); Apr; pBR322 origin; araC; C-terminal polyhistidine tag; rrnB transcription termination region | 21 |
| pBAD-yfaL | pBAD carrying yfaL gene from E. coli K-12 | This study |
| pBAD-aga16B | pBAD carrying aga16B gene from S. degradans | This study |
| pBAD-mRFP1 | pBAD carrying mRFP1; expression in the cytoplasm | This study |
| pATLIC vector | Derivative of pBAD-yfaL; deletion of the passenger domain sequence of yfaL and addition of TEV protease recognition site | This study |
| pATLICsec vector | Derivative of pATLIC vector; addition of predicted cleavage site of YfaL | This study |
| pATLIC-NABH | pATLIC carrying NABH gene from S. degradans | This study |
| pATLIC-NABH | pATLICsec carrying NABH gene from S. degradans | This study |
| pATLIC*-mRFP1 | pATLIC carrying mRFP1 gene; lacking the translocator domain and expression in the periplasm | This study |
| pATLIC-mRFP1 | pATLIC carrying mRFP1 gene | This study |
| pATLIC-aga50A | pATLIC carrying aga50A gene from S. degradans | This study |
| pATLIC-aga16B | pATLIC carrying aga16B gene from S. degradans | This study |
| pATLIC-aga86C | pATLIC carrying aga86C gene from S. degradans | This study |
| pATLIC-aga50D | pATLIC carrying aga50D gene from S. degradans | This study |
| pATLIC-aga86E | pATLIC carrying aga86E gene from S. degradans | This study |
| pATLIC(m29-948)-mRFP1 | pATLIC carrying mRFP1 gene; deletion of 29-948 region of YfaL | This study |
| pATLIC(m29-785)-mRFP1 | pATLIC carrying mRFP1 gene; deletion of 29-785 region of YfaL | This study |
| pATLIC(m29-699)-mRFP1 | pATLIC carrying mRFP1 gene; deletion of 29-699 region of YfaL | This study |
| pATLIC(m29-692)-mRFP1 | pATLIC carrying mRFP1 gene; deletion of 29-695 region of YfaL | This study |
| Primers | ||
| yfaL_F | 5′-GAAGGAGATATAAGGATGCGGATTATCTTTCTACGCAAG-3′ | This study |
| yfaL_R | 5′-ATGATGGTGATGGTGCCATTTCACCGTCATCGACAAA-3′ | This study |
| aga16B_F | 5′-GAAGGAGATATAAGGATGGATTGGGACGGAATTCCTGTC-3′ | This study |
| aga16B_R | 5′-ATGATGGTGATGGTGACTGCCACCATTACCTGGGG-3′ | This study |
| mRFP1_F | 5′-GAAGGAGATATAAGGATGGCTTCCTCCGAAGACGTTATC-3′ | This study |
| mRFP1_R | 5′-ATGATGGTGATGGTGAGCACCGGTGGAGTGACG-3′ | This study |
| pATLIC948_F | 5′-CCCGGGCCAACGACCGAAAACCTTTACTTCCAGGCTTACCAGCCGGTGTTGAATGC-3′ | This study |
| pATLIC785_F | 5′-CCCGGGCCAACGACCGAAAACCTTTACTTCCAGCTTTCCAACGTGACGGTTAATGGC-3′ | This study |
| pATLIC699_F | 5′-GGCGGCGGGGGTGGGTCGTCACTGTGGGTTGGTGATGG-3′ | This study |
| pATLIC695_F | 5′-CACCACCACCATCATCACGGTGATGTTGCTGATATCCTTCCTTAT-3′ | This study |
| pATLIC29_R | 5′-GTTGGCCCGGGCGCGACACCGTTAGCAGAGAAAA-3′ | This study |
| mRFP129_F | 5′-CGGTGTCGCGCCCGCTTCCTCCGAAGACGTTATC-3′ | This study |
| mRFP1948_R | 5′-CGGTCGTTGGCCCAGCACCGGTGGAGTGACG-3′ | This study |
| mRFP1785_R | 5′-CGGTCGTTGGCCCAGCACCGGTGGAGTGACG-3′ | This study |
| mRFP1699_R | 5′-CCCGCCGCCCCCAGCACCGGTGGAGTGACG-3′ | This study |
| mRFP1692_R | 5′-GTGGTGGTGCCCAGCACCGGTGGAGTGACG-3′ | This study |
| mRFP1(stop)_R | 5′-CGGTCGTTGGCCCTTAAGCACCGGTGGAGTGAC-3′ | This study |
| NABH29-_F | 5′-CGGTGTCGCGCCCAGCGATTCAAAAGTAAATAAAAAATTG-3′ | This study |
| NABH785(tev)_R | 5′-CGGTCGTTGGCCCTACTGCTCCGGAATCGCCTG-3′ | This study |
| NAHB695(sec)_R | 5′-GTGGTGGTGCCCTACTGCTCCGGAATCGCCTG-3′ | This study |
| aga50A29_F | 5′-CGGTGTCGCGCCCGATAAAGACGAGCCGCAAGC-3′ | This study |
| aga50A785_R | 5′-CGGTCGTTGGCCCCTCGCCAAAACGTCGACTATAT-3′ | This study |
| aga16B29_F | 5′-CGGTGTCGCGCCCGATTGGGACGGAATTCCTGTC-3′ | This study |
| aga16B785_R | 5′-CGGTCGTTGGCCCGTTGCTAAGCGTGAACTTATCTAG-3′ | This study |
| aga86C29_F | 5′-CGGTGTCGCGCCCGGGGGCGGTAAATCCTCCAC-3′ | This study |
| aga86C785_R | 5′-CGGTCGTTGGCCCCTGCATAGGACGAGTAATTGAG-3′ | This study |
| aga50D29_F | 5′-CGGTGTCGCGCCCGGTGCAATTGGAGGTCTCGT-3′ | This study |
| aga50D785_R | 5′-CGGTCGTTGGCCCTTTGCTGCCTAGCCTTTCGG-3′ | This study |
| aga86E29_F | 5′-CGGTGTCGCGCCCGCCGATTATGTAATCGAAGCGG-3′ | This study |
| aga86E785_R | 5′-CGGTCGTTGGCCCTCTATTTGGCTCAGAAGTAAATTCC-3′ | This study |
The LIC sequences are underlined. Apr, ampicillin resistance.
