

Abstract
Pancreatic beta-cells, which secrete the hormone insulin, are the key arbiters of glucose homeostasis. Defective beta-cell numbers and/or function underlie essentially all major forms of diabetes and must be restored if diabetes is to be cured. Thus, the identification of the molecular regulators of beta-cell mass and a better understanding of the processes of beta-cell differentiation and proliferation may provide further insight for the development of new therapeutic targets for diabetes. This review will focus on the principal hormones and nutrients, as well as downstream signalling pathways regulating beta-cell mass in the adult. Furthermore, we will also address more recently appreciated regulators of beta-cell mass, such as microRNAs.
1. Introduction
The endocrine cells of the pancreas, located in the islets of Langerhans, are responsible for blood glucose homeostasis, secreting hormones with differing and even opposing effects on blood glucose disposal. Beta-cells, the most numerous islet cells, secrete the hormone insulin which reduces blood glucose levels by increasing peripheral uptake of glucose and by suppressing release of glucose from the liver. Conversely, islet alpha-cells secrete the hormone glucagon which can increase blood glucose levels. Glucagon mainly acts on the liver where it promotes glycogenolysis, releasing glucose from breakdown of glycogen stores and gluconeogenesis. Optimal control of blood glucose levels depends on delicate changes in insulin production and secretion by the pancreatic beta-cells and on their capacity for large increases in secretion after meals, requiring large stores of insulin. It is of critical importance that islets maintain an adequate beta-cell mass in response to various changes.
Recent evidence has revealed that beta-cell replication plays a central role in maintaining adult beta-cell mass [1]. In addition, rates of beta-cell proliferation change dynamically according to metabolic demand throughout life [2]. However, replication of pre-existing beta-cells is not the only available mechanism for generating new beta-cells. In fact, a reasonable body of evidence supports the existence of four other potentially important contributors to adult beta-cell mass regulation: (i) differentiation from stem cells precursors, (ii) transdifferentiation from a non-beta-cell differentiated precursor, (iii) whole islet neogenesis on the plus side and apoptosis on the negative, and (iv) changes in beta-cell size [3, 4]. However, the relative contribution of these processes in maintaining and expanding beta-cell mass is at present not well defined and varies between species [5–7]. During adult life, the beta-cell mass may have to adapt in the face of increased demands due to increases in body mass, pregnancy, or even loss of insulin sensitivity of peripheral tissues. If such compensatory adaptation is inadequate, then glucose homeostasis will be compromised and result in chronically elevated blood glucose, or diabetes [8, 9].
It is well known that beta-cells proliferate extensively during late embryonic development, but the rate of replication slows during postnatal life. During adult life, beta-cell proliferation is detected between 0.5% and 2% [10] gradually declining with age [11]. Remarkably, this low rate of baseline proliferation can be increased significantly in response to pregnancy or obesity and is regarded as an adaptive mechanism in response to increasing systemic insulin demand.
Although important roles of insulin [12] and glucose [13] in beta-cell compensation have been suggested, the mechanism underlying this process is not well understood.
In recent years, various groups have identified microRNAs (miRNAs) small molecules of noncoding RNA that are able to regulate protein expression that contribute to beta-cell dysfunction and diabetes onset [14–18]. However, the role of these miRNAs is not yet fully understood.
Type 2 diabetes (T2D) is characterized by hyperglycaemia resulting from impaired insulin secretion and/or impaired insulin action in peripheral tissues [19]. T2D constitutes one of the greatest pandemics of our time, with 220 million people currently diagnosed [20], and 439 million people expected to be affected by 2030 [21]. Importantly, there is substantial evidence that beta-cell dysfunction plays a major role in the pathology of T2D. For this reason, great efforts are being made in order to develop new therapeutic strategies, such as beta-cell replacement or regenerative medicine.
However, despite progress, most diabetic patients will still die prematurely as a direct result of their disease, its complications, or sometimes even its treatments. In fact, although one may hope that GLP-1 analogues and improved lifestyle may eventually translate into a slowing of T2D progress, clinical trials data have been generally disappointing and confirm that the disease continues to progress [22–25]. To date, no effective treatments for T1D based on slowing or reversing the natural history of the disease exist. Thus we must rely on treatments that can maintain or restore blood glucose levels, and evidences that disease progression can be significantly arrested are scanty. Thus, all T1D and over time most T2D patients will require exogenous insulin (or in rare cases an islet transplant). It is not all negative, and short-term benefits (usually progression from prediabetes to overt diabetes) have been reported in small studies for metformin and acarbose with the common factor likely to be weight loss. The similar success of supervised weight loss programs supports this view. Trials with the PPARg ligand rosiglitazone have also suggested that the progression of T2D may be retarded, but as the drug has been withdrawn, due to an increased risk of cardiovascular disease, this remains interesting rather than clinically useful. In the largest study of T2D to date (UKPDS-UK Prospective Diabetes Study (UKPDS) Group), treatments did not slow progression of patients to additional drugs in order to maintain blood glucose levels at target levels [26].
Furthermore, beta-cells from donor pancreases are in such short supply that transplants can be provided only to a limited number of patients. Ultimately, to cure diabetes, missing beta-cells that must be replaced, and in practical terms this would need to be done from within or a limitless source of beta-like cells must be developed for transplants. One way forward is to “create” beta-cells from alternative cell sources (neogenesis, transdifferentiation, stem cells, etc.). Such an approach requires further knowledge of the mechanisms that regulate pancreatic beta-cell mass.
This review briefly outlines current knowledge of significant factors/nutrients regulating beta-cells mass, and their signal transduction pathways, with greater focus on postnatal regulation and the role of a new class of beta-cell mass regulators: the microRNAs.
2. Postnatal Beta-Cell Regulation
How Do Beta-Cells Maintain Their Normal Mass? —
Beta-cell mass is increased by neogenesis (differentiation from beta-cell precursor cells), proliferation, and cell hypertrophy (increased cell size) and is decreased by beta-cell death, primarily through apoptosis, and beta-cell atrophy (decreased cell size, Figure 1). Several studies have revealed that the primary mechanism by which new beta-cells arise during adulthood is through proliferation of existing beta-cells rather than neogenesis [7, 27]. Thus, organisms born with reduced beta-cell mass have fewer beta-cells available to enter the cell cycle later in life. Under normal circumstances during adulthood, beta-cells are a slowly renewing population, with steady low levels of proliferation and apoptosis. However, beta-cell mass continuously expands over the lifespan of an organism [28], likely due to age-related increases in body weight and insulin resistance. A large number of hormones and nutrients have been shown to affect beta-cell mass, as extensively reviewed by Nielsen et al. [29]. As a result, the number of molecular pathways that have been implicated in beta mass regulation is also large. Many studies have employed animal models, and it is important to note that significant differences between species have been observed [30].
Figure 1.
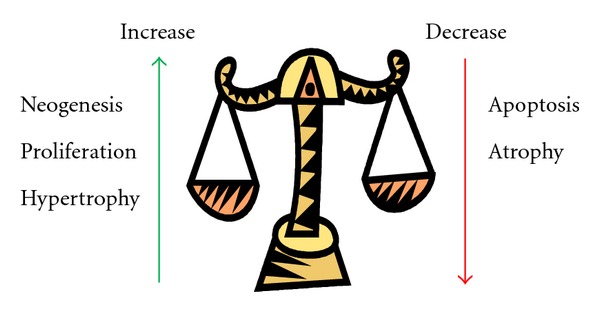
Beta-cell mass equilibrium. Beta-cell proliferation, neogenesis, and hypertrophy (enlarged cell size) increase beta-cell mass, while apoptosis and atrophy (reduced cell size) decrease beta-cell mass.
2.1. Regulation of Beta-Cell Mass by Hormones, Growth Factors, and other Factors
Beta-cell mass regulation is modulated by various environmental factors and nutrients including glucose, insulin, amino acids, fatty acids, and various other growth factors/hormones, such as IGF-I, IGF-II, glucagon-like peptide-1 (GLP-1) 1, glucagon, gastroinhibitory peptide (GIP), somatostatin (SST), HGF and betacellulin, gastrin, cholecystokinin (CCK), growth hormone (GH), prolactin (PRL), placental lactogen (PL), and leptin, amongst others (Table 1). These growth factors and nutrients can affect a variety of beta-cell functions—suppress/stimulate beta-cell replication, survival, mass expansion, and differentiation through different intracellular pathways discussed later.
Table 1.
Hormones, growth factors, and other factors regulating beta-cell mass.
| Inhibitors | Stimulators | |
|---|---|---|
| Metabolites | Glucose, FFA | Glucose, FFA, amino acids |
| Cytokines | IL-1, IFN-g, TNF-a, leptin | GH, PRL, PL |
| Growth factor family | HGF | IGF-I, IGF-II, insulin, TGF-a, betacellulin, HB-EGF, aFGF, VEGF, PDGF, HGF |
| Placental hormones | placental lactogen, prolactin | |
| Glucagon family | GLP-1, GIP, glucagon | |
| Somatostatin family | Somatostatin | |
| CGRP family | IAPP/amylin | |
| Gastrin family | Gastrin, CCK | |
| TGF-b family | TGF-b, follistatin | Activin A |
| Neurotrophins | NGF, NT-3 | |
| Neurotransmitters | (Nor)epinephrine | Acetylcholine |
| Lectins | Reg/INGAP/PSP/PTP | |
| Adhesion molecules | Integrin a6b1, Cx43, Cx36 | |
| Drugs | Diazoxide | Nicotinamide, SU |
| Toxins | Streptozotocin, alloxan |
Modified from Nielsen et al. [29]. aFGF, acidic fibroblast growth factor; CCK, cholecystokinin; Cx. Connexin; HB-EGF, heparin-binding EGF-like protein; HGF, hepatocyte growth factor/scatter factor; IAPP, islet amyloid polypeptide; IFN, interferon; IL, interleukin; INGAP, islet neogenesis- associated peptide; NGF, nerve growth factor; NT-3, neurotrophin-3; PDGF, platelet-derived growth factor; PSP, pancreatic stone protein; PTP, pancreatic thread protein; SU, sulfonylurea; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Glucose —
Glucose is thought to be the most important determinant of beta-cell mass equilibrium [31–36]. Glucose is not only the pathological hallmark of diabetes, but it is also a potential contributor to further decline in beta-cell mass through what has been termed glucotoxicity. Thus, in the face of evolving demand for insulin, if adaptation (functional or numeric) fails, then blood glucose levels rise, sequentially resulting in IGT and then T2D. Once glucose levels exceed the safe threshold, then further beta-cell death will likely arise [37].
It has been reported that hyperglycaemia for a short time in rats induces beta-cells to enter the cell cycle accompanied by ~50% increase in beta-cell number by neogenesis [33, 38] and suppression of beta-cell apoptosis [33, 36]. In another study using prolonged periods of glucose infusion, an increase of beta-cell mass was also reported, although the origin of these cells has been disputed. While some authors describe proliferation of new beta-cells formed by neogenesis of precursor cells [36], other groups report an increase in beta-cell replication and hypertrophy as well as neogenesis, leading to sustained effects on beta-cell mass even after glucose infusion is stopped [35, 39].
To support the mitogen effect of glucose on beta-cells, Porat and colleagues demonstrated in an animal model an increased replication rate of beta-cells correlated with an increased blood glucose level. Moreover, this effect was mediated by a specific enzyme involved in glucose metabolism (glycolysis) in beta-cells, glucokinase (GCK). In fact, it was demonstrated that GCK has a key role in beta-cell proliferation through membrane depolarization [40].
In addition, glucose has been shown to promote beta-cell survival by suppressing a constitutive apoptotic program in vitro [41]. Moreover, it was reported that mild hyperglycaemia can affect beta-cell phenotype [37] with a progressive loss of beta-cell differentiation markers (Pdx-1, BETA2/NeuroD, NKX6.1, and HNF-1β) and genes involved in glucose sensing (GLUT-2 and glucokinase) [37, 42]. On the other hand, some genes that are normally suppressed in beta-cells (e.g., c-Myc and its gene target, lactate dehydrogenase-A) are increased. This proapoptotic effect of glucose may also involve altered calcium homeostasis [43], activation of caspases by cytokines, such as IL-1β which is secreted by human islets in the presence of high glucose and leads to beta-cell apoptosis [44] or Fas-Fas-Ligand interactions [44], reactive oxygen species [45], and c-Myc [46, 47]. Intriguingly, c-Myc has been shown to trigger apoptosis through all of these pathways in other systems, including in particular the activation of some caspases (caspase 8 and 9), Fas-Fas-Ligand signalling, and reactive oxygen species (reviewed in [48]).
Therefore, paradoxically, it appears that glucose may have both an inhibitory and stimulating effect on beta-cell apoptosis. However, it appears that both the level and duration of hyperglycaemia are crucial in determining the fate of the beta-cell. For example, prolonged hyperglycaemia appears to have a proapoptotic effect, a process referred to as glucose toxicity. Chronic hyperglycemia may thus be injurious to beta-cells and contribute to the development of T2D [49]. Increased beta-cell apoptosis and reduced beta-cell mass have been observed in humans with T2D also, in keeping with the preclinical data in [6, 50–53]. This difference in beta-cell mass between T2D patients and matched controls is particularly evident when those are matched for obesity (obese patients without T2D have increased beta-cell mass compared to lean controls).
Insulin —
The effect of insulin on beta-cell mass has not been completely clarified. Various authors have shown in various in vivo studies that insulin alone can stimulate beta-cell mass, whereas other authors have found that it promotes growth only in the presence of hyperglycaemia [34, 36, 54–56].
In a physiological condition, it seems reasonable that during a period of increased insulin demand, insulin itself could stimulate beta-cell growth, creating a positive feedback loop. Further studies have suggested a positive role of insulin in cell regeneration [57, 58].
Genetically altered mice in which the insulin receptor was knocked out exhibited a decreased beta-cell mass in adults and diabetes onset [59]. Insulin resistance, leading to hyperinsulinemia, stimulates an increase in beta-cell mass.
In contrast, as previously described [60, 61], insulin can also impose negative effect on beta-cell mass and/or proliferation. For example, rats with insulinomas resulting in hyperinsulinemia and hypoglycaemia were found to have smaller beta-cells, as well as reduced beta-cell numbers (in the islets without tumour), suggesting that a “sensor” mechanisms exist that can co-ordinate beta-cell mass across islets and possibly even distal grafts [62]. Increased rates of beta-cell apoptosis were evident [60, 61], but interestingly, beta-cell mass regenerated within days of tumour excision [63]. This demonstrates that excess insulin with a negative feedback is able to inhibit beta-cell growth.
These observations indicate that insulin can control beta-cell population dynamics, although the mechanism is still unclear.
