

Abstract
ExsA is a transcriptional activator of the Pseudomonas aeruginosa type III secretion system (T3SS) and a member of the AraC/XylS protein family. Each of the 10 ExsA-dependent promoter regions that define the T3SS regulon has two adjacent binding sites for monomeric ExsA. Whereas the promoter-proximal sites (binding site 1) contain highly conserved GnC and TGnnA sequences that are separated by ∼10 bp, the promoter-distal sites (binding site 2) share no obvious sequence similarity to each other or to the binding site 1 consensus. In the present study, we used footprinting with Fe-BABE (a protein-labeling reagent that can be conjugated to cysteine residues) to demonstrate that the two ExsA monomers bind to the PexsC, PexsD, PexoT, and PpcrG promoters in a head-to-tail orientation. The footprinting data further indicate that the conserved GnC and TGnnA sequences constitute binding site 1. When bound to site 1, the first helix-turn-helix (HTH) motif of ExsA interacts with the conserved GnC sequence, and the second HTH interacts at or near the TGnnA sequences. Genetic data using the PexoT promoter indicate that residues L198 and T199 in the first HTH motif of ExsA contact the guanine in the GnC sequence and that residue K202, also in the first HTH motif, contacts the cytosine. Likewise, evidence is presented that residues Q248, Y250, T252, and R257 located in the second HTH motif contribute to the recognition of the TGnnA sequence. These combined data define interactions of ExsA with site 1 on the PexoT promoter and provide insight into the nature of the interactions involved in recognition of binding site 2.
INTRODUCTION
The Gram-negative opportunistic pathogen Pseudomonas aeruginosa is a significant cause of morbidity and mortality in immunocompromised patients (32, 33). A primary virulence factor of P. aeruginosa is a type III secretion system (T3SS). The T3SS consists of a large macromolecular complex that is assembled in the bacterial cell envelope and functions by translocating effector proteins into eukaryotic host cells (1, 16, 21, 23, 37). The translocated effectors, which include ExoS, ExoT, ExoU, and ExoY, generally promote virulence through inhibition of phagocytosis, modulation of host inflammatory responses, and host cell killing (2, 16, 36). Mutants lacking a functional T3SS are severely attenuated for virulence in animal infection models (20, 21, 23, 37), and a role for the T3SS has been implicated in human disease severity and progression (35). Several studies have examined therapeutic interventions targeting the T3SS, including inhibition of translocase activity (37), effector activity (25), and T3SS gene transcription (15). The latter approach involved isolation of small-molecule inhibitors of ExsA, the primary transcriptional regulator of T3SS gene expression.
ExsA is a member of the AraC/XylS family of transcription factors that activates 10 promoters controlling expression of the T3SS effectors and their chaperones, the secretion and translocation machinery, and regulators of T3SS gene expression (10, 44). ExsA-dependent transcription is coupled to T3SS secretory activity by a partner-switching mechanism involving ExsD, ExsC, and ExsE (10, 44). ExsD functions as an antiactivator by binding to ExsA and inhibiting ExsA-dependent transcription (7, 27, 39). ExsC is a type III secretion chaperone that can form complexes with either ExsD or ExsE, a secreted substrate of the T3SS (9, 45). Under noninducing conditions for T3SS gene expression (e.g., high-Ca2+ growth medium), the T3SS machinery is secretion incompetent, resulting in intracellular accumulation of ExsE and preferential formation of ExsE•ExsC and ExsD•ExsA complexes. Inducing signals, which include Ca2+-limiting growth conditions and contact of P. aeruginosa with host cells, trigger T3SS-dependent secretion and/or translocation of ExsE into host cells (34, 40, 41). The resulting decrease in ExsE concentration in the bacterial cytoplasm triggers ExsC partner switching whereby formation of the ExsC•ExsD complex is favored and free ExsA is available to activate T3SS gene expression (9, 45).
The ExsA consensus-binding sequence (AaAAAnwmMygrCynnnmYTGayAk; underlining indicates the three highly conserved elements, and uppercase and lowercase correlate with the degree of sequence conservation) is similarly positioned in each of the 10 ExsA-dependent promoter regions and contains three conserved elements, i.e., an adenine-rich region, a GnC sequence, and a TGnnA sequence, the centers of each being separated by ∼10 bp (6). ExsA-dependent promoter regions contain two adjacent binding sites for monomeric ExsA, which are centered at the −41 (site 1) and −65 (site 2) positions relative to the transcriptional start site (6). Genetic analyses of the PexoT promoter suggested that the GnC and TGnnA sequences constitute binding site 1. Binding of ExsA to the promoter located upstream of exoT (PexoT) occurs via a monomer assembly pathway whereby monomeric ExsA first binds to site 1 and then recruits a second ExsA monomer to the lower-affinity site 2 (6). Efficient filling of site 2 is dependent upon the adenine-rich region (5 nucleotides) located at the boundary between binding sites 1 and 2 and on self-association between ExsA monomers, which is mediated by the amino-terminal domain of ExsA (6, 8, 22). While the determinants for binding site 1 are fairly well defined, binding sites 1 and 2 share no recognizable sequence similarity, and the elements within binding site 2 that are required for ExsA binding are addressed in an accompanying paper (8a).
Most members of the AraC/XylS family, including ExsA, consist of two distinct domains connected by a flexible linker. The amino-terminal domain is poorly conserved among family members but is generally involved in oligomerization and/or ligand binding (13, 26). The amino terminus of ExsA mediates the self-interaction of the two monomers when bound to promoter DNA and is also the target for binding by the ExsD antiactivator (8). The carboxy-terminal domain of AraC/XylS family members consists of two helix-turn-helix (HTH) DNA-binding motifs. The high-resolution crystal structure of the AraC/XylS family member MarA bound to target DNA provides a structural model for DNA binding by AraC/XylS proteins (31). Each MarA HTH motif has a recognition helix (helices 3 and 6) that inserts into adjacent major grooves on the same face of the DNA and makes base-specific contacts with target DNA (31). The high level of conservation in the DNA-binding domains of AraC/XylS family members suggests a common mechanism for recognition and binding to target DNA.
In the present study, we performed footprinting experiments with Fe-BABE (a protein-labeling reagent that can be conjugated to cysteine residues) to define the promoter regions recognized by ExsA at binding sites 1 and 2 and found that two ExsA monomers bind to the PexoT, PexsC, PexsD, and PpcrG promoters in a head-to-tail orientation. Using genetic screens, we determined that the two HTH motifs of an ExsA monomer interact with the conserved GnC and TGnnA sequences in binding site 1 and identified amino acid residues located within the recognition helices of the HTH motifs that participate in the recognition of the GnC and TGnnA sequences. Based on these findings, we propose that specific recognition of binding site 1 at each of the ExsA-dependent promoters involves a common mechanism.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and sample preparation.
Escherichia coli DH5α was used for all cloning and was maintained on Luria-Bertani (LB) agar plates with gentamicin (15 μg/ml) or ampicillin (100 μg/ml) as necessary. P. aeruginosa strains were cultured on Vogel-Bonner minimal medium (VBMM) agar plates with 100 μg/ml gentamicin as required (42). To measure β-galactosidase activity, P. aeruginosa strains were cultured to an absorbance (A600) of 1.0 in Trypticase soy broth (TSB) supplemented with 100 mM monosodium glutamate, 1% glycerol, and 2 mM EGTA. In some experiments, the P. aeruginosa strains were cultured in the presence of arabinose (as indicated in the figure legends) to elevate the expression of ExsA and the ExsA alanine substitution mutants from the arabinose-inducible expression vectors. β-Galactosidase activity was determined using ONPG (ortho-nitrophenyl-galactopyranoside) as previously described (9). The reported values for the β-galactosidase assays denote the average of at least three independent experiments, and error bars represent the standard error of the mean (SEM). Statistical analyses (two-tailed unpaired t tests) were performed using Prism 5 (GraphPad Software, Inc., La Jolla, CA).
Whole-cell lysate samples were prepared by pelleting 1.25 ml of cell culture (A600 = 1.0), suspending the pellet in 0.25 ml of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, and sonicating for 10 s. Samples were analyzed by SDS-PAGE followed by immunoblotting using antibodies directed against ExsA and developed using enhanced chemiluminescent fluorescence detection reagents (Thermo Scientific, Rockford, IL).
Mutagenesis.
Random mutagenesis of exsA was performed using the Diversify PCR random mutagenesis kit (Clontech, Mountain View, CA) and oligonucleotide primers flanking exsA (primers 29590679 and 42148424) (see Table S2 in the supplemental material). The resulting PCR products were digested with XbaI and SacI and ligated into the corresponding restriction sites in the pJN105 arabinose-inducible expression vector (28). E. coli DH5α was transformed with the resulting ligation mixture, and a plasmid library was generated by pooling colonies from LB agar plates containing gentamicin. The resulting plasmid library was introduced into the PA103 exsA::Ω mini-CTX-PexoT(C−45A)-lacZ reporter strain by electroporation, and screening was performed on VBMM agar plates containing 20 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside).
Libraries of exsA alleles carrying random mutations targeted within recognition helix 1 or 2 were generated using a two-step PCR as previously described with the following modifications (6). In the first step, megaprimers were generated by PCR using a common reverse primer (primer 42148424) and a specific forward primer randomized at the nucleotide position corresponding to codon L198, T199, T200, K202, E203, L204, Q248, S249, Y250, T252, Q253, or S254 (as indicated in Table S2 in the supplemental material). The megaprimers were gel purified (Qiagen, Inc.), mixed at an equimolar ratio for RH1 (L198, T199, T200, K202, E203, and L204) or RH2 (Q248, S249, Y250, T252, Q253, and S254), and used in subsequent PCRs with a common forward primer (primer 29590679). The resulting PCR products were cloned into the XbaI-SacI restriction sites of pJN105 and introduced into E. coli DH5α by standard transformation. A plasmid library derived from ∼7,000 colonies was isolated as described above, introduced into the indicated reporter strains, and screened on VBMM plates with 20 μg/ml X-Gal.