CGSC, E. coli Genetic Stock Center; ATCC, American Type Culture Collection.
Sequence analysis and domain recognition.
The protein sequences having similarity to YfaL of E. coli (Uniprot no. P45508) were retrieved from the NCBI GenBank using BLASTP (1). The multiple-sequence alignment was carried out with the ClustalW program (32), and the domain organization of YfaL was determined with the Pfam database and the Conserved Domain Database (CDD) (15, 24). The signal sequence analysis was performed with the SignalP 3.0 server (4).
Cloning, expression, and purification of the YfaL translocator domain.
We cloned the yfaL gene of E. coli DH5α into a modified pBAD vector (pBAD-yfaL) having a six-histidine tag at the carboxy terminus for affinity chromatography purification (21). E. coli BW25113 harboring pBAD-yfaL was grown in 1 liter of LB medium with ampicillin (100 μg/ml) and induced with 0.2% (wt/vol) l-(+)-arabinose (Sigma) at an optical density at 600 nm (OD600) of 0.6 for 12 h at 37°C. The cells were harvested by centrifugation (3,000 × g for 30 min at 4°C), resuspended in 0.1 M Tris-HCl buffer (pH 8.0), and disrupted by sonication for 15 min at 4°C. The crude cell extract was centrifuged at 10,000 × g for 50 min and 4°C to remove the supernatant. The resulting pellet was resuspended in 8 M urea in 20 mM Tris-HCl (pH 8.0) containing 1% (vol/vol) Triton X-100 and centrifuged at 10,000 × g for 50 min at 4°C. The resulting supernatant was then placed on a histidine affinity column (HiTrap HP; GE Healthcare) equilibrated with 20 mM Tris-HCl buffer (pH 8.0) containing 1% (vol/vol) Triton X-100 in an LP system (Bio-Rad). The rate of sample loading and column elution was maintained at 3.0 ml/min by the LP system. The translocator domain of YfaL was eluted by a linear gradient of imidazole (0 to 1 M) included in the same buffer. The amino-terminal sequencing of the purified translocator domain was performed at the Korea Basic Science Institute.
Construction of the LIC vector.
We designed the surface display system by adopting a ligation-independent cloning (LIC) strategy for rapid and reliable cloning (2, 30). Using pBAD-yfaL as a backbone vector, we included a SmaI restriction site at the LIC site, while the predicted passenger domain region of YfaL was removed by PCR with primers listed in Table 1. The constructed LIC vector was designated pATLIC. For controlled release of the displayed protein, the tobacco etch virus (TEV) cleavage site (ENLYFQ) was also added in the cloning site prior to the carboxy terminus of the displayed protein by PCR with primers which have overhangs containing the TEV coding sequence (30). The LIC-ready pATLIC vector was prepared by SmaI (New England BioLabs) digestion (at 37°C for 2 h) followed by T4 DNA polymerase (New England BioLabs) reaction with dATP (Promega) for 30 min at 37°C. The linear LIC-ready vectors were stored at −20°C for further experimentation.
Cloning and expression of various proteins in the pATLIC system.
To examine the size variability and functionality of displayed proteins, we cloned the monomeric red fluorescent protein (mRFP1) (25.3 kDa) (7) and the β-agarases (Aga) and neoagarobiose hydrolase (NABH) of S. degradans 2-40T (Aga50A, 84.8 kDa; Aga16B, 62.3 kDa; Aga86C, 83.7 kDa; Aga50D, 88.3 kDa; Aga86E, 143 kDa; and NABH, 41.5 kDa) (13, 16, 18). We removed the predicted signal sequences in the N termini of Aga50A (positions 1 to 23), Aga16B (1 to 19), Aga86C (1 to 33), Aga50D (1 to 46), and Aga86E (1 to 29) for cloning of those genes into pATLIC. Target genes were amplified using LA-Taq polymerase (TaKaRa) by PCR with primers having overhangs overlapping with the LIC cloning region (the SmaI site) (sequences of primers are given in Table 1). The PCR products were treated with T4 DNA polymerase and dTTP (Promega) for 30 min at 37°C. The linear LIC-ready vector was prepared by SmaI restriction of pATLIC. The vector and insert were mixed at a 1:2 molar ratio and incubated for 30 min at room temperature before direct transformation into E. coli DH5α competent cells prepared by the TSS method (9). All recombinant clones were confirmed by DNA sequencing. Recombinant cells harboring pATLIC of various genes were induced with a final concentration of 0.0002 to 0.2% (wt/vol) l-(+)-arabinose at an OD600 of 0.6. The optimum inducer concentration for the maximum display was determined by the activity of displayed Aga16B. The activity of Aga16B was determined by the amount of reducing sugars released from agarose using the dinitrosalicylic acid (DNS) method (36). Cells were cultured for 24 h at 16°C after induction and harvested by centrifugation (3,000 × g for 10 min at 4°C). The cell pellet was washed with ice-cold 10 mM NaCl and stored at −20°C for a functional analysis.
