

Abstract
Single-point mutation in genome, for example, single-nucleotide polymorphism (SNP) or rare genetic mutation, is the change of a single nucleotide for another in the genome sequence. Some of them will produce an amino acid substitution in the corresponding protein sequence (missense mutations); others will not. This paper focuses on genetic mutations resulting in a change in the amino acid sequence of the corresponding protein and how to assess their effects on protein wild-type characteristics. The existing methods and approaches for predicting the effects of mutation on protein stability, structure, and dynamics are outlined and discussed with respect to their underlying principles. Available resources, either as stand-alone applications or webservers, are pointed out as well. It is emphasized that understanding the molecular mechanisms behind these effects due to these missense mutations is of critical importance for detecting disease-causing mutations. The paper provides several examples of the application of 3D structure-based methods to model the effects of protein stability and protein-protein interactions caused by missense mutations as well.
1. Introduction
Human DNA is not identical among individuals, and this causes natural differences among races and ethnic populations, and also among healthy individuals and individuals susceptible to disease. On the DNA sequence level, the differences could be large or small, the smallest being a difference in a single-nucleotide. If such a difference occurs in some fraction of the population, but not in a single case, the difference is termed single nucleotide polymorphism (SNP) [1, 2]. Some of the SNPs occur at the noncoding, while other SNPs happen in the coding regions [3]. The SNPs occurring at the noncoding region does not affect the gene product, that is, the protein sequence is not changed, such types of SNPs are termed silent mutations [4]. However, silent mutation could also be found in the coding region because each amino acid is coded by more than one codon. Thus, even the mutation changes the codon, though it is still possible that the protein sequence is not affected. However, a silent mutation still could affect the function of the cell by altering the gene's expression and regulation.
On the other side of the spectrum are nonsynonymous SNPs (nsSNPs), which cause changes in protein sequence. The most dramatic change is induced by the nonsense mutations, which result in a premature stop codon and produce truncated, usually nonfunctional proteins [4]. Missense mutation, on the other hand, is a change of a single amino acid into another. Such a mutation could be polymorphism if it is observed in significant fraction of the population, or it could be a rare missense mutation if found in an individual or small group of people, as, for example, in a family. In both cases, on protein level, these mutations are termed single-point mutations and they are the primary focus of this paper.
Missense mutations and, in general, nsSNPs were extensively investigated in the past to reveal their plausible effects on protein stability [5–10], protein-protein interactions [11], the characteristics of the active site [5, 6], and many others [12–20]. In parallel, significant efforts were invested to catalog naturally occurring genetic differences, those found in general population and presumably harmless as the SNP database [21–23] and those known to be disease associated as the Online Mendelian Inheritance in Man (OMIM) [24–26]. The OMIM includes the full-text description of disease phenotypes and genes, mapping, molecular genetics, PubMed references, and many other features [26, 27]. OMIM is currently provided by the U.S. National Center for Biotechnology Information (NCBI) [28] and edited by Dr. Victor A. McKusick at John Hopkins University. By 2011, more than 21,000 entries including data and over 13,100 established gene loci and phenotypic descriptions are contained in OMIM Entrez database [28]. SNP database is used to organize and systematize the huge amount of information of gene sequencing. So far, several SNP databases have been developed such as the dbSNP database [21, 29, 30], the Human Genome Variation Database (HGVbase) [22], the Human Gene Mutation Database (HGMD) [31], and the TopoSNP database [23, 32–35].
The existence of such databases combined with available biochemical data of the effects of single-point mutations on protein stability and interactions prompted the development of in silico methods to predict the effects of mutations on the wild-type characteristics of the corresponding proteins or assemblages. Currently, the approaches can be classified into several categories: first principle methods, which calculate the folding or binding free energy change based on detailed atomic models [36–46]; methods based on statistical potentials [47–55] and utilizing known protein structures in the Protein Data Bank [56]; methods using empirical potential combining both physical force fields and free parameters fitted with experimental data [57–65]; machine learning approaches, which are trained against known experimental databases, and then used to predict the effect of the newly found mutations [66–72].
2. Overview of Plausible Effects Induced by Genetic Differences
Genetic differences can potentially affect the function of the cell in a variety of ways, which can be broadly classified into several categories outlined below.
2.1. Active Sites, Reaction Kinetics, and the Reaction Parameters
If a mutation occurs in an active site, then it should be considered lethal since such substitution will affect critical components of the biological reaction, which, in turn, will alter the normal protein function [73, 74]. At the same time, the biochemical reaction is very sensitive to the precise geometry of the active sites for both of the reactants and products; therefore, any conformational change altering the active sites will also affect the biochemical reaction; however, conservative mutations are not expected to perturb protein function by much. Thus, even if the mutation does not occur at the active site, but quite close to it, the characteristics of the catalytic groups will be perturbed [5, 6, 75]. In such a case, the mutation may not completely abolish the biochemical reaction but can change the kinetics of the reaction [76]. Moreover, the biochemical reaction strongly relies on a particular (optimum) cellular environment such as pH, salt concentration, and temperature. Thus in the living cells, the proteins' behavior is controlled by these cellular environments [77, 78]. Changing the reaction rate, the pH, or salt and temperature dependencies away from the native parameters can lead to a malfunctioning protein. The isoelectric point (pI) is a very important parameter that refers to the pH at which the net charge of the protein is zero. Recently, it was demonstrated that five missense mutations involving charged groups in the sodium iodide symporter (NIS) gene, which generates a protein called iodide transporter and is associated with iodide transport defect, can cause an obvious pI shift and influence the electrostatic interactions in the trans-membrane domains of the NIS protein. Even more, these substitutions will probably, in turn, affect the protein stability, protein trafficking, and iodide transport activity [79].
2.2. Kinetics of Protein Folding, Protein Stability, Flexibility, and Aggregation
Protein folding is the process of converting the linear unfolded polypeptide into the native 3D structure driven by the gradient of potential energy [80, 81]. The importance of kinetics of protein folding is manifested by the fact that protein miss-folding is involved in many diseases [12]. An amino acid substitution at a critical folding position can prevent the forming of the folding nucleus, which makes the remainder of the structure rapidly condense [12]. Protein stability is also a key characteristic of a functional protein [5, 6, 71, 82–86], and as such, a mutation on a native protein amino acid can considerably affect its stability [76, 77, 87] through perturbing conformational constraints (e.g., substituting a small side chain residue to a large one and vice versa, resulting in backbone strain or overpacking) or physicochemical effects (substitutions between hydrophilic residues and hydrophobic residues, burial of charged residues, the disruption of hydrogen bonds, loss of hydrogen bonds, of S–S bonds) [88]. It was shown that 80% of missense mutations associated with disease are amino acid substitutions that affect the stability of proteins by several kcal/mol [84]. In addition, the missense mutation can also alter the protein flexibility [5, 89, 90]. When a protein is carrying its function, frequently the reaction requires a small or large conformational change to occur that is specific for the particular biochemical reaction. Thus, if a mutation makes the protein more rigid or flexible compared to the native structure, then it will affect the protein's function [91, 92]. Additionally, conformational flexibility is the main mechanism affecting protein aggregation propensity [93], thus the influence on protein flexibility could cause protein aggregation and formation of fibrils [94].
2.3. Interactions between Protein-Protein, Protein-DNA, Protein-RNA, and Protein-Membrane
If a missense mutation occurs at hot-spots of the binding interface that are crucial in contributing to the interaction [95, 96], then the binding affinity would be dramatically affected due to geometrical constrains and/or energetic effects [7, 97]. For instance, when substituting a small side chain for a bulky side chain in a narrow binding pocket, the entrance of the partner group will be blocked and the binding process will be completely or partially prevented [6, 98–101]. Similarly, a mutation at the protein-DNA interface can affect DNA regulation [13–15]. A mutation occurring at the protein-membrane interface can affect the signal processes across the membrane, protein association with the membrane, and function of various channels and pumps [16, 17].
2.4. Subcellular Localization and Protein Expression
Subcellular localization is a very important factor, which provides a specific environment for protein function, protein interactions, protein activity in signaling pathways, and many other features. Transporting a protein to the correct compartment allows it to form the necessary wild-type interactions with its biological partners and take part in the corresponding biological networks like signaling and metabolic pathways. Otherwise, mislocalizing the protein in a wrong subcellular compartment will have harmful effects on the other proteins which function there [20]. Typically, a mutation affecting the subcellular localization is a mutation that occurs at a signaling region. For example, missense mutations in Otopetrin 1 affects the subcellular location and causes nonsyndromic otoconia agenesis and a subsequent balance defect in mice [102]. Fanconi anemia is a genetic disease associated with the missense mutations in FANCA protein. These missense mutations affect the subcellular localization of the FANCA protein and make it unable to relocate to the nucleus and activate the FA/BRCA pathway [103].
