

Abstract
c-Myc is an oncogene transcription factor that causes cancer in many settings, including solid tumors that arise in the context of organized tissue structures. Given that disruption of tissue architecture frequently occurs in cancer, there is considerable interest in how cell organization impacts oncogene function. A previous report found that organization of mammary epithelial cells into defined 3-dimensional structures renders them insensitive to the effects of retrovirus-mediated overexpression of Myc, leading to the notion that organization tempers the sensitivity of individual cells to Myc activity. In this article, we report that epithelial cell organization does not profoundly alter Myc activity but, instead, suppresses Myc by modulating its expression. We show that the morphogenesis of mammary epithelial cells into organized acinar structures in vitro is accompanied by widespread changes in gene expression patterns, including a substantial decrease in the expression of Myc. Concomitant with the decrease in endogenous Myc expression, we observe a decrease in transcription from retroviral vectors during morphogenesis and find that Myc transgene expression in acini is much lower than in unorganized cells. This decrease in Myc transgene activity is responsible for the apparent recalcitrance of organized cells to ectopic Myc, as adenovirus-mediated expression of Myc in organized structures potently induces apoptosis. These observations reveal that organization does not alter the inherent response of epithelial cells to Myc and suggest that other tumor suppression mechanisms, apart from structure, antagonize Myc in the development of solid tumors.
Introduction
The c-Myc oncogene encodes a basic helix–loop–helix leucine zipper transcription factor that features prominently in cancer (1). Capable of acting as both a transcriptional activator and a repressor, Myc modulates critical cell-fate decisions by controlling the expression of genes required for cell growth and proliferation (2). In nontransformed cells, forced expression of Myc results in ectopic cell-cycle entry and leads to the potent induction of apoptosis (3): Accordingly, Myc-induced cancer typically requires a collaborating oncogenic event in which the apoptotic response is attenuated, either by loss of proapoptotic factors (e.g., Bim; ref. 4) or by overexpression of proteins that block programmed cell death (e.g., Bcl-2; ref. 5).
Much of what we know about how Myc drives tumorigenesis comes from in vitro studies carried out in cell-culture systems that lack the 3-dimensional (3D) organization typical of tissues and organs. Although these systems have been very informative, it is doubtful that 2-dimensional (2D) cultures fully recapitulate all of the regulatory events that exist within an organized tissue, and considerable interest has emerged in developing 3D culture systems that more accurately reflect events that occur during the emergence of solid tumors such as breast cancer (6). Indeed, studies in 3D culture are particularly important in understanding how Myc contributes to breast cancer, because more than one third of all breast cancers show elevated Myc expression and because overexpression of Myc in ductal carcinomas and invasive breast cancer correlates with a loss of epithelial organization and tumor progression (7-10).
One particularly powerful 3D culture system involves growing nontransformed human mammary epithelial MCF10A cells under conditions in which they form structures that resemble the organization of epithelial cells within the terminal ductal lobule units (TDLU) of the breast, where ductal carcinomas originate (11-13). In this model, MCF10A cells are plated on Matrigel—a proteinaceous mixture that acts as a surrogate for the extracellular matrix (ECM)—and undergo morphogenesis to form structures that resemble hollow acini within the TDLU (14). Initially, MCF10A cells grown on Matrigel proliferate to form a solid sphere of cells with an established polarity. As the cultures develop, cells within the center of each sphere selectively undergo apoptosis and cell proliferation halts within the structures, resulting in the development of mature acini that are organized to have an outer layer of quiescent epithelial cells surrounding a hollow luminal core (14, 15). Recently, Partanen and colleagues (16) used this system to probe Myc function and made the surprising discovery that response of MCF10A cells to ectopic Myc activation depends on their organizational state (16). In immature cultures, or cultures in which organization is perturbed by loss of the polarity regulator LKB1, activation of Myc induces proliferation and apoptosis, as expected. In mature, organized, structures, however, activation of retrovirally expressed Myc fails to efficiently induce either response, leading to the notion that multicellular organization is a tumor-suppressor mechanism that tempers properties of Myc (17).