Functional analysis of the displayed agarases.
Functional activities of five different types of displayed agarases (Aga50A, Aga16B, Aga86C, Aga50D, and Aga86E) were examined on LB agar plates containing 0.2% l-(+)-arabinose as an inducer. After incubation for 24 h at 25°C, the activity of agarases was detected as the hollow zone around colonies after staining with 5% iodine solution. Whole-cell catalysis by the displayed proteins was carried out as follows. The cells displaying agarases were collected after induction at 16°C for 24 h and concentrated 50-fold into the reaction mixture (20 mM Tris-HCl, pH 8.0) with 1.0% agarose for 24 h at 25°C. The activity of NABH was measured with 0.1% neoagarobiose (NB) as a substrate in the same way. After reaction at 25°C for 12 h, the reaction products were analyzed with thin-layer chromatography (TLC) in the solvent system of n-butanol–ethanol–water (3:2:2, vol/vol) and visualized with 10% (vol/vol) H2SO4 and 0.2% naphthoresorcinol in ethanol by heating (12).
Differential cell fractionation for detection of cell surface display.
Differential cell fractionation was performed according to the method described by Li et al. (22). The cells displaying proteins were collected, diluted to approximately 108 CFU/ml, washed in 10 mM NaCl, and resuspended in phosphate-buffered saline (PBS) containing 1 mM EDTA and lysozyme (20-μg/ml final concentration). After 2 h of incubation, the cell suspension was lysed by sonication (approximately 30 to 40% of full power for 2 s) on ice. The whole-cell lysate was centrifuged at 10,000 × g for 10 min at 4°C to remove unbroken cells and large debris. The clarified cell lysate was centrifuged at 100,000 × g for 1 h using an ultracentrifuge (Optima L-90K; Beckman). The resulting supernatant contains the total soluble (cytoplasmic and periplasmic) fraction. The pellet (total membrane fraction) was resuspended in PBS containing 0.01 mM MgCl2 and 2% (vol/vol) Triton X-100 and incubated at room temperature for 20 min. Equal volumes of each fractionated sample were prepared for measuring red fluorescence intensity.
Protease accessibility test for the surface-displayed protein.
To demonstrate the surface display, we used the protease accessibility test for the displayed protein (37). We collected the cells displaying Aga16B and concentrated them 50-fold into PBS buffer. Trypsin stock solution (1 mg/ml trypsin) was added to the suspension to yield a final concentration of 50 μg/ml. The cells were incubated in the reaction mixture for 1 h at 37°C, and the reaction was stopped by washing the cells three times with PBS. Using untreated cells as a control, the trypsin-treated cells were incubated in the reaction mixture (20 mM Tris-HCl, pH 8.0) containing 1.0% agarose for 3 h at 25°C and analyzed by TLC as described above.
Controlled release of the displayed proteins.
A 50-ml portion of cell culture displaying NABH was collected after induction at 16°C for 24 h and resuspended in 1 ml reaction mixture (25 mM Tris-HCl, pH 8.0) containing 50 U of TEV protease (TurboTEV; Eton Bioscience). The reaction mixture was incubated at 4°C for 16 h. The cell-free supernatants were obtained by centrifugation at 10,000 × g for 15 min (4°C), and the activity of the released NABH in the supernatants was examined.
Enumeration of the number of displayed proteins.
The total number of displayed and released proteins was measured by fluorescence of mRFP1. The cell-associated fluorescence was measured by using a Victor3 spectrophotometer (Perkin-Elmer) with excitation at 590 nm (20-nm bandwidth) and emission at 616 nm (8.5-nm bandwidth) in a 96-well plate containing 1 ml of cells displaying mRFP1 (approximately 109 CFU/ml). The background fluorescence of cells was subtracted to obtain the relative fluorescence units (RFU).
RESULTS AND DISCUSSION
Designing a surface display system based on YfaL of E. coli.
Identification of the passenger or translocator domain boundary in YfaL is critical for designing a surface display system because the passenger domain region can be replaced with heterologous proteins. According to the multiple-sequence alignment of YfaL with other autotransporters and domain organization analysis with the Pfam database (Pfam no. PF03797) and the Conserved Domain Database (CDD no. cl02365 and cd01344), we deduced the putative passenger, linker, and translocator domains of YfaL. The signal peptide was predicted as the amino-terminal 28 residues by the SignalP 3.0 sever (4). We designated the region from position 29 to 785 (78 kDa) in YfaL as a passenger domain to be replaced with any protein of interest. The pertactin-like linker domain (CDD no. cd01344) matched the region from position 789 to 916 in YfaL. By these observations, we assigned the region from position 789 to 916 in YfaL as the putative linker between the passenger domain (positions 29 to 785) and translocation β-barrel domain (positions 917 to 1250) (Fig. 1A). Almost all AIDA-I type ATs are known to have conserved regions (e.g., the pertactin-like domain region) including repetitive sequence motifs between the passenger and translocator β-barrel domains (14, 34). The conserved linker region between the passenger and translocator β-barrel domains of BrkA is known to play a crucial role in the translocation or secretion of the passenger domain (11, 26). However, a recent study showed that just a β-barrel domain containing an N-terminally hydrophilic α-helix of ATs is sufficient for cell surface display in E. coli (25). We assumed that a pertactin-like region in YfaL may have a similar function as a linker region in BrkA. We tested the functionality of the predicted linker region (positions 789 to 916) for translocation by the linker region deletion mutants in terms of functional display of a reporter protein (mRFP1). The deletion mutant (missing the region from position 24 to 948) displayed only 33% of the fluorescence of the other mutants having a predicted linker region (see Fig. S1 in the supplemental material). Although the functionality of the linker region was not obvious, we kept the linker region for facilitating translocation of the passenger domain.