Tables S1 and S2 in the supplemental material show the oligonucleotide primers used for site-directed mutagenesis of exsA and the PexoT promoter. A previously described two-step PCR method was used to generate mutants (6). The exsA mutants were cloned as XbaI-SacI restriction fragments in the arabinose-inducible expression vector pJN105 (28). Promoter mutants were cloned as HindIII-EcoRI fragments in mini-CTX-lacZ (19) and integrated into the PA103 chromosome as previously described (3, 19) or used as PCR templates to generate DNA probes for electrophoretic mobility shift assay (EMSA) or footprinting experiments.
Fe-BABE conjugation and footprinting.
To generate the cysteine-less derivative of the exsAHis allele (pET16b exsAHisΔcys), each of the seven cysteine residues in exsA was changed to alanine using the QuikChange multisite-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions and the oligonucleotide primers indicated in Table S2 in the supplemental material. Novel cysteine residues were then introduced into pET16b exsAHisΔcys at glutamate 193 (E193C), methionine 196 (M196C), glycine 244 (G244C), or serine 246 (S246C) using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies) with the primers indicated in Table S2.
ExsAHisΔcys M196C and ExsAHisΔcys S246C were expressed in E. coli and purified by Ni2+-nitrilotriacetic acid (NTA) chromatography as previously described for ExsA (6). Prior to Fe-BABE conjugation, purified ExsAHisΔcys M196C and ExsAHisΔcys S246C were exchanged into conjugation buffer (20 mM MOPS [morpholinepropanesulfonic acid; pH 8.0], 200 mM NaCl, 1 mM EDTA, and 5% glycerol) using a PD-10 column (GE Healthcare). A 10-fold molar excess of Fe-BABE reagent (Pierce) was then added, and reaction mixtures were incubated in the dark for 2 h at 25°C. Conjugated ExsA was then exchanged into conjugation buffer, diluted with an equal volume of glycerol, and stored at −20°C.
DNA probes were generated by PCR using the primers and methodology for single end labeling with 32P as previously described (6). Binding reaction mixtures (20 μl) containing 1 nM promoter probe in DNA binding buffer (10 mM Tris [pH 8.0], 50 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, and 5% glycerol), 2.5 ng/μl poly(2′-deoxyinosinic-2′-deoxycytidylic acid) (Sigma), 100 ng/μl bovine serum albumin (New England BioLabs), and 100 nM ExsA were incubated for 15 min at 25°C. Cleavage reactions were initiated by the addition of 2.5 μl ascorbate solution (40 mM sodium ascorbate, 10 mM EDTA) for 1 min followed by the addition of 2.5 μl H2O2 solution (40 mM H2O2, 10 mM EDTA) for 2 min. Cleavage was terminated by adding 5 μl 100 mM thiourea, and the DNA was precipitated with sodium acetate and ethanol. The precipitate was suspended in formamide loading buffer (95% formamide, 10 mM NaOH, 0.05% xylene cyanol, 0.05% bromophenol blue), and samples were subjected to electrophoresis on a 1× Tris-borate-EDTA (TBE) sequencing gel as previously described (6). Imaging was performed using an FLA-7000 phosphorimager (Fujifilm) and Multigauge v3.0 software (Fujifilm).
EMSA.
The indicated alanine substitution mutants were PCR amplified from their respective pEB124 expression vectors (see Table S1 in the supplemental material) using primers 42308574 and 80815243 (Table S2). The resulting PCR products were cloned into the NdeI and BamHI restriction sites in the expression vector pET16b. The proteins were expressed in E. coli Tuner (DE3) as previously described (6), and whole-cell extracts were prepared by harvesting 1.7 × 1010 cells, suspending in 250 μl ExsA buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 1 mM dithiothreitol, and 0.5% Tween 20) containing protease inhibitor cocktail tablets (Complete Mini, EDTA-free; Roche Diagnostics, Indianapolis, IN), and lysing the cells by sonication. The cell lysate was cleared by centrifugation (16,000 × g, 30 min, 4°C), and the supernatant was immediately aliquoted and flash frozen in liquid nitrogen. The cell extracts were thawed, diluted in DNA binding buffer, and subjected to EMSA experiments as previously described (6) and anti-ExsA immunoblotting to determine the level of specific DNA-binding activity for each alanine substitution mutant relative to wild-type (wt) ExsA. Imaging was performed using an FLA-7000 phosphoimager (Fujifilm) and Multigage v3.0 software (Fujifilm) for data analyses.
Missing-nucleoside footprinting.
The missing-nucleoside footprinting experiments were performed according to a protocol adapted from Hayes and Tullius (17). PCR primers (50 pmol) were end labeled using 20 U polynucleotide kinase (New England BioLabs) and 120 μCi [γ-32P]ATP (Perkin Elmer). Reactions were applied to NucAway columns (Ambion) to remove unincorporated [γ-32P]ATP. End-labeled PexsD, PexoT, PexsC, and PpcrG promoter probes were generated in a PCR using labeled (above) and unlabeled primer (20 pmol), and the PCR products were gel purified (Qiagen). Nucleosides were randomly excised by treating the probes (16 μl in 1× DNA binding buffer) with 1 μl each of the following solutions for 1 min: 160 mM sodium ascorbate, 3.2 mM Fe(II)-EDTA [(NH4)2Fe(II)(SO4)2 · 6H2O], 6.4 mM EDTA, and 4.8% H2O2. The reactions were quenched by adding 2 μl 100 mM thiourea. Probes were ethanol precipitated, dissolved in 10 mM Tris-Cl (pH 8.0), 0.1 mM-EDTA, and used in an EMSA reaction as previously described (6). The unbound and ExsA-bound probes were excised as a gel slice. Probes were eluted from the gel slices by incubating for 16 h at 37°C in 0.5 M ammonium acetate, 1 mM EDTA (pH 7.5), 0.1% SDS, and 10 mM MgCl2. Eluted probes were ethanol precipitated, washed, dried, suspended in formamide loading buffer, and analyzed on a 6% sequencing gel.
RESULTS
Two ExsA monomers bind to T3SS promoter regions in a head-to-tail orientation.
AraC/XylS family members are characterized by the presence of two HTH DNA-binding motifs (Fig. 1A). Each binding motif contains a recognition helix that contacts the target DNA. Amino acid residues that directly contact the target DNA have been determined for a number of AraC/XylS family members and fall within discrete regions of the recognition helices (Fig. 1A) (4, 5, 11, 14, 24, 31). To define the interaction of ExsA with target DNA, we generated structural models based on the crystal structures of MarA and Rob bound to target DNA (Fig. 1B and C) (24, 31). Both models predicted that the HTH recognition helices of ExsA (RH1 and RH2) interact with promoter sequences located ∼10 bases apart. This observation is consistent with the previous identification of two highly conserved sequences (GnC and TGnnA) which are separated by ∼10 bp and present in binding site 1 of all ExsA-dependent promoter regions (6).
Fig 1.

(A) ClustalW alignment of the DNA-binding domains from selected AraC family members (AraC, MelR, RhaS, XylS, Rob, MarA, and ExsA). Residues that form the hydrophobic core of the DNA-binding domains are boxed in gray, and residues experimentally shown to make base-specific contacts with the promoter DNA are boxed in black. The predicted α-helices are indicated by rounded rectangles and based upon the MarA crystal structure (31). The predicted recognition helices for the two helix-turn-helix motifs are labeled RH1 and RH2, respectively. RH1 and RH2 in ExsA consist of residues L198 to Y209 and Q248 to F259, respectively. (B and C) Tertiary structure prediction of the ExsA DNA-binding domain bound to target DNA based upon the crystal structures of MarA (B) and Rob (C). The DNA binding domain of ExsA (residues 159 to 278) was threaded onto the crystal structure of MarA (residues 1 to 127) or Rob (residues 1 to 121) using the PHYRE 0.2 server (www.sbg.bio.ic.ac.uk/∼phyre/), and the predicted structures were visualized using the PyMOL molecular graphics system (www.pymol.org). Residues located within the “turn” regions of RH1 and RH2 (M196 and S246) that were changed to cysteine for the Fe-BABE footprinting experiments in Fig. 2 are indicated.
To determine where RH1 and RH2 interact with the promoter region, we employed Fe-BABE footprinting. Fe-BABE is a protein-labeling reagent that can be conjugated to cysteine residues. When conjugated to a DNA-binding protein, the immobilized Fe-BABE generates hydroxyl radicals that cleave target DNA in the immediate vicinity (within 7 to 16 Å) of the interaction site (12, 30). To adapt this system to ExsA, we first generated a cysteine-less exsA allele (exsAHisΔcys) by changing the seven native cysteine residues (C15, C50, C102, C121, C139, C261, and C271) to alanine. As shown in Fig. 2A, ExsAHisΔcys activates expression of a PexsD-lacZ transcriptional reporter to levels comparable to those seen for wt ExsA. Based on the modeled structure of the ExsA DNA-binding domain, we then identified amino acid residues positioned within the “turn” regions adjacent to RH1 (E193 and M196) or RH2 (G244 and S246) (Fig. 1A to C) and individually changed each to a cysteine. When expressed from a plasmid, all four cysteine substitution mutants complemented an exsA mutant for expression of the PexsD-lacZ reporter, albeit with varying degrees of efficiency (Fig. 2A). ExsAHisΔcys M196C and ExsAHisΔcys S246C were chosen for subsequent experiments, as they possessed the highest levels of activity and were stably expressed in P. aeruginosa (Fig. 2A).
Fig 2.