Protein expression is a subcomponent of gene expression and commonly used to denote the measurement of the protein concentration in a particular cell or tissue. Missense mutations can affect DNA-transcription factors resulting in altering the expression of the corresponding protein. Altering the wild-type protein expression in the compartment where it is designed to function will disrupt the normal cell cycle and in turn may cause diseases [20]. Recently, functional analysis of pancreatitis-associated missense mutations was performed in the pancreatic secretory trypsin inhibitor (SPINK1) gene, which encodes pancreatic secretory trypsin inhibitor (PSTI). It was shown that one of the disease-causing missense mutations R65Q reduced protein expression by almost 60%, and four other pathogenic missense mutations G48E, D50E, Y54H, and R67C caused complete or almost complete loss of PSTI expression [104]. By excluding the possibility that reduced transcription or unstable mRNA can lead to reduced protein expression, it was surmised that these disease-causing missense mutations probably cause intracellular retention of their respective mutant proteins. This is suggestive of a potential unifying pathological mechanism underlying both the signal peptide and mature peptide mutations [104].
In this section, we presented the plausible effects which mutations can cause. In fact, mutations often affect the normal protein function by the combined molecular effects listed above [5, 6, 8]. For Instance, in the studies of genotype-phenotype correlations of TGFBI (transforming growth factor, beta-induced) mutations, it was shown that a missense mutation V613G strongly destabilizes the wild-type protein keratoepithelin by 3.1 kcal/mol; additionally, the same mutation might also result in an improper folding due to the backbone structure of the substituted gly is not restricted by the presence of a side chain, thus can adopt any conformation and lead to a misfolded protein. At the same time, it was shown that V613G also facilitates formation of beta-sheet structure of TGFBI which is known to favor amyloid formation [105]. Similarly, another study performed in silico investigation on 18 missense mutations in electron transfer flavoprotein (ETF) associated with multiple acyl-coa dehydrogenase deficiency (MADD), and it was found that these 18 missense mutations can be classified into two groups by their molecular effects: altering protein folding and assembling, affecting the catalytic activity of functional sites, and disrupting interactions with their biological partner, that is, dehydrogenases in this case [106].
3. Methods and Approaches to Predict the Effects of Mutations
Current efforts in this field are aimed at predicting the deleterious mutations since such predictions can be used for diagnostics and drug design. The features used to make such predictions can be classified into three categories: (a) amino acids properties, such as size, side chain polarity, side chain flexibility, and its ability to form a hydrogen bond and other geometrical considerations; (b) 3D protein structural properties such as protein stability, affinity of receptor-ligand complex, and structural flexibility; (c) evolutionary properties like sequence conservation and phylogenetic trees. It is almost impossible to review these approaches one by one since most of the current methodology is using a combination of these features [27]. Table 1 shows several examples for application of molecular modeling methods, free of charge for academia, to study the molecular mechanisms of missense mutations affecting wild-type properties of proteins. Comparison of their performance is provided in references [65, 107]. In the following paragraphs, we explain in detail some of the available resources.
Table 1.
| Methods | Short Summary | Examples (references)* | Some tools based on this method |
|---|---|---|---|
| Molecular dynamics | The trajectories of molecules are determined at atomic level by numerically solving the Newton's equation of motion | (i) Thrombosis-related R2-FV haplotype: D2194G, Coagulation Factor V, domain C2 [8] (ii) Parahemophilia, Factor V new brunswick: A221V, Coagulation Factor V, domain A [9] (iii) FPLD, R482W; Lamin A/C [159] (iv) Intellectual Disability: H101Q; CLIC2 protein [10] (v) Snyder-Robin syndrome: G56S, V132G, I150T; spermine synthase; [5] |
Eris [112, 132, 133] Tinker [158] GROMACS [160] |
|
| |||
| Molecular mechanics | Using molecular mechanics force field and optimization to model molecular systems | (i) 21-Hydroxylase-Deficiency: R132C, R149C, M283V, E431K; CYP450; C21 [161] (ii) Cancer: A159V, A161V, N235I, N239Y, T256I, S269I; p53 [162] (iii) Intellectual Disability: H101Q; CLIC2 protein; [10] (iv) Mutability of human spermine synthase: all amino acids substitution at disease associated missense mutation sites G56, V132, and I150; human spermine synthase [6] (v) Studying effects of nsSNPs on protein-protein interactions: nsSNPs in OMIM and non-OMIM; 264 protein-protein complexes with known nsSNPs located at the interface; [11] |
FoldX [63, 64] |
|
| |||
| Monte Carlo simulation | Applying Monte Carlo sampling to predict preferred conformational states | (i) Noonan syndrome: D61Y, Tyrosine phosphatase SHP-2 [163] | IMC [164] |
|
| |||
| Electrostatic calculation | Calculating electrostatics energy and pKa/ionized states changes/electrostatic stability upon the missense mutations | (i) Snyder-Robinson Syndrome:; G56S, V132G, I150T human spermine synthase [5] (ii) Thrombosis-related R2-FV haplotype: D2194G, Coagulation Factor V, domain C2 [8] (iii) Noonan syndrome: D61Y, Tyrosine phosphatase SHP-2 [163] (iv) Studying effects of nsSNPs on protein-protein interactions: nsSNPs in OMIM and non-OMIM; 264 protein-protein complexes with known nsSNPs located at the interface; [11] |
DelPhi [165] MCCE [166–168] pKD [169] |
|
| |||
| Evolutionary properties | Based on structure and sequence analysis, for example, highly conserved residues in a protein family | (i) Homocystinuria: 204 mutations; cystathionine beta synthase; [170] | SNPs3D [138] PolyPhen [86] |
|
| |||
| Machine learning | learn the behavior of a system based on training datasets | (i) Snyder-Robinson Syndrome: G56S, V132G, I150T; human spermine synthase; [5] (ii) Gastrointestinal stromal tumors: 19 mutations; KIT receptor [171] |
I-Mutant 2.0/3.0 [71, 72, 134] |
|
| |||
| Graph methods | A branch of discrete mathematics. In protein science, this method is used to analyze the topological details of proteins with known structure | (i) Cancer: Y220C, R273H, R273C, R282W, and G245S; p53 protein; [172] (ii) Predicting the structural effects of nsSNPs: 506 disease-associated nsSNPs; [173] |
Bongo [173] |
|
| |||
| Statistical Potential | Based on the knowledge of statistical mechanics such as inverse Boltzmann law, ΔG = −kT log [gij(r)] | (i) Human X-linked Agammaglobulinemia (XLA): 16 missense mutations; Bruton's tyrosine kinase (Btk); [174, 175] (ii) Severe form of phenylketonuria: G46S; human phenylalanine hydroxylase (hPAH); [176] |
DFIRE [55, 177, 178] PoPMuSiC-2.0 [179, 180] CUPSAT [181–183] |
|
| |||
| The BellKor collaborative filtering (CF) algorithm | Model relations of the known data points and the parameters of the model are learnt by the training database | (i) Using the known ΔΔG value to predict the ΔΔG value of other missense mutations at the same substitution site; 4803 mutants were used; [184] | Pro-Maya [184] |
It is essential to identify the most informative features among the features mentioned above for making successful predictions. Such a necessity inspired several works among which a recent study evaluating 32 features using their mutual information together with the functional effects of the amino acid substitutions, as measured by in vivo assays. Sequentially, a greedy algorithm was performed to identify a subset of highly informative features [108]. Finally, it was concluded that two features describing the solvent accessibility of “wild-type” and “mutant” amino-acid residues and another feature of evolutionary properties based on superfamily-level multiple alignments produce the best accuracy [109]. Another investigation developed a formalism and a computational method based on a structural model and phylogenetic information to indicate the effects of amino acid substitution on protein functions. With such a protocol, approximately 26%–32% of naturally occurring missense mutations were predicted to affect the protein functions [110].
The amino acid properties are often considered an important characteristic, which could play a crucial role in protein folding, stability, interaction of protein-protein complexes, and protein function, although sometimes they may be misleading [11]. The side chain properties such as volume, polarity, acidity, basicity, conformational flexibility and the ability to form a hydrogen-bond and salt bridge, are distinguishable. Therefore, the compatibility of a substitution at the dominant allele could be used to make the prediction as it was done in a recent study [111], which combined amino acid properties and structural information to identify deleterious mutations by analyzing the effects on protein stability.
An alternative approach to assess the effect of mutation on protein stability is to evaluate the change of folding free energy ΔG(folding). The difference between ΔG(folding) of the wild-type protein and the mutant, typically described as ΔΔG(folding), is a measure of the effect of mutation on protein stability [5, 6, 64, 65, 107, 112]. If the change in ΔΔG(folding) is negative, then the prediction is that the mutation will destabilize the protein. In contrast, if the calculated change is positive, the mutation is expected to stabilize the protein. The same considerations are valid in the case of predicting the effect on receptor-ligand binding [113]. Numerous investigations were reported in the past to reveal the change in the stability of the native structure [71, 82–86], the macromolecular interactions [11], or altering the wild-type (WT) hydrogen-bond network, in terms of affecting the stability [5, 114, 115]. Currently, several distinctive approaches to predict the protein stability and affinity changes due to mutations have been developed and they can be classified into four categories: (a) first principle methods that using the detailed atomic models to calculate the folding/binding free energy changes caused by mutations [36–46]—these approaches are scientifically sound, but are quite computationally expensive and may not be the best choice in the cases of large sets of mutations [116]; (b) methods based on the statistical potentials [47, 48] were shown to be successful in predicting the change of protein stability upon the mutations [49–55]; (c) Methods utilizing empirical potential, combining both physical force fields, and free parameters fitted with experimental data [57–62]; (d) machine learning methods, utilizing a training database [66–70].