Although the idea that organization presents a barrier to tumorigenesis is not without precedent (6, 18, 19), we recently found that disruption of MCF10A acinar organization by knockdown of the polarity regulator Scribble disables Myc-induced apoptosis by interfering with induction of Bim (18, 20). This result is contradictory to the notion that organization inhibits Myc function and led us to speculate that some property of maturing acini, other than organization-induced antagonism of Myc activity, is responsible for the resistance of mature acini to Myc activation. In this study, we show that morphogenesis of MCF10A acinar structures in vitro is associated with a profound reduction in the expression of both endogenous and retrovirally transduced exogenous Myc and that it is the inability to express Myc at high levels in maturing 3D cultures that is responsible for the apparent differences in how organized versus unorganized cells respond to Myc. We conclude that Myc functions similarly in acini with immature versus mature organization and that regulation of Myc expression is critical for acinar morphogenesis.
Materials and Methods
Vectors
Myc and Myc-mutant sequences were created by site-directed mutagenesis (Quikchange II Site Directed Mutagenesis Kit from Stratagene) of pCGN-HA-Myc (21), PCR amplified, and subcloned as BglII-EcoRI fragments into MSCV-PIGΔRI (22) to create the MLP Myc retroviral vectors. The pBabeMycER vector has been described previously (23). pBabeMyc constitutively expresses Myc with amino-terminal 6xHis and Flag tags. Adenoviral Myc vectors were constructed by recombining a PCR amplicon containing Myc or Myc-mutant sequences, flanked by attB sites, into pDONR207 using the Gateway system (Invitrogen) to initially generate entry clones. These clones were then recombined with pAd/CMV/V5-DEST (Invitrogen) to create the pAd/CMV/Myc adenoviral vectors. LacZ staining of acini infected with adenovirus from pAd/CMV/lacZ was used as a control for infection and titering (24). Primer sequences are available on request.
Cell culture
Retroviral vectors were transfected into 293-GPG [gift from Richard Mulligan (Harvard Medical School, Boston, MA) in 1999] cells for packaging and pseudotyping as described (25). MCF10A cells (obtained from the American Type Cell Culture in 1999) were maintained in growth medium [Dulbecco’s modified Eagle medium (DMEM)/F12, 5% horse serum, 20 ng/mL epidermal growth factor (EGF), 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin, 500 U/mL penicillin, and 500 μg/mL streptomycin] and seeded at a density of 250,000 cells on each 10-cm dish for retroviral infection. Pools of cells were selected with 2.5 μg/mL puromycin for 24 hours postinfection before seeding on growth factor–reduced Matrigel with assay medium (DMEM/F12, 2% horse serum, 20 ng/mL EGF, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin, 500 U/mL penicillin, and 500 μg/mL streptomycin), as described previously (26, 27). MCF10A cells transduced with pBabeMycER were treated, where noted, with 1 μmol/L tamoxifen or vehicle control (Sigma). For adenoviral infections, a multiplicity of infection of 10 plaque-forming units per cell was used for infection of 3D acini on day 18. Infected cells were assayed for gene expression, or by immunofluorescence, 40 hours later.
RNA analysis
For reverse transcription followed by quantitative PCR (qRT-PCR), total RNA from 2D and 3D cell cultures was harvested with TRIzol (Invitrogen). For each sample, 2 μg of RNA was reverse-transcribed with random hexamers and MultiScribe Reverse Transcriptase (Applied Biosystems) and then quantified by quantitative PCR (qPCR) in triplicate. For Northern blots, 15 μg of total RNA for each sample was resolved on a 1% glyoxal gel (NorthernMax-Gly Kit from Ambion), transferred to a Hybond-N+ membrane (GE Healthcare), and cross-linked by UV light before hybridization. For microarrays, 5 μg total RNA was reverse-transcribed using a T7-oligo(dT) primer for first-strand cDNA synthesis followed by RNase H treatment and second-strand cDNA synthesis. In vitro transcription with T7 RNA polymerase was used to amplify and biotinylate cRNA probes. Biotinylated cRNAs were purified, fragmented, and hybridized to Affymetrix HG-U133A arrays at 45°C for 16 hours, washed and stained in a Fluidics Station 450, and then scanned by an Affymetrix GeneChip Scanner 3000.