Fe-BABE footprints of the ExsA RH1- and RH2-Fe-BABE conjugants bound to the PexsC, PpcrG, PexsD, and PexoT promoter probes. (A) The PA103 exsA::Ω strain carrying a PexsD-lacZ reporter fusion was transformed with either a vector control or plasmids expressing wt ExsA, ExsA lacking all seven of the native cysteine residues (Δcys), or ExsA Δcys with unique cysteines introduced at positions E193, M196, G244, or S246. Transformants were grown under inducing conditions for T3SS gene expression and assayed for β-galactosidase activity. Whole-cell lysates (normalized by cell number) were immunoblotted with ExsA antiserum to examine steady-state expression levels. (B) Silver-stained SDS-polyacrylamide gel of wt ExsAHis and the ExsAHis M196C and ExsAHis S246C mutants following purification by Ni2+-affinity chromatography (1 μg each). Note that wt ExsA was further purified using a heparin column. The migration points of molecular weight standards are indicated on the left side of the gel. (C) The indicated promoter probes labeled with 32P on the forward strand were incubated in the absence (−) or presence of the RH1- or RH2-Fe-BABE conjugants for 15 min at 25°C as indicated. Binding reactions were subjected to Fe-BABE cleavage, separated by electrophoresis, and visualized by phosphorimaging. Maxam-Gilbert sequencing ladders (A+G) were included for orientation, and the positions of the GnC and TGnnA sequences in ExsA binding site 1 are indicated. (D) Summary of the Fe-BABE footprinting data for the forward (Fig. 2C) and reverse (data not shown) strands. The −10 hexamers (boxed), conserved GnC and TGnnA sequences (bold with larger typeface), adenine-rich region (gray box), and position of ExsA binding sites 1 and 2 are indicated. The regions of PexsC, PexsD, and PexoT previously shown to be protected from DNase I cleavage by ExsA are underlined (6). The protected region of PpcrG is based upon the DNase I footprint presented in Fig. S1 in the supplemental material. The cleavage events generated by the RH1- and RH2-Fe-BABE conjugants are indicated with filled and open circles, respectively. The open and filled circles located above and below the nucleotide sequence represent the Fe-BABE footprints for the forward and reverse strands, respectively.
The ExsAHisΔcys M196C and ExsAHisΔcys S246C proteins were expressed in E. coli, partially purified by Ni+2 affinity chromatography (Fig. 2B), and conjugated with Fe-BABE at the unique cysteine residue (referred to as the RH1 and RH2 conjugants, respectively). The RH1- or RH2-Fe-BABE conjugants were then incubated with 32P end-labeled probes (∼200 bp) derived from the ExsA-dependent PexsC, PpcrG, PexsD, and PexoT promoters and subjected to Fe-BABE-mediated cleavage. Cleavage patterns by the RH1-Fe-BABE conjugant were similar for all four promoters tested with cleavage sites located near the −75 and −50 regions on both the top (Fig. 2C and D, filled circles) and bottom (Fig. 2D) strands of the DNA. The one anomaly was an absence of cleavage on the bottom strand at the −50 region of the PexsC promoter. The cleavage patterns generated by the RH2-Fe-BABE conjugant were more variable. At binding site 1, the RH2-generated cleavage patterns were similar for each promoter with cleavage of the top strand centered at −40 and cleavage of the bottom strand centered at −33 and −43 (Fig. 2C and D, open circles). Cleavage by the RH2 conjugant at binding site 2, however, was observed only for the PexsC and PpcrG promoters, where it was centered at −59 and −61 on the top and bottom strands, respectively. The combined RH1 and RH2 cleavage patterns are consistent with our previous data showing the presence of two binding sites for monomeric ExsA on the PexsC, PexsD, and PexoT promoters (6). In addition, the cleavage patterns in binding site 1 indicate that RH1 interacts near the GnC sequence and that RH2 interacts near the TGnnA sequence (Fig. 2D). It should be noted that the Fe-BABE cleavage patterns were not expected to perfectly coincide with the GnC and TGnnA sequences because the Fe-BABE is conjugated to the “turn” regions located adjacent to RH1 or RH2 (Fig. 1B and C). Finally, the cleavage patterns at the PexsC and PpcrG promoters indicate that the two ExsA monomers bind in a head-to-tail configuration (i.e., in the same orientation) because the RH1 conjugant cleaves upstream of the RH2 conjugant at both binding sites 1 and 2.
Recognition helices 1 and 2 of ExsA make specific contacts with the GnC and TGnnA regions of the PexoT promoter.
To determine which amino acids within RH1 and RH2 contact the conserved GnC and TGnnA sequences in binding site 1 of the PexoT promoter, we screened a library of PCR-mutagenized exsA alleles for gain-of-function mutants that could activate PexoT-lacZ transcriptional reporters carrying mutations within the GnC or TGnnA sequences (Fig. 3A). In the first experiment, we utilized a PexoT(C−45A)-lacZ reporter which carries a GnA substitution for the conserved GnC sequence. As previously reported, activation of the PexoT(C−45A)-lacZ reporter by wt ExsA is reduced ∼14-fold compared to the wt PexoT-lacZ reporter (6) (Fig. 3B). The mutagenized ExsA expression library was introduced into the PA103 exsA::Ω PexoT(C−45A)-lacZ reporter strain, and ∼20,000 colonies were screened for increased β-galactosidase activity on VBMM agar plates containing X-Gal. This approach repeatedly identified a single substitution in RH1 (lysine 202 changed to an arginine [K202R]) that resulted in a 4-fold increase in expression from the PexoT(C−45A)-lacZ reporter and a 3.8-fold reduction in PexoT-lacZ expression (Fig. 3B). Immunoblotting of whole-cell lysates with ExsA antiserum confirmed that the steady-state expression level of ExsA K202R was similar to that of wt ExsA (Fig. 3B, inset). The finding is consistent with the results of the Fe-BABE footprinting data showing that RH1 interacts near the GnC sequence and suggests that K202 in RH1 contacts the −45 cytosine in the PexoT promoter. Note that each of the contacts identified throughout this study will be defined relative to the sequence of the coding (top) strand of the DNA. Although the genetic data cannot differentiate whether the contact is occurring with the coding or noncoding strand, an experiment to address this ambiguity is presented later (see Fig. 9).
Fig 3.
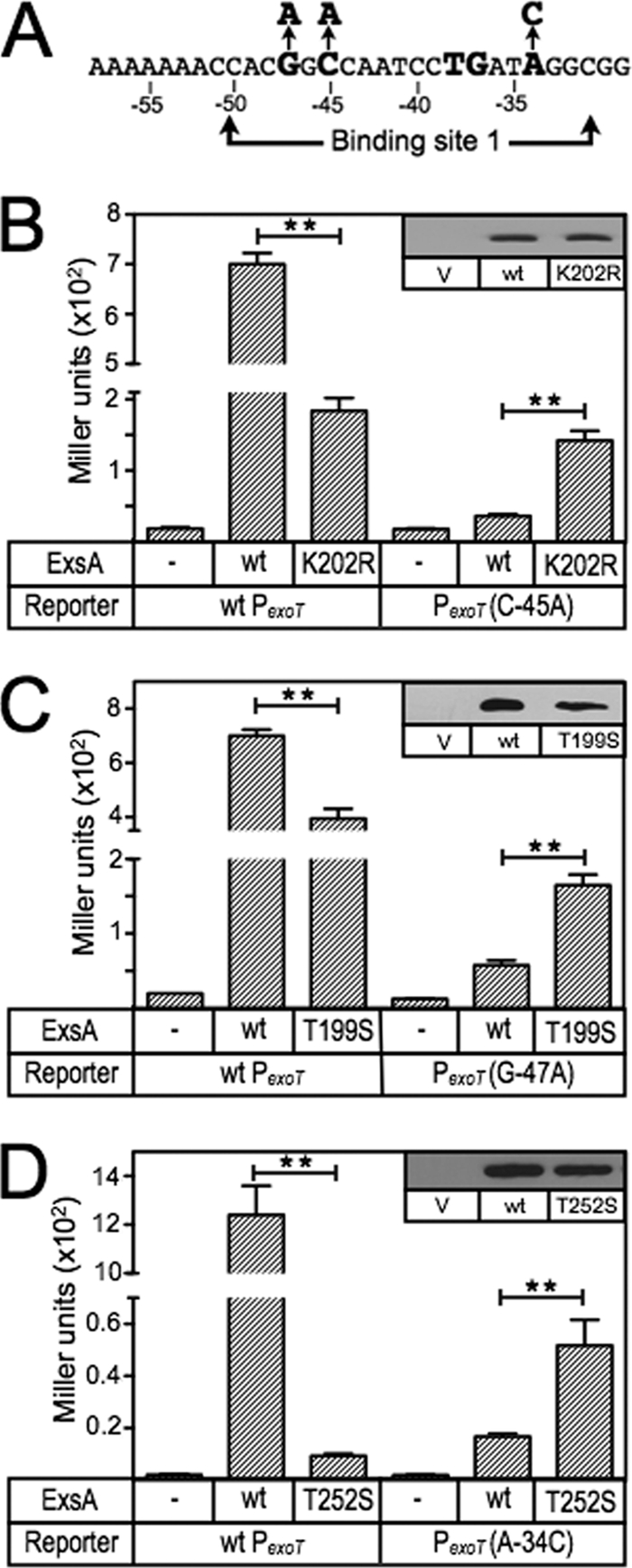
ExsA gain-of-function mutants with altered promoter specificity. (A) Diagram of the nucleotide substitutions in the PexoT(C−45A)-lacZ, PexoT(G−47A)-lacZ, and PexoT(A−34C)-lacZ mutant reporters that were used for the gain-of-function screen. (B to D) The PA103 exsA::Ω strain carrying either the wild-type PexoT-lacZ reporter or a mutant reporter (PexoT(C−45A)-lacZ [B], PexoT(G−47A)-lacZ [C], or PexoT(A−34C)-lacZ [D]) was transformed with either a vector (V) control (−), an ExsA expression plasmid (wt), or a plasmid expressing ExsA with the indicated amino acid substitutions (K202R, T199S, or T252S). Transformants were cultured under inducing conditions for T3SS gene expression (in panel D, the strains were cultured in the presence of 0.05% arabinose to enhance detection of T252S-dependent activity) and assayed for β-galactosidase activity. The reported values for the β-galactosidase assays (Miller units) throughout this study represent the average of at least three independent experiments, and error bars represent the standard error of the mean (SEM); ** P < 0.01. Anti-ExsA immunoblots demonstrating the steady-state expression levels of ExsA and the K202R, T199S, and T252S mutants are shown as insets.