The 3D structure of proteins can be used not only for energy calculations, as described above, but to map mutations onto it and to use geometrical considerations to predict the effects of mutations [117]. Recently, such an approach, the alpha-shape method from computational geometry, was used to divide all nsSNP sites into three categories: (a) Type P: nsSNPs located in a pocket or a void; (b) Type S: nsSNPs occurred on a convex region; and (c) Type I: nsSNP sites are completely buried inside the protein. It was found that 88% of pathogenic nsSNPs are of type P and rarely of type I [32]. Along the same line, 3D structures were used in combinations with machine learning (SVM) and random forest methods. It was demonstrated that these methods outperformed the SIFT algorithm developed by Ng and Henikoff [118], and was indicated that incorporating structural information is crucial to make an accurate prediction if no sufficient evolutionary information is available [119]. Based on the 3D structures, the solvent-accessibility term is also an important feature, which is often used for investigating the effects of missense mutations. It has been shown that using a solvent-accessibility term, the Cβ density, and a score derived from homologous sequences will make the most accurate prediction [120]. Recent studies took into account several protein structural parameters such as solvent accessibility, location within beta strands, or active sites to predict the effects on nsSNPs. It was found that approximately 70% of the disease-associated mutations are buried and solvent inaccessible [121–124] and that such mutations have strong effects on protein structure, folding, stability, and normal function [121, 123].
Another important feature reviewed here is evolutionary properties. Among homologous proteins, the highly conserved residues are generally considered to be critical for protein stability, interaction, and function. One of the evolutionary approaches, which assumes that residues located at a highly conserved position are most likely crucial, is to extract conservation scores from a multiple sequence alignment of homologous proteins. Another widely used computational technique is named the “evolutionary trace” method [125–127]. It uses phylogenetic information based on homologous sequences to rank residues according to evolutionary importance based on their conserved residues in the protein family. After that, such evolutionary conserved residues are mapped on the representative structure. In addition, a group of conserved residues could occur at the interface of a protein-protein complex. Based on the extraction of functionally important residues, an approach was developed utilizing the evolutionary trace method to identify active sites and functional interfaces of proteins based on their available structures. The method was tested on SH2 and SH3 modular signaling domains and the DNA binding domain of the hormone receptors. It was demonstrated this method can delineate the functional epitope and identify the essential residues for binding specificity [128].
In order to train machine learning algorithms properly and to have a benchmarking case, appropriate databases are required. A particular example is the Catalytic Site Atlas database [129], which collects 177 original hand-annotated entries and 2608 homologous entries and covers about 30% of all enzyme in the Protein Data Bank [56]. At the same time, the computational methods of predicting the functional residues were also well-developed [125, 130, 131]. A selection of structure and sequence-based features was used to indicate an amino acid polymorphism effect on protein function, and it was found that ~26%–32% of the naturally occurring nsSNPs will affect the protein's function [110].
4. Webservers for Analyzing the Effects of Mutations
In past years, several methods were implemented into webservers to predict the effects on protein stability due to mutations. The Eris webserver is based on Medusa force field [132], and it was benchmarked on 595 mutants with available experimental data resulting in RMSD 2.4 kcal/mol between the predicted ΔΔGcal (folding) and corresponding experimental values (ΔΔGexp (folding)) [112, 132, 133]. The FoldX is perhaps the most popular web server [63] for predicting the folding free energy changes due to the mutations, and it is based on the empirical potentials [64]. The I-Mutant 2.0/3.0 is Support Vector Machine-based (SVM: a machine learning method) webserver utilizing the 3D structural or sequential information to predict protein stability change upon single-point mutations [71, 72, 134]. Other webservers include the Site Directed Mutator (SDM) [54] and the Mupro method [135].
In parallel, there are webservers predicting the effects of mutations on protein-protein interaction. The COILCHECK is an interactive webserver, which measures the strength of interactions between two helices involved in coiled coil structures utilizing nonbonded and electrostatic interactions and the presence of hydrogen bonds and salt bridges. It can be used to assess the strength of coiled coil regions, to recognize weak and strong regions, to rationalize the phenotypic behavior of single mutations and to design mutation experiments [136]. Recently, DrugScorePPI was reported, which is a fast and accurate computational approach to predict impacts on binding affinity by the change of the binding free energy upon alanine mutations at protein-protein interfaces. The primary motivation of developing this webserver is to identify hotspot residues at protein-protein interfaces, which will guide both biological experiments and the development of protein-protein interaction modulators [137].
There are many webservers Which are designed to predict if the mutation is pathogenic or not without providing information about the magnitude of expected energy changes. The SNPs3D [138] is a primary resource and database, which provides various disease/gene relationships at the molecular level. This server has three modules: (a) identifying the gene candidates involved in a specific disease; (b) relationships between the sets of candidate genes; and (c) analyzing the possible effects of nsSNPs on normal protein function. It is very convenient for the users to quickly obtain the available information and so develop models of gene-pathway-disease interaction. Another online predictor of molecular and structural effects of protein-coding variants was recently developed, the SNPeffect 4.0 [139]. It uses sequence- and structure-based bioinformatics tools such as aggregation prediction (TANGO) [140], amyloid prediction (WALTZ) [141], chaperone-binding prediction (LIMBO) [142], and protein stability analysis (FoldX) [63] to predict the effect of SNPs. In addition, it also contains the information of effects on catalytic sites, posttranslational modifications, and all known human protein variants from Uniprot. At the same time, SNPeffect allows users to submit custom protein variants for analyzing the SNP effects and plot correlations between phenotypic features for a user-selected set of variants [139]. The dbSNP database in NCBI lists over 9 million SNPs in the human genome but includes very limited annotation information. To fill this gap, the LS-SNP was developed to annotate the nsSNPs [83]. It can map nsSNPs onto protein sequences, functional pathways, and comparative protein structure models and predicts the positions where nsSNPs cause the effects. The results can be used to find out the functional SNP candidates within a gene, haplotype, or pathway, and also in understanding the molecular mechanisms responsible for functional effects of nsSNPs [83]. At the same time, a protocol based on Sorting Intolerant From Tolerant (SIFT) [118] was reported to predict if a missense mutation will affect the protein function. To assess the effects of a missense mutation, SIFT utilizes evolutionary properties of the protein and considers the substitutions at the conserved positions which may affect protein function. Thus, SIFT makes a prediction on effects of all possible substitutions at each position in the protein sequence by using sequence homology [143]. The Polyphen (Polymorphism Phenotype) is a tool that predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations. It combines a variety of features such as sequences, evolutionary properties, and structural information to predict if an nsSNP will affect the protein function and performs optimally if the structural information is available. More than 11000 nsSNPs are annotated by this webserver [86]. A new version of Polyphen, namely, Polyphen-2, was recently released [144]. Its features include high quality multiple sequence alignment pipeline and probabilistic classifier based on a machine-learning method, and it is optimized for high-throughput analysis of the next-generation sequencing data [144]. After the development of SIFT and Polyphen, the Parepro (Predicating the amino acid replacement probability) was created, based on two independent databases HumVar and NewHumVar. The predictions are if an nsSNP will be either deleterious or will have no effect on protein function. Compared to SIFT and Polyphen, Parepro achieved a higher Matthews correlation coefficient (MCC) and overall accuracy (Q2) when predications were made using a 20-fold cross validation test on the HumVar dataset [145]. StSNP is a webserver referencing the data from dbSNP in NCBI, the gene and protein database from Entrez, the protein structures from the PDB, and pathway information from KEGG and makes an effort to provide combined, integrated reports about nsSNPs. Researchers can use the metabolic pathways in StSNP to examine the likely relationship between the disease-related pathways and particular nsSNPs, and link the disease with the current available molecular structure data [146]. AUTO-MUTE is a knowledge-based computational mutagenesis used to predict the disease potential of human nsSNPs. In this study, 1790 neutral and disease-associated human nsSNPs on 243 diverse human protein structures were used. With a trained model, this method achieves 76% cross-validation accuracy [147].
5. Application of Structure-Based Methods to Predict the Effects of Mutations on Protein Stability and Protein-Protein Interactions
In this section, we outline several examples of utilizing structural information to predict the effects of mutations on wild-type characteristics of proteins and protein complexes.
5.1. Application of Molecular Dynamics (MD) Simulation for Predicting the Effects of Mutations
Coagulation factor V (FV) is the precursor of an essential procoagulant cofactor that accelerates FXa-catalyzed prothrombin activation in the coagulation system. It is a large glycoprotein containing several domains, A1-A2-B-A3-C1-C2 [8]. A missense mutation D2194G in its C2 domain was shown to cause low expression level and to have plausible effect on stability of the corresponding protein. To investigate the molecular mechanism of D2194G affecting the wild type of the corresponding protein, MD simulations were carried out on both of the WT and mutant structure to reveal the flexibility change upon this mutation [8]. The program CHARMm [148] was used, and the total simulation time was 900 ps. The root mean square fluctuations (RMSFs) for the α-carbon atoms of the C2 domain per residue were calculated for series of snapshots. The comparison for the WT and mutant structures is shown in Figure 1(a). It was concluded that the regions 2075–2085 and 2140–2150 in both WT structure and mutant structures are flexible. The loop 2042–2053 (Figure 1(b)) which is close to the mutation site, is more flexible in the mutant structure. At the same time, loop 2060–2067 became more flexible in the mutant as well, and this effect was attributed to the increased mobility of the loop 2042–2053. The substitution of Asp for Gly will lead to a big cavity and the nonflexible C-terminus (Tyr2196) inserts itself into the domain and attempts to fill out this cavity and to compensate for the missing negative charge of the mutant. These events could be the reason for the enhancing flexibility of the loop 2042–2053.