Free Fatty Acids —
Another proposed stimulus for beta-cell mass regulation has been free fatty acids (FFAs). It is well documented that chronic high levels of FFAs, which are often accompanied by obesity, contributes to the pathophysiology of insulin resistance and T2D [64, 65].
It is reported that acute FFA exposure stimulates insulin secretion, while prolonged FFA exposure decreases glucose-stimulated insulin secretion [66, 67] and induces insulin resistance and beta-cell dysfunction in both animal models [68] and humans [69–72].
Animal studies have been conflicting with some arguing that FFAs have a positive trophic effect on beta-cells, whilst others suggest the contrary. However, precise molecular mechanisms linking FFA to beta-cell dysfunction have yet to be fully elucidated.
It was reported that a prolonged in vitro exposure of isolated islets or insulin-secreting cell lines to elevated levels of fatty acids is associated with inhibition of glucose-induced insulin secretion [67, 73, 74], impairment of insulin gene expression [75–78], and induction of cell death by apoptosis both in vitro [79–88] and in vivo studies [80].
Importantly, in vitro [76, 88, 89] and in vivo [90, 91] studies have provided evidence that lipotoxicity only occurs in the presence of concomitantly elevated glucose levels. A number of studies have shown that fatty acids can induce beta-cell death by apoptosis in the presence of high glucose [88]. In vitro, saturated fatty acids induce beta-cell apoptosis [82, 86, 88], whereas unsaturated fatty acids are usually protective [81, 82, 88].
Recently, it was observed by various authors that oxidative stress is implicated in the pathogenesis of beta-cell dysfunction induced by FFAs. It has been suggested that increased reactive oxygen species (ROS) levels are the important trigger for beta-cell dysfunction. Under diabetic conditions, ROS is increased in many tissues and organs and cause various forms of tissue damage in patients with diabetes. It is considered that enhanced ROS generation may act as a link between FFA and beta-cell dysfunction [92].
Glucagon-Like Peptide-1 —
Glucagon-like peptide-1 (GLP-1) is produced and secreted in response to fat and glucose from intestinal L-cells, located in the distal ileum and colon.
Preclinical studies reveal potential benefits of GLP-1 receptor agonist treatment in individuals with T2D, which include the promotion of beta-cell proliferation [93] and reduced beta-cell apoptosis [94]. These preclinical results indicate that GLP-1 could be beneficial in treating patients with T2D.
However, the role of endogenous GLP-1 in stimulating beta-cell proliferation is less clear. Data regarding the regenerative role of GLP-1 and related agonists are controversial.
However, there is strong evidence to suggest that GLP-1 receptor activation can protect beta-cells from apoptosis [94–97] and equally convincing evidence of a beta-cell mitogenic effect of GLP-1 [93, 94, 98, 99].
It was reported that exogenous GLP-1 treatment enhances beta-cell replication in normoglycaemic rats [100] and mice [101–105].
However, while there are reports of GLP-1-mediated islet neogenesis [98, 106–109], recent statements by researchers examining the in vivo effects of GLP-1 call into question a neogenic response [110–112].
GLP-1 treatment stimulates beta-cell replication in multiple rodent models of obesity-induced diabetes, including both intact [113, 114] and defective leptin signalling [94, 115–117] models. In addition, some authors have demonstrated that exogenous GLP-1 treatment promotes beta-cell replication in models of beta-cell regeneration and obesity-induced diabetes [118, 119].
Moreover, GLP-1 prevents beta-cell death in mouse models of beta-cell loss, in particular, in both unstressed mice [102] and in streptozotocin-induced beta-cell toxicity [95, 120]. In summary, exogenous GLP-1 treatment stimulates beta-cell proliferation in multiple rodent species and models of beta-cell mitogenesis. Reports of human beta-cell proliferation are, however, sparse. Data to support a role for endogenous GLP-1 signalling in beta-cell proliferation are less clear.
Glucose-Dependent Insulinotropic Polypeptide —
Glucose-dependent insulinotropic polypeptide (GIP) is a gut hormone synthesized and secreted from intestinal K-cells, which reside in the duodenum and jejunum.
It was previously reported that if GIP promotes beta-cell proliferation during obesity, then GIP inhibition should reduce beta-cell mass [121].
However, it is unclear whether the primary effect of GIP antagonism is on the adipocyte or the beta-cell. Few studies implicate GIP in in vivo beta-cell proliferation [119, 122]. However, one report demonstrated that GIP treatment increases islet size and number in the Leptin ob/ob mouse [123]. The latter study did not discriminate between beta-cell proliferation, apoptosis, or islet neogenesis. In a human patient, elevated fasting plasma GIP correlated with islet cell hyperplasia [124]. Despite the limited in vivo evidence, in vitro experiments demonstrate that GIP enhances beta-cell mitogenesis [125–128].
Furthermore, the role of GIP in the reduction of beta-cell apoptosis in vivo is more clearly defined than its role in beta-cell proliferation.
Various studies have also demonstrated that exogenous GIP treatment prevents beta-cell apoptosis during severe obesity [123, 129] and streptozotocin-induced diabetes [120, 129].
Insulin-Like Growth Factor I and II —
The role of insulin-like growth factor I (IGF-I) and insulin-like growth factor II (IGF-II) in beta-cell mass regulation has long been accepted [130–132].
IGF-I has been shown to increase the number of replicating beta-cells in rodent islets by up to 6% of the islet cell population [133–135] and to induce differentiated pancreatic beta-cell growth [133]. However, IGF-I appears only to be effective at inducing beta-cell proliferation under physiologically relevant glucose concentration range: 6 mM to 18 mM [136]. Thus, IGF-I potentiates the mitogenic effect of glucose on beta-cell proliferation.
Interestingly, overexpression of IGF-I in beta-cells is associated with increased beta-cell proliferation, but not mass [137]. In contrast, transgenic mice overexpressing IGF-II exhibit an increase in beta-cell mass due to augmented proliferation [138]. Taken together, there is clear evidence that the IGF molecules are important factors in beta-cell proliferation and mass.
Seemingly paradoxically, beta-cell-specific IGF-I knockout mice showed a delayed onset of T2D induced by a high-fat diet, accompanied by enlarged pancreatic islets, increased insulin mRNA levels, and preserved sensitivity to insulin [139]. This is likely due to secondary increases in insulin and others. Moreover, given the dominant role of circulating IGFs on tissue mass homeostasis in other systems, the significance of this is uncertain. Concerning IGF-II, it has been reported that mice overexpressing this hormone display an increase in beta-cell mass, hyperinsulinemia, and increased glucose-stimulated insulin secretion [140]. These results suggest that IGF-II may have some potential as an islet growth factor.
Cholecystokinin —
Cholecystokinin (CCK) is able to stimulate beta-cell proliferation in vivo, as reported by different authors. In fact, various studies have demonstrated that the administration of CCK-8 after streptozotocin treatment in rats expands beta-cell mass via proliferation, in association with fasting plasma insulin increasing, and fasting plasma glucose reduction [141].
At the same time, it was reported that CCK enhances rat beta-cell proliferation ex vivo. In fact, neonatal rat beta-cells proliferate in culture after CCK-8 treatment [142]. Similarly, in a rat model, loss of CCK resulted in decreased islet size and reduced beta-cell mass through increased beta-cell death [143]. However, overexpression of Cck in mouse and human islets has no effect on replication [143].
Parathyroid Hormone-Related Protein —
A recent study has demonstrated that acute and systemic administration of the first 36 amino acids of parathyroid hormone-related protein (PTHrP (1–36)) in rodents can stimulate beta-cell proliferation and enhance beta-cell mass in vivo, without negatively affecting beta-cell function or survival [144]. Previous studies have demonstrated that RIP-PTHrP transgenic mice producing the full-length PTHrP (1–139) in beta-cells show an increase in beta-cell mass, proliferation and islet number, and improvement of glucose homeostasis [145–147]. In addition, in vitro studies (in rodent insulinoma cell lines and primary human beta-cells) have shown that PTHrP (1–36) is sufficient to enhance not only proliferation, but also function, increasing insulin mRNA and protein, and augmenting glucose-stimulated insulin secretion [148–150].
However, it is notable that PTHrP knockout mice have normal beta-cell mass as compared to controls, suggesting that PTHrP may not be crucial for normal beta-cell mass in vivo. However, the explanation of this result should be found on the possibility that the PTHrP was absent in islets but it could be produced in other islet cell types, resulting in a paracrine compensatory mechanism [151].
These findings suggest that PTHrP may provide a possible target for gene therapeutic strategies designed to increase beta-cell mass and function.
Ghrelin —
Ghrelin is a stomach-derived hormone involved in glucose metabolism and has been shown to regulate cellular differentiation and proliferation in various systems. In beta-cells, ghrelin treatment has been shown to increase expression of insulin and PDX1, as well as beta-cell replication in a diabetes model [152]. This suggests a potential to increase beta-cell mass and function.
Moreover, it has been recently reported that both acylated- and unacylated-ghrelin can on the one hand promote cell proliferation while on the other inhibit apoptosis of beta-cells, including human pancreatic islets induced by serum starvation and/or cytokine's stimulation [153, 154].
Menin —
Menin is a tumour suppressor encoded by the Men1 gene. Mice with heterozygous deletion for Men1 develop pancreatic hyperplasia [155] that coincides with hyperinsulinemia and hypoglycaemia [156], with an increased islet cell growth through a progressive reduction in expression of some cell growth suppressors, such as p18 and p27 [157]. It was reported that the regulation of p18 and p27 by menin is through histone methylation [158, 159]. Moreover, mutation of Men1 can induce acceleration in S-phase entry and consequently an enhancement of cell proliferation in pancreatic islets [160]. Recently, it has been reported that menin is also able to increase caspase-8 expression [161] and that caspase-8 is critical for the maintenance of beta-cell mass under physiological conditions [162].
More recently, menin has been demonstrated to play an important role during pregnancy, but this will be discussed below.
Leptin —
Leptin is one of the most important cytokines produced by the ob gene and secreted by adipose tissue, and it plays an important role in controlling food intake/satiety and body energy balance (51) through the leptin-receptor (Ob-R).
Ob-R is also present in pancreatic tissue, and it has been proposed that leptin may increase beta-cell proliferation of insulin-secreting cell line [163].
Leptin has also been reported to prevent pancreatic beta-cells from inducible apoptosis, and this may partially account for islet hypertrophy in obese rodents and patients. Leptin may exert its antiapoptotic effects on pancreatic beta-cells. This was associated with a reduction of triglyceride accumulation, an inhibition of nitric oxide production, and a restoration of Bcl-2 expression in the face of fatty acid insult. This may be a possible mechanism for its antiapoptotic effect [164–166].
In the past, it was reported that leptin acted via the JAK/STAT and MAPK pathways and has been shown to increase beta-cell proliferation in cell line studies and foetal pancreatic tissue [167, 168].
However, recent findings indicate that leptin mainly acts through the PI3K-AKT signalling pathway in beta-cell proliferation [169–171]. All these results suggest that leptin has a protective role on beta-cell function. However, the effect of leptin on apoptosis has been conflicting. In vitro, prolonged exposure to leptin has been associated with impaired beta-cell function, and increased rates of apoptosis [172].
Furthermore, in contrast to the observations in rodent animal, leptin is likely to have a deleterious effect on human islet function [173]. For instance, it was reported that in isolated islets from mice, leptin was able to increase cell number through the suppression of apoptosis [164] meanwhile in human islets in vitro leptin impairs insulin secretion and induces apoptosis of beta-cells in the presence of 20 mM glucose via activation of c-JNK [174].
Overall, leptin is likely to exert diverse effects in the regulation of pancreatic beta-cell function and proliferation. Therefore, further research is still required to clarify its distinct role in various conditions.
Visfatin —
Visfatin is an adipokine secreted by adipose tissue [175], elevated in T2D [176]. This novel compound can mimic the effects of insulin, by binding insulin-receptor, and exerts protective effects on pancreatic beta-cells acting via the PI3K/AKT and MAPK pathways [175]. Through these pathways, visfatin would appear to have the potential to regulate plasma glucose levels, as well as beneficial effects on beta-cell mass.
Overall, visfatin may have a protective effect on pancreatic beta-cell mass, but further research is necessary to clarify its distinct roles.
Placental Hormones —
These hormones (especially placental lactogen and prolactin) are principally responsible for the changes in beta-cell mass during pregnancy (reviewed in [177, 178]). These hormones stimulate beta-cell proliferation in isolated islets, and beta-cell mass is reduced 26–42% in receptor deficient mice [179] as described in a later.
Connexin —
Beta-cell-cell interactions are mediated by connexins (Cxs). These proteins form the gap junction domains of cell membranes. Cxs have been shown to affect cell proliferation and survival in different cell types [180–182]. In particular, it was reported that the Cx36 isoform is the only one expressed in rodent beta-cells [183]. A recent study performed by Klee et al. [184], using Connexins-inducible/knockout transgenic mouse models, reported a key role in beta-cell mass regulation by demonstrating that the native Cx36 does not alter islet size or insulin content, whereas the Cx43 isoform increases both parameters, and Cx32 has a similar effect only when combined with the GH.
Bone Morphogenetic Proteins —
As recently reported, there are some new mediators that are responsible of proliferation/apoptosis of beta-cells. Bone morphogenetic proteins (BMPs) are growth factors involved in the pancreas mass homeostasis both during embryonic development and adult life [185, 186]. The binding of these molecules, in particular BMP1 and BMP4, to their specific receptors induce, the phosphorylation and nuclear translocation of SMADs. In addition, there were identified different target genes able to regulate different transcription factors (ID1, ID2, ID3, ID4) and proteins involved both in the proliferation and apoptosis processes (including GK, IRS, E2F, SMAD7/9, HES-1). This is the reason why BMPs have a double effect, both proliferative and proapoptotic.
2.2. Signalling Pathways in Beta-Cell Mass Homeostasis
An ever expanding literature suggests that the above-described hormonal and nonhormonal factors are able to regulate beta-cell mass by cell cycle regulatory proteins and a range of key signalling pathways. Of particular interest in the light of recent data are mitogenic and apoptotic signalling pathways regulated by the transcription factor c-Myc [46], downstream targets involved in G1/S transition [187–195], and various relevant pathways, including PI3K-AKT and MAPK.
In particular, the PI3K-AKT pathway is an important mediator of beta-cell mass increase [5, 136, 196]. In fact, PI3K-AKT is known to play a major role in preventing c-Myc apoptosis in many cell types [197] and to promote hypertrophy, hyperplasia, and neogenesis [5].