Fig 1.

Schematic diagrams for the domain organization of autotransporter (A) and the display system designed in this study (B). (A) Domain organizations of AIDA-I and YfaL of E. coli. The annotated domain names from the Pfam and CDD databases are shown in a separate box. The predicted cleavage sites of passenger domains are indicated by asterisks (between Ser846 and Ala847 for AIDA-I and between Asp696 and Ile697 for YfaL). (B) The designed surface display system, including a TEV cleavage site (pATLIC) (top) or a predicted cleavage site (pATLICsec) (bottom).
Construction of a ligation-independent cloning vector for cell surface display.
We designed a surface display LIC vector having a truncated YfaL missing the passenger domain region (positions 29 to 785) under the control of arabinose-inducible promoter (araBAD promoter). Because the LIC can be applicable to a high-throughput method without restriction and ligation reaction in cloning procedures, the resultant LIC system might be adopted for a library construction for screening engineered proteins by surface display (19). The constructed LIC vector carried a truncated YfaL that is composed of a predicted signal peptide (residues 1 to 28), a putative linker (residues 786 to 916), and a translocator domain (residues 917 to 1250) without the passenger domain. The cloning site exists between the signal peptide and the putative linker (Fig. 1B, top). We also incorporated the TEV protease cleavage site (ENLYFQ) that is fused with the carboxy terminus of the displayed protein for releasing the protein into the medium by TEV cleavage (30). We designated the LIC vector pATLIC. According to the typical LIC procedure (2), we used a 1:2 molar ratio of insert and vector in a total of 30 ng of DNA for transformation of competent cells (transformation efficiency of ∼107 CFU/μg plasmid DNA) and obtained about 120 colonies (the number of colonies of the vector-only sample was 8). The success rate for cloning was about 90% (18 of 20 colonies). This efficiency of the pATLIC system was comparable to those of known LIC systems (5, 6), showing potential for rapid and reliable cloning. An overview of the construction of the pATLIC vector and the typical cloning procedure is shown in Fig. S2 in the supplemental material.
Demonstration of surface display by enzymatic activity, protease accessibility test, and probing localization.
We confirmed the cell surface display by (i) the functionality of the agarase enzyme, (ii) a protease accessibility test of enzymatic activity, and (iii) probing localization by differential cell fractionation of mRFP1. Because agar is a polymer unable to be diffused across membranes of E. coli, β-agarases have been suggested as reporter genes for identification of secretion signal sequences (38). Since only displayed or secreted agarases can show the enzymatic activity and since the aga16B gene coding the β-agarase (Aga16B) from S. degradans is known to secrete into medium (type II protein secretion signal) (13), we cloned Aga16B without its genuine signal peptide into the pATLIC vector (pATLIC-aga16B) and tested the functionality of agarase. To exclude false positives caused by the leakage of enzymes from cell lysis, we used a strain overexpressing active Aga16B in the cytoplasm (pBAD-aga16B) as a negative control. When we patched colonies of E. coli cells harboring pATLIC-aga16B and pBAD-aga16B on LB agar [containing 0.2% l-(+)-arabinose for induction of cloned genes], only cells harboring pATLIC-aga16B showed extracellular agarase activity in the plate (Fig. 2A).
Fig 2.