Fig 9.

Missing-nucleoside footprints of the PexsC and PpcrG promoter regions. PexsC (A) or PpcrG (B) promoter probes labeled with 32P on the forward or reverse strand were treated with hydroxyl radicals, incubated with ExsAHis, and subjected to nondenaturing electrophoresis in a typical EMSA reaction. The unbound band and the ExsA-bound band corresponding to shift product 2 (representing ExsA bound to both sites 1 and 2) were eluted from the gel, separated by denaturing electrophoresis, and visualized by phosphorimaging. Maxam-Gilbert sequencing ladders (A+G) were included for orientation. The locations of the GnC and TGnnA sequences in binding site 1 and binding site 2 are indicated. (C) Summary of the footprinting data for the forward and reverse strands. The conserved GnC and TGnnA sequences (bold with larger typeface), adenine-rich region (gray box), and positions of ExsA binding sites 1 and 2 are indicated. Nucleotides indicated with arrows are those present in the unbound samples and required for maximal ExsA binding. (D) Proposed model for the binding of ExsA to site 1 in the PexoT promoter region. The amino acid residues that are proposed to contribute to base-specific recognition of the coding or noncoding strand of the PexoT promoter region are indicated.
The above approach involved random mutagenesis of the entire ExsA coding sequence. Since only a single gain-of-function mutant was identified, we used directed mutagenesis to increase coverage of RH1 by generating a library of exsA alleles randomized at each of six positions (residues 198 to 200 and 202 to 204) within RH1 (Fig. 1A). Residues 201 and 205 were excluded from the library, as they form part of the conserved hydrophobic core of the HTH motif, and residues 206 to 208 were excluded because the structural models predicted that each of the side chains was buried within the structure of ExsA (Fig. 1A). The RH1-targeted library was screened as described above, and again only the K202R substitution mutant demonstrated a gain of function using the PexoT(C−45A)-lacZ reporter.
To identify amino acids that interact with the guanine of the GnC sequence, we repeated the screen using the RH1-targeted library and a reporter carrying an AnC substitution (PexoT(G−47A)-lacZ) (Fig. 3A) (6). Activation of the PexoT(G−47A)-lacZ reporter by wt ExsA is reduced ∼12-fold compared to the wt PexoT-lacZ reporter (6) (Fig. 3C). One substitution in RH1 (T199S) resulted in a significant increase (2.9-fold) in β-galactosidase expression from the PexoT(G−47A)-lacZ reporter and a corresponding 1.7-fold decrease in the expression of the wt reporter. These combined results suggest that residues T199 and K202 in RH1 interact with the conserved GnC sequence.
The Fe-BABE footprinting data and the finding that RH1 contacts the GnC sequence suggested that RH2 might contact the TGnnA sequence centered 10 bp downstream. To identify amino acids that interact with the adenine of the TGnnA sequence in the PexoT promoter, we generated a targeted library of ExsA alleles randomized at the six positions (residues 252 to 254 and 256 to 258) in RH2 predicted to make base-specific contacts with the promoter (Fig. 1A). Amino acids that form the conserved hydrophobic core of the HTH motif (residues 251 and 255) or predicted to be buried in the structure of the HTH motif (residues 248 to 250) were excluded from the library (Fig. 1A). The initial screens were complicated by the fact that Q253 changes to valine, leucine, or isoleucine resulted in a superactivation phenotype such that expression from both the wt PexoT-lacZ and PexoT(A−34C)-lacZ reporters was ∼10-fold higher than with wt ExsA (data not shown). For this reason, we regenerated the RH2-targeted library but excluded Q253. Using this new library, we identified a T252S gain-of-function mutant within RH2 that resulted in a 3-fold increase in expression of the PexoT(A−34C)-lacZ reporter (Fig. 3D) and reduced expression (130-fold) of the wt PexoT-lacZ reporter. In contrast, activation of the PexoT(A−34C)-lacZ reporter by wt ExsA was reduced ∼70-fold compared to that of the wt PexoT-lacZ reporter (6) (Fig. 3D). These data suggest that RH2 contacts the TGnnA sequence in binding site 1 of the PexoT promoter.
Loss-of-contact analyses identify base-specific contacts mediated by T199 and K202.
To further examine the roles of residues T199, K202, and T252, we employed a genetic loss-of-contact approach, previously used to define base-specific contacts for a number of AraC/XylS proteins for which structural information is unavailable (4, 11, 18). The approach is based on the rationale that a DNA-binding protein with an alanine substitution at a residue involved in a critical base-specific contact will be insensitive to the identity of that base. For instance, our data would predict an ExsA T199A mutant to be significantly impaired for activation of the wt PexoT-lacZ reporter due to loss of the contact between residue T199 in RH1 and the guanine at position −47. The residual activation of the PexoT-lacZ reporter by the T199A mutant, therefore, should be insensitive to the identity of the base at position −47. Conversely, residual activation by T199A would remain sensitive to promoter mutations that disrupt unrelated base-specific contacts.
For these analyses, we used a previously generated panel of mutant PexoT-lacZ reporters with nucleotide substitutions at the conserved GnC (G−47A and C−45A) and TGnnA (T−38A, G−37C, and A−34T) positions in binding site 1 (summarized in Fig. 4A) (6). Each of these substitutions results in a significant reduction in ExsA-dependent activity (Fig. 4B). The PexoT(C−50G)-lacZ reporter has a substitution at the noncritical −50 position and served as a negative control. We also introduced nucleotide substitutions at positions −39, −36, and −35, which represent conserved but more degenerative nucleotides in the ExsA-binding consensus site (6). Whereas the substitutions at positions −35 and −36 resulted in a significant defect in activation by wt ExsA, the −39 mutation had little effect on reporter activity. Based on the latter finding, therefore, we expected both the PexoT(C−50G)-lacZ and PexoT(G−39A)-lacZ reporters to serve as negative controls.
Fig 4.
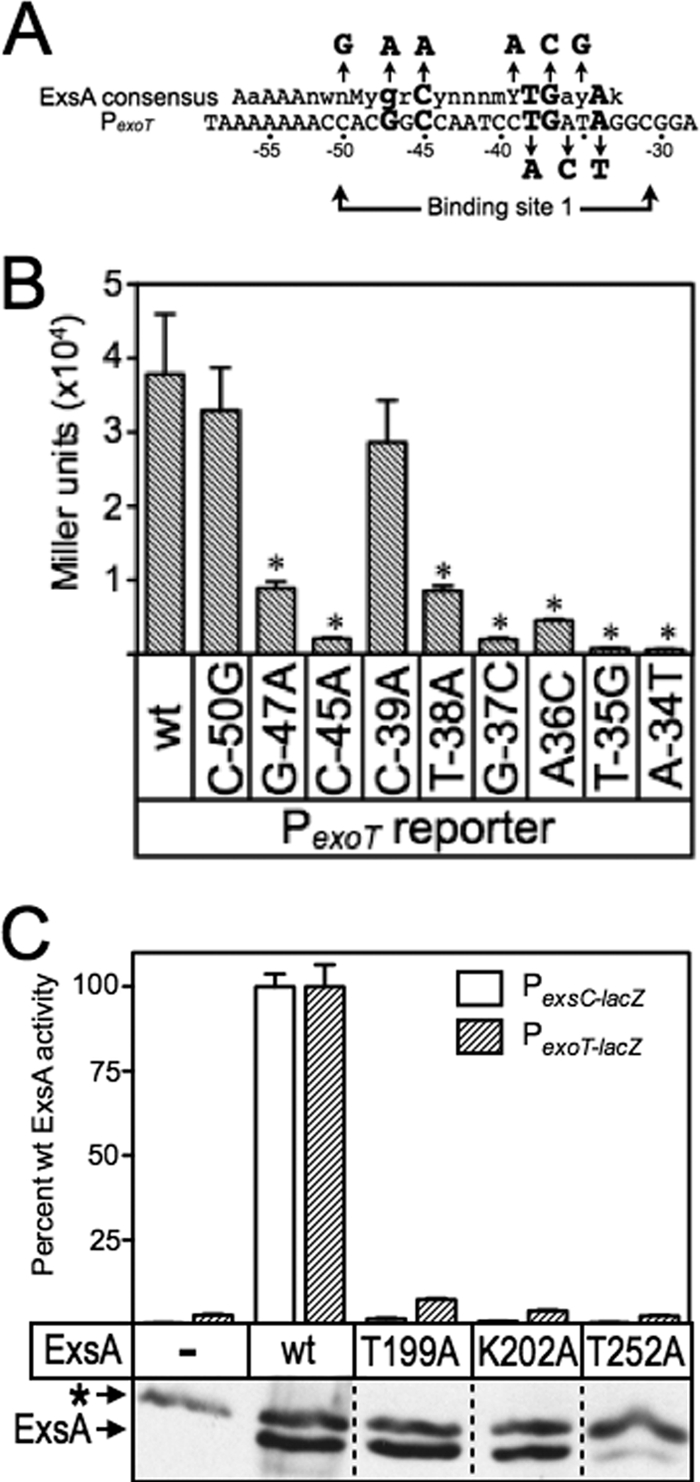
(A) Map of the PexoT promoter showing the ExsA consensus binding sequence, and the conserved GnC and TGnnA sequences (bold with larger typeface) located within binding site 1. The nucleotide substitutions for each of the mutant PexoT reporters used in the genetic loss-of-contact experiments are indicated with arrows. (B) The PA103 exsA::Ω strain carrying the mutant PexoT-lacZ reporters and expressing wt ExsA from an arabinose-inducible expression vector was grown in the presence of EGTA and 0.1% arabinose and assayed for β-galactosidase activity. *, P < 0.05. (C) The PA103 exsA::Ω strain carrying either a PexsC-lacZ (open bars) or a PexoT-lacZ (hatched bars) transcriptional reporter was transformed with vectors expressing wt ExsA or the indicated ExsA alanine substitution mutants. The resulting strains were cultured under inducing (+EGTA) conditions for T3SS gene expression with 0.1% arabinose and assayed for β-galactosidase activity. The reported values are the percent activity for each alanine substitution mutant relative to wt ExsA. Immunoblots demonstrating the steady-state expression levels of ExsA and the alanine scanning mutants are shown as insets. The asterisk indicates a cross-reactive band that served as a loading control.