Phosphatidylinositol 3-Kinase/AKT Pathway —
There are many signal transduction pathways involved in the regulation of beta-cell mass, with phosphatidylinositol 3-kinase (PI3K) and AKT signalling being one of the best defined [198]. This pathway is activated by factors, such as glucose, insulin, IGFs, and GLP-1 [136, 199], and is able to regulate beta-cell size, proliferation, and apoptosis. Following ligand-receptor binding, insulin receptor substrate (IRS) is phosphorylated and is able to interact with various downstream targets. The most important target of IRS is PI3K, which in turn can activate PDK1 (via a secondary messenger) [200], resulting in the activation of AKT [200, 201]. AKT plays diverse roles in the beta-cell, including activation of glycogen synthase kinase-3 (GSK3, described later), and phosphorylation and nuclear exclusion of Forkhead transcription factor O1 (FOXO1) [202], ultimately leading to cell growth/replication and suppression of apoptosis [191] (described in Figure 2).
This is further evidenced in IRS2 −/− mice, whereby beta-cell proliferation was achieved by ablation of one allele of FOXO1 [203].
It was reported that PDX1 transcription is regulated positively by forkhead transcription factor A2 (FOXA2) but negatively by FOXO1 [204, 205]. In fact, these 2 forkhead transcription factors share common DNA-binding sites in the Pdx1 promoter, FOXO1 competes with FOXA2 for the same binding site resulting in the inhibition of Pdx1 transcription [204]. Moreover, nuclear translocation of FOXO1 excludes PDX1 from the nucleus, or vice versa [204, 206]. In addition, constitutively active nuclear FOXO1 has been shown to inhibit beta-cell proliferation in the face of insulin resistance [207]. This serves as a second mechanism by which FOXO1 inhibits beta-cell proliferation.
Recently, studies of beta-cells exposed to low nutrition revealed an additional beneficial role of FOXO1 activation—beta-cell proliferation without any changes in apoptosis compared to the control group [208]. Moreover, this study revealed that PI3K and MAPK signalling pathways are dampened and that the induction of CyclinD1 expression by activated FOXO1 in low nutrition is responsible for the improved proliferation of beta-cells [208].
Various studies using animal models to evaluate the role of different molecules on the PI3K/AKT pathway and beta-cell mass regulation have been performed.
Both IRS1 and 2 are critical for beta-cell growth and survival (previously mentioned), and they occupy a key position between the insulin receptor and downstream signaling elements. IRS1 −/− knockout mice develop severe insulin resistance [209, 210] and compensate for insulin resistance by increasing beta-cell mass [211]. Although IRS2 −/− mice are born with only a slightly reduced beta-cell mass, they later develop diabetes due to a marked increase in spontaneous apoptosis and reduced survival of beta-cells [209]. It, therefore, appears that IRS2 plays an important role in maintaining beta-cell homeostasis.
Furthermore, the role of AKT has been studied in beta-cells using genetically altered mice in which AKT is knocked out or overexpressed. AKT knockout mice displayed an increase of blood glycaemia and insulin resistance, although there have been conflicting reports on the effect on compensatory beta-cell growth [212]. Overexpressing AKT in transgenic mice led to a significant increase in beta-cell mass [5, 213]. In addition, AKT has been shown to promote beta-cell survival in vivo [214], suggesting an important role for AKT in beta-cell mass regulation.
The role of FoxO1 on signal transduction was further evidenced in different mouse models. It was demonstrated that constitutively active form of FoxO1 protects β cells from oxidative-stress-induced damage [197], and using another mouse model, it was demonstrated that deletion of one allele of FoxO1 resulted in a marked increase in the number, but not the size [209].
The role of PDX1 in beta-cell biology has been predominately examined in knockout studies. However, inactivating mutations in one Pdx-1 allele have yielded conflicting results, although a “critical” level of PDX-1 is thought to be required in order to maintain beta-cell mass that [215, 216].
Figure 2.
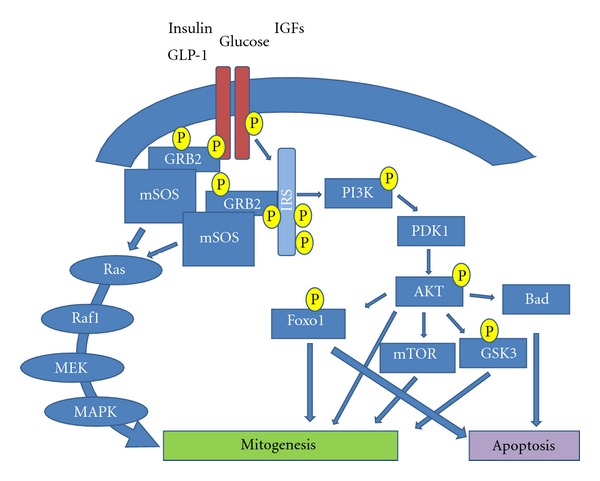
Schematic representation of PI3K/Akt and MAPK signalling. Mechanisms of some growth factors modulate beta-cell replication through proliferation or apoptosis. These factors on one hand stimulate PI3K, AKT, MAPK, and increase beta-cell proliferation. On the other hand, stimulate AKT, FOXO1, and other proapoptotic molecules resulting in induction of apoptosis.
GSK3 —
Glycogen synthase kinase-3 (GSK3), a constitutively active serine/threonine kinase, was the first substrate shown to be phosphorylated by AKT (8).
Mice overexpressing a constitutively active form of GSK3 in beta-cells display reduced levels of PDX-1 protein [217]. In vitro studies confirmed the role of GSK3 in regulating PDX-1 protein stability, for example, in MIN6 cells, GSK3 has been shown to phosphorylate PDX-1 leading to its proteasomal degradation. These results highlighted a new mechanism, whereby signalling through the PI3K/AKT/GSK3 pathway regulates beta-cell growth [217]. In addition to the modulation of PDX-1 stability, GSK3 can also regulate beta cell proliferation through phosphorylation and degradation of cell cycle proteins, such as cyclin D1 and/or p27 [218, 219]. Other targets of GSK3 include numerous transcription factors involved in the cell cycle including c-Myc and c-Jun [220]. Therefore, in response to different growth factors, AKT prevents GSK3 phosphorylation of cyclin D, thereby promoting cell cycle progression and subsequent mitogenesis (Figure 2).
Mitogen-Activated Protein-Kinase (MAPK) Pathway —
IRS can activate the RAF-MEK-ERK pathway (aka the mitogen-activated protein-kinase (MAPK) pathway, Figure 2) which plays a crucial role in development, cellular differentiation, and mitogenesis [221]. Firstly, IRS engages the adaptor molecule growth factor receptor-bound protein-2 (Grb2) via its SH2 domain [222, 223]. In turn, Grb2 binds to the murine Son-Of-Sevenless-1 protein (mSOS), which is able to activate RAS protein [222, 223]. It then, associates with RAF serine kinase [224], which subsequently phosphorylates mitogen activated protein kinase-kinase (MEK) resulting in the activation of ERK/MAPK [223]. Activated ERK/MAPK can then activate other protein kinases or migrate to the nucleus to activate transcription factors, such as c-JUN, c-MYC and c-FOS, important in the cell cycle [225].
However, the RAS/MAPK pathway can also be activated independently of IRS via direct binding of Grb2 to the receptor's SH2 domain [222, 223].
Janus Kinase/Signal Transducers and Activators of Transcription Pathway —
Another important pathway in beta-cell biology is the JAK/STAT pathway. Both growth hormone (GH) and prolactin have been shown to mediate their effects via this pathway [226, 227]. Once activated, Janus kinases (JAKs) are able to phosphorylate signal transducers and activators of transcriptions (STATs) which enter the nucleus to regulate transcription of target genes. In particular, JAK2 and STAT-5 have been shown to regulate beta-cell growth and survival [226–228], as well as to reduce apoptosis [229]. More recently, it was recognized that the JAK/STAT pathway is regulated by Suppressors of cytokine signaling (SOCS). These suppressor proteins inhibit STAT phosphorylation by binding to phosphorylated JAKs, or compete with STATs for phosphotyrosine binding sites on cytokine receptors. Furthermore, SOCs facilitate the degradation of JAKs [230] (Figure 3). Its effects have resulted in reduced beta-cell proliferation in a sex-dependent manner [231].
Figure 3.
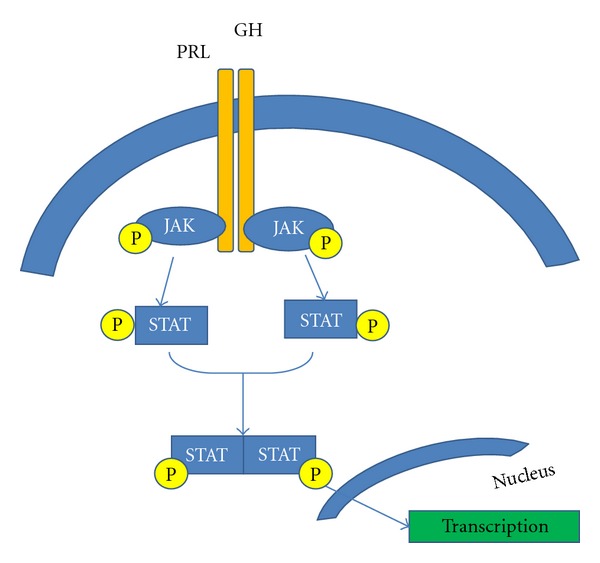
Schematic representation of JAK/STAT signalling. Mechanisms through prolactin and growth hormone modulate beta-cell proliferation. These hormones via prolactin receptor activate JAK, which is able to phosphorylate STAT. After phosphorylation, two molecules of STAT bind together and translocate into the nuclei where elicit their transcriptional function.
c-Myc —
The cellular proto-oncogene, c-Myc, encodes the protein c-Myc whose key biological function is to promote cell cycle progression. However, this enigmatic protein appears to be a key player in various other biological processes, such as differentiation, cell death, and angiogenesis.
Recent studies by several laboratories, including our own, support the notion that c-Myc may have a central role in the regulation of beta-cell mass required for replication (during the G1/S transition), cell growth, and apoptosis [227].
It is likely that blood glucose levels influence the regulation and expression of c-Myc in beta-cells. In fact, it was observed that expression of c-Myc was stimulated by high glucose in a Ca2+-dependent manner and also by cAMP [232]. High glucose also increases c-Myc mRNA levels in rat islets in vitro [233]. Therefore, taken together, these studies support the opinion that high blood glucose levels may activate beta-cell replication through c-Myc expression.
Besides, the actual mechanism by which hyperglycaemia activates c-Myc is fully clear. It is known that cell signalling of c-Myc pass through cyclin E-CDK2 activity early in the G1 phase of the cell cycle, an essential event in c-Myc-induced G1-S progression [234, 235].
In particular, it is well known that CCND2 (which encodes cyclin D2) and CDK4 are direct target genes of c-MYC [236, 237]. Expression of CCND2 and CDK4 leads to sequestration of p27, the CDK inhibitor, in cyclin D2–CDK4 complexes [236]. The subsequent degradation of p27 has been shown to involve two other c-MYC target genes, CUL-1 and CKS [237]. By preventing the binding of p27 to cyclin E-CDK2 complexes, c-MYC allows inhibitor-free cyclin E-CDK2 complexes to become accessible to phosphorylation by cyclin activating kinase (CAK). Increased CDK2 and CDK4 activities would result in Rb hyperphosphorylation and subsequent release of E2F from Rb.
However, the final result of c-Myc activation on beta-cell is peculiar and differs in various tissues. For example, in contrast of skin, the predominant effect of activating c-MYC in pancreatic beta-cells of adult transgenic mice (cMYCERTAM) is apoptosis and not proliferation, resulting in development of diabetes [46, 238].
However, concerning cell signalling mechanism involved in cMYC-induced apoptosis, D cyclins do not appear to be required for c-MYC-induced apoptosis in vitro [239], indicating that these two major functions of c-MYC (proliferation and apoptosis) may involve, at least in part, distinct sets of downstream mediators.
Calcineurin/Nuclear Factor of Activated T-Cells —
More recently a new important pathway able to regulate beta-cell mass and function has been identified. The key components of this pathway are Ca2+/calmodulin-dependent protein phosphatase 2-b (or calcineurin-b) and nuclear factor of activated T cells (NFAT).
In beta-cells calcineurin is activated by an increase of cytosolic Ca2+, once activated, it can dephosphorylate NFAT transcription factors, which translocate into the nucleus where bind to consensus NFAT binding elements on specific gene promoters resulting in the activation of gene transcription [240]. On the contrary, some specific NFAT kinase can phosphorylate NFAT proteins and then inactivate and exporting them from nucleus.
In particular, it was reported in transgenic NFAT activated mice that NFAT in beta-cells can induce proliferation activating transcription of genes coding for cyclin D1, cyclin D2, and CDK4 [159].
More recently, another possible way to regulate the beta-cell mass has been reported. In fact, it was observed that calcineurin/NFAT pathway is implicated in the glucose regulation of IRS-2 gene expression. In particular, NFAT activated can bind to IRS-2 promoter enhancing its transcription and it was prevented by specific inhibition of NFAT [241]. This positive effect on beta-cell survival observed was also previously described [242]. Unfortunately, until now, the mechanisms involved in the regulation of beta-cell cycle by calcineurin are partially understood.
The important role of calcineurin/NFAT on beta-cell proliferation was highlighted using different transgenic models. Using mice with beta-cell-specific deletion of the Calcineurin phosphatase regulatory subunit, Calcineurin b1 (Cnb1), has shown a reduction of beta-cell function, proliferation and mass, and it coincided with reduced expression of normal regulators of beta-cell proliferation. Similarly, long-term activation of calcineurin induces impaired glucose tolerance by alterations in beta-cell mass, and the activation of calcineurin signalling negatively affects proliferation and survival of beta-cells [243].
On the contrary, in normal adult beta-cells, conditional NFAT activation has shown to promote expression of cell-cycle regulators resulting in an increase of beta-cell proliferation and mass, resulting in hyperinsulinaemia [159].
2.3. Cell Cycle/Molecular Pathways of Beta-Cell Proliferation
The critical point in the beta-cell cycle seems to be the G1S “checkpoint.” At this point, the cell is committed to DNA replication, and it is regulated by the retinoblastoma protein (Rb). Rb is critical because it has the ability to stall the cell cycle. Rb itself is inactivated by a group of serine threonine protein kinases, the cyclin-dependant kinases (CDKs) including CDK4/6 and CDK2, which in turn are negatively regulated by cyclin-dependent kinase inhibitors (CDKIs).
Rb is a member of a family of pocket proteins which is able to block G1S transition by binding to E2F transcriptional activator proteins. When liberated from Rb, E2F transcription factor elicits its activity in controlling the transition from G1 to S phase.