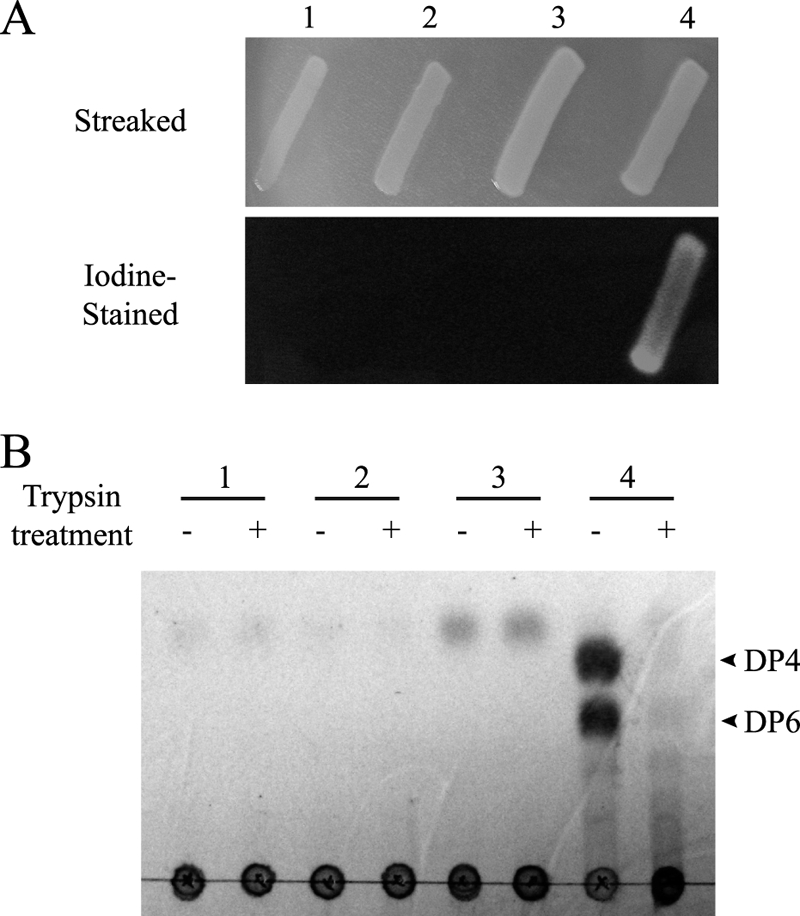
Demonstration of the surface display system by agarase activity and protease accessibility test. (A) Functional activity analysis of the displayed Aga16B. Top, E. coli BW25113 harboring pBAD-yfaL (lane 1), pATLIC void vector (lane 2), pBAD-aga16B for cytosolic expression (lane 3), or pATLIC-aga16B (lane 4) was patched on an agar plate. The pitting of agar by the functionally displayed Aga16B was detected by iodine staining of the plate. pBAD-aga16B was used as a control to exclude false positives caused by the leakage of β-agarase from cell lysis. (B) Protease accessibility test. The agarase (Aga16B) activity of trypsin-treated cells was analyzed by whole-cell catalysis as described in Materials and Methods. Lane 1, pBAD-yfaL; lane 2, pATLIC void vector; lane 3, pBAD-aga16B; lane 4, pATLIC-aga16B. The agar hydrolysis products (DP4, neoagarotetraose; DP6, neoagarohexaose) of Aga16B appeared only in untreated of pATLIC-aga16B cells.
We further verified the surface display by the protease accessibility test (37) by treating the displayed agarase with trypsin. After whole cells were treated with trypsin, the displayed β-agarase activity of the cells disappeared (Fig. 2B). To validate the protease accessibility test by checking whether periplasmic proteins can be accessed by trypsin treatment, we used mRFP1 cloned in pATLIC missing the translocator domain (pATLIC-mRFP1 with AT deletion) as a control and found that trypsin cannot access periplasmic proteins (see Fig. S3 in the supplemental material).
To probe the cellular location of proteins expressed by the pATLIC system, we carried out differential fractionation of cells displaying mRFP1 combined with the protease accessibility test. As shown in Fig. 3, the majority of the whole-cell fluorescence was distributed in the total membrane fraction of cells displaying mRFP1 (pATLIC-mRFP1), while no fluorescence was detected in the membrane fraction of cells expressing cytosolic mRFP1 (pBAD-mRFP1). When cells displaying mRFP1 were treated with trypsin, the fluorescence of the total membrane faction disappeared (data not shown). These results confirm that the protein is not only integrated in the outer membrane but presented to the surface of E. coli.
Fig 3.

Differential cell fractionation of E. coli BW25113 harboring pBAD-mRFP1 (cytosolic mRFP1) or pATLIC-mRFP1 (displayed mRFP1). The relative percentages of red fluorescence intensities for the whole cell (WC), cytoplasm (CP), and total membrane (TM) are shown for cytosolic (pBAD-mRFP1) and displayed (pATLIC-mRFP1) mRFP1.
Functional display and characterization of various agarases.
We cloned four other β-agarases (Aga50A, Aga86C, Aga50D, and Aga86E) from S. degradans into the pATLIC vector (pATLIC-aga50A, -aga86C, -aga50D, and -aga86E, respectively). These agarases belong to glycoside hydrolase (GH) families 50 and 86 (GH50 and -86). We determined the optimal expression condition, changing the inducer [l-(+)-arabinose] concentration from 0% to 0.2% and the growth temperature. Greater amounts of overexpressed proteins were obtained at higher inducer concentrations, but the maximum functional display reached approximately 0.02% of l-(+)-arabinose (see Fig. S4 in the supplemental material). During the expression of various proteins in the surface of E. coli, the pATLIC system had no influence on host cell viability even at the late stationary phase (Fig. 4; see Fig. S4 in the supplemental material).
Fig 4.

The growth of host cells (E. coli BW25113) displaying various proteins was measured by OD600. E. coli BW25113 harboring pBAD void vector (♦), pATLIC void vector (■), pATLIC-agarases (▲), pATLIC-NABH (●), or pATLIC-mRFP1 (×) was grown for 24 h at 16°C. An asterisk indicates the time point of induction with l-(+)-arabinose.