Figure 1.

(a) The structural and flexibility differences between the simulated WT and mutant structures. The black line represents the RMSF of the WT structure and the red line represents the mutant protein. (b) 3D structure of the C2 domain of the WT FV. The S–S bond is marked in yellow and the loop 2042–2053 is indicated by the arrow.
5.2. Application of Energy Calculation for Predicting the Effects of Mutations in Human Spermine Synthase
In this section, we describe the molecular mechanism of three missense mutations in human spermine synthase (SMS) causing Snyder-Robin Syndrome (SRS) [149–151] to demonstrate application of structure-based methods and energy calculation to predict the effects on protein stability and protein-protein interaction [5].
SMS (OMIM: 300105) is an enzyme converting spermidine (SPD) into spermine (SPM) both of which are two polyamines controlling normal mammalian cell growth and development [152–155]. The importance of SMS for the normal function is illustrated by the fact that three clinical missense mutations, c.267G > A (p.G56S) [150], c.496T > G (p.V132G) [151], and I150T [5], on SMS will cause an X-Linked mental retardation disorder named SRS (OMIM: 309583). At the same time, the 3D structures of human SMS with either the substrates SPD or product SPM have been experimentally determined [156]. The 3D structure of SMS with the substrates SPD and product MTA (PDB ID: 3C6K) is shown in Figure 2. SMS contains two subunits forming a dimer, and each subunit includes two terminal domains: the N-terminal domain which plays a key role in dimerization and the C-terminal domain which includes the active site. The importance of dimerization for SMS function was also demonstrated by series of deletion experiments in vitro [156]. Additionally, two missense mutations G56S and V132G are located at the dimer interface, while the other missense mutation I150T occurred at the C-terminal domain and quite close to the active sites.
Figure 2.
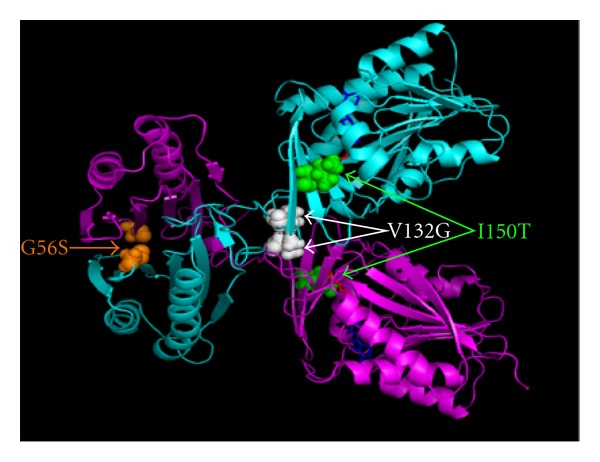
3D structure of human SMS with three missense mutation sites. Two subunits were represented by ribbon in cyan and magenta. Three mutation sites were shown with sphere representation: G56S in orange, V132G in white and I150T in green. The substrates of SPD and MTA were shown in red sticks and blue sticks, respectively.
These three mutants were made in silico by SCAP, a program in JACKAL package [157], based on the native 3D SMS structure. Then, the TINKER package was used to perform the energy minimization and calculation [158]. It was shown that the missense mutation G56S will strongly decrease the dimer affinity by nearly 14 kcal/mol, but the other two have no impact on it. With the analysis based on the 3D structure, it was concluded the reason that G56S strongly decreases the dimerization is because the side chain of Ser in the mutant is pointing to the dimer interface, and there is no enough room to harbor this side chain (Figure 3(a)). In contrast, while the mutation V132G is located at the dimer interface as well, the side chain of Val in the native structure does not point towards the interface and is close to large cavity (Figure 3(b)); thus this substitution can be accommodated easily without introducing any steric constrains. The third mutation, I150T, is very far away from the dimer interface (Figure 2); thus it is not supposed to affect the dimerization.
Figure 3.

Effects on dimerization. (a) G56S: we superimposed WT structure (presented with two chains in white) and mutant structure (presented with Only one chain in green). Cyan stick represented Gly in the WT structure and magenta stick represented Ser in the mutant structure; (b) V132G: Only the region around the mutation site was shown in the figure. We superimpose the WT structure (presented with two chains in white) and mutant (presented with only one chain in green). The orange stick represented Val in the WT structure and red stick represented Gly in the mutant structure.
With regards the folding energy calculation, all these missense mutations are predicted to destabilize the protein monomer by 2.8 kcal/mol (G56S), 1.1 kcal/mol (V132G), and 3.5 kcal/mol (I150T), respectively. Figure 4(a) gives the comparison of the native structure and mutant structure and is zoomed into the mutation site G56S. In the mutant structure, we can see this mutation occurs in a sharp turn, and the substitution with almost any other amino acid will introduce strain. Figure 4(b) shows the superposition of the native structure and mutant, zoomed in the mutation site V132G. It is clear that the side chain of Val points to the interior, thus the substitution with Gly will leave a big cavity inside the monomer, which in turn will affect the stability. In addition, considering the physicochemical property feature, Val and Gly have different hydrophobicity. The destabilization by I150T is mainly attributed the totally different physicochemical properties between Ile, which is a hydrophobic residue, and Thr, which is a hydrophilic residue.
Figure 4.

Effects on monomer stability. (a) G56S: N-terminal domain of both WT monomer (white) and mutant monomer (green) are superimposed. Cyan stick represented Gly in the WT structure and magenta represented Ser in the mutant structure; (b) V132G: C-terminal domain of both WT monomer (white) and mutant monomer (green) are superimposed. We use stick and ball representation in orange to represent Val in the WT structure and in red to represent Gly in the mutant structure.
Thus, combining the 3D structure, physicochemical properties of amino acids, and energy calculations, it was shown that one can successfully predict molecular effects due to these three missense mutations. Such an analysis helps better understand how these missense mutations affect the SMS function and in turn reveal the molecular origin of SRS.
6. Conclusion
In this paper, we outlined the current state-of-the-art methods in the field of computational modeling of effects of nsSNPs and rare missense mutations. Available resources are pointed out along with short description of their functionality and accuracy. The basic concepts and major research directions are described and their advantages and disadvantages discussed.
Acknowledgments
The authors thank Nicholas Smith for proof reading the paper. The work of Z. Zhang and E. Alexov was supported in part by NIH, NLM grant, Grant no. 1R03LM009748. Z. Zhang thanks “Chateaubriand Fellowship,” which is supported by the Embassy of France in the USA.