Mice deleted for E2F-1 display defective pancreatic growth and islet dysfunction [187]. In E2F-1 and E2F-2 double knockout mice, both exocrine and endocrine glands had atrophied by 3 months of age, a feature not seen in single knockouts at the same time point. Diabetes subsequently followed, along with an increased rate of apoptosis [182].
The cyclin-dependent kinase CDK4 and 6 are activated by Cyclin D (in particular, D1 and D2) whereas CDK2 is activated by Cyclin E. Both Cyclins D1 and D2 are essential for normal postnatal islet growth. In adult islets, global deletion of cyclin D2 fails to stimulate adequate compensatory upregulation of cyclin D1 within islets and drastically damages postnatal beta-cell proliferation, islet mass, and decreases glucose tolerance [7, 189]. The importance of cyclin D1 has been highlighted using transgenic mice that overexpress cyclin D1. In this animal model, an increase in both beta-cell mass and proliferation was shown [190].
CDK4 has also been shown to be a key regulator of beta-cell cycle [244]. Knockout studies have shown a very specific phenotype, with beta-cell hypoplasia and severe diabetes [191, 194, 245], whilst studies using an activating mutation in CDK4, rendering it resistant to p16, have resulted in pancreatic hyperplasia, which demonstrated normal physiology [191, 192]. Other studies have shown up to a three-fold increase in beta-cell proliferation with CDK4 overexpression [191, 193].
The CDKs are inhibited by two groups of CDK inhibitors (CDKIs) which are also expressed in islets, INK4 proteins (p16, p15, p18, p19 inhibit CDK 4 and CDK6), and CIP/KIP proteins (p21, p27 and p57). Whilst INK4 family proteins promote cell cycle arrest, p21 and p27 are integrated into the cyclin D/Cdk4 complex that is able to arrest cell cycle progression through dephosphorylation of Rb protein [246], on the other hand, they are able to inhibit cyclin E/A CDK2 complex resulting in a block of phosphorylation of Rb protein [247].
Various studies have looked at the effects of CDKIs on islets. In p21−/− mice, islets were phenotypically and metabolically comparable to their wildtype counterparts, possibly due to upregulation of p57, whereas loss of function of p57 has been associated with hyperinsulinaemia at infancy [248]. Actively inhibiting the effect of p18, using knockout mice resulted in beta-cell hyperplasia of up to 40% that appears to be CDK4 dependent [245].
In contrast, p27 has been shown to partially reverse the islet phenotype independent of CDK4. Overexpression of p27 in beta-cells of transgenic mice impairs beta-cell proliferation, resulting in decreased beta-cell mass and diabetes [195]. Furthermore, p27−/− mice increases beta-cell proliferation doubling their beta-cell mass at birth, and this expansion was accompanied by increased insulin secretion [249].
3. Role of MicroRNAs on the Beta-Cell Mass Regulation
MicroRNAs (miRNAs) are small 19–23 nucleotide noncoding RNA molecules that act as posttranscriptional regulators of different genes involved in various cellular processes, such as apoptosis, differentiation, and proliferation. This regulation pass through Generally, miRNAs can regulate protein synthesis either by repressing translation and/or by degradation through deadenylation of mRNA targets [250, 251].
The role of miRNAs in beta-cell mass regulation is not yet fully understood. The most studied miRNA in this contest is miR-375. It was reported that its overexpression attenuates proliferation of beta-cells and glucose-induced insulin secretion [16–18]. Indeed, ectopic expression of miR-375 in diabetic pancreatic beta-cells results in increased susceptibility to fatty acid induced apoptosis [16]. Moreover, it was reported that in ob/ob mice in which miR-375 was deleted, they develop a marked decrease in beta-cell mass, which results in a severe insulin-deficient diabetes not found in ob/ob mice [15]. Therefore, it is becoming clear that miR-375 targets a suit of genes that negatively regulate cell growth and proliferation and that aberrant loss of this miRNA leads to dramatic reduction of beta-cell mass [15].
Furthermore, it was reported that overexpressions of both miR-34a and miR-146a are involved in programmed cell death, in particular, in apoptotic processes. In contrast, reducing miR-34a or miR-146a levels did not affect cell survival.
In fact, prolonged beta-cell exposure to palmitate (FFA) and proinflammatory cytokines changes/increases the expression of miR-34a and miR-146a, and treatment with anti-miR34a or anti-miR148 diminished the number of death cells in the presence of apoptotic stimulus.
The effect on the regulation of apoptosis may be due to the capacity of miR-34 to control the expression of the antiapoptotic protein Bcl2 [252], meanwhile miR-146a may control the apoptotic process through the regulation of the NF-kB pathway [253].
4. Beta-Cell Mass Plasticity
In addition to maintaining beta-cell mass under normal circumstances, as just discussed, an organism must also be able to alter its beta-cell mass in accordance with insulin's requirements. In particular conditions, such as pregnancy and obesity, beta-cell mass enlargement is observed [2]. However, when compensatory beta-cell mass expansion is inadequate, gestational diabetes in the case of pregnancy and T2D in the case of obesity are the results.
Although the majority of humans do not become diabetic in these circumstances, a significant part of the population is predisposed to beta-cell failure, for currently unknown reasons. The precise mechanism of beta-cell mass maintenance and expansion, that is, proliferation, neogenesis, or increase in size, has been elucidated only in part [178, 254, 255].
4.1. Reversible Beta-Cell Mass Expansion during Pregnancy
Different studies in rodents found a two- to five-fold increase in beta-cell mass during gestation [255, 256] and demonstrated an involvement of both beta-cell hypertrophy [178, 256, 257] and proliferation [256, 257]. Van Assche et al. [254] have reported that in humans during pregnancy the beta-cell mass increased 2.4-fold compared with nonpregnant women. Unfortunately, human data are limited to few studies using a small number of subjects. Butler et al. [258] have reported an approximately 1.4-fold increase in beta-cell mass using 18 women. In contrast to rodents, beta-cell mass expansion in humans was achieved by the formation of new islets or islet neogenesis [258].
The main stimuli for beta-cell proliferation during pregnancy are placental lactogen (PL), prolactin (PRL), growth hormone (GH) [178, 259], and serotonin [260]. Postpartum, in rodents, beta-cell mass returns to normal levels within 10 days through increased beta-cell apoptosis, decreased beta-cell proliferation, and reduced beta-cell size [255].
Placental Lactogen —
Placental lactogen (PL) hormone has been implicated as the primary factor responsible for the enhanced islet mass and function that occur during pregnancy [178, 259]. PL interacts with receptors in the PRL/GH receptor family, stimulating the JAK-2/STAT-5 intracellular signalling pathway [261, 262].
Prior studies performed in vitro and over the short term suggested that PL is a more powerful islet mitogen than GH or PRL [178, 263]. These data were confirmed in in vivo studies using transgenic mice expressing mouse PL-1 under the control of the rat insulin promoter (RIP-mPL1) [259, 264]. The expansion of beta-cell mass in RIP-mPL1 was attributed principally to a two-fold increase in beta-cell proliferation and a 20% increase in beta-cell size (hypertrophy). Interestingly, islet number was not significantly increased when compared to wild-type mice [259]. These findings were supported by a PL receptor knockout study, which showed a significant reduction in beta-cell mass [265]. Therefore, PL could offer a novel therapeutic target to treat diabetes.
Prolactin —
Prolactin (PRL), a hormone, acts through its specific receptor, prolactin-receptor (PRLR) to induce beta-cell proliferation in vitro [263]. Targeted deletion of the PRLR reduces beta-cell mass and mildly impairs insulin secretion [265]. The pivotal role of the PRLR for beta-cell adaptation during pregnancy was demonstrated using pregnant mice heterozygous for the PRLR null mutant. These mice exhibited reduced beta-cell proliferation, and impaired glucose tolerance [266]. It has been extensively demonstrated in different in vitro studies that PRL binding to PRLR is able to activate different signaling pathways such as STAT5, MAPK, and IRS/PI3K resulting in enhancing beta-cell mass during pregnancy [267–269]. It is currently unclear whether PRL and/or PL stimulate these pathways in vivo.
Menin —
More recently, menin (a tumour suppressor encoded by the Men1 gene) has been linked to the regulation of beta-cell proliferation during pregnancy [157]. Expression of menin, p18, and p27 are reduced during pregnancy in maternal islets, thus leading to islet mass expansion to meet the increased metabolic demand. After birth, menin returns to normal. Interestingly, menin is regulated by prolactin [157], supporting menin as an important mediator of beta-cell proliferation during pregnancy. It has also been proposed that, during pregnancy, activation of JAK2 and AKT, in response to prolactin, leads to increased p21 expression, whereas menin and p18 expression are suppressed. It is well known that AKT and p21 induce beta-cell proliferation, meanwhile the downregulation of menin and p18 allows for enhanced beta-cell proliferation [270].
Serotonin —
A recent study by Kim et al. [260] shows that beta-cells, like serotoninergic neurons, are able to synthesize, store, and secrete serotonin (5-HT) as well as express the specific serotonin receptors (5-HTR2B and 5-HTR1D). These authors reported that, in pregnant mice, 5-HTR2Bexpression increased significantly during midgestation (day 6 to 16) and normalized at the end of gestation, whereas 5-HTR1D expression increased at the end of gestation (day 17) and postpartum. Notably, increased 5-HTR2Bexpression closely correlated with the period of increased beta-cell proliferation, and increased 5-HTR1Dexpression correlated with the cessation of beta-cell proliferation and regression of beta-cell mass. Moreover, it was reported that in pregnant mice stimulation with high concentrations of prolactin induces a strong increase in the serotonin expression compared with nonpregnant control mice.
These results suggest that during pregnancy lactogenic signalling induces serotonin expression and synthesis in islets able to stimulate beta-cell proliferation through the 5-HTR2B pathway. Shortly before parturition, expression of 5-HTR2B decreases and 5-HTR1D expression increases, resulting in the reduction of beta-cell proliferation and beta-cell mass. In this way, beta-cell mass returns rapidly to prepregnancy levels.
4.2. Compensation for Body Mass
Excessive increase in body weight leads to obesity. Obesity is linked/correlated with insulin resistance and associated with compensatory physiological response at the level of beta-cell mass increase. As previously described, when compensatory beta-cell mass expansion is inadequate, T2D in the case of obesity is consequential.
Diet-induced obesity results in insulin resistance and beta-cell mass expansion in humans and mice. In nondiabetic animal models of obesity, for example, the C57Bl/6 mouse strain which is notoriously susceptible to these effects, there is a 2.2-fold increase in beta-cell mass and proliferation after 4 months of a high-fat diet versus a control diet [271]. Meanwhile, in the Zucker diabetic fatty (ZDF) fa/fa rat, beta-cell mass increased four folds compared with lean controls [79]. There is also evidence for increased beta-cell mass in nondiabetic obese humans [51].
However, these mice eventually become diabetic and lose their beta-cell mass due to increased beta-cell apoptosis and reduced beta-cell proliferation.
In genetic models of obesity and insulin resistance, there is a compensatory expansion of beta-cell mass. For example, in mice lacking a functional leptin receptor (db/db mice), beta-cell mass exhibits two-fold beta-cell mass increase [115]. The Zucker rat (fa/fa) also has a homozygous mutation in the gene encoding the leptin receptor. ZDF rats exhibit increased beta-cell mass and increased beta-cell proliferation prior to the onset of diabetes, but increased beta-cell apoptosis prevents them from adequately expanding their beta-cell mass after the onset of diabetes, despite continued high rates of beta-cell proliferation [79]. This phenotype contrasts with that observed in nondiabetic Zucker fatty (ZF) rats, which possess the same mutation as ZDF rats and also become obese and insulin resistant but do not develop diabetes due to sufficient beta-cell mass expansion through increased beta-cell proliferation, neogenesis, and hypertrophy [79].
5. Conclusions
On the basis of available research findings that we have discussed above, we can propose that (i) postnatal beta-cell mass sizes in response to changing metabolic demands, (ii) is carried out by an interaction of beta-cell replication (proliferation and/or neogenesis) and apoptosis, and (iii) this process is regulated by different growth factors/nutrients that interact between them.
Actually, our present knowledge to the understanding of proliferation, neogenesis, and apoptosis is still incomplete.
The balance between apoptosis and replication seems to be pivotal in a right beta-cell mass maintenance. Failure in this balance results in beta-cell dysfunction and consequently diabetes onset. Because of the worldwide diffusion of this pathology, new therapies able to restore the original beta-cell mass and its functionality are under continuous development. Islet transplantation is already under clinical investigation but for the foreseeable future it will be very restricted in application due to the limited supplies of human cadaveric islets for transplantation. As a result, there is intense interest in developing other sources of new beta-cells and identification of new molecules able to restore beta-cell mass in diabetics.
References
- 1.Dor Y, Melton DA. How important are adult stem cells for tissue maintenance? Cell Cycle. 2004;3(9):1104–1106. [PubMed] [Google Scholar]
- 2.Rhodes CJ. Type 2 diabetes—a matter of β-cell life and death? Science. 2005;307(5708):380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 3.Bonner-Weir S. Islet growth and development in the adult. Journal of Molecular Endocrinology. 2000;24(3):297–302. doi: 10.1677/jme.0.0240297. [DOI] [PubMed] [Google Scholar]
- 4.Duvillié B, Currie C, Chrones T, et al. Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to β-cell hyperplasia in mutant mice lacking insulin. Endocrinology. 2002;143(4):1530–1537. doi: 10.1210/endo.143.4.8753. [DOI] [PubMed] [Google Scholar]
- 5.Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet β cell expression of constitutively active Akt1/PKBα induces striking hypertrophy, hyperplasia, and hyperinsulinemia. Journal of Clinical Investigation. 2001;108(11):1631–1638. doi: 10.1172/JCI13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 7.Georgia S, Bhushan A. β cell replication is the primary mechanism for maintaining postnatal β cell mass. Journal of Clinical Investigation. 2004;114(7):963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathis D, Vence L, Benoist C. β-cell death during progression to diabetes. Nature. 2001;414(6865):792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 9.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 10.Swenne I, Andersson A. Effect of genetic background on the capacity for islet cell replication in mice. Diabetologia. 1984;27(4):464–467. doi: 10.1007/BF00273912. [DOI] [PubMed] [Google Scholar]
- 11.Ackermann AM, Gannon M. Molecular regulation of pancreatic β-cell mass development, maintenance, and expansion. Journal of Molecular Endocrinology. 2007;38(1-2):193–206. doi: 10.1677/JME-06-0053. [DOI] [PubMed] [Google Scholar]
- 12.Okada T, Chong WL, Hu J, et al. Insulin receptors in β-cells are critical for islet compensatory growth response to insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8977–8982. doi: 10.1073/pnas.0608703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS-2 are required for compensatory β cell hyperplasia in response to high-fat diet-induced insulin resistance. Journal of Clinical Investigation. 2007;117(1):246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez-Valverde SL, Taft RJ, Mattick JS. MicroRNAs in β-cell biology, insulin resistance, diabetes and its complications. Diabetes. 2011;60(7):1825–1831. doi: 10.2337/db11-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poy MN, Hausser J, Trajkovski M, et al. miR-375 maintains normal pancreatic α- and β-cell mass. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(14):5813–5818. doi: 10.1073/pnas.0810550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Xu X, Liang Y, et al. miR-375 enhances palmitate-induced lipoapoptosis in insulin-secreting NIT-1 cells by repressing myotrophin (V1) protein expression. International Journal of Clinical and Experimental Pathology. 2010;3(3):254–264. [PMC free article] [PubMed] [Google Scholar]
- 17.Ouaamari AE, Baroukh N, Martens GA, Lebrun P, Pipeleers D, Van Obberghen E. MiR-375 targets 3′l-phosphoinositide-dependent protein Kinase-1 and regulates glucose-induced biological responses in pancreatic β-Cells. Diabetes. 2008;57(10):2708–2717. doi: 10.2337/db07-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia HQ, Pan Y, Peng J, Lu GX. Over-expression of miR375 reduces glucose-induced insulin secretion in Nit-1 cells. Molecular Biology Reports. 2011;38(5):3061–3665. doi: 10.1007/s11033-010-9973-9. [DOI] [PubMed] [Google Scholar]
- 19.McCarthy MI. Genomics, type 2 diabetes, and obesity. The New England Journal of Medicine. 2010;363(24):2339–2350. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization. Diabetes Fact Sheet no. 312. 2011, http://www.who.int/mediacentre/factsheets/fs312/en/index.html.