Although Aga86C and Aga86E from S. degradans previously failed to be functionally expressed in E. coli (13), we could functionally display Aga86C and Aga86E in this study. The reaction products of these two agarases were characterized by whole-cell catalysis using the displayed cells. The displayed enzymes (pATLIC-aga86C and -aga86E) completely liquefied solid agarose after 24 h at 25°C. By analysis of the reaction products by TLC (Fig. 5A), we showed that these agarases have different activity profiles on substrates having various degrees of polymerization (DP). As shown in Fig. 5B, all proteins in the pATLIC system showed overexpression in SDS-PAGE, but the enzymatic activity was not proportional to the amount of overexpressed protein (see Fig. S4 in the supplemental material). While pATLIC-aga50D displayed β-agarase activity as previously reported (18), pATLIC-aga50A initially showed no activity on agarose. After changing the substrate from agarose to agaro-oligosaccharides for Aga50A, we found that pATLIC-aga50A had activity on neoagarotetraose (DP4) and neoagarohexaose (DP6), converting them into neoagarobiose (DP2). Consequently, we could display all five agarases of S. degradans in active forms and showed for the first time the activity profiles of three agarases (Aga50A, Aga86C, and Aga86E) that had failed to be actively expressed in E. coli.
Fig 5.
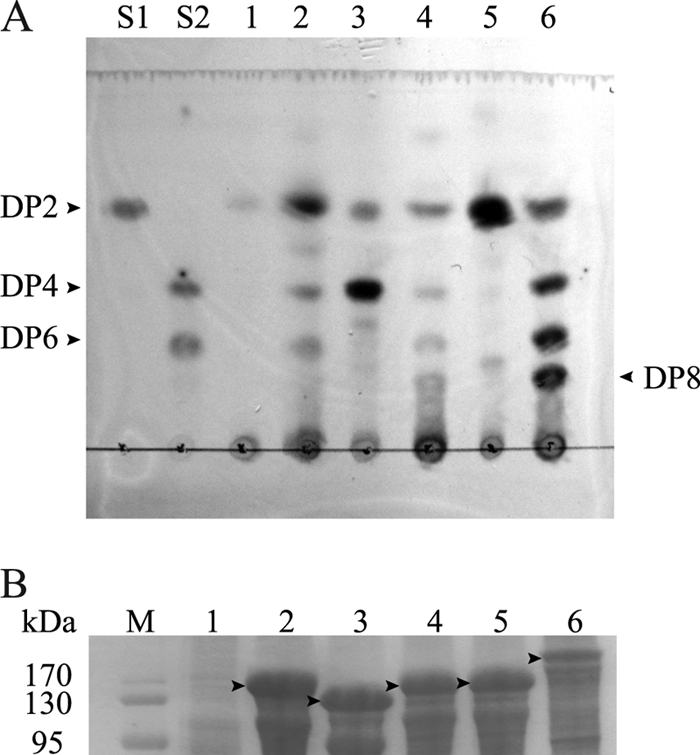
Functional analysis of the surface-displayed β-agarases from Saccharophagus degradans 2-40. (A) TLC chromatogram of the reaction products on agaro-oligosaccharides (pATLIC-aga50A) or agarose (other displayed agarases). Lane S1, DP2 (neoagarobiose); lane S2, DP4 (neoagarotetraose) and DP6 (neoagarohexaose); lane 1, reaction products of pATLIC void vector; lane 2, pATLIC-aga50A; lane 3, pATLIC-aga16B; lane 4, pATLIC-aga86C; lane 5, pATLIC-aga50D; lane 6, pATLIC-aga86E. (B) The arrowheads in the SDS-PAGE indicate the overexpressed β-agarases fused to the translocator domain in the crude extract (lane M, molecular mass marker; other lane numbers are the same as for panel A).
Effect of the size and structural variability of displayed proteins on the pATLIC system.
It is known that the size of the displayed protein is a critical factor for an efficient surface display, and there are often harmful effects on host cells in surface display systems other than the AT system (28). Because the size of the deduced passenger domain in YfaL of E. coli was around 75 kDa, we assumed that the pATLIC system would have be able to display variable sizes of heterologous proteins. We successfully displayed mRFP1 (25.3 kDa) (7), NABH (41.5 kDa), and five agarases (62.3 to 143 kDa). The size of pATLIC is great enough to hold a heterologous protein 1/3-fold (25.3 kDa) to 2-fold (143 kDa) the size of the native passenger domain. The structural folds of the displayed proteins were somewhat variable, but all were globular. mRFP1 is a β-barrel fold (7). The agarases are hydrolase folds representing β-jellyroll (GH16; Aga16B) (http://www.cazy.org/) (8), (β/α)8 barrel (GH50; Aag50A and Aga50D) (http://www.cazy.org/) (8), TIM-(α/β) barrel (GH86; Aga86C and Aga86E) (http://supfam.org/) (35), and five-bladed β-propeller (GH117; NABH) folds. The size and structural versatility of the displayed proteins imply that the pATLIC system can adopt various passenger proteins.
Releasing displayed proteins in the pATLIC system.
The passenger domain in ATs is often cleaved and released from the translocator domain (11). Although the mechanism is not clearly understood, it has been proposed that endo- or exogenous proteolytic activity is involved in the release of the passenger domain from ATs (11). YfaL is also known to process and release the passenger domain, as described by Marani et al. (23), but the exact cleavage site has been unknown. We predicted the cleavage site (between Asp696 and Ile697) in YfaL by sequence comparison to the known cleavage site of the AIDA-I type (31) and confirmed it by amino-terminal amino acid sequencing of the purified truncated 55-kDa translocator domain. To test this cleavage site in YfaL, we constructed a new LIC vector (pATLICSEC) including the putative cleavage site (Fig. 1B). We tested the functionality (cleavage of the displayed protein) of the pATLICSEC with the above agarases. Despite prolonged expression of the pATLICSEC-agarases in E. coli under various conditions, we could not detect the released agarases in the medium in terms of their activity. This observation implies that the release of the passenger domain of YfaL may require the original passenger domain itself or unknown exogenous factors.