References
- 1.Taillon-Miller P, Gu Z, Li Q, Hillier L, Kwok PY. Overlapping genomic sequences: a treasure trove of single-nucleotide polymorphisms. Genome Research. 1998;8(7):748–754. doi: 10.1101/gr.8.7.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mooney S. Bioinformatics approaches and resources for single nucleotide polymorphism functional analysis. Briefings in Bioinformatics. 2005;6(1):44–56. doi: 10.1093/bib/6.1.44. [DOI] [PubMed] [Google Scholar]
- 3.Hagmann M. Human genome. A good SNP may be hard to find. Science. 1999;285(5424):21–22. doi: 10.1126/science.285.5424.21a. [DOI] [PubMed] [Google Scholar]
- 4.Strachan T, Read AP. Human Molecular Genetics . 1999. [Google Scholar]
- 5.Zhang Z, Teng S, Wang L, Schwartz CE, Alexov E. Computational analysis of missense mutations causing Snyder-Robinson syndrome. Human Mutation. 2010;31(9):1043–1049. doi: 10.1002/humu.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Norris J, Schwartz C, Alexov E. In silico and in vitro investigations of the mutability of disease-causing missense mutation sites in spermine synthase. PLoS One. 2011;6(5) doi: 10.1371/journal.pone.0020373. Article ID e20373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akhavan S, Miteva MA, Villoutreix BO, et al. A critical role for Gly25 in the B chain of human thrombin. Journal of Thrombosis and Haemostasis. 2005;3(1):139–145. doi: 10.1111/j.1538-7836.2004.01086.x. [DOI] [PubMed] [Google Scholar]
- 8.Miteva MA, Brugge JM, Rosing J, Nicolaes GAF, Villoutreix BO. Theoretical and experimental study of the D2194G mutation in the C2 domain of coagulation factor V. Biophysical Journal. 2004;86(1, part 1):488–498. doi: 10.1016/S0006-3495(04)74127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steen M, Miteva M, Villoutreix BO, Yamazaki T, Dahlback B. Factor V new brunswick: Ala221Val associated with FV deficiency reproduced in vitro and functionally characterized. Blood. 2003;102(4):1316–1322. doi: 10.1182/blood-2003-01-0116. [DOI] [PubMed] [Google Scholar]
- 10.Witham S, Takano K, Schwartz C, Alexov E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins. 2011;79(8):2444–2454. doi: 10.1002/prot.23065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teng S, Madej T, Panchenko A, Alexov E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophysical Journal. 2009;96(6):2178–2188. doi: 10.1016/j.bpj.2008.12.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobson CM. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 13.Venkatesan RN, Treuting PM, Fuller ED, et al. Mutation at the polymerase active site of mouse DNA polymerase δ increases genomic instability and accelerates tumorigenesis. Molecular and Cellular Biology. 2007;27(21):7669–7682. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elles LMS, Uhlenbeck OC. Mutation of the arginine finger in the active site of Escherichia coli DbpA abolishes ATPase and helicase activity and confers a dominant slow growth phenotype. Nucleic Acids Research. 2008;36(1):41–50. doi: 10.1093/nar/gkm926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright JD, Lim C. Mechanism of DNA-binding loss upon single-point mutation in p53. Journal of Biosciences. 2007;32(5):827–839. doi: 10.1007/s12038-007-0083-3. [DOI] [PubMed] [Google Scholar]
- 16.Kwa LG, Wegmann D, Brugger B, Wieland FT, Wanner G, Braun P. Mutation of a single residue, β-glutamate-20, alters protein-lipid interactions of light harvesting complex II. Molecular Microbiology. 2008;67(1):63–77. doi: 10.1111/j.1365-2958.2007.06017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kariya Y, Tsubota Y, Hirosaki T, et al. Differential regulation of cellular adhesion and migration by recombinant laminin-5 forms with partial deletion or mutation within the G3 domain of α3 chain. Journal of Cellular Biochemistry. 2003;88(3):506–520. doi: 10.1002/jcb.10350. [DOI] [PubMed] [Google Scholar]
- 18.Tiede S, Cantz M, Spranger J, Braulke T. Missense mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTA) in a patient with mucolipidosis II induces changes in the size and cellular distribution of GNPTG. Human Mutation. 2006;27(8):830–831. doi: 10.1002/humu.9443. [DOI] [PubMed] [Google Scholar]
- 19.Krumbholz M, Koehler K, Huebner A. Cellular localization of 17 natural mutant variants of ALADIN protein in triple A syndrome—shedding light on an unexpected splice mutation. Biochemistry and Cell Biology. 2006;84(2):243–249. doi: 10.1139/o05-198. [DOI] [PubMed] [Google Scholar]
- 20.Hanemann CO, D’Urso D, Gabreels-Festen AAWM, Muller HW. Mutation-dependent alteration in cellular distribution of peripheral myelin protein 22 in nerve biopsies from Charcot-Marie-Tooth type 1A. Brain. 2000;123(5):1001–1006. doi: 10.1093/brain/123.5.1001. [DOI] [PubMed] [Google Scholar]
- 21.Sherry ST, Ward MH, Kholodov M, et al. DbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fredman D, Munns G, Rios D, et al. HGVbase: a curated resource describing human DNA variation and phenotype relationships. Nucleic Acids Research. 2004;32:D516–D519. doi: 10.1093/nar/gkh111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stitziel NO, Binkowski TA, Tseng YY, Kasif S, Liang J. topoSNP: a topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic Acids Research. 2004;32(Database issue):D520–D522. doi: 10.1093/nar/gkh104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online mendelian inheritance in man (OMIM) Human Mutation. 2000;15(1):57–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 25.Hamosh A, Scott AF, Amberger J, Bocchini C, Valle D, McKusick VA. Onlined mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Research. 2002;30(1):52–55. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Research. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teng S, Michonova-Alexova E, Alexov E. Approaches and resources for prediction of the effects of non-synonymous single nucleotide polymorphism on protein function and interactions. Current Pharmaceutical Biotechnology. 2008;9(2):123–133. doi: 10.2174/138920108783955164. [DOI] [PubMed] [Google Scholar]
- 28.Sayers EW, Barrett T, Benson DA, et al. Database resources of the national center for biotechnology information. Nucleic Acids Research. 2011;39(Database issue) supplement 1:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Research. 2000;28(1):352–355. doi: 10.1093/nar/28.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Research. 1999;9(8):677–679. [PubMed] [Google Scholar]
- 31.Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Research. 1998;26(1):285–287. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stitziel NO, Tseng YY, Pervouchine D, Goddeau D, Kasif S, Liang J. Structural location of disease-associated single-nucleotide polymorphisms. Journal of Molecular Biology. 2003;327(5):1021–1030. doi: 10.1016/s0022-2836(03)00240-7. [DOI] [PubMed] [Google Scholar]
- 33.Altman RB. PharmGKB: a logical home for knowledge relating genotype to drug response phenotype. Nature Genetics. 2007;39(4, article 426) doi: 10.1038/ng0407-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Packer BR, Yeager M, Burdett L, et al. SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic acids research. 2006;34:D617–D621. doi: 10.1093/nar/gkj151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bidwell J, Keen L, Gallagher G, et al. Cytokine gene polymorphism in human disease: on-line databases. Genes and Immunity. 1999;1(1):3–19. doi: 10.1038/sj.gene.6363645. [DOI] [PubMed] [Google Scholar]
- 36.Bash PA, Singh UC, Langridge R, Kollman PA. Free energy calculations by computer simulation. Science. 1987;236(4801):564–568. doi: 10.1126/science.3576184. [DOI] [PubMed] [Google Scholar]
- 37.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282(5389):740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 38.Khare SD, Caplow M, Dokholyan NV. FALS mutations in Cu, Zn superoxide dismutase destabilize the dimer and increase dimer dissociation propensity: a large-scale thermodynamic analysis. Amyloid. 2006;13(4):226–235. doi: 10.1080/13506120600960486. [DOI] [PubMed] [Google Scholar]
- 39.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C. Testing homology modeling on mutant proteins: predicting structural and thermodynamic effects in the Ala→Val mutants of T4 lysozyme. Folding and Design. 1996;1(1):1–12. doi: 10.1016/S1359-0278(96)00006-5. [DOI] [PubMed] [Google Scholar]
- 41.Lee C, Levitt M. Accurate prediction of the stability and activity effects of site-directed mutagenesis on a protein core. Nature. 1991;352(6334):448–451. doi: 10.1038/352448a0. [DOI] [PubMed] [Google Scholar]
- 42.Miyazawa S, Jernigan RL. Protein stability for single substitution mutants and the extent of local compactness in the denatured state. Protein Engineering. 1994;7(10):1209–1220. doi: 10.1093/protein/7.10.1209. [DOI] [PubMed] [Google Scholar]
- 43.Pitera JW, Kollman PA. Exhaustive mutagenesis in silico: multicoordinate free energy calculations on proteins and peptides. Proteins. 2000;41(3):385–397. [PubMed] [Google Scholar]
- 44.Prevost M, Wodak SJ, Tidor B, Karplus M. Contribution of the hydrophobic effect to protein stability: analysis based on simulations of the Ile-96→Ala mutation in barnase. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(23):10880–10884. doi: 10.1073/pnas.88.23.10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tidor B. Simulation analysis of the stability mutant R96h of T4 lysozyme. Biochemistry. 1991;30(13):3217–3228. doi: 10.1021/bi00227a009. [DOI] [PubMed] [Google Scholar]
- 46.Vorobjev YN, Hermans J. ES/IS: estimation of conformational free energy by combining dynamics simulations with explicit solvent with an implicit solvent continuum model. Biophysical Chemistry. 1999;78(1-2):195–205. doi: 10.1016/s0301-4622(98)00230-0. [DOI] [PubMed] [Google Scholar]
- 47.Ben-Naim A. Statistical potentials extracted from protein structures: are these meaningful potentials? Journal of Chemical Physics. 1997;107(9):3698–3706. [Google Scholar]