- 21.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Research and Clinical Practice. 2010;87(1):4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Levy J, Atkinson AB, Bell PM, McCance DR, Hadden DR. Beta-cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10-year follow-up of the belfast diet study. Diabetic Medicine. 1998;15(4):290–296. doi: 10.1002/(SICI)1096-9136(199804)15:4<290::AID-DIA570>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 23.Turner R. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) The Lancet. 1998;352(9131):837–853. [PubMed] [Google Scholar]
- 24.Turner R. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) The Lancet. 1998;352(9131):854–865. [PubMed] [Google Scholar]
- 25.Shamoon H, Duffy H, Fleischer N, et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 26.Richter B, Bandeira-Echtler E, Bergerhoff K, Clar C, Ebrahim SH. Rosiglitazone for type 2 diabetes mellitus. Cochrane Database of Systematic Reviews (Online) 2007;(3, article CD006063) doi: 10.1002/14651858.CD006063.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 28.Montanya E, Nacher V, Biarnes M, Soler J. Linear correlation between β-cell mass and body weight throughout the lifespan in Lewis rats. Role of β-cell hyperplasia and hypertrophy. Diabetes. 2000;49(8):1341–1346. doi: 10.2337/diabetes.49.8.1341. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen JH, Galsgaard ED, Møldrup A, et al. Regulation of β-cell mass by hormones and growth factors. Diabetes. 2001;50(1, supplement):S25–S29. doi: 10.2337/diabetes.50.2007.s25. [DOI] [PubMed] [Google Scholar]
- 30.Butler AE, Janson J, Soeller WC, Butler PC. Increased β-cell apoptosis prevents adaptive increase in β-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52(9):2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 31.Gagliardino JJ, Del Zotto H, Massa L, Flores LE, Borelli MI. Pancreatic duodenal homeobox-1 and islet neogenesis-associated protein: a possible combined marker of activateable pancreatic cell precursors. Journal of Endocrinology. 2003;177(2):249–259. doi: 10.1677/joe.0.1770249. [DOI] [PubMed] [Google Scholar]
- 32.Steil GM, Trivedi N, Jonas JC, et al. Adaptation of β-cell mass to substrate oversupply: enhanced function with normal gene expression. American Journal of Physiology. 2001;280(5):E788–E796. doi: 10.1152/ajpendo.2001.280.5.E788. [DOI] [PubMed] [Google Scholar]
- 33.Bernard C, Berthault MF, Saulnier C, Ktorza A. Neogenesis vs. apoptosis as main components of pancreatic β cell mass changes in glucose-infused normal and mildly diabetic adult rats. FASEB Journal. 1999;13(10):1195–1205. doi: 10.1096/fasebj.13.10.1195. [DOI] [PubMed] [Google Scholar]
- 34.Bernard C, Thibault C, Berthault MF, et al. Pancreatic β-cell regeneration after 48-h glucose infusion in mildly diabetic rats is not correlated with functional improvement. Diabetes. 1998;47(7):1058–1065. doi: 10.2337/diabetes.47.7.1058. [DOI] [PubMed] [Google Scholar]
- 35.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38(1):49–53. doi: 10.2337/diab.38.1.49. [DOI] [PubMed] [Google Scholar]
- 36.Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic β-cell mass in vivo in adult rats. Endocrinology. 2003;144(6):2717–2727. doi: 10.1210/en.2002-221112. [DOI] [PubMed] [Google Scholar]
- 37.Laybutt DR, Glandt M, Xu G, et al. Critical reduction β-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. The Journal of Biological Chemistry. 2003;278(5):2997–3005. doi: 10.1074/jbc.M210581200. [DOI] [PubMed] [Google Scholar]
- 38.Swenne I. The role of glucose on the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic B-cells. Diabetes. 1982;31(9):754–760. doi: 10.2337/diab.31.9.754. [DOI] [PubMed] [Google Scholar]
- 39.Topp BG, McArthur MD, Finegood DT. Metabolic adaptations to chronic glucose infusion in rats. Diabetologia. 2004;47(9):1602–1610. doi: 10.1007/s00125-004-1493-5. [DOI] [PubMed] [Google Scholar]
- 40.Porat S, Weinberg-Corem N, Tornovsky-Babaey S, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metabolism. 2011;13(4):440–449. doi: 10.1016/j.cmet.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoorens A, van de Casteele M, Klöppel G, Pipeleers D. Glucose promotes survival of rat pancreatic β cells by activating synthesis of proteins which suppress a constitutive apoptotic program. Journal of Clinical Investigation. 1996;98(7):1568–1574. doi: 10.1172/JCI118950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jonas JC, Sharma A, Hasenkamp W, et al. Chronic hyperglycemia triggers loss of pancreatic β cell differentiation in an animal model of diabetes. The Journal of Biological Chemistry. 1999;274(20):14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- 43.Grill V, Björklund A. Overstimulation and β-cell function. Diabetes. 2001;50(supplement 1):S122–S124. doi: 10.2337/diabetes.50.2007.s122. [DOI] [PubMed] [Google Scholar]
- 44.Maedler K, Sergeev P, Ris F, et al. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. Journal of Clinical Investigation. 2002;110(6):851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka Y, Tran POT, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic β cells against oxidative stress in a model of glucose toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(19):12363–12368. doi: 10.1073/pnas.192445199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in β cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109(3):321–334. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 47.Laybutt DR, Weir GC, Kaneto H, et al. Overexpression of c-Myc in β-cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression, and diabetes. Diabetes. 2002;51(6):1793–1804. doi: 10.2337/diabetes.51.6.1793. [DOI] [PubMed] [Google Scholar]
- 48.Pelengaris S, Khan M. The many faces of c-MYC. Archives of Biochemistry and Biophysics. 2003;416(2):129–136. doi: 10.1016/s0003-9861(03)00294-7. [DOI] [PubMed] [Google Scholar]
- 49.Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and β-cell failure in type 2 diabetes mellitus. Journal of Pediatric Endocrinology and Metabolism. 2003;16(1):5–22. doi: 10.1515/jpem.2003.16.1.5. [DOI] [PubMed] [Google Scholar]
- 50.Clark A, Wells CA, Buley ID, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Research. 1988;9(4):151–159. [PubMed] [Google Scholar]
- 51.Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Survey and Synthesis of Pathology Research. 1985;4(2):110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- 52.Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45(1):85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 53.Yoon KH, Ko SH, Cho JH, et al. Selective β-cell loss and α-cell expansion in patients with type 2 diabetes mellitus in Korea. Journal of Clinical Endocrinology and Metabolism. 2003;88(5):2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 54.Andersson A. The influence of hyperglycaemia, hyperinsulinaemia and genetic background on the fate of intrasplenically implanted mouse islets. Diabetologia. 1983;25(3):269–272. doi: 10.1007/BF00279942. [DOI] [PubMed] [Google Scholar]
- 55.Guillen C, Navarro P, Robledo M, Valverde AM, Benito M. Differential mitogenic signaling in insulin receptor-deficient fetal pancreatic β-cells. Endocrinology. 2006;147(4):1959–1968. doi: 10.1210/en.2005-0831. [DOI] [PubMed] [Google Scholar]
- 56.Koiter TR, Wijkstra S, van der Schaaf-Verdonk GCJ, Moes H, Schuiling GA. Pancreatic beta-cell function and islet-cell proliferation: effect of hyperinsulinaemia. Physiology and Behavior. 1995;57(4):717–721. doi: 10.1016/0031-9384(94)00290-8. [DOI] [PubMed] [Google Scholar]
- 57.Movassat J, Saulnier C, Portha B. β-cell mass depletion precedes the onset of hyperglycaemia in the GK rat, a genetic model of non-insulin-dependent diabetes mellitus. Diabete et Metabolisme. 1995;21(5):365–370. [PubMed] [Google Scholar]
- 58.Guz Y, Nasir I, Teitelman G. Regeneration of pancreatic β cells from intra-islet precursor cells in an experimental model of diabetes. Endocrinology. 2001;142(11):4956–4968. doi: 10.1210/endo.142.11.8501. [DOI] [PubMed] [Google Scholar]
- 59.Otani K, Kulkarni RN, Baldwin AC, et al. Reduced β-cell mass and altered glucose sensing impair insulin-secretory function in βIRKO mice. American Journal of Physiology. 2004;286(1):E41–E49. doi: 10.1152/ajpendo.00533.2001. [DOI] [PubMed] [Google Scholar]
- 60.Blume N, Skouv J, Larsson LI, Hoist JJ, Madsen OD. Potent inhibitory effects of transplantable rat glucagonomas and insulinomas on the respective endogenous islet cells are associated with pancreatic apoptosis. Journal of Clinical Investigation. 1995;96(5):2227–2235. doi: 10.1172/JCI118278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flatt PR, Tan KS, Bailey CJ. Effects of transplantation and resection of a radiation-induced rat insulinoma on glucose homeostasis and the endocrine pancreas. British Journal of Cancer. 1986;54(4):685–692. doi: 10.1038/bjc.1986.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(13):7475–7480. doi: 10.1073/pnas.131192998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miyaura C, Chen L, Appel M, et al. Expression of reg/PSP, a pancreatic exocrine gene: relationship to changes in islet β-cell mass. Molecular Endocrinology. 1991;5(2):226–234. doi: 10.1210/mend-5-2-226. [DOI] [PubMed] [Google Scholar]
- 64.Giacca A, Xiao C, Oprescu AI, Carpentier AC, Lewis GF. Lipid-induced pancreatic β-cell dysfunction: focus on in vivo studies. American Journal of Physiology. 2011;300(2):E255–E262. doi: 10.1152/ajpendo.00416.2010. [DOI] [PubMed] [Google Scholar]
- 65.Poitout V. Glucolipotoxicity of the pancreatic β-cell: myth or reality? Biochemical Society Transactions. 2008;36(5):901–904. doi: 10.1042/BST0360901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. Journal of Clinical Investigation. 1994;93(2):870–876. doi: 10.1172/JCI117042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou YP, Grill V. Long term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. Journal of Clinical Endocrinology and Metabolism. 1995;80(5):1584–1590. doi: 10.1210/jcem.80.5.7745004. [DOI] [PubMed] [Google Scholar]
- 68.Oprescu AI, Bikopoulos G, Naassan A, et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56(12):2927–2937. doi: 10.2337/db07-0075. [DOI] [PubMed] [Google Scholar]
- 69.Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic β- cell function in obese nondiabetic humans but not in individuals with type 2. Diabetes. 2000;49(3):399–408. doi: 10.2337/diabetes.49.3.399. [DOI] [PubMed] [Google Scholar]
- 70.Carpentier A, Mittelman SD, Lamarche B, Bergman RN, Giacca A, Lewis GF. Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. American Journal of Physiology. 1999;276(6):E1055–E1066. doi: 10.1152/ajpendo.1999.276.6.E1055. [DOI] [PubMed] [Google Scholar]
- 71.Kashyap S, Belfort R, Gastaldelli A, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52(10):2461–2474. doi: 10.2337/diabetes.52.10.2461. [DOI] [PubMed] [Google Scholar]
- 72.Xiao C, Giacca A, Carpentier A, Lewis GF. Differential effects of monounsaturated, polyunsaturated and saturated fat ingestion on glucose-stimulated insulin secretion, sensitivity and clearance in overweight and obese, non-diabetic humans. Diabetologia. 2006;49(6):1371–1379. doi: 10.1007/s00125-006-0211-x. [DOI] [PubMed] [Google Scholar]
- 73.Sako Y, Grill VE. A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology. 1990;127(4):1580–1589. doi: 10.1210/endo-127-4-1580. [DOI] [PubMed] [Google Scholar]
- 74.Elks ML. Chronic perifusion of rat islets with palmitate suppresses glucose- stimulated insulin release. Endocrinology. 1993;133(1):208–214. doi: 10.1210/endo.133.1.8319569. [DOI] [PubMed] [Google Scholar]
- 75.Gremlich S, Bonny C, Waeber G, Thorens B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. The Journal of Biological Chemistry. 1997;272(48):30261–30269. doi: 10.1074/jbc.272.48.30261. [DOI] [PubMed] [Google Scholar]
- 76.Briaud I, Harmon JS, Kelpe CL, Segu VBG, Poitout V. Lipotoxicity of the pancreatic β-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50(2):315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. The Journal of Biological Chemistry. 2003;278(32):30015–30021. doi: 10.1074/jbc.M302548200. [DOI] [PubMed] [Google Scholar]
- 78.Hagman DK, Hays LB, Parazzoli SD, Poitout V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. The Journal of Biological Chemistry. 2005;280(37):32413–32418. doi: 10.1074/jbc.M506000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of β-cell mass compensation for insulin resistance and β-cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47(3):358–364. doi: 10.2337/diabetes.47.3.358. [DOI] [PubMed] [Google Scholar]
- 80.Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced β cell apoptosis: a link between obesity and diabetes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(5):2498–2502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 2001;50(8):1771–1777. doi: 10.2337/diabetes.50.8.1771. [DOI] [PubMed] [Google Scholar]
- 82.Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on β-cell turnover and function. Diabetes. 2001;50(1):69–76. doi: 10.2337/diabetes.50.1.69. [DOI] [PubMed] [Google Scholar]
- 83.Lupi R, Dotta F, Marselli L, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51(5):1437–1442. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- 84.Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic β-cells (INS-1) The Journal of Biological Chemistry. 2002;277(51):49676–49684. doi: 10.1074/jbc.M208756200. [DOI] [PubMed] [Google Scholar]
- 85.Piro S, Anello M, Di Pietro C, et al. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism. 2002;51(10):1340–1347. doi: 10.1053/meta.2002.35200. [DOI] [PubMed] [Google Scholar]
- 86.Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic β-cell turnover and function. Diabetes. 2003;52(3):726–733. doi: 10.2337/diabetes.52.3.726. [DOI] [PubMed] [Google Scholar]
- 87.Maestre I, Jordán J, Calvo S, et al. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the β-cell line INS-1. Endocrinology. 2003;144(1):335–345. doi: 10.1210/en.2001-211282. [DOI] [PubMed] [Google Scholar]