Immunoblotting
MCF10A cells were harvested by scraping (2D) or trypsinization (3D) and washed in PBS before lysis in radioimmunoprecipitation assay buffer and shearing with a 25-G needle as described (16, 27). For each blot, 40 to 50 μg of total protein was resolved by SDS-PAGE, and Western blotting analysis was done using either electrochemiluminescence (Pierce Biotechnology) or the Odyssey Imaging System (LI-COR). The following antibodies were used: Bim (Cell Signaling #2819), Myc (9E10; Calbiochem #OP10), Actin (AC15; Sigma #A1978), E-cadherin (Becton Dickinson Biosciences #610181), anti-rabbit IgG-HRP (GE Healthcare #NA934V), anti-Mouse-IgG-HRP (GE Healthcare #NA931V), and anti-Mouse-IRDye-800CW (LI-COR #926-32212). SUMO-Myc standards were purified from Escherichia coli expressing pSUMO-WT-Myc using Ni-NTA beads and quantified by Coomassie blue staining.
Immunofluorescence
Immunofluorescence analysis of MCF10A cells was done as described previously (27). Nuclei were counterstained with Hoechst 33342 and mounted using ProLong Antifade Kit (Invitrogen). The following antibodies were used: cleaved caspase-3 (CC3; Cell Signaling #9661), Ki67 (Invitrogen #18-0191Z), Myc (N262; Santa Cruz Biotechnology #sc-764), anti-rabbit IgG AlexaFluor488 (Invitrogen #A11008), anti-rabbit IgG AlexaFluor568 (Invitrogen #A11036), goat anti-mouse IgG (H + L) F(ab′)2 fragment (Jackson Immunoresearch 115-006-006), and goat anti-rabbit IgG (H + L) Fab fragment (Jackson Immunoresearch #111-007-003). For quantification, at least 100 acini per sample were counted. Acini containing 3 or more cells with Ki67 or CC3 immunofluorescence were counted as positive for proliferation or apoptosis, respectively.
Microarray analysis
Six hybridizations from 2D MCF10A cells (3 from day 2 in culture and 3 from day 5 in culture) and 8 hybridizations from 3D MCF10A acini (3 from day 8 in culture and 5 from day 16 in culture) were analyzed with Affymetrix Gene Expression Console software using robust multiarray average for normalization. Significance analysis of microarrays and a false discovery rate (FDR) of 0.0001 were used for analysis of differentially expressed genes in 2D culture versus 3D culture (28). A highly conservative FDR (0.001) was selected to allow us to conclude with confidence which genes are differentially expressed between the 2 culture conditions. To analyze Myc target genes, the 1,160 Myc target genes in the Myc Target Gene Database (2), which were also present on the Affymetrix HG-U133A array, were hierarchically clustered using Pearson correlation distance and the complete linkage method. Gene expression data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus database and are accessible through accession number GSE26148.
Supplementary methods
Further information on materials and methods is provided in the Supplementary Material.
Results and Discussion
Myc expression prevents formation of normal 3D acinar morphology
Recently, Partanen and colleagues (16) used a tamoxifen-inducible form of Myc (Myc ER) to show that activation of Myc in developing MCF10A acini potently induces apoptosis and disrupts acinar morphogenesis, whereas activation at later stages, after acini mature, fails to induce any of the phenotypic consequences normally associated with Myc expression.
To independently confirm and extend these observations, we used retroviral vectors to constitutively express Myc—wild-type (WT) and a panel of Myc mutants—in MCF10A cells, and asked what effect chronic Myc expression had on acinar development (Figs. 1 and 2). These studies produced results that were very similar to those reported by Partanen and colleagues (16). MCF10A cells transduced with an “empty vector” (MLP) control underwent a characteristic morphogenesis process that resulted, by day 16, in mature acini with regular structure and a hollow lumen that was cleared by apoptosis early in development (Fig. 1A). In contrast, MCF10A cells transduced to express WT Myc (MLP Myc) were highly disorganized (Fig. 1A) and showed profoundly elevated levels of apoptosis, as measured by immunostaining for CC3 (Fig. 1A; quantification and additional images are shown in Supplementary Figs. S1A and S2). Furthermore, as expected (26, 27), these cells showed enhanced levels of ectopic proliferation, as measured by staining for Ki67 (Fig. 1A; quantification and additional images are shown in Supplementary Figs. S1B and S3A and B). Chronic expression of Myc in this setting was accompanied by 5-fold higher levels of CC3 staining at day 16 in the mature acini (Supplementary Fig. S1A), a time at which the lumen had been completely cleared of acini transduced with the MLP control. Consistent with the high level of apoptosis we observed, chronic Myc expression was also associated with induction of the proapoptotic protein Bim (Fig. 1A; Supplementary Fig. S3C), indicating that downstream processes used by Myc to drive apoptosis (15) are intact in these cells. We conclude that, as previously reported (16, 20), Myc possesses an ability to induce both proliferation and apoptosis when expressed before, and during early, acinar morphogenesis.