- 48.Thomas PD, Dill KA. Statistical potentials extracted from protein structures: how accurate are they? Journal of Molecular Biology. 1996;257(2):457–469. doi: 10.1006/jmbi.1996.0175. [DOI] [PubMed] [Google Scholar]
- 49.Gilis D, Rooman M. Stability changes upon mutation of solvent-accessible residues in proteins evaluated by database-derived potentials. Journal of Molecular Biology. 1996;257(5):1112–1126. doi: 10.1006/jmbi.1996.0226. [DOI] [PubMed] [Google Scholar]
- 50.Gilis D, Rooman M. Predicting protein stability changes upon mutation using database-derived potentials: solvent accessibility determines the importance of local versus non-local interactions along the sequence. Journal of Molecular Biology. 1997;272(2):276–290. doi: 10.1006/jmbi.1997.1237. [DOI] [PubMed] [Google Scholar]
- 51.Gilis D, Rooman M. PoPMuSiC, an algorithm for predicting protein mutant stability changes. Application to prion proteins. Protein Engineering. 2000;13(12):849–856. doi: 10.1093/protein/13.12.849. [DOI] [PubMed] [Google Scholar]
- 52.Hoppe C, Schomburg D. Prediction of protein thermostability with a direction- and distance-dependent knowledge-based potential. Protein Science. 2005;14(10):2682–2692. doi: 10.1110/ps.04940705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ota M, Isogai Y, Nishikawa K. Knowledge-based potential defined for a rotamer library to design protein sequences. Protein Engineering. 2001;14(8):557–564. doi: 10.1093/protein/14.8.557. [DOI] [PubMed] [Google Scholar]
- 54.Topham CM, Srinivasan N, Blundell TL. Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Engineering. 1997;10(1):7–21. doi: 10.1093/protein/10.1.7. [DOI] [PubMed] [Google Scholar]
- 55.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Science. 2002;11(11):2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Research. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bordner AJ, Abagyan RA. Large-scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins. 2004;57(2):400–413. doi: 10.1002/prot.20185. [DOI] [PubMed] [Google Scholar]
- 58.Domingues H, Peters J, Schneider KH, et al. Improving the refolding yield of interleukin-4 through the optimization of local interactions. Journal of Biotechnology. 2000;84(3):217–230. doi: 10.1016/s0168-1656(00)00327-8. [DOI] [PubMed] [Google Scholar]
- 59.Munoz V, Serrano L. Development of the multiple sequence approximation within the AGADIR Model of α-helix formation: comparison with zimm-bragg and lifson-roig formalisms. Biopolymers. 1997;41(5):495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 60.Takano K, Ota M, Ogasahara K, Yamagata Y, Nishikawa K, Yutani K. Experimental verification of the “stability profile of mutant protein” (SPMP) data using mutant human lysozymes. Protein Engineering. 1999;12(8):663–672. doi: 10.1093/protein/12.8.663. [DOI] [PubMed] [Google Scholar]
- 61.Verma D, Jacobs DJ, Livesay DR. Predicting the melting point of human C-type lysozyme mutants. Current Protein and Peptide Science. 2010;11(7):562–572. doi: 10.2174/138920310794109210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villegas V, Viguera AR, Aviles FX, Serrano L. Stabilization of proteins by rational design of α-helix stability using helix/coil transition theory. Folding and Design. 1996;1(1):29–34. [PubMed] [Google Scholar]
- 63.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Research. 2005;33(2):W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. Journal of Molecular Biology. 2002;320(2):369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Z, Wang L, Gao Y, Zhang J, Zhenirovskyy M, Alexov E. Predicting folding free energy changes upon single point mutations. Bioinformatics. 2012;28(5):664–671. doi: 10.1093/bioinformatics/bts005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Capriotti E, Fariselli P, Casadio R. A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics. 2004;20(supplement 1):i63–i68. doi: 10.1093/bioinformatics/bth928. [DOI] [PubMed] [Google Scholar]
- 67.Casadio R, Compiani M, Fariselli P, Vivarelli F. Predicting free energy contributions to the conformational stability of folded proteins from the residue sequence with radial basis function networks. Proceedings International Conference on Intelligent Systems for Molecular Biology. 1995;3:81–88. [PubMed] [Google Scholar]
- 68.Frenz CM. Neural network-based prediction of mutation-induced protein stability changes in staphylococcal nuclease at 20 residue positions. Proteins. 2005;59(2):147–151. doi: 10.1002/prot.20400. [DOI] [PubMed] [Google Scholar]
- 69.Joachims T. Learning to Classify Text Using Support Vector Machines. Springer; 2002. [Google Scholar]
- 70.Masso M, Vaisman II. Accurate prediction of stability changes in protein mutants by combining machine learning with structure based computational mutagenesis. Bioinformatics. 2008;24(18):2002–2009. doi: 10.1093/bioinformatics/btn353. [DOI] [PubMed] [Google Scholar]
- 71.Capriotti E, Fariselli P, Calabrese R, Casadio R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics. 2005;21(supplement 2):ii54–ii58. doi: 10.1093/bioinformatics/bti1109. [DOI] [PubMed] [Google Scholar]
- 72.Capriotti E, Fariselli P, Casadio R. I-mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Research. 2005;33(2):W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamada Y, Banno Y, Yoshida H, et al. Catalytic inactivation of human phospholipase D2 by a naturally occurring Gly901Asp mutation. Archives of Medical Research. 2006;37(6):696–699. doi: 10.1016/j.arcmed.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 74.Stevanin G, Hahn V, Lohmann E, et al. Mutation in the catalytic domain of protein kinase C γ and extension of the phenotype associated with spinocerebellar ataxia type 14. Archives of Neurology. 2004;61(8):1242–1248. doi: 10.1001/archneur.61.8.1242. [DOI] [PubMed] [Google Scholar]
- 75.Takamiya O, Seta M, Tanaka K, Ishida F. Human factor VII deficiency caused by S339C mutation located adjacent to the specificity pocket of the catalytic domain. Clinical and Laboratory Haematology. 2002;24(4):233–238. doi: 10.1046/j.1365-2257.2002.00449.x. [DOI] [PubMed] [Google Scholar]
- 76.Koukouritaki SB, Poch MT, Henderson MC, et al. Identification and functional analysis of common human flavin-containing monooxygenase 3 genetic variants. Journal of Pharmacology and Experimental Therapeutics. 2007;320(1):266–273. doi: 10.1124/jpet.106.112268. [DOI] [PubMed] [Google Scholar]
- 77.de Cristofaro R, Carotti A, Akhavan S, et al. The natural mutation by deletion of Lys9 in the thrombin A-chain affects the PKa value of catalytic residues, the overall enzyme’s stability and conformational transitions linked to Na+ binding. The FEBS Journal. 2006;273(1):159–169. doi: 10.1111/j.1742-4658.2005.05052.x. [DOI] [PubMed] [Google Scholar]
- 78.Alexov E. Numerical calculations of the pH of maximal protein stability: the effect of the sequence composition and three-dimensional structure. European Journal of Biochemistry. 2004;271(1):173–185. doi: 10.1046/j.1432-1033.2003.03917.x. [DOI] [PubMed] [Google Scholar]
- 79.Fujiwara H, Tatsumi KI, Tanaka S, Kimura M, Nose O, Amino N. A novel V59E missense mutation in the sodium iodide symporter gene in a family with iodide transport defect. Thyroid. 2000;10(6):471–474. doi: 10.1089/thy.2000.10.471. [DOI] [PubMed] [Google Scholar]
- 80.Dill KA, Fiebig KM, Chan HS. Cooperativity in protein-folding kinetics. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(5):1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dill KA, Ozkan SB, Weikl TR, Chodera JD, Voelz VA. The protein folding problem: when will it be solved? Current Opinion in Structural Biology. 2007;17(3):342–346. doi: 10.1016/j.sbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Ye Y, Li Z, Godzik A. Modeling and analyzing three-dimensional structures of human disease proteins. Pacific Symposium on Biocomputing. 2006:439–450. [PubMed] [Google Scholar]
- 83.Karchin R, Diekhans M, Kelly L, et al. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21(12):2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- 84.Wang Z, Moult J. SNPs, protein structure, and disease. Human Mutation. 2001;17(4):263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- 85.Wang Z, Moult J. Three-dimensional structural location and molecular functional effects of missense SNPs in the T cell receptor Vβ domain. Proteins. 2003;53(3):748–757. doi: 10.1002/prot.10522. [DOI] [PubMed] [Google Scholar]
- 86.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Research. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ode H, Matsuyama S, Hata M, et al. Computational characterization of structural role of the non-active site mutation M36I of human immunodeficiency virus type 1 protease. Journal of Molecular Biology. 2007;370(3):598–607. doi: 10.1016/j.jmb.2007.04.081. [DOI] [PubMed] [Google Scholar]
- 88.Shirley BA, Stanssens P, Hahn U, Pace CN. Contribution of hydrogen bonding to the conformational stability of ribonuclease T1. Biochemistry. 1992;31(3):725–732. doi: 10.1021/bi00118a013. [DOI] [PubMed] [Google Scholar]
- 89.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(19):6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Young MA, Gonfloni S, Superti-Furga G, Roux B, Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 2001;105(1):115–126. doi: 10.1016/s0092-8674(01)00301-4. [DOI] [PubMed] [Google Scholar]
- 91.Tang KES, Dill KA. Native protein fluctuations: the conformational-motion temperature and the inverse correlation of protein flexibility with protein stability. Journal of Biomolecular Structure and Dynamics. 1998;16(2):397–411. doi: 10.1080/07391102.1998.10508256. [DOI] [PubMed] [Google Scholar]
- 92.Song ES, Daily A, Fried MG, Juliano MA, Juliano L, Hersh LB. Mutation of active site residues of insulin-degrading enzyme alters allosteric interactions. The Journal of Biological Chemistry. 2005;280(18):17701–17706. doi: 10.1074/jbc.M501896200. [DOI] [PubMed] [Google Scholar]
- 93.Valerio M, Colosimo A, Conti F, et al. Early events in protein aggregation: molecular flexibility and hydrophobicity/charge interaction in amyloid peptides as studied by molecular dynamics simulations. Proteins. 2005;58(1):110–118. doi: 10.1002/prot.20306. [DOI] [PubMed] [Google Scholar]