To screen for loss-of-contact phenotypes, we introduced wt ExsA or the T199A, K202A, and T252A expression plasmids into an exsA::Ω mutant carrying the wt PexoT-lacZ or PexsC-lacZ reporters. The resulting strains were cultured under inducing conditions for T3SS gene expression (low Ca2+) in the absence or presence of arabinose and assayed for β-galactosidase activity. Each of the alanine substitution mutants demonstrated a significant reduction in activation of the wt PexoT-lacZ and PexsC-lacZ reporters when cultured in the absence of arabinose (see Table S3 in the supplemental material). Growth in the presence of arabinose, however, resulted in a significant increase in reporter activity by the T199A and K202A mutants (Fig. 4C; also see Table S3). The subsequent loss-of-contact analyses, therefore, were performed in the presence of 0.1% arabinose to maximize the sensitivity of the assay. In contrast to what was seen for the T199A and K202A mutants, the activity of the T252A mutant was not significantly recovered by growth in the presence of arabinose. Examination of the steady-state expression levels of wt ExsA and the T199A, K202A, and T252A mutants offers an explanation for this, since the T252A mutant was poorly expressed (Fig. 4C). This finding was unexpected because the T252S substitution identified in the gain-of-function screen was stably expressed (Fig. 3D).
Although activation of the wt PexoT-lacZ reporter by the T199A and K202A mutants was significantly impaired, both mutants retained residual activity above the vector alone (see Table S3 in the supplemental material). This residual activity allowed us to compare T199A- and K202R-dependent activation of the wt PexoT-lacZ reporter (normalized to 100% activity) (Fig. 5) to each of the mutant PexoT-lacZ reporters. Reporters with base-pair substitutions in the conserved −45, −38, −37, and −34 positions led to a further decrease in activation by the T199A mutant. This decrease in activity is consistent with loss of a second base-specific contact that further impairs DNA binding (Fig. 5). In contrast, PexoT-lacZ reporters with substitutions at the noncritical −50 position or at the conserved −47 guanine had activity comparable to that of the wt PexoT-lacZ reporter (Fig. 5). An important prediction is that wt ExsA should be sensitive to the G−47A substitution relative to the T199A mutant. Comparison of Fig. 4 and 5 shows that the T199A mutant activates both the wt PexoT-lacZ and PexoT(G−47A)-lacZ reporters to similar levels. In contrast, activation of the PexoT(G−47A)-lacZ reporter by wt ExsA is reduced 4-fold relative to that of the wt PexoT-lacZ. The findings indicate that the T199A mutant is insensitive to the identity of the base at the −47 position and supports our conclusion that T199A makes a base-specific contact with the −47 guanine.
Fig 5.
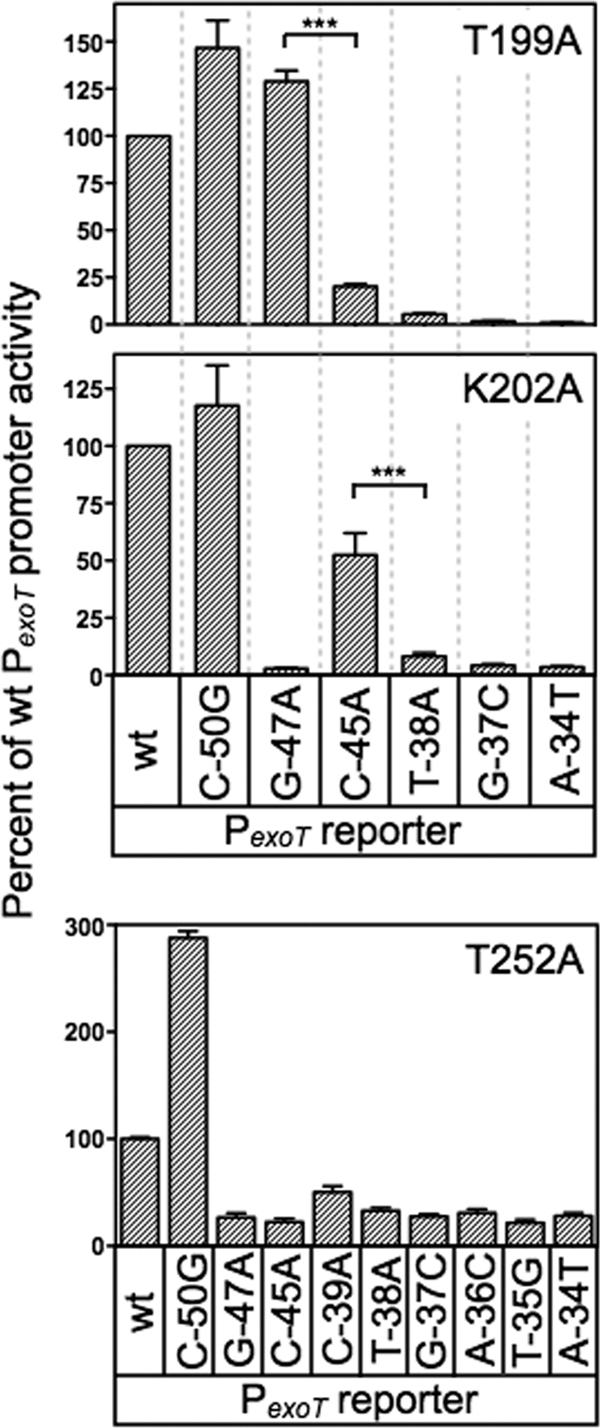
Genetic loss-of-contact analyses of the T199A, K202A, and T252A mutants. The T199A, K202A, and T252A expression plasmids were introduced into the PA103 exsA::Ω strain carrying the indicated mutant PexoT-lacZ reporters. Transformants were cultured in the presence of EGTA and 0.1% arabinose and assayed for β-galactosidase activity. The reported values are the percent activity at the mutant promoter normalized to the activity of each alanine substitution mutant at the wt PexoT-lacZ reporter; ***, P < 0.001.
A similar result was seen for K202, predicted to contact cytosine −45 by the gain-of-function screen. In the loss-of-contact assay, the K202A mutant was relatively insensitive to a substitution at the −45 position in the PexoT-lacZ reporter (50% activity) (Fig. 5). Importantly, activation by K202A was sensitive to mutations at other critical nucleotide positions in the promoter. As expected, wt ExsA is very sensitive to the C−45A substitution in PexoT (20-fold reduction compared to wt PexoT) relative to the K202R mutant, which demonstrated only a 2-fold reduction (Fig. 4B and 5). These combined findings provide strong genetic evidence that ExsA bound to site 1 on the PexoT promoter utilizes residues T199 and K202 to contact the −47 guanine and −45 cytosine, respectively.
The loss-of-contact approach was also used to examine residue T252, predicted by the gain-of-function screen to contact the adenine at position −34. In contrast to our findings for T199A and K202A, none of the mutant PexoT-lacZ reporters with substitutions in the GnC and TGnnA sequences demonstrated a loss-of-contact phenotype for the T252A mutant (Fig. 5). We considered the possibility that residue T252 contacts another position in the ExsA consensus binding sequence by using PexoT-lacZ reporters with substitutions at the less highly conserved −36 and −35 positions and again failed to detect a loss-of-contact phenotype. Curiously, the negative controls demonstrated opposite properties. Whereas activation of the PexoT(C−50G)-lacZ reporter by T252A was 3-fold higher than that of the wt PexoT-lacZ reporter, the activity of the PexoT(C−39A)-lacZ reporter was significantly reduced. While an explanation for this is not evident, it should be noted that the T252A mutant is severely impaired for activation of the PexoT-lacZ reporter (see Table S3 in the supplemental material). We suspect that the defective nature of the T252A mutant results in hypersensitivity to promoter mutations that only modestly alter binding affinity. In summary, the loss-of-contact findings are not supportive of a role for T252 in base-specific recognition of the TGnnA sequence, but the T252S gain-of-function phenotype argues otherwise. The interpretation we favor is that T252 participates in base-specific recognition of the TGnnA sequence but that the low residual activity of the T252A mutant precludes detection of loss-of-contact phenotypes.
Alanine scanning mutagenesis of RH1 and RH2.
While the loss-of-contact approach can be used to confirm base-specific interactions, it can also be used to discover unknown interactions. To determine whether additional residues contribute to ExsA activity, we performed alanine-scanning mutagenesis of the remaining residues that constitute RH1 and RH2. Seven alanine substitutions were introduced into RH1 and eight substitutions in RH2 using site-directed mutagenesis (Fig. 6A and B). As in the gain-of-function screen described above, residues F201 and F205 in RH1, and F251 and Y255 in RH2, were excluded from the analyses because they form the hydrophobic core of the HTH motifs.