- 88.El-Assaad W, Buteau J, Peyot ML, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology. 2003;144(9):4154–4163. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- 89.Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V. Inhibition of insulin gene expression by long-term exposure of pancreatic β cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metabolism. 2000;49(4):532–536. doi: 10.1016/s0026-0495(00)80021-9. [DOI] [PubMed] [Google Scholar]
- 90.Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP. Antecedent hyperglycemia, not hyperlipidemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes. 2001;50(7–12):2481–2486. doi: 10.2337/diabetes.50.11.2481. [DOI] [PubMed] [Google Scholar]
- 91.Briaud I, Kelpe CL, Johnson LM, Tran POT, Poitout V. Differential effects of hyperlipidemia on insulin secretion in islets of langerhans from hyperglycemic versus normoglycemic rats. Diabetes. 2002;51(3):662–668. doi: 10.2337/diabetes.51.3.662. [DOI] [PubMed] [Google Scholar]
- 92.Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic β-cells exposed to free fatty acids. The Journal of Biological Chemistry. 2003;278(22):19709–19715. doi: 10.1074/jbc.M209709200. [DOI] [PubMed] [Google Scholar]
- 93.Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic β-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52(1):124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- 94.Farilla L, Hongxiang H, Bertolotto C, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143(11):4397–4408. doi: 10.1210/en.2002-220405. [DOI] [PubMed] [Google Scholar]
- 95.Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drueker DJ. Glucagon-like peptide-1 receptor signaling modulates β cell apoptosis. The Journal of Biological Chemistry. 2003;278(1):471–478. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- 96.Hui H, Nourparvar A, Zhao X, Perfetti R. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology. 2003;144(4):1444–1455. doi: 10.1210/en.2002-220897. [DOI] [PubMed] [Google Scholar]
- 97.Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144(12):5149–5158. doi: 10.1210/en.2003-0323. [DOI] [PubMed] [Google Scholar]
- 98.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both β-cell replication and neogenesis, resulting in increased β-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48(12):2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- 99.Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Cζ activation mediates glucagon-like peptide-1—induced pancreatic β-cell proliferation. Diabetes. 2001;50(10):2237–2243. doi: 10.2337/diabetes.50.10.2237. [DOI] [PubMed] [Google Scholar]
- 100.Perfetti R, Zhou JIE, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology. 2000;141(12):4600–4605. doi: 10.1210/endo.141.12.7806. [DOI] [PubMed] [Google Scholar]
- 101.Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic β-cell mass. Diabetes. 2006;55(5):1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- 102.Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. β-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes. 2005;54(2):482–491. doi: 10.2337/diabetes.54.2.482. [DOI] [PubMed] [Google Scholar]
- 103.Park S, Dong X, Fisher TL, et al. Exendin-4 uses Irs2 signaling to mediate pancreatic β cell growth and function. The Journal of Biological Chemistry. 2006;281(2):1159–1168. doi: 10.1074/jbc.M508307200. [DOI] [PubMed] [Google Scholar]
- 104.Song WJ, Schreiber WE, Zhong E, et al. Exendin-4 stimulation of cyclin A2 in β-cell proliferation. Diabetes. 2008;57(9):2371–2381. doi: 10.2337/db07-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58(6):1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tourrel C, Bailbé D, Meile MJ, Kergoat M, Portha B. Glucagon-like peptide-1 and exendin-4 stimulate β-cell neogenesis in streptozotocin-treated newborn rats resulting in persistently improved glucose homeostasis at adult age. Diabetes. 2001;50(7):1562–1570. doi: 10.2337/diabetes.50.7.1562. [DOI] [PubMed] [Google Scholar]
- 107.Abraham EJ, Leech CA, Lin JC, Zulewski H, Habener JF. Insulinotropic hormone glucagon-like peptide-1 differentiation of human pancreatic islet-derived progenitor cells into insulin-producing cells. Endocrinology. 2002;143(8):3152–3161. doi: 10.1210/endo.143.8.8973. [DOI] [PubMed] [Google Scholar]
- 108.Xu G, Kaneto H, Lopez-Avalos MD, Weir GC, Bonner-Weir S. GLP-1/exendin-4 facilitates β-cell neogenesis in rat and human pancreatic ducts. Diabetes Research and Clinical Practice. 2006;73(1):107–110. doi: 10.1016/j.diabres.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 109.Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates β-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes. 2003;52(3):741–750. doi: 10.2337/diabetes.52.3.741. [DOI] [PubMed] [Google Scholar]
- 110.Carpenter T, Trautmann ME, Baron AD, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. The New England Journal of Medicine. 2005;353(20):2192–2194. doi: 10.1056/NEJM200511173532017. [DOI] [PubMed] [Google Scholar]
- 111.Cummings DE. Gastric bypass and nesidioblastosis—too much of a good thing for islets? The New England Journal of Medicine. 2005;353(3):300–302. doi: 10.1056/NEJMe058170. [DOI] [PubMed] [Google Scholar]
- 112.Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. The New England Journal of Medicine. 2005;353(3):249–254. doi: 10.1056/NEJMoa043690. [DOI] [PubMed] [Google Scholar]
- 113.Takiguchi S, Takata Y, Funakoshi A, et al. Disrupted cholecystokinin type-A receptor (CCKAR) gene in OLETF rats. Gene. 1997;197(1-2):169–175. doi: 10.1016/s0378-1119(97)00259-x. [DOI] [PubMed] [Google Scholar]
- 114.Wang Y, Guo XH. Effects of GLP-1 treatment on protection of B cells in Otsuka Long-Evans Tokushima fatty rats. Beijing da Xue Xue Bao. 2006;38(4):375–380. [PubMed] [Google Scholar]
- 115.Wang Q, Brubaker P. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45(9):1263–1273. doi: 10.1007/s00125-002-0828-3. [DOI] [PubMed] [Google Scholar]
- 116.Green BD, Lavery KS, Irwin N, et al. Novel glucagon-like peptide-1 (GLP-1) analog (Val8)GLP-1 results in significant improvements of glucose tolerance and pancreatic β-cell function after 3-week daily administration in obese diabetic (ob/ob) mice. Journal of Pharmacology and Experimental Therapeutics. 2006;318(2):914–921. doi: 10.1124/jpet.105.097824. [DOI] [PubMed] [Google Scholar]
- 117.Kim JG, Baggio LL, Bridon DP, et al. Development and characterization of a glucagon-like peptide 1-albumin conjugate the ability to activate the glucagon-like peptide 1 receptor in vivo. Diabetes. 2003;52(3):751–759. doi: 10.2337/diabetes.52.3.751. [DOI] [PubMed] [Google Scholar]
- 118.Li Y, Shi C, Lv Q, et al. GLP-1 C-terminal structures affect its blood glucose lowering-function. Journal of Peptide Science. 2008;14(7):777–785. doi: 10.1002/psc.997. [DOI] [PubMed] [Google Scholar]
- 119.Mu J, Petrov A, Eiermann GJ, et al. Inhibition of DPP-4 with sitagliptin improves glycemic control and restores islet cell mass and function in a rodent model of type 2 diabetes. European Journal of Pharmacology. 2009;623(1–3):148–154. doi: 10.1016/j.ejphar.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 120.Maida A, Hansotia T, Longuet C, Seino Y, Drucker DJ. Differential importance of glucose-dependent insulinotropic polypeptide vs glucagon-like peptide 1 receptor signaling for beta cell survival in mice. Gastroenterology. 2009;137(6):2146–2157. doi: 10.1053/j.gastro.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 121.Gault VA, Irwin N, Green BD, et al. Chemical ablation of gastric inhibitory polypeptide receptor action by daily (Pro3)GIP administration improves glucose tolerance and ameliorates insulin resistance and abnormalities of islet structure in obesity-related diabetes. Diabetes. 2005;54(8):2436–2446. doi: 10.2337/diabetes.54.8.2436. [DOI] [PubMed] [Google Scholar]
- 122.Cheng Q, Law PK, De Gasparo M, Leung PS. Combination of the dipeptidyl peptidase IV inhibitor LAF237 [(S)-1-[(3-hydroxy-1-adamantyl)ammo]acetyl-2-cyanopyrrolidine] with the angiotensin II type 1 receptor antagonist valsartan [N-(1-oxopentyl)-N- [[2′-(1H-tetrazol-5-yl)-[1,1′-biphenyl]-4-yl]methyl]-L-valine] enhances pancreatic islet morphology and function in a mouse model of type 2 diabetes. Journal of Pharmacology and Experimental Therapeutics. 2008;327(3):683–691. doi: 10.1124/jpet.108.142703. [DOI] [PubMed] [Google Scholar]
- 123.Irwin N, Clarke GC, Green BD, et al. Evaluation of the antidiabetic activity of DPP IV resistant N-terminally modified versus mid-chain acylated analogues of glucose-dependent insulinotropic polypeptide. Biochemical Pharmacology. 2006;72(6):719–728. doi: 10.1016/j.bcp.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 124.Kidd GS, Donowitz M, O’Dorisio T, Cataland S, Newman F. Mild chronic watery diarrhea-hypokalemia syndrome associated with pancreatic islet cell hyperplasia. Elevated plasma and tissue levels of gastric inhibitory polypeptide and successful management with nicotinic acid. The American Journal of Medicine. 1979;66(5):883–888. doi: 10.1016/0002-9343(79)91147-1. [DOI] [PubMed] [Google Scholar]
- 125.Friedrichsen BN, Neubauer N, Lee YC, et al. Stimulation of pancreatic β-cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. Journal of Endocrinology. 2006;188(3):481–492. doi: 10.1677/joe.1.06160. [DOI] [PubMed] [Google Scholar]
- 126.Ehses JA, Casilla VR, Doty T, et al. Glucose-dependent insulinotropic polypeptide promotes β-(INS-1) cell survival via cyclic adenosine monophosphate-mediated caspase-3 inhibition and regulation of p38 mitogen-activated protein kinase. Endocrinology. 2003;144(10):4433–4445. doi: 10.1210/en.2002-0068. [DOI] [PubMed] [Google Scholar]
- 127.Trümper A, Trümper K, Hörsch D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in β(INS-1)-cells. Journal of Endocrinology. 2002;174(2):233–246. doi: 10.1677/joe.0.1740233. [DOI] [PubMed] [Google Scholar]
- 128.Trümper A, Trümper K, Trusheim H, Arnold R, Göke B, Hörsch D. Glucose-dependent insulinotropic polypeptide is a growth factor for β (INS-1) cells by pleiotropic signaling. Molecular Endocrinology. 2001;15(9):1559–1570. doi: 10.1210/mend.15.9.0688. [DOI] [PubMed] [Google Scholar]
- 129.Widenmaier SB, Kim SJ, Yang GK, et al. A GIP receptor agonist exhibits β-cell anti-apoptotic actions in rat models of diabetes resulting in improved β-cell function and glycemic control. PLoS ONE. 2010;5(3, article e9590) doi: 10.1371/journal.pone.0009590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hogg J, han VKM, Clemmons DR, Hill DJ. Interactions of nutrients, insulin-like growth factors (IGFs) and IGF-binding proteins in the regulation of DNA synthesis by isolated fetal rat islets of Langerhans. Journal of Endocrinology. 1993;138(3):401–412. doi: 10.1677/joe.0.1380401. [DOI] [PubMed] [Google Scholar]
- 131.Sieradzki J, Fleck H, Chatterjee AK, Schatz H. Stimulatory effect of insulin-like growth factor-I on [3H]thymidine incorporation, DNA content and insulin biosynthesis and secretion of isolated pancreatic rat islets. Journal of Endocrinology. 1988;117(1):59–62. doi: 10.1677/joe.0.1170059. [DOI] [PubMed] [Google Scholar]
- 132.Swenne I, Hill DJ, Strain AJ, Milner RDG. Growth hormone regulation of somatomedin C/insulin-like growth factor I production and DNA replication in fetal rat islets in tissue culture. Diabetes. 1987;36(3):288–294. doi: 10.2337/diab.36.3.288. [DOI] [PubMed] [Google Scholar]
- 133.Swenne I. Pancreatic beta-cell growth and diabetes mellitus. Diabetologia. 1992;35(3):193–201. doi: 10.1007/BF00400917. [DOI] [PubMed] [Google Scholar]
- 134.Nielsen JH, Linde S, Welinder BS, Billestrup N, Madsen OD. Growth hormone is a growth factor for the differentiated pancreatic β-cell. Molecular Endocrinology. 1989;3(1):165–173. doi: 10.1210/mend-3-1-165. [DOI] [PubMed] [Google Scholar]
- 135.Billestrup N, Moldrup A, Serup P, Mathews LS, Norstedt G, Nielsen JH. Introduction of exogenous growth hormone receptors augments growth hormone-responsive insulin biosynthesis in rat insulinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(18):7210–7214. doi: 10.1073/pnas.87.18.7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hügl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic β-cell growth is glucose-dependent synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS- 1 cells. The Journal of Biological Chemistry. 1998;273(28):17771–17779. doi: 10.1074/jbc.273.28.17771. [DOI] [PubMed] [Google Scholar]
- 137.George M, Ayuso E, Casellas A, Costa C, Devedjian JC, Bosch F. β cell expression of IGF-I leads to recovery from type 1 diabetes. Journal of Clinical Investigation. 2002;109(9):1153–1163. doi: 10.1172/JCI12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Petrik J, Pell JM, Arany E, et al. Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology. 1999;140(5):2353–2363. doi: 10.1210/endo.140.5.6732. [DOI] [PubMed] [Google Scholar]
- 139.Lu Y, Herrera PL, Guo Y, et al. Pancreatic-specific inactivation of IGF-I gene causes enlarged pancreatic islets and significant resistance to diabetes. Diabetes. 2004;53(12):3131–3141. doi: 10.2337/diabetes.53.12.3131. [DOI] [PubMed] [Google Scholar]
- 140.Devedjian JC, George M, Casellas A, et al. Transgenic mice overexpressing insulin-like growth factor-II in β cells develop type 2 diabetes. Journal of Clinical Investigation. 2000;105(6):731–740. doi: 10.1172/JCI5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kuntz E, Pinget M, Damgé C. Cholecystokinin octapeptide: a potential growth factor for pancreatic beta cells in diabetic rats. Journal of the Pancreas. 2004;5(6):464–475. [PubMed] [Google Scholar]
- 142.Brelje TC, Sorenson RL. Role of prolactin versus growth hormone on islet B-cell proliferation in vitro: implications for pregnancy. Endocrinology. 1991;128(1):45–57. doi: 10.1210/endo-128-1-45. [DOI] [PubMed] [Google Scholar]