- 94.Board PG, Pierce K, Coggan M. Expression of functional coagulation factor XIII in Escherichia coli . Thrombosis and Haemostasis. 1990;63(2):235–240. [PubMed] [Google Scholar]
- 95.Ozbabacan SEA, Gursoy A, Keskin O, Nussinov R. Conformational ensembles, signal transduction and residue hot spots: application to drug discovery. Current Opinion in Drug Discovery and Development. 2010;13(5):527–537. [PubMed] [Google Scholar]
- 96.Dixit A, Torkamani A, Schork NJ, Verkhivker G. Computational modeling of structurally conserved cancer mutations in the RET and MET kinases: the impact on protein structure, dynamics, and stability. Biophysical Journal. 2009;96(3):858–874. doi: 10.1016/j.bpj.2008.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones R, Ruas M, Gregory F, et al. A CDKN2A mutation in familial melanoma that abrogates binding of p16 INK4a to CDK4 but not CDK6. Cancer Research. 2007;67(19):9134–9141. doi: 10.1158/0008-5472.CAN-07-1528. [DOI] [PubMed] [Google Scholar]
- 98.Rignall TR, Baker JO, McCarter SL, et al. Effect of single active-site cleft mutation on product specificity in a thermostable bacterial cellulase. Applied Biochemistry and Biotechnology A. 2002;98-100:383–394. doi: 10.1385/abab:98-100:1-9:383. [DOI] [PubMed] [Google Scholar]
- 99.van Wijk R, Rijksen G, Huizinga EG, Nieuwenhuis HK, van Solinge WW. HK Utrecht: missense mutation in the active site of human hexokinase associated with hexokinase deficiency and severe nonspherocytic hemolytic anemia. Blood. 2003;101(1):345–347. doi: 10.1182/blood-2002-06-1851. [DOI] [PubMed] [Google Scholar]
- 100.Hardt M, Laine RA. Mutation of active site residues in the chitin-binding domain ChBDChiA1 from chitinase A1 of Bacillus circulans alters substrate specificity: use of a green fluorescent protein binding assay. Archives of Biochemistry and Biophysics. 2004;426(2):286–297. doi: 10.1016/j.abb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 101.Ortiz MA, Light J, Maki RA, Assa-Munt N. Mutation analysis of the pip interaction domain reveals critical residues for protein-protein interactions. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):2740–2745. doi: 10.1073/pnas.96.6.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim E, Hyrc KL, Speck J, et al. Missense mutations in Otopetrin 1 affect subcellular localization and inhibition of purinergic signaling in vestibular supporting cells. Molecular and Cellular Neuroscience. 2011;46(3):655–661. doi: 10.1016/j.mcn.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Castella M, Pujol R, Callen E, et al. Origin, functional role, and clinical impact of fanconi anemia fanca mutations. Blood. 2011;117(14):3759–3769. doi: 10.1182/blood-2010-08-299917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boulling A, le Marechal C, Trouve P, Raguenes O, Chen JM, Ferec C. Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. European Journal of Human Genetics. 2007;15(9):936–942. doi: 10.1038/sj.ejhg.5201873. [DOI] [PubMed] [Google Scholar]
- 105.Niel-Butschi F, Kantelip B, Iwaszkiewicz J, et al. Genotype-phenotype correlations of TGFBI p.Leu509Pro, p.Leu509Arg, p.Val613Gly, and the allelic association of p.Met502Val-p.Arg555Gln mutations. Molecular Vision. 2011;17:1192–11202. [PMC free article] [PubMed] [Google Scholar]
- 106.Henriques BJ, Bross P, Gomes CM. Mutational hotspots in electron transfer flavoprotein underlie defective folding and function in multiple acyl-CoA dehydrogenase deficiency. Biochimica et Biophysica Acta. 2010;1802(11):1070–1077. doi: 10.1016/j.bbadis.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 107.Khan S, Vihinen M. Performance of protein stability predictors. Human Mutation. 2010;31(6):675–684. doi: 10.1002/humu.21242. [DOI] [PubMed] [Google Scholar]
- 108.Battiti R. Using mutual information for selecting features in supervised neural net learning. IEEE Transactions on Neural Networks. 1994;5(4):537–550. doi: 10.1109/72.298224. [DOI] [PubMed] [Google Scholar]
- 109.Karchin R, Kelly L, Sali A. Improving functional annotation of non-synonomous SNPs with information theory. Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing. 2005:397–408. doi: 10.1142/9789812702456_0038. [DOI] [PubMed] [Google Scholar]
- 110.Chasman D, Adams RM. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. Journal of Molecular Biology. 2001;307(2):683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- 111.Yue P, Moult J. Identification and analysis of deleterious human SNPs. Journal of Molecular Biology. 2006;356(5):1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 112.Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nature Methods. 2007;4(6):466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 113.Cai J, Cai LQ, Hong Y, Zhu YS. Functional characterisation of a natural androgen receptor missense mutation (N771H) causing human androgen insensitivity syndrome. doi: 10.1111/j.1439-0272.2011.01219.x. submitted to Andrologia. [DOI] [PubMed] [Google Scholar]
- 114.Hunt DM, Saldanha JW, Brennan JF, et al. Single nucleotide polymorphisms that cause structural changes in the cyclic AMP receptor protein transcriptional regulator of the tuberculosis vaccine strain Mycobacterium bovis BCG alter global gene expression without attenuating growth. Infection and Immunity. 2008;76(5):2227–2234. doi: 10.1128/IAI.01410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nicolao P, Carella M, Giometto B, et al. Missense polymorphism in the human carboxypeptidase E gene alters enzymatic activity. Human Mutation. 2001;18(2):120–131. doi: 10.1002/humu.1161. [DOI] [PubMed] [Google Scholar]
- 116.Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Accounts of Chemical Research. 2000;33(12):889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 117.Cavallo A, Martin ACR. Mapping SNPs to protein sequence and structure data. Bioinformatics. 2005;21(8):1443–1450. doi: 10.1093/bioinformatics/bti220. [DOI] [PubMed] [Google Scholar]
- 118.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bao L, Cui Y. Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information. Bioinformatics. 2005;21(10):2185–2190. doi: 10.1093/bioinformatics/bti365. [DOI] [PubMed] [Google Scholar]
- 120.Saunders CT, Baker D. Evaluation of structural and evolutionary contributions to deleterious mutation prediction. Journal of Molecular Biology. 2002;322(4):891–901. doi: 10.1016/s0022-2836(02)00813-6. [DOI] [PubMed] [Google Scholar]
- 121.Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends in Genetics. 2000;16(5):198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- 122.Sunyaev SR, Lathe WC, III, Ramensky VE, Bork P. SNP frequencies in human genes: an excess of rare alleles and differing modes of selection. Trends in Genetics. 2000;16(8):335–337. doi: 10.1016/s0168-9525(00)02058-8. [DOI] [PubMed] [Google Scholar]
- 123.Sunyaev S, Ramensky V, Koch I, Lathe W, III, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Human Molecular Genetics. 2001;10(6):591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 124.Dimmic MW, Sunyaev S, Bustamante CD. Inferring SNP function using evolutionary, structural, and computational methods. Pacific Symposium on Biocomputing. 2005:382–384. [PubMed] [Google Scholar]
- 125.Landgraf R, Xenarios I, Eisenberg D. Three-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins. Journal of Molecular Biology. 2001;307(5):1487–1502. doi: 10.1006/jmbi.2001.4540. [DOI] [PubMed] [Google Scholar]
- 126.Lichtarge O, Sowa ME. Evolutionary predictions of binding surfaces and interactions. Current Opinion in Structural Biology. 2002;12(1):21–27. doi: 10.1016/s0959-440x(02)00284-1. [DOI] [PubMed] [Google Scholar]
- 127.Innis CA, Shi J, Blundell TL. Evolutionary trace analysis of TGF-β and related growth factors: implications for site-directed mutagenesis. Protein Engineering. 2000;13(12):839–847. doi: 10.1093/protein/13.12.839. [DOI] [PubMed] [Google Scholar]
- 128.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. Journal of Molecular Biology. 1996;257(2):342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 129.Porter CT, Bartlett GJ, Thornton JM. The catalytic site atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Research. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chelliah V, Chen L, Blundell TL, Lovell SC. Distinguishing structural and functional restraints in evolution in order to identify interaction sites. Journal of Molecular Biology. 2004;342(5):1487–1504. doi: 10.1016/j.jmb.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 131.Pazos F, Sternberg MJE. Automated prediction of protein function and detection of functional sites from structure. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(41):14754–14759. doi: 10.1073/pnas.0404569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ding F, Dokholyan NV. Emergence of protein fold families through rational design. PLoS Computational Biology. 2006;2(7):0725–0733. doi: 10.1371/journal.pcbi.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yin S, Ding F, Dokholyan NV. Modeling backbone flexibility improves protein stability estimation. Structure. 2007;15(12):1567–1576. doi: 10.1016/j.str.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 134.Capriotti E, Fariselli P, Rossi I, Casadio R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinformatics. 2008;9(supplement 2, article S6) doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cheng JL, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62(4):1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- 136.Alva V, Syamala Devi DP, Sowdhamini R. COILCHECK: an interactive server for the analysis of interface regions in coiled coils. Protein and Peptide Letters. 2008;15(1):33–38. doi: 10.2174/092986608783330314. [DOI] [PubMed] [Google Scholar]