Figure 1.
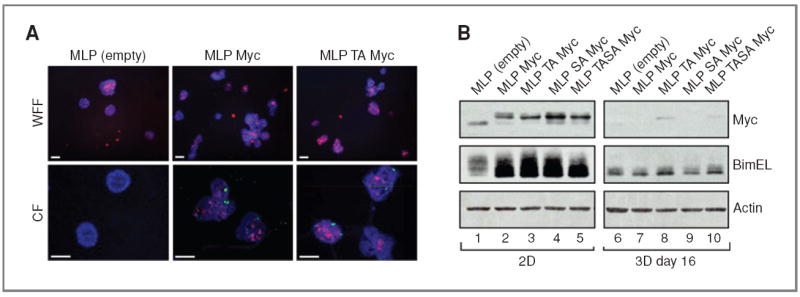
The effect of Myc expression on apoptosis, morphology, and Bim regulation during the formation of MCF10A acini. A, wide-field fluorescence (WFF) and confocal (CF) images of cleaved CC3 (red, both WFF and CF) and Ki67 (green, CF only) immunofluorescence in day 16 acini expressing an empty vector (MLP), Myc (MLP Myc), or T58A Myc (MLP TA Myc). Nuclei were counterstained with Hoechst 33342 (blue). Scale bars, 50 μm. B, Western blot showing Myc and Bim expression relative to actin in MCF10A cell monolayers (2D) and MCF10A acini (3D; day 16) expressing an empty vector (MLP), Myc (MLP Myc), T58A Myc (MLP TA Myc), S62A Myc (MLP SA Myc), or T58A S62A Myc (MLP TASA Myc). The extra long (EL) isoform of Bim is revealed in these blots.
Figure 2.
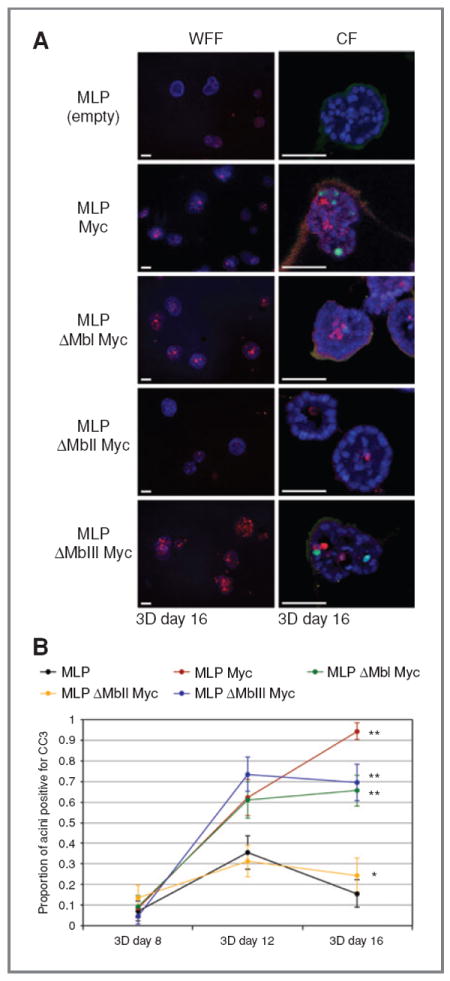
Myc-induced apoptosis in developing MCF10A acini requires MbII. A, wide-field fluorescence (WFF) and confocal (CF) images of CC3 (red, both WFF and CF) and Ki67 (green, CF only) immunofluorescence in day 16 acini expressing an empty vector (MLP), Myc (MLP Myc