Fig 6.

Alanine scanning mutagenesis of RH1 and RH2. The PA103 exsA::Ω strain carrying either a PexsC-lacZ (open bars) or PexoT-lacZ (hatched bars) transcriptional reporter was transformed with a vector expressing wt ExsA or the indicated alanine substitution mutants in RH1 (A) and RH2 (B). The resulting strains were grown in the presence of EGTA and 0.1% arabinose and assayed for β-galactosidase activity. The reported values are the percent activity for each alanine substitution mutant relative to wt ExsA. ***, P < 0.001; **, P < 0.01. Anti-ExsA immunoblots demonstrating the steady-state expression levels of ExsA and the alanine-substituted mutants are shown below each panel. The asterisk indicates a cross-reactive band that served as a loading control.
Most of the alanine-substituted mutants activated both the PexoT-lacZ and PexsC-lacZ transcriptional reporters to similar levels, although some modest differences (≤2-fold) existed, with L204A, Q253A, S254A, and R258A being most notable (Fig. 6A and B; also see Table S3 in the supplemental material). The alanine substitution mutants were classified into three primary groups based on activity relative to wt ExsA: (i) mutants with alanine substitutions at positions 203, 206, and 207 in RH1 (Fig. 6A) and at positions 253 and 254 in RH2 (Fig. 6B) retained >50% of the activity of wt ExsA and were excluded from further consideration; (ii) mutants with alanine substitutions at positions T200, L204, V208, S249, and R258 had activities in the range of 10 to 49%; and (iii) mutants with alanine substitutions at positions L198, Q248, Y250, R256, and R257 were the most severely impaired, with <10% of the activity of wt ExsA. A trivial explanation for reduced activation by the mutant proteins is that the alanine substitutions render the proteins unstable. To address this possibility, we measured the steady-state expression levels using α-ExsA immunoblots. With the exception of the L204A mutant, however, each of the mutant proteins was expressed at levels similar to that of wt ExsA (Fig. 6A and B).
The loss-of-contact approach was applied to each of the alanine substitution mutants that retained <50% activity relative to wt ExsA (Fig. 7). The only alanine substitution mutant in RH1 to demonstrate loss of base specificity was L198A, whereby activation of the PexoT-lacZ reporter was relatively insensitive to a nucleotide substitution at the −47 position (Fig. 7) and wt ExsA was more sensitive to the G47A substitution relative to the L198A mutant (Fig. 4B). Base-specific interactions for the T200A, L204A, and V208A mutants were not detected (see Fig. S2A in the supplemental material). From this we conclude that the primary specificity determinants in RH1 are residues L198 and T199, which contribute to base-specific recognition of the guanine at position −47, and K202, which interacts with the cytosine at position −45. Two alanine substitution mutants in RH2 demonstrated potential interactions with or near the TGnnA sequence. The Y250A mutant was somewhat insensitive to the substitution at position 38 and the R257A mutant was completely insensitive to the substitution at position −37 (Fig. 7). Base-specific interactions for the Q248A, S249A, R256A, and R258A mutants were not detected (see Fig. S2B in the supplemental material).
Fig 7.
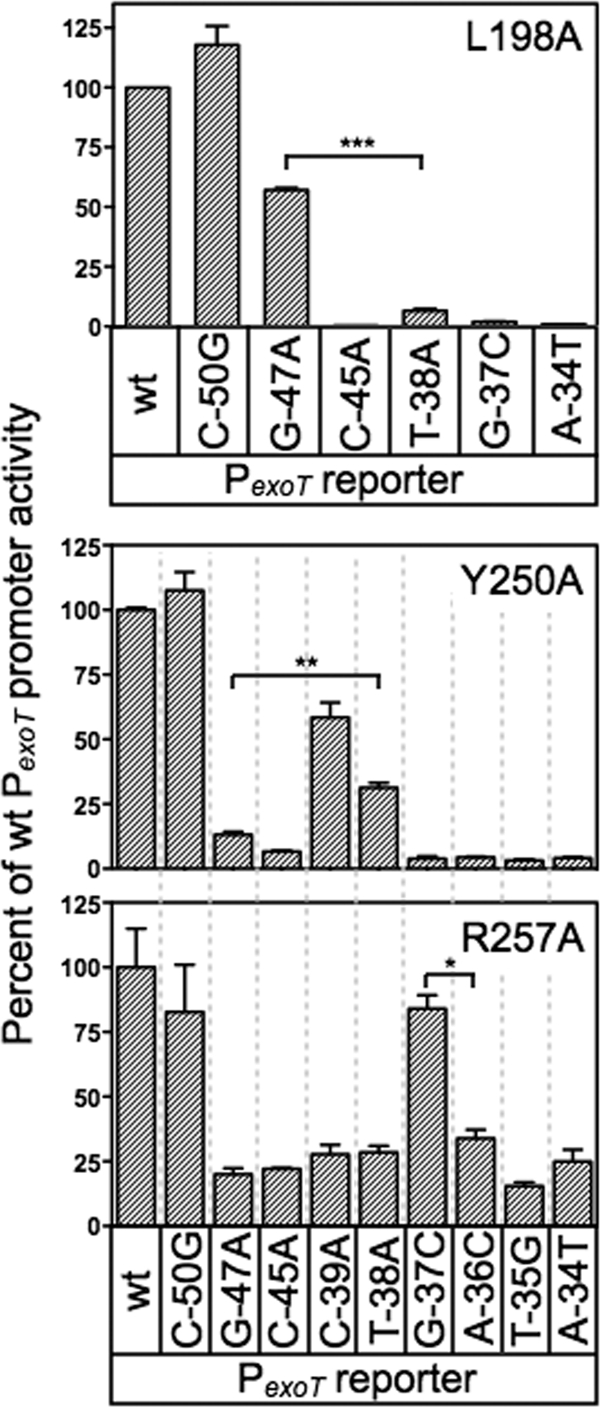
Additional base-specific contacts mediated by residues in RH1 and RH2. The PA103 exsA::Ω strains expressing either L198A, Y250A, or R257A were assayed for reporter activity using the panel of mutant PexoT-lacZ reporter described in Fig. 4A. The strains were cultured in the presence of EGTA and 0.1% arabinose and assayed for β-galactosidase activity. The reported values are the percent activity at the mutant promoter compared to the activity at the wt PexoT-lacZ reporter. **, P < 0.01; *, P < 0.05.
DNA-binding activity of selected alanine scanning mutants.
The L198A, T199A, K202A, Y250A, T252A, and R257A mutants each demonstrated a significant reduction in expression of the PexoT-lacZ and PexsC-lacZ reporters, and potential base-specific interactions with the PexoT promoter. While these findings were consistent with a primary defect in DNA-binding activity, impaired recruitment of RNA polymerase could also account for the reduced activity of the mutant proteins. To address this question whole-cell extracts were prepared from E. coli expressing either wt ExsA or the indicated alanine substitution mutants. The Q248A mutant, defective for activation of the PexoT-lacZ and PexsC-lacZ reporters but lacking a loss-of-contact phenotype, was included as a control. The extracts were then normalized for ExsA content (Fig. 8, bottom), incubated with a radiolabeled PexsC promoter probe and a nonspecific control probe, and analyzed by electrophoretic mobility shift assays. As shown in Fig. 8, top, purified ExsAHis and the E. coli extracts containing either wt ExsA or the Q248A mutant bound specifically to the PexsC promoter probe, resulting in the formation of shift product 2 which represents ExsA bound to binding sites 1 and 2. None of the remaining extracts, however, possessed specific DNA-binding activity. Subsequent titrations of the extracts indicate that the DNA-binding activity of the L198A, T199A, K202A, Y250A, and R257A mutants is at least 12-fold lower than that of wt ExsA. These findings indicate that reduced activation of the PexoT-lacZ and PexsC-lacZ reporters by these mutants results from a primary defect in DNA-binding activity. This finding, however, does not exclude the possibility that some mutants also possess defects in recruitment of RNA polymerase that further contribute to reduced expression of the PexoT-lacZ and PexsC-lacZ reporters in vivo. In fact, impaired recruitment of RNA polymerase most likely accounts for the phenotype of the Q248A mutant, which has DNA-binding activity similar to that of wt ExsA but a 50-fold defect in activation of the PexoT-lacZ reporter (see Table S3 in the supplemental material).
Fig 8.
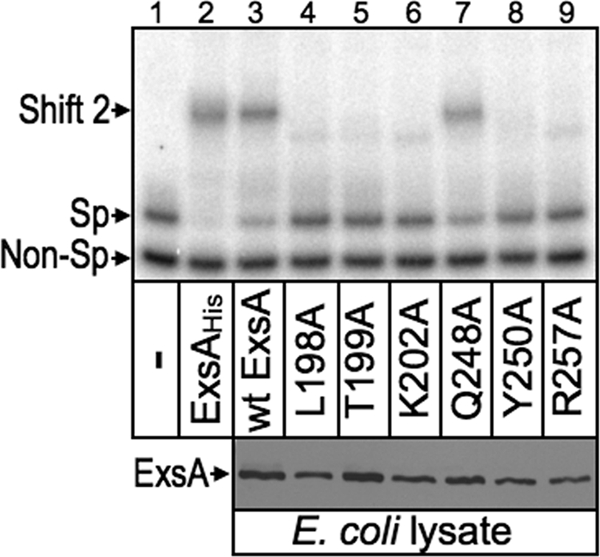
DNA binding activity of selected alanine substitution mutants. For the EMSA experiment (top), an equimolar mixture of nonspecific (Non-Sp, derived from the coding region of pscF) and specific (Sp, derived from the wt PexsC reporter) radiolabeled probes (0.05 nM each) was incubated for 15 min at 25°C in the absence (−) or presence of 40 nM purified ExsAHis, or 2 μl of whole-cell lysate prepared from E. coli expressing wt ExsA or the indicated alanine substitution mutants. Samples were analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. Anti-ExsA immunoblots (bottom) indicate that the E. coli lysates used in the EMSA experiment contain comparable levels of wt ExsA and the indicated alanine substitution mutants.