- 137.Kruger DM, Gohlke H. DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Research. 2010;38(supplement 2):W480–W486. doi: 10.1093/nar/gkq471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7, article no. 166 doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de Baets G, van Durme J, Reumers J, et al. Snpeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Research. 2012;40:D935–D939. doi: 10.1093/nar/gkr996. submitted to Nucleic Acids Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology. 2004;22(10):1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 141.Maurer-Stroh S, Debulpaep M, Kuemmerer N, et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nature Methods. 2010;7(3):237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- 142.van Durme J, Maurer-Stroh S, Gallardo R, Wilkinson H, Rousseau F, Schymkowitz J. Accurate prediction of DnaK-peptide binding via homology modelling and experimental data. PLoS Computational Biology. 2009;5(8) doi: 10.1371/journal.pcbi.1000475. Article ID e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4(7):1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 144.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tian J, Wu N, Guo X, Guo J, Zhang J, Fan Y. Predicting the phenotypic effects of non-synonymous single nucleotide polymorphisms based on support vector machines. BMC Bioinformatics. 2007;8, article 450 doi: 10.1186/1471-2105-8-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Uzun A, Leslin CM, Abyzov A, Ilyin V. Structure SNP (StSNP): a web server for mapping and modeling nsSNPs on protein structures with linkage to metabolic pathways. Nucleic Acids Research. 2007;35:W384–392. doi: 10.1093/nar/gkm232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Masso M, Vaisman II. Knowledge-based computational mutagenesis for predicting the disease potential of human non-synonymous single nucleotide polymorphisms. Journal of Theoretical Biology. 2010;266(4):560–568. doi: 10.1016/j.jtbi.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 148.Brooks BR, Brooks CL, III, Mackerell AD, Jr., et al. CHARMM: the biomolecular simulation program. Journal of Computational Chemistry. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cason AL, Ikeguchi Y, Skinner C, et al. X-linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. European Journal of Human Genetics. 2003;11(12):937–944. doi: 10.1038/sj.ejhg.5201072. [DOI] [PubMed] [Google Scholar]
- 150.de Alencastro G, McCloskey DE, Kliemann SE, et al. New SMS mutation leads to a striking reduction in spermine synthase protein function and a severe form of Snyder-Robinson X-linked recessive mental retardation syndrome. Journal of Medical Genetics. 2008;45(8):539–543. doi: 10.1136/jmg.2007.056713. [DOI] [PubMed] [Google Scholar]
- 151.Becerra-Solano LE, Butler J, Castaneda-Cisneros G, et al. A missense mutation, p.V132G, in the X-linked spermine synthase gene (SMS) causes Snyder-Robinson syndrome. American Journal of Medical Genetics A. 2009;149(3):328–335. doi: 10.1002/ajmg.a.32641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gerner EW, Meyskens FL., Jr. Polyamines and cancer: old molecules, new understanding. Nature Reviews Cancer. 2004;4(10):781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 153.Ikeguchi Y, Bewley MC, Pegg AE. Aminopropyltransferases: function, structure and genetics. Journal of Biochemistry. 2006;139(1):1–9. doi: 10.1093/jb/mvj019. [DOI] [PubMed] [Google Scholar]
- 154.Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61(9):880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Geerts D, Koster J, Albert D, et al. The polyamine metabolism genes ornithine decarboxylase and antizyme 2 predict aggressive behavior in neuroblastomas with and without MYCN amplification. International Journal of Cancer. 2010;126(9):2012–2024. doi: 10.1002/ijc.25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu H, Min J, Zeng H, et al. Crystal structure of human spermine synthase: implications of substrate binding and catalytic mechanism. The Journal of Biological Chemistry. 2008;283(23):16135–16146. doi: 10.1074/jbc.M710323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xiang Z, Honig B. Extending the accuracy limits of prediction for side-chain conformations. Journal of Molecular Biology. 2001;311(2):421–430. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 158.Ponder JW. Tinker-Software Tools for Molecular Design. 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rajendran V, Purohit R, Sethumadhavan R. In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in lamin A/C protein. doi: 10.1007/s00726-011-1108-7. submitted to Amino Acids. [DOI] [PubMed] [Google Scholar]
- 160.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. Journal of Computational Chemistry. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 161.Minutolo C, Nadra AD, Fernandez C, et al. Structure-based analysis of five novel disease-causing mutations in 21-hydroxylase-deficient patients. PLoS One. 2011;6(1) doi: 10.1371/journal.pone.0015899. Article ID e15899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tan Y, Luo R. Structural and functional implications of p53 missense cancer mutations. PMC Biophysics. 2009;2(1, article 5) doi: 10.1186/1757-5036-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lee WH, Raas-Rotschild A, Miteva MA, et al. Noonan syndrome type I with PTPN11 3 bp deletion: structure-function implications. Proteins. 2005;58(1):7–13. doi: 10.1002/prot.20296. [DOI] [PubMed] [Google Scholar]
- 164.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. Journal of Molecular Biology. 1994;235(3):983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 165.Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. Journal of Physical Chemistry B. 2001;105(28):6507–6514. [Google Scholar]
- 166.Alexov EG, Gunner MR. Calculated protein and proton motions coupled to electron transfer: electron transfer from QA- to QB in bacterial photosynthetic reaction centers. Biochemistry. 1999;38(26):8253–8270. doi: 10.1021/bi982700a. [DOI] [PubMed] [Google Scholar]
- 167.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating PKas in proteins. Biophysical Journal. 2002;83(4):1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Song Y, Mao J, Gunner MR. MCCE2: improving protein PKa calculations with extensive side chain rotamer sampling. Journal of Computational Chemistry. 2009;30(14):2231–2247. doi: 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tynan-Connolly BM, Nielsen JE. pKD: re-designing protein PKa values. Nucleic Acids Research. 2006;34:W48–W51. doi: 10.1093/nar/gkl192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wei Q, Wang L, Wang Q, Kruger WD, Dunbrack RL., Jr. Testing computational prediction of missense mutation phenotypes: functional characterization of 204 mutations of human cystathionine beta synthase. Proteins. 2010;78(9):2058–2074. doi: 10.1002/prot.22722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rajasekaran R, Sethumadhavan R. Exploring the cause of drug resistance by the detrimental missense mutations in KIT receptor: computational approach. Amino Acids. 2010;39(3):651–660. doi: 10.1007/s00726-010-0486-6. [DOI] [PubMed] [Google Scholar]
- 172.Abdur Rauf SM, Ismael M, Sahu KK, et al. A graph theoretical approach to the effect of mutation on the flexibility of the DNA binding domain of p53 protein. Chemical Papers. 2009;63(6):654–661. [Google Scholar]
- 173.Cheng TMK, Lu YE, Vendruscolo M, Lio P, Blundell TL. Prediction by graph theoretic measures of structural effects in proteins arising from non-synonymous single nucleotide polymorphisms. PLoS Computational Biology. 2008;4(7) doi: 10.1371/journal.pcbi.1000135. Article ID e1000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Danielian S, El-Hakeh J, Basilico G, et al. Bruton tyrosine kinase gene mutations in Argentina. Human Mutation. 2003;21(4):p. 451. doi: 10.1002/humu.9131. [DOI] [PubMed] [Google Scholar]
- 175.Iqbal H, Mir A, Faryal R. Molecular modeling of mutant kinase domain of Btk, a Tec family member, for structure prediction. African Journal of Biotechnology. 2011;10(17):3274–3289. [Google Scholar]
- 176.Leandro J, Simonsen N, Saraste J, Leandro P, Flatmark T. Phenylketonuria as a protein misfolding disease: the mutation pG46S in phenylalanine hydroxylase promotes self-association and fibril formation. Biochimica et Biophysica Acta. 2011;1812(1):106–120. doi: 10.1016/j.bbadis.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 177.Yang Y, Zhou Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins. 2008;72(2):793–803. doi: 10.1002/prot.21968. [DOI] [PubMed] [Google Scholar]
- 178.Yang Y, Zhou Y. Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely related all-atom statistical energy functions. Protein Science. 2008;17(7):1212–1219. doi: 10.1110/ps.033480.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dehouck Y, Grosfils A, Folch B, Gilis D, Bogaerts P, Rooman M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics. 2009;25(19):2537–2543. doi: 10.1093/bioinformatics/btp445. [DOI] [PubMed] [Google Scholar]
- 180.Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011;12, article 151 doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Research. 2006;34:W239–W242. doi: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Parthiban V, Gromiha MM, Hoppe C, Schomburg D. Structural analysis and prediction of protein mutant stability using distance and torsion potentials: role of secondary structure and solvent accessibility. Proteins. 2007;66(1):41–52. doi: 10.1002/prot.21115. [DOI] [PubMed] [Google Scholar]
- 183.Parthiban V, Gromiha MM, Abhinandan M, Schomburg D. Computational modeling of protein mutant stability: analysis and optimization of statistical potentials and structural features reveal insights into prediction model development. BMC Structural Biology. 2007;7, article 54 doi: 10.1186/1472-6807-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wainreb G, Wolf L, Ashkenazy H, Dehouck Y, Ben-Tal N. Protein stability: a single recorded mutation aids in predicting the effects of other mutations in the same amino acid site. Bioinformatics. 2011;27(23):3286–3292. doi: 10.1093/bioinformatics/btr576. [DOI] [PMC free article] [PubMed] [Google Scholar]