- 143.Lavine JA, Raess PW, Davis DB, et al. Contamination with E1A-positive wild-type adenovirus accounts for species-specific stimulation of islet cell proliferation by CCK: a cautionary note. Molecular Endocrinology. 2010;24(2):464–467. doi: 10.1210/me.2009-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Williams K, Abanquah D, Joshi-Gokhale S, et al. Systemic and acute administration of parathyroid hormone-related peptide(1–36) stimulates endogenous beta cell proliferation while preserving function in adult mice. Diabetologia. 2011;54(11):2867–2877. doi: 10.1007/s00125-011-2260-z. [DOI] [PubMed] [Google Scholar]
- 145.Fujinaka Y, Sipula D, Garcia-Ocaña A, Vasavada RC. Characterization of mice doubly transgenic for parathyroid hormone-related protein and murine placental lactogen: a novel role for placental lactogen in pancreatic β-cell survival. Diabetes. 2004;53(12):3120–3130. doi: 10.2337/diabetes.53.12.3120. [DOI] [PubMed] [Google Scholar]
- 146.Vasavada RC, Cavaliere C, D’Ercole AJ, et al. Overexpression of parathyroid hormone-related protein in the pancreatic islets of transgenic mice causes islet hyperplasia, hyperinsulinemia, and hypoglycemia. The Journal of Biological Chemistry. 1996;271(2):1200–1208. doi: 10.1074/jbc.271.2.1200. [DOI] [PubMed] [Google Scholar]
- 147.Porter SE, Sorenson RL, Dann P, Garcia-Ocana A, Stewart AF, Vasavada RC. Progressive pancreatic islet hyperplasia in the islet-targeted, parathyroid hormone-related protein-overexpressing mouse. Endocrinology. 1998;139(9):3743–3751. doi: 10.1210/endo.139.9.6212. [DOI] [PubMed] [Google Scholar]
- 148.Kondegowda NG, Joshi-Gokhale S, Harb G, et al. Parathyroid hormone-related protein enhances human β-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes. 2010;59(12):3131–3138. doi: 10.2337/db09-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Villanueva-Peñacarrillo ML, Cancelas J, De Miguel F, et al. Parathyroid hormone-related peptide stimulates DNA synthesis and insulin secretion in pancreatic islets. Journal of Endocrinology. 1999;163(3):403–408. doi: 10.1677/joe.0.1630403. [DOI] [PubMed] [Google Scholar]
- 150.Zhang B, Hosaka M, Sawada Y, et al. Parathyroid hormone-related protein induces insulin expression through activation of MAP kinase-specific phosphatase-1 that dephosphorylates c-Jun NH2-terminal kinase in pancreatic beta-cells. Diabetes. 2003;52(11):2720–2730. doi: 10.2337/diabetes.52.11.2720. [DOI] [PubMed] [Google Scholar]
- 151.Vandegraaff N, et al. Growth factors and beta cell replication. The International Journal of Biochemistry & Cell Biology. 2006;38(5-6):931–950. doi: 10.1016/j.biocel.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 152.Irako T, Akamizu T, Hosoda H, et al. Ghrelin prevents development of diabetes at adult age in streptozotocin-treated newborn rats. Diabetologia. 2006;49(6):1264–1273. doi: 10.1007/s00125-006-0226-3. [DOI] [PubMed] [Google Scholar]
- 153.Granata R, Settanni F, Trovato L, et al. Unacylated as well s acylated ghrelin promotes cell survival and inhibit apoptosis in HIT-T15 pancretic β-cells. Journal of Endocrinological Investigation. 2006;29(9):RC19–RC22. doi: 10.1007/BF03347367. [DOI] [PubMed] [Google Scholar]
- 154.Granata R, Settanni F, Biancone L, et al. Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic β-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-kinase/Akt signaling. Endocrinology. 2007;148(2):512–529. doi: 10.1210/en.2006-0266. [DOI] [PubMed] [Google Scholar]
- 155.Sayo Y, Murao K, Imachi H, et al. The multiple endocrine neoplasia type 1 gene product, menin, inhibits insulin production in rat insulinoma cells. Endocrinology. 2002;143(6):2437–2440. doi: 10.1210/endo.143.6.8950. [DOI] [PubMed] [Google Scholar]
- 156.Crabtree JS, Scacheri PC, Ward JM, et al. Of mice and MEN1: insulinomas in a conditional mouse knockout. Molecular and Cellular Biology. 2003;23(17):6075–6085. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic β-cell proliferation. Annual Review of Cell and Developmental Biology. 2006;22:311–338. doi: 10.1146/annurev.cellbio.22.010305.104425. [DOI] [PubMed] [Google Scholar]
- 159.Heit JJ, Apelqvist A, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic β-cell growth and function. Nature. 2006;443(7109):345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 160.Schnepp RW, Chen YX, Wang H, et al. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Research. 2006;66(11):5707–5715. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.La P, Yang Y, Karnik SK, et al. Menin-mediated caspase 8 expression in suppressing multiple endocrine neoplasia type 1. The Journal of Biological Chemistry. 2007;282(43):31332–31340. doi: 10.1074/jbc.M609555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liadis N, Salmena L, Kwan E, et al. Distinct in vivo roles of Caspase-8 in β-cells in physiological and diabetes models. Diabetes. 2007;56(9):2302–2311. doi: 10.2337/db06-1771. [DOI] [PubMed] [Google Scholar]
- 163.Islam MS, Morton NM, Hansson A, Emilsson V. Rat insulinoma-derived pancreatic β-cells express a functional leptin receptor that mediates a proliferative response. Biochemical and Biophysical Research Communications. 1997;238(3):851–855. doi: 10.1006/bbrc.1997.7399. [DOI] [PubMed] [Google Scholar]
- 164.Okuya S, Tanabe K, Tanizawa Y, Oka Y. Leptin increases the viability of isolated rat pancreatic islets by suppressing apoptosis. Endocrinology. 2001;142(11):4827–4830. doi: 10.1210/endo.142.11.8494. [DOI] [PubMed] [Google Scholar]
- 165.Shimabukuro M, Wang MY, Zhou YT, Newgard CB, Unger RH. Protection against lipoapoptosis of β cells through leptin-dependent maintenance of Bcl-2 expression. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9558–9561. doi: 10.1073/pnas.95.16.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brown JEP, Dunmore SJ. Leptin decreases apoptosis and alters BCL-2: bax ratio in clonal rodent pancreatic beta-cells. Diabetes/Metabolism Research and Reviews. 2007;23(6):497–502. doi: 10.1002/dmrr.726. [DOI] [PubMed] [Google Scholar]
- 167.Islam MS, Sjöholm A, Emilsson V. Fetal pancreatic islets express functional leptin receptors and leptin stimulates proliferation of fetal islet cells. International Journal of Obesity. 2000;24(10):1246–1253. doi: 10.1038/sj.ijo.0801370. [DOI] [PubMed] [Google Scholar]
- 168.Tanabe K, Okuya S, Tanizawa Y, Matsutani A, Oka Y. Leptin induces proliferation of pancreatic β cell line MIN6 through activation of mitogen-activated protein kinase. Biochemical and Biophysical Research Communications. 1997;241(3):765–768. doi: 10.1006/bbrc.1997.7894. [DOI] [PubMed] [Google Scholar]
- 169.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature Reviews Molecular Cell Biology. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 170.Kubota N, Terauchi Y, Tobe K, et al. Insulin receptor substrate 2 plays a crucial role in β cells and the hypothalamus. Journal of Clinical Investigation. 2004;114(7):917–927. doi: 10.1172/JCI21484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huypens PR. Leptin and adiponectin regulate compensatory beta cell growth in accordance to overweight. Medical Hypotheses. 2007;68(5):1134–1137. doi: 10.1016/j.mehy.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 172.Maedler K, Sergeev P, Ehses JA, et al. Leptin modulates β cell expression of IL-1 receptor antagonist and release of IL-1β in human islets. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(21):8138–8143. doi: 10.1073/pnas.0305683101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Guldstrand M, Ahrén B, Adamson U. Improved β-cell function after standardized weight reduction in severely obese subjects. American Journal of Physiology. 2003;284(3):E557–E565. doi: 10.1152/ajpendo.00325.2002. [DOI] [PubMed] [Google Scholar]
- 174.Maedler K, Schulthess FT, Bielman C, et al. Glucose and leptin induce apoptosis in human β-cells and impair glucose-stimulated insulin secretion through activation of c-Jun N-terminal kinases. FASEB Journal. 2008;22(6):1905–1913. doi: 10.1096/fj.07-101824. [DOI] [PubMed] [Google Scholar]
- 175.Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: a protein secreted by visceral fat that Mimics the effects of insulin. Science. 2005;307(5708):426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 176.Chen MP, Chung FM, Chang DM, et al. Elevated plasma level of visfatin/pre-B cell colony-enhancing factor in patients with type 2 diabetes mellitus. Journal of Clinical Endocrinology and Metabolism. 2006;91(1):295–299. doi: 10.1210/jc.2005-1475. [DOI] [PubMed] [Google Scholar]
- 177.Nielsen JH, Svensson C, Galsgaard ED, Møldrup A, Billestrup N. Beta cell proliferation and growth factors. Journal of Molecular Medicine. 1999;77(1):62–66. doi: 10.1007/s001090050302. [DOI] [PubMed] [Google Scholar]
- 178.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: β-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Hormone and Metabolic Research. 1997;29(6):301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 179.Elghazi L, Cras-Méneur C, Czernichow P, Scharfmann R. Role for FGFR2IIIb-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(6):3884–3889. doi: 10.1073/pnas.062321799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Freidin M, Asche S, Bargiello TA, Bennett MVL, Abrams CK. Connexin 32 increases the proliferative response of Schwann cells to neuregulin-1 (Nrg1) Proceedings of the National Academy of Sciences of the United States of America. 2009;106(9):3567–3572. doi: 10.1073/pnas.0813413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lim MCC, Maubach G, Zhuo L. TGF-β1 down-regulates connexin 43 expression and gap junction intercellular communication in rat hepatic stellate cells. European Journal of Cell Biology. 2009;88(12):719–730. doi: 10.1016/j.ejcb.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 182.Naus CCG, Bechberger JF, Zhang Y, et al. Altered gap junctional communication, intercellular signaling, and growth in cultured astrocytes deficient in connexin43. Journal of Neuroscience Research. 1997;49(5):528–540. doi: 10.1002/(SICI)1097-4547(19970901)49:5<528::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 183.Serre-Beinier V, Le Gurun S, Belluardo N, et al. Cx36 preferentially connects β-cells within pancreatic islets. Diabetes. 2000;49(5):727–734. doi: 10.2337/diabetes.49.5.727. [DOI] [PubMed] [Google Scholar]
- 184.Klee P, Lamprianou S, Charollais A, et al. Connexin implication in the control of the murine beta-cell mass. Pediatric Research. 2011;70(2):142–147. doi: 10.1203/PDR.0b013e318220f106. [DOI] [PubMed] [Google Scholar]
- 185.Winnier G, Blessing M, Labosky PA, Hogan BLM. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes and Development. 1995;9(17):2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 186.Hua H, Zhang YQ, Dabernat S, et al. BMP4 regulates pancreatic progenitor cell expansion through Id2. The Journal of Biological Chemistry. 2006;281(19):13574–13580. doi: 10.1074/jbc.M600526200. [DOI] [PubMed] [Google Scholar]
- 187.Fajas L, Annicotte JS, Miard S, Sarruf D, Watanabe M, Auwerx J. Impaired pancreatic growth, β cell mass, and β cell function in E2F1-7(−/−) mice. Journal of Clinical Investigation. 2004;113(9):1288–1295. doi: 10.1172/JCI18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Iglesias A, Murga M, Laresgoiti U, et al. Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. Journal of Clinical Investigation. 2004;113(10):1398–1407. doi: 10.1172/JCI18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kushner JA, Ciemerych MA, Sicinska E, et al. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Molecular and Cellular Biology. 2005;25(9):3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang X, Gaspard JP, Mizukami Y, Li J, Graeme-Cook F, Chung DC. Overexpression of cyclin D1 in pancreatic β-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes. 2005;54(3):712–719. doi: 10.2337/diabetes.54.3.712. [DOI] [PubMed] [Google Scholar]
- 191.Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nature Genetics. 1999;22(1):44–54. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 192.Marzo N, Mora C, Fabregat ME, et al. Pancreatic islets from cyclin-dependent kinase 4/R24C (Cdk4) knockin mice have significantly increased beta cell mass and are physiologically functional, indicating that Cdk4 is a potential target for pancreatic beta cell mass regeneration in Type 1 diabetes. Diabetologia. 2004;47(4):686–694. doi: 10.1007/s00125-004-1372-0. [DOI] [PubMed] [Google Scholar]
- 193.Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF. Induction of beta-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes. 2004;53(1):149–159. doi: 10.2337/diabetes.53.1.149. [DOI] [PubMed] [Google Scholar]
- 194.Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Molecular and Cellular Biology. 1999;19(10):7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Uchida T, Nakamura T, Hashimoto N, et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nature Medicine. 2005;11(2):175–182. doi: 10.1038/nm1187. [DOI] [PubMed] [Google Scholar]
- 196.Jetton TL, Lausier J, LaRock K, et al. Mechanisms of compensatory β-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes. 2005;54(8):2294–2304. doi: 10.2337/diabetes.54.8.2294. [DOI] [PubMed] [Google Scholar]
- 197.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385(6616):544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 198.Lingohr MK, Dickson LM, Wrede CE, McCuaig JF, Myers Jr. MG, Rhodes CJ. IRS-3 inhibits IRS-2-mediated signaling in pancreatic β-cells. Molecular and Cellular Endocrinology. 2003;204(1-2):85–99. doi: 10.1016/s0303-7207(03)00124-2. [DOI] [PubMed] [Google Scholar]
- 199.Wang Q, Li L, Xu E, Wong V, Rhodes C, Brubaker PL. Glucagon-like peptitle-1 regulates proliferation and apoptosis via activation of protein kinase B in pancreatic INS-1 beta cells. Diabetologia. 2004;47(3):478–487. doi: 10.1007/s00125-004-1327-5. [DOI] [PubMed] [Google Scholar]
- 200.Cohen P. The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Philosophical Transactions of the Royal Society B. 1999;354(1382):485–495. doi: 10.1098/rstb.1999.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Belham C, Wu S, Avruch J. Intracellular signalling: PDK1—a kinase at the hub of things. Current Biology. 1999;9(3):R93–R96. doi: 10.1016/s0960-9822(99)80058-x. [DOI] [PubMed] [Google Scholar]