Strand-specific contacts by RH1 and RH2.
The data presented above define several base-specific contacts with the PexoT promoter region but could not differentiate whether the contacted nucleosides are located on the top or bottom strands of the DNA. To address this question we used the missing-nucleoside footprinting approach (17). For this technique nucleosides are randomly excised from end-labeled promoter probes through brief exposure to hydroxyl radicals. The treated probes are then used in an EMSA reaction, and the ExsA-bound and unbound probes are excised and separated on a sequencing gel to identify nucleosides important for ExsA binding. Comparison of the ExsA-bound and unbound samples yielded complementary data whereby the most prominent bands in the unbound samples were reduced or absent from the ExsA bound samples for both the PexsC and PpcrG promoter probes (Fig. 9). The identity of the bands in the unbound samples indicates that binding of ExsA to site 1 requires the −38 thymine and −39 guanosine on the top strand and the −34 thymine on the bottom strand. Although the pattern was not as clear with regard to the GnC sequence, the data for the PpcrG promoter suggest that ExsA binding requires the guanosines at positions −47 and −45 on the top and bottom strands, respectively. This interpretation is consistent with the molecular modeling data shown in Fig. S3 in the supplemental material, which predict that residues L198 and T199 in RH1 interact with the −47 guanosine on one strand and that K202 interacts with the −45 cytosine on the opposite strand. Despite repeated attempts to footprint the PexoT and PexsD promoter probes using the missing-nucleoside approach, our efforts were unsuccessful, and we suspect that this reflects a sensitivity issue related to the low yield of shift product 2. Previous EMSA experiments using PexsC and PpcrG promoter probes found that the yield of shift product 2 (i.e., ExsA bound to both sites 1 and 2) is much higher than when using the PexoT and PexsD promoter probes (6).
DISCUSSION
In the present study, we tested the hypothesis that the RH1 and RH2 recognition helices of a single ExsA monomer interact with the GnC and TGnnA sequences in binding site 1. Using Fe-BABE footprinting, we found that the ExsA monomers are in the same orientation (i.e., head-to-tail) when bound to sites 1 and 2 on the PexoT, PexsC, PexsD, and PpcrG promoter probes (Fig. 2C). This binding configuration is also seen with other AraC family members that function as class II activators, including AraC, MelR, ToxT, and XylS, although variations on this theme can occur at different promoters (11, 14, 26, 43). A previous study found that cooperative binding by ExsA and efficient filling of site 2 are dependent upon self-association between the ExsA monomers (8). ExsA self-association occurs through interactions mediated by the amino-terminal domain. The crystal structure of the AraC dimerization and ligand binding domain indicates that dimerization occurs in a head-to-head orientation (38). Assuming that ExsA monomers also associate in a head-to-head orientation, the amino-terminal domain of one ExsA monomer would have to be rotated 180° relative to the carboxy-terminal DNA-binding domain to allow both monomers to bind in a head-to-tail orientation. This is consistent with a previous study suggesting that the amino- and carboxy-terminal domains of ExsA are connected by a flexible linker region located between residues 149 and 157 (8). That study found that amino- and carboxyl-terminal domains could be separated at the predicted linker region and still retain function. Whereas the amino-terminal domain mediates ExsA self-association and ExsD binding, the carboxy-terminal domain retains DNA-binding activity and the ability to recruit RNA polymerase to the promoter. This linker region likely confers the flexibility required to dimerize in a head-to-head orientation and still bind DNA in a head-to-tail orientation.
In addition to establishing the orientation of the bound ExsA monomers, the Fe-BABE footprinting data also defined the general location of the RH1- and RH2-mediated contacts at binding site 1 (Fig. 2D). Cleavage by the RH1 conjugant was located 1 to 5 bp upstream of the GnC sequence on the top strand of the promoter probes and 3 to 7 bp upstream of the GnC sequence on the bottom strands. Although the RH1 cleavage patterns could alternatively be described relative to the adenine-rich region (i.e., immediately downstream), three pieces of data indicate that RH1 contacts the GnC sequence as opposed to the adenine-rich region. First, the modeled structure of ExsA bound to DNA indicates that the “turn” region is located amino terminal to RH1 (see Fig. S3A in the supplemental material). For this reason, we expected and observed that the RH1 conjugant cleaves on the upstream (5′) side of the GnC sequence (Fig. 2). Second, the gain-of-function mutants and genetic loss-of-contact data indicate that RH1 makes base-specific contacts with the GnC sequence (Fig. 3 and 4). Finally, the midpoints of RH1 and RH2 cleavage on the top strand of the promoter are separated by ∼11 bp at binding site 1. This spacing is typical of promoter elements recognized by the two recognition helices of a single AraC/XylS subunit (11, 29).
Cleavage by the RH2 conjugant at binding site 1 coincided with the TG portion of the TGnnA sequence on the top strand of the promoter probes and at two locations positioned 2 to 4 bp upstream and downstream of the TGnnA sequence on the bottom strand (Fig. 2C). The two areas of cleavage on the bottom strand are separated by almost a full helical turn of the DNA, or ∼34 Å. This finding might indicate that the residue conjugated with Fe-BABE (S246) is positioned equidistant from both areas of cleavage, since the outer limit that the Fe-BABE generated hydroxyl radicals can diffuse before being neutralized by water is ∼17 Å (12, 30). Another possible explanation for the bipartite cleavage pattern is that the previously observed ExsA-dependent bending of the promoters (8) brings both regions close enough to permit cleavage by the RH2 conjugant.
The cleavage pattern generated by the RH2 conjugant indicates that RH2 does not closely interact with binding site 2 at the PexsD and PexoT promoters. The MarA and Rob crystal structures bound to target DNA demonstrate two distinct binding modes, which could explain the lack of interactions detected for ExsA at binding site 2 (24, 31). Whereas both recognition helices of MarA are fully inserted into adjacent major grooves on the DNA, only recognition helix 1 of Rob engages the major groove of the binding site, while recognition helix 2 lies on the surface of the DNA through contacts with the phosphodiester backbone. Such a binding arrangement might position the RH2 conjugant far enough away from the DNA backbone to eliminate observable cleavage. Chemical footprinting and mutational data show that a dimer of AraC uses both modes to bind the araI site (5, 18, 29). Whereas one molecule of the AraC dimer binds the upstream araI1 sites like MarA, the other molecule binds the downstream araI2 site like Rob. In an analogous scenario, our data indicate that ExsA binds to site 1 in all of the promoters like MarA, with both helices fully inserted into the DNA. Binding to site 2 at the PexsD and PexoT promoters, however, may more closely resemble the binding properties of Rob.
Having determined that ExsA interacts at or near the GnC and TGnnA sequences, we utilized gain-of-function screens and loss-of-contact analyses to identify amino acid residues in RH1 and RH2 involved in making base-specific contact with DNA. Combined data from those studies make for a compelling case that residues L198, T199, and K202 in RH1 are the primary, and possibly only, residues involved in base-specific recognition of the GnC sequence (Fig. 3 and 5). The finding for T199A is in agreement with previous studies of AraC, MelR, RhaS, XylS, Rob, and MarA, each reporting that the residue equivalent to T199 participates in base-specific interactions with target DNA (Fig. 1A). Similarly, the equivalent of residue K202 in Rob and MarA also interacts with target DNA. Based on our findings, we asked whether the MarA-DNA complex structure could be used to more precisely model the interaction of ExsA with the PexoT promoter at binding site 1. MarA was chosen because binding of an ExsA monomer to binding site 1 of the PexoT promoter bends the DNA to a similar angle as seen in the MarA-DNA complex structure (8, 31). Assuming that the −47 guanine is the nucleotide closest to residue T199, we indeed found that L198 is also positioned favorably to interact with the −47 guanine and that K202 is oriented toward the −45 bp with the side chain pointing to the opposing strand (see Fig. S3A in the supplemental material). These findings are consistent with the missing-nucleoside footprinting data at the PpcrG promoter (Fig. 9).
In comparison to RH1, interpreting the gain-of-function and loss-of-contact data for RH2 was not as straightforward. The only gain-of-function mutant identified in RH2 was T252S, specific for the −34 adenine of the TGnnA sequence. The T252A mutant, however, was poorly expressed and failed to demonstrate a base-specific interaction in the loss-of-contact analyses. Although there is no evidence that the corresponding residue participates in base-specific interactions for other AraC/XylS family members, the equivalent of ExsA residue 253 in XylS and MarA is important for DNA recognition (Fig. 1A) (11, 31). An alternative explanation for the T252S gain-of-function phenotype is that T252 normally functions to stabilize a neighboring residue involved in base-specific interactions. Under this scenario, however, a loss-of-contact phenotype would still be expected through secondary effects on the neighboring residue. Nevertheless, a role for protein stability is indicated through the observation that the conservative T252S substitution had no effect on protein stability, whereas the T252A substitution resulted in a significant decrease in steady-state expression levels. Based upon our combined data, we conclude that T252 is involved in a base-specific contact with the −34 adenine but cannot distinguish whether the effect of the T252A substitution is direct or indirect.