We added a TEV cleavage site in front of the linker region in pATLIC that is fused with the carboxy terminus of a displayed protein. We attempted the controlled release of the displayed proteins from the cells using Aga16B from S. degradans. Under the optimal display conditions, 50 ml of cells harboring pATLIC-aga16B (1 × 109 CFU/ml) was treated with TEV protease. By comparison of the enzymatic activity of released Aga16B to the whole-cell activity, we found that 96% of the activity of the initially displayed Aga16B was released from the cells by TEV treatment (Fig. 6A).
Fig 6.

Controlled release of the displayed proteins by TEV protease. (A) Released Aga16B from the cells displaying Aga16B by TEV digestion. The relative activity of the amount of reducing sugars produced in whole-cell activity of untreated cells (UC), treated cells (TC), and the supernatant isolated from the cells displaying Aga16B after the TEV treatment (SU) is shown. (B) TLC analysis of the reaction mixtures for NABH-displaying cells and the released NABH. Lane 1, 3,6-anhydro-l-galactose (l-AHG); lane 2, d-galactose (d-Gal); lane 3, neoagarobiose (DP2); lane 4, reaction products of cells displaying pATLIC-NABH (no TEV protease treatment); lane 5, reaction products of cells displaying pATLIC-NABH (treated with TEV protease); lane 6, reaction products of the supernatant isolated from the cells displaying pATLIC-NABH after TEV protease pretreatment.
In contrast to Aga16B, the displayed neoagarobiose hydrolase (NABH) showed no activity, because NABH is known to function as a dimer (16). Because the enzymatic activity would be recovered by TEV cleavage if the displayed protein could be released from the cells and conform to a dimer, we used NABH as another demonstration of controlled release. After treating the cells displaying NABH with the TEV protease, the NABH activity was detected only in the cell-free supernatant (Fig. 6B), while the culture supernatant of NABH-displaying cells prior to TEV digestion or cells after digestion showed no activity of NABH (see Fig. S5 in the supplemental material). This observation coincided with our assumption that NABH would recover its activity as a dimer in solution after successful release from the cells.
Enumerating the number of displayed proteins on the surface of E. coli.
It might be interesting to know how many proteins can be displayed on the surface of a cell. To investigate the maximum number of displayed proteins on the cell surface of E. coli, we used a reporter protein, monomeric red fluorescent protein (mRFP1).
One milliliter of cells harboring pATLIC-mRFP1 (1 × 109 CFU) was determined to display approximately 2.6 μg of mRFP1 by the RFU curve of mRFP1 (see Fig. S6 in the supplemental material). We calculated that 2.6 × 10−15 g of mRFP1 was displayed in a single cell, which corresponds to 6.1 × 104 copies of mRFP1 per cell based on the equation [(0.0023 × RFm + 0.2505) × NA]/(MWmRFP1 × Ctotal), where RFm is the measured red fluorescence intensity, NA is Avogadro's number, MWmRFP1 is the molecular weight of mRFP1, and Ctotal is the total cell number.
Considering the surface area of a single E. coli cell (4.42 μm2) and the diameters of the β-barrels of ATs with ellipsoid shape described by major and minor axes of approximately 27 Å and 19 Å (3), these numbers of displayed molecules indicated that approximately 5.6% of the surface of a E. coli cell is occupied with the displayed proteins.
Conclusion.
We describe a cell surface display built in the LIC vector system (pATLIC) using a new scaffold protein, YfaL of E. coli. The YfaL system based on AT has the following unique features: (i) heterologous proteins having versatile sizes and structures can be functionally displayed, (ii) functional cell surface display shows no detrimental effect on cell growth, and (iii) the displayed protein can be released from the cell by TEV treatment under appropriate conditions.
The controlled release of protein can be utilized for recovery of recombinant proteins that have problematic expression in the cytoplasm. For instance, if all displayed proteins were cleaved by the TEV treatment, we can release about 2.6 μg/ml of the recombinant mRFP1 in the medium. Once we fused an affinity tag (e.g., a six-histidine tag) to the recombinant protein during the cloning, the displayed protein could be easily recovered from the medium after the controlled release.
Based on these properties and the LIC strategy, the system will be useful for many biotechnological applications requiring high-throughput cloning and library construction, such as screening of functionally improved activity of evolved enzymes in protein engineering. The display system is now under the control of an arabinose-inducible promoter, but we will improve the system to control the number of displayed cells by using fine-tunable promoters. We also will consider the aid of accessory factors such as periplasmic chaperones like Skp (33) and the outer membrane β-barrel assembly machinery (Bam complex) (17) to increase the number of functionally displayed proteins over the levels of protein expression.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intelligent Synthetic Biology Center (2011-0031953) and the Advanced Biomass R&D Center of Korea (2011-0031353) and grants funded by the Korean government (MEST) and GS Caltex Inc. (R1101691).