- 202.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? Journal of Cell Science. 2001;114(16):2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 203.Kitamura YI, Kitamura T, Kruse JP, et al. FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metabolism. 2005;2(3):153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 204.Kitamura T, Nakae J, Kitamura Y, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. Journal of Clinical Investigation. 2002;110(12):1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lee CS, Sund NJ, Vatamaniuk MZ, Matschinsky FM, Stoffers DA, Kaestner KH. Foxa2 controls Pdx1 gene expression in pancreatic β-cells in vivo. Diabetes. 2002;51(8):2546–2551. doi: 10.2337/diabetes.51.8.2546. [DOI] [PubMed] [Google Scholar]
- 206.Kawamori D, Kaneto H, Nakatani Y, et al. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. The Journal of Biological Chemistry. 2006;281(2):1091–1098. doi: 10.1074/jbc.M508510200. [DOI] [PubMed] [Google Scholar]
- 207.Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in β cell compensation to insulin resistance. Journal of Clinical Investigation. 2006;116(3):775–782. doi: 10.1172/JCI24967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ai J, Duan J, Lv X, et al. Overexpression of FoxO1 causes proliferation of cultured pancreatic β cells exposed to low nutrition. Biochemistry. 2010;49(1):218–225. doi: 10.1021/bi901414g. [DOI] [PubMed] [Google Scholar]
- 209.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 210.Araki E, Lipes MA, Patti ME, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372(6502):186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 211.Hennige AM, Ozcan U, Okada T, et al. Alterations in growth and apoptosis of insulin receptor substrate-1-deficient β-cells. American Journal of Physiology. 2005;289(2):E337–E346. doi: 10.1152/ajpendo.00032.2004. [DOI] [PubMed] [Google Scholar]
- 212.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. The Journal of Biological Chemistry. 2001;276(42):38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 213.Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα . Nature Medicine. 2001;7(10):1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- 214.Dickson LM, Rhodes CJ. Pancreatic β-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? American Journal of Physiology. 2004;287(2):E192–E198. doi: 10.1152/ajpendo.00031.2004. [DOI] [PubMed] [Google Scholar]
- 215.Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. Journal of Clinical Investigation. 2004;114(6):828–836. doi: 10.1172/JCI21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Brissova M, Blaha M, Spear C, et al. Reduced PDX-1 expression impairs islet response to insulin resistance and worsens glucose homeostasis. American Journal of Physiology. 2005;288(4):E707–E714. doi: 10.1152/ajpendo.00252.2004. [DOI] [PubMed] [Google Scholar]
- 217.Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA. Mice with beta cell overexpression of glycogen synthase kinase-3β have reduced beta cell mass and proliferation. Diabetologia. 2008;51(4):623–631. doi: 10.1007/s00125-007-0914-7. [DOI] [PubMed] [Google Scholar]
- 218.Surjit M, Lal SK. Glycogen synthase kinase-3 phosphorylates and regulates the stability of p27kip1 protein. Cell Cycle. 2007;6(5):580–588. doi: 10.4161/cc.6.5.3899. [DOI] [PubMed] [Google Scholar]
- 219.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes and Development. 1998;12(22):3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. The Journal of Biological Chemistry. 2006;281(10):6395–6403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- 221.Downward J. Ras signalling and apoptosis. Current Opinion in Genetics and Development. 1998;8(1):49–54. doi: 10.1016/s0959-437x(98)80061-0. [DOI] [PubMed] [Google Scholar]
- 222.Benito M, Valverde AM, Lorenzo M. IGF-I: a mitogen also involved in differentiation processes in mammalian cells. International Journal of Biochemistry and Cell Biology. 1996;28(5):499–510. doi: 10.1016/1357-2725(95)00168-9. [DOI] [PubMed] [Google Scholar]
- 223.Kadowaki T, et al. Signal transduction mechanism of insulin and insulin-like growth factor-1. Endocrine Journal. 1996;43(supplement):S33–S41. doi: 10.1507/endocrj.43.suppl_s33. [DOI] [PubMed] [Google Scholar]
- 224.Avruch J, Zhang XF, Kyriakis JM. Raf meets ras: completing the framework of a signal transduction pathway. Trends in Biochemical Sciences. 1994;19(7):279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 225.Khoo S, Cobb MH. Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(11):5599–5604. doi: 10.1073/pnas.94.11.5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Smit LS, Meyer DJ, Billestrup N, Norstedt G, Schwartz J, Carter-Su C. The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Molecular Endocrinology. 1996;10(5):519–533. doi: 10.1210/mend.10.5.8732683. [DOI] [PubMed] [Google Scholar]
- 227.Chow JC, Ling PR, Qu Z, et al. Growth hormone stimulates tyrosine phosphorylation of JAK2 and STAT5, but not insulin receptor substrate-1 or SHC proteins in liver and skeletal muscle of normal rats in vivo. Endocrinology. 1996;137(7):2880–2886. doi: 10.1210/endo.137.7.8770909. [DOI] [PubMed] [Google Scholar]
- 228.Galsgaard ED, Friedrichsen BN, Nielsen JH, Møldrup A. Expression of dominant-negative STAT5 inhibits growth hormone- and prolactin-induced proliferation of insulin-producing cells. Diabetes. 2001;50(1, supplement):S40–S41. doi: 10.2337/diabetes.50.2007.s40. [DOI] [PubMed] [Google Scholar]
- 229.Jensen J, Galsgaard ED, Karlsen AE, Lee YC, Nielsen JH. STAT5 activation by human GH protects insulin-producing cells against interleukin-1β, interferon-γ and tumour necrosis factor-α-induced apoptosis independent of nitric oxide production. Journal of Endocrinology. 2005;187(1):25–36. doi: 10.1677/joe.1.06086. [DOI] [PubMed] [Google Scholar]
- 230.Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 231.Lindberg K, Rønn SG, Tornehave D, et al. Regulation of pancreatic β-cell mass and proliferation by SOCS-3. Journal of Molecular Endocrinology. 2005;35(2):231–243. doi: 10.1677/jme.1.01840. [DOI] [PubMed] [Google Scholar]
- 232.Jonas JC, Laybutt DR, Steil GM, et al. High glucose stimulates early response gene c-Myc expression in rat pancreatic β cells. The Journal of Biological Chemistry. 2001;276(38):35375–35381. doi: 10.1074/jbc.M105020200. [DOI] [PubMed] [Google Scholar]
- 233.Elouil H, Cardozo AK, Eizirik DL, Henquin JC, Jonas JC. High glucose and hydrogen peroxide increase c-Myc and haeme-oxygenase 1 mRNA levels in rat pancreatic islets without activating NFκB. Diabetologia. 2005;48(3):496–505. doi: 10.1007/s00125-004-1664-4. [DOI] [PubMed] [Google Scholar]
- 234.Steiner P, Philipp A, Lukas J, et al. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO Journal. 1995;14(19):4814–4826. doi: 10.1002/j.1460-2075.1995.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Berns K, Hijmans EM, Bernards R. Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene. 1997;15(11):1347–1356. doi: 10.1038/sj.onc.1201280. [DOI] [PubMed] [Google Scholar]
- 236.Bouchard C, Thieke K, Maier A, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO Journal. 1999;18(19):5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hermeking H, Rago C, Schuhmacher M, et al. Identification of CDK4 as a target of c-MYC. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(5):2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pelengaris S, Khan M. Oncogenic co-operation in β-cell tumorigenesis. Endocrine-Related Cancer. 2001;8(4):307–314. doi: 10.1677/erc.0.0080307. [DOI] [PubMed] [Google Scholar]
- 239.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate Myc-induced proliferation via sequestration of p27Kip1 and p21Cip1 . EMBO Journal. 1999;18(19):5310–5320. doi: 10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes and Development. 2003;17(18):2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 241.Demozay D, Tsunekawa S, Briaud I, Shah R, Rhodes CJ. Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet β-cells. Diabetes. 2011;60(11):2892–2902. doi: 10.2337/db11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Soleimanpour SA, Crutchlow MF, Ferrari AM, et al. Calcineurin signaling regulates human islet β-cell survival. The Journal of Biological Chemistry. 2010;285(51):40050–40059. doi: 10.1074/jbc.M110.154955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bernal-Mizrachi E, Cras-Méneur C, Ye BR, Johnson JD, Permutt MA. Transgenic overexpression of active calcineurin in β-cells results in decreased β-cell mass and hyperglycemia. PLoS ONE. 2010;5(8, article e11969) doi: 10.1371/journal.pone.0011969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Fatrai S, Elghazi L, Balcazar N, et al. Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55(2):318–325. doi: 10.2337/diabetes.55.02.06.db05-0757. [DOI] [PubMed] [Google Scholar]
- 245.Pei XH, Bai F, Tsutsui T, Kiyokawa H, Xiong Y. Genetic evidence for functional dependency of p18Ink4c on Cdk4. Molecular and Cellular Biology. 2004;24(15):6653–6664. doi: 10.1128/MCB.24.15.6653-6664.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Rane SG, Reddy EP. Cell cycle control of pancreatic beta cell proliferation. Front Biosci. 2000;5:D1–19. doi: 10.2741/rane. [DOI] [PubMed] [Google Scholar]
- 247.Sherr CJ. The INK4a/ARF network in tumour suppression. Nature Reviews Molecular Cell Biology. 2001;2(10):731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 248.Kassem SA, Ariel I, Thornton PS, et al. p57KIP2 expression in normal islet cells and in hyperinsulinism of infancy. Diabetes. 2001;50(12):2763–2769. doi: 10.2337/diabetes.50.12.2763. [DOI] [PubMed] [Google Scholar]
- 249.Georgia S, Bhushan A. p27 regulates the transition of β-cells from quiescence to proliferation. Diabetes. 2006;55(11):2950–2956. doi: 10.2337/db06-0249. [DOI] [PubMed] [Google Scholar]
- 250.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 251.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 252.Lovis P, Roggli E, Laybutt DR, et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic β-cell dysfunction. Diabetes. 2008;57(10):2728–2736. doi: 10.2337/db07-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Roggli E, Britan A, Gattesco S, et al. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic β-cells. Diabetes. 2010;59(4):978–986. doi: 10.2337/db09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. British Journal of Obstetrics and Gynaecology. 1978;85(11):818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 255.Scaglia L, Smith FE, Bonner-Weir S. Apoptosis contributes to the involution of β cell mass in the post partum rat pancreas. Endocrinology. 1995;136(12):5461–5468. doi: 10.1210/endo.136.12.7588296. [DOI] [PubMed] [Google Scholar]
- 256.Parsons JA, Bartke A, Sorenson RL. Number and size of islets of langerhans in pregnant, human growth hormone- expressing transgenic, and pituitary dwarf mice: effect of lactogenic hormones. Endocrinology. 1995;136(5):2013–2021. doi: 10.1210/endo.136.5.7720649. [DOI] [PubMed] [Google Scholar]
- 257.Rieck S, White P, Schug J, et al. The transcriptional response of the islet to pregnancy in mice. Molecular Endocrinology. 2009;23(10):1702–1712. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53(10):2167–2176. doi: 10.1007/s00125-010-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Vasavada RC, Garcia-Ocaña A, Zawalich WS, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. The Journal of Biological Chemistry. 2000;275(20):15399–15406. doi: 10.1074/jbc.275.20.15399. [DOI] [PubMed] [Google Scholar]
- 260.Kim H, Toyofuku Y, Lynn FC, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nature Medicine. 2010;16(7):804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocrine Reviews. 1998;19(3):225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- 262.Stout LE, Svensson AM, Sorenson RL. Prolactin regulation of islet-derived INS-1 cells: characteristics and immunocytochemical analysis of STAT5 translocation. Endocrinology. 1997;138(4):1592–1603. doi: 10.1210/endo.138.4.5089. [DOI] [PubMed] [Google Scholar]
- 263.Brelje TC, Scharp DW, Lacy PE, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology. 1993;132(2):879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 264.Cozar-Castellano I, Weinstock M, Haught M, Velázquez-Garcia S, Sipula D, Stewart AF. Evaluation of β-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21 cip. Diabetes. 2006;55(1):70–77. [PubMed] [Google Scholar]
- 265.Freemark M, Avril I, Fleenor DON, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology. 2002;143(4):1378–1385. doi: 10.1210/endo.143.4.8722. [DOI] [PubMed] [Google Scholar]
- 266.Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of β-cell mass during pregnancy. Endocrinology. 2009;150(4):1618–1626. doi: 10.1210/en.2008-1003. [DOI] [PubMed] [Google Scholar]
- 267.Brelje TC, Stout LE, Bhagroo NV, Sorenson RL. Distinctive roles for prolactin and growth hormone in the activation of signal transducer and activator of transcription 5 in pancreatic islets of Langerhans. Endocrinology. 2004;145(9):4162–4175. doi: 10.1210/en.2004-0201. [DOI] [PubMed] [Google Scholar]
- 268.Amaral MEC, Cunha DA, Anhê GF, et al. Participation of prolactin receptors and phosphatidylinositol 3-kinase and MAP kinase pathways in the increase in pancreatic islet mass and sensitivity to glucose during pregnancy. Journal of Endocrinology. 2004;183(3):469–476. doi: 10.1677/joe.1.05547. [DOI] [PubMed] [Google Scholar]
- 269.Amaral MEC, Ueno M, Carvalheira JB, et al. Prolactin-signal transduction in neonatal rat pancreatic islets and interaction with the insulin-signaling pathway. Hormone and Metabolic Research. 2003;35(5):282–289. doi: 10.1055/s-2003-41303. [DOI] [PubMed] [Google Scholar]
- 270.Hughes E, Huang C. Participation of Akt, menin, and p21 in pregnancy-induced β-cell proliferation. Endocrinology. 2011;152(3):847–855. doi: 10.1210/en.2010-1250. [DOI] [PubMed] [Google Scholar]
- 271.Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48(1):58–67. doi: 10.1007/s00125-004-1605-2. [DOI] [PubMed] [Google Scholar]