Attempts to isolate gain-of-function mutants specific for the thymine or guanine of the conserved TGnnA sequence were unsuccessful. One possible reason is that the specific promoter substitutions used in the screens (PexoT(T−38A)-lacZ and PexoT(G−37C)-lacZ) were not exhaustive and may not have been permissive for isolating gain-of-function mutants. Another important point to consider is that ExsA-dependent activation of the PexoT-lacZ reporter is dependent upon occupation of both binding sites 1 and 2 (6). For this reason, we expected gain-of-function mutants to be difficult to isolate, since the mutant proteins would have to bind both the native sequence at binding site 2 and the mutant sequence at site 1. In fact, compared to activation of the wt PexoT-lacZ reporter by wt ExsA, neither the T199S, K202R, nor T252S mutant actually demonstrated increased activation of its cognate reporters compared to the wt PexoT-lacZ reporter (Fig. 3B to D). The simplest interpretation of this finding is that the gain of function for each mutant protein at binding site 1 is offset by reduced function at site 2 and, when combined, results in a net reduction in overall function compared to wt ExsA. An equally plausible explanation, however, is that reduced activation by the T199S, K202R, and/or T252S mutant results solely from suboptimal interactions at binding site 2.
The Y250A and R257A mutants each demonstrated a significant defect in DNA binding activity and a potential interaction with the TGnnA sequence in the loss-of-contact experiments (Fig. 7 and 8). The Y250 residue is predicted to interact with position −38 (thymine) in the PexoT promoter. Involvement of Y250 is consistent with previous studies demonstrating that the equivalent of residue Y250 in MelR, XylS, and MarA also participates in base-specific recognition of target DNA (Fig. 1A). Missing-nucleoside footprinting data indicated that Y250 interacts with the coding strand (Fig. 9), a finding that is supported by the modeling data presented in Fig. S3B in the supplemental material, which also indicate that the Y250 side chain is orientated toward the −38 thymine. The loss-of-contact data for R257 were suggestive of an interaction with the −37 position (Fig. 7). Assuming that Y250 and T252 interact with the −38 and −34 positions, however, it is difficult to explain how residue R257, located at the far end of the recognition helix (Fig. 1A), could interact with the −37 position. The modeling data also indicate that R257 is located too far away from the −37 position to participate in a direct interaction (data not shown). The model does suggest that R257 could interact with residue Q253, which is a prime position to bind to the −37 site. The Q253A mutant, however, had only a modest defect in activation of the wt PexoT-lacZ and PexsC-lacZ reporters. We conclude that the MarA-DNA complex is only a fair model of the interactions between RH2 and the TGnnA sequence. Ultimately, structural studies will be required to fully understand the nature of the contacts occurring at the TGnnA sequence.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health (RO1-AI055042 to T.L.Y.).
We thank Gary Gussin, Mark Urbanowski, and David Weiss for critical insight and suggestions.
Footnotes
Published ahead of print 9 March 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Apodaca G, et al. 1995. Characterization of Pseudomonas aeruginosa-induced MDCK cell injury: glycosylation-defective host cells are resistant to bacterial killing. Infect. Immun. 63:1541–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barbieri JT, Sun J. 2004. Pseudomonas aeruginosa ExoS and ExoT. Rev. Physiol. Biochem. Pharmacol. 152:79–92 [DOI] [PubMed] [Google Scholar]
- 3. Becher A, Schweizer HP. 2000. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29:948–950, 952 [DOI] [PubMed] [Google Scholar]
- 4. Bhende PM, Egan SM. 1999. Amino acid-DNA contacts by RhaS: an AraC family transcription activator. J. Bacteriol. 181:5185–5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brunelle A, Schleif R. 1989. Determining residue-base interactions between AraC protein and araI DNA. J. Mol. Biol. 209:607–622 [DOI] [PubMed] [Google Scholar]
- 6. Brutinel ED, Vakulskas CA, Brady KM, Yahr TL. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 68:657–671 [DOI] [PubMed] [Google Scholar]
- 7. Brutinel ED, Vakulskas CA, Yahr TL. 2010. ExsD inhibits expression of the Pseudomonas aeruginosa type III secretion system by disrupting ExsA self-association and DNA binding activity. J. Bacteriol. 192:1479–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brutinel ED, Vakulskas CA, Yahr TL. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191:3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a. Brutinel ED, King JM, Marsden AE, Yahr TL. 2012. The distal ExsA-binding site in Pseudomonas aeruginosa type III secretion system promoters is the primary determinant for promoter-specific properties. J. Bacteriol. 194:2564–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dasgupta N, Lykken GL, Wolfgang MC, Yahr TL. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53:297–308 [DOI] [PubMed] [Google Scholar]
- 10. Diaz MR, King JM, Yahr TL. 2011. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front. Microbiol. 2:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dominguez-Cuevas P, Marin P, Marques S, Ramos JL. 2008. XylS-Pm promoter interactions through two helix-turn-helix motifs: identifying XylS residues important for DNA binding and activation. J. Mol. Biol. 375:59–69 [DOI] [PubMed] [Google Scholar]
- 12. Ebright YW, Chen Y, Pendergrast PS, Ebright RH. 1992. Incorporation of an EDTA-metal complex at a rationally selected site within a protein: application to EDTA-iron DNA affinity cleaving with catabolite gene activator protein (CAP) and Cro. Biochemistry 31:10664–10670 [DOI] [PubMed] [Google Scholar]
- 13. Egan SM. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 184:5529–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grainger DC, Belyaeva TA, Lee DJ, Hyde EI, Busby SJ. 2003. Binding of the Escherichia coli MelR protein to the melAB promoter: orientation of MelR subunits and investigation of MelR-DNA contacts. Mol. Microbiol. 48:335–348 [DOI] [PubMed] [Google Scholar]
- 15. Grier MC, et al. 2010. N-Hydroxybenzimidazole inhibitors of ExsA MAR transcription factor in Pseudomonas aeruginosa: in vitro anti-virulence activity and metabolic stability. Bioorg. Med. Chem. Lett. 20:3380–3383 [DOI] [PubMed] [Google Scholar]
- 16. Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7:654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayes JJ, Tullius TD. 1989. The missing nucleoside experiment: a new technique to study recognition of DNA by protein. Biochemistry 28:9521–9527 [DOI] [PubMed] [Google Scholar]
- 18. Hendrickson W, Schleif R. 1985. A dimer of AraC protein contacts three adjacent major groove regions of the araI DNA site. Proc. Natl. Acad. Sci. U. S. A. 82:3129–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72 [DOI] [PubMed] [Google Scholar]
- 20. Holder IA, Neely AN, Frank DW. 2001. PcrV immunization enhances survival of burned Pseudomonas aeruginosa-infected mice. Infect. Immun. 69:5908–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holder IA, Neely AN, Frank DW. 2001. Type III secretion/intoxication system important in virulence of Pseudomonas aeruginosa infections in burns. Burns 27:129–130 [DOI] [PubMed] [Google Scholar]
- 22. Hovey AK, Frank DW. 1995. Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 177:4427–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kurahashi K, et al. 1999. Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J. Clin. Invest. 104:743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon HJ, Bennik MH, Demple B, Ellenberger T. 2000. Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nat. Struct. Biol. 7:424–430 [DOI] [PubMed] [Google Scholar]
- 25. Lee VT, et al. 2007. Pseudolipasin A is a specific inhibitor for phospholipase A2 activity of Pseudomonas aeruginosa cytotoxin ExoU. Infect. Immun. 75:1089–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin RG, Rosner JL. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4:132–137 [DOI] [PubMed] [Google Scholar]
- 27. McCaw ML, Lykken GL, Singh PK, Yahr TL. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46:1123–1133 [DOI] [PubMed] [Google Scholar]
- 28. Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203 [DOI] [PubMed] [Google Scholar]
- 29. Niland P, Huhne R, Muller-Hill B. 1996. How AraC interacts specifically with its target DNAs. J. Mol. Biol. 264:667–674 [DOI] [PubMed] [Google Scholar]
- 30. Rana TM, Meares CF. 1991. Transfer of oxygen from an artificial protease to peptide carbon during proteolysis. Proc. Natl. Acad. Sci. U. S. A. 88:10578–10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rhee S, Martin RG, Rosner JL, Davies DR. 1998. A novel DNA-binding motif in MarA: the first structure for an AraC family transcriptional activator. Proc. Natl. Acad. Sci. U. S. A. 95:10413–10418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richards MJ, Edwards JR, Culver DH, Gaynes RP. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510–515 [DOI] [PubMed] [Google Scholar]
- 33. Richards MJ, Edwards JR, Culver DH, Gaynes RP. 1999. Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit. Care Med. 27:887–892 [DOI] [PubMed] [Google Scholar]
- 34. Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 102:8006–8011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roy-Burman A, et al. 2001. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis. 183:1767–1774 [DOI] [PubMed] [Google Scholar]
- 36. Sato H, Frank DW. 2004. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53:1279–1290 [DOI] [PubMed] [Google Scholar]
- 37. Sawa T, et al. 1999. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 5:392–398 [DOI] [PubMed] [Google Scholar]
- 38. Soisson SM, MacDougall-Shackleton B, Schleif R, Wolberger C. 1997. Structural basis for ligand-regulated oligomerization of AraC. Science 276:421–425 [DOI] [PubMed] [Google Scholar]
- 39. Thibault J, Faudry E, Ebel C, Attree I, Elsen S. 2009. Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 284:15762–15770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urbanowski ML, Brutinel ED, Yahr TL. 2007. Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 75:4432–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urbanowski ML, Lykken GL, Yahr TL. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc. Natl. Acad. Sci. U. S. A. 102:9930–9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vogel HJ, Bonner DM. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97–106 [PubMed] [Google Scholar]
- 43. Withey JH, DiRita VJ. 2006. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol. Microbiol. 59:1779–1789 [DOI] [PubMed] [Google Scholar]
- 44. Yahr TL, Wolfgang MC. 2006. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 62:631–640 [DOI] [PubMed] [Google Scholar]
- 45. Zheng Z, et al. 2007. Biochemical characterization of a regulatory cascade controlling transcription of the Pseudomonas aeruginosa type III secretion system. J. Biol. Chem. 282:6136–6142 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.