Footnotes
Published ahead of print 17 February 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2. Aslanidis C, de Jong PJ. 1990. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18:6069–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnard TJ, Dautin N, Lukacik P, Bernstein HD, Buchanan SK. 2007. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat. Struct. Mol. Biol. 14:1214–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783–795 [DOI] [PubMed] [Google Scholar]
- 5. Berrow NS, et al. 2007. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 35:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cabrita LD, Dai W, Bottomley SP. 2006. A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol. 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campbell RE, et al. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99:7877–7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cantarel BL, et al. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung CT, Niemela SL, Miller RH. 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. U. S. A. 86:2172–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daugherty PS. 2007. Protein engineering with bacterial display. Curr. Opin. Struct. Biol. 17:474–480 [DOI] [PubMed] [Google Scholar]
- 11. Dautin N, Bernstein HD. 2007. Protein secretion in gram-negative bacteria via the autotransporter pathway. Annu. Rev. Microbiol. 61:89–112 [DOI] [PubMed] [Google Scholar]
- 12. Duckworth M, Yaphe W. 1970. Thin-layer chromatographic analysis of enzymic hydrolysates of agar. J. Chromatogr. 49:482–487 [DOI] [PubMed] [Google Scholar]
- 13. Ekborg NA, et al. 2006. Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl. Environ. Microbiol. 72:3396–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emsley P, Charles IG, Fairweather NF, Isaacs NW. 1996. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381:90–92 [DOI] [PubMed] [Google Scholar]
- 15. Finn RD, et al. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ha SC, et al. 2011. Crystal structure of a key enzyme in the agarolytic pathway, alpha-neoagarobiose hydrolase from Saccharophagus degradans 2-40. Biochem. Biophys. Res. Commun. 412:238–244 [DOI] [PubMed] [Google Scholar]
- 17. Ieva R, Bernstein HD. 2009. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:19120–19125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim HT, et al. 2010. Overexpression and molecular characterization of Aga50D from Saccharophagus degradans 2-40: an exo-type beta-agarase producing neoagarobiose. Appl. Microbiol. Biotechnol. 86:227–234 [DOI] [PubMed] [Google Scholar]
- 19. Kim YS, Jung HC, Pan JG. 2000. Bacterial cell surface display of an enzyme library for selective screening of improved cellulase variants. Appl. Environ. Microbiol. 66:788–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klauser T, Kramer J, Otzelberger K, Pohlner J, Meyer TF. 1993. Characterization of the Neisseria Iga beta-core. The essential unit for outer membrane targeting and extracellular protein secretion. J. Mol. Biol. 234:579–593 [DOI] [PubMed] [Google Scholar]
- 21. Lee J, Kim SH. 2009. High-throughput T7 LIC vector for introducing C-terminal poly-histidine tags with variable lengths without extra sequences. Protein Expr. Purif. 63:58–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li L, Kang DG, Cha HJ. 2004. Functional display of foreign protein on surface of Escherichia coli using N-terminal domain of ice nucleation protein. Biotechnol. Bioeng. 85:214–221 [DOI] [PubMed] [Google Scholar]
- 23. Marani P, et al. 2006. New Escherichia coli outer membrane proteins identified through prediction and experimental verification. Protein Sci. 15:884–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marchler-Bauer A, et al. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marin E, Bodelon G, Fernandez LA. 2010. Comparative analysis of the biochemical and functional properties of C-terminal domains of autotransporters. J. Bacteriol. 192:5588–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliver DC, Huang G, Nodel E, Pleasance S, Fernandez RC. 2003. A conserved region within the Bordetella pertussis autotransporter BrkA is necessary for folding of its passenger domain. Mol. Microbiol. 47:1367–1383 [DOI] [PubMed] [Google Scholar]
- 27. Roux A, Beloin C, Ghigo JM. 2005. Combined inactivation and expression strategy to study gene function under physiological conditions: application to identification of new Escherichia coli adhesins. J. Bacteriol. 187:1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rutherford N, Mourez M. 2006. Surface display of proteins by gram-negative bacterial autotransporters. Microb. Cell. Fact. 5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Samuelson P, Gunneriusson E, Nygren PA, Stahl S. 2002. Display of proteins on bacteria. J. Biotechnol. 96:129–154 [DOI] [PubMed] [Google Scholar]
- 30. Stols L, et al. 2002. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25:8–15 [DOI] [PubMed] [Google Scholar]
- 31. Suhr M, Benz I, Schmidt MA. 1996. Processing of the AIDA-I precursor: removal of AIDAc and evidence for the outer membrane anchoring as a beta-barrel structure. Mol. Microbiol. 22:31–42 [DOI] [PubMed] [Google Scholar]
- 32. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagner JK, Heindl JE, Gray AN, Jain S, Goldberg MB. 2009. Contribution of the periplasmic chaperone Skp to efficient presentation of the autotransporter IcsA on the surface of Shigella flexneri. J. Bacteriol. 191:815–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wells TJ, Totsika M, Schembri MA. 2010. Autotransporters of Escherichia coli: a sequence-based characterization. Microbiology 156:2459–2469 [DOI] [PubMed] [Google Scholar]
- 35. Wilson D, Madera M, Vogel C, Chothia C, Gough J. 2007. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res. 35:D308–D313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao Z, Storms R, Tsang A. 2004. Microplate-based filter paper assay to measure total cellulase activity. Biotechnol. Bioeng. 88:832–837 [DOI] [PubMed] [Google Scholar]
- 37. Yang TH, Pan JG, Seo YS, Rhee JS. 2004. Use of Pseudomonas putida EstA as an anchoring motif for display of a periplasmic enzyme on the surface of Escherichia coli. Appl. Environ. Microbiol. 70:6968–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang WW, Sun L. 2007. Cloning, characterization, and molecular application of a beta-agarase gene from Vibrio sp. strain V134. Appl. Environ. Microbiol. 73:2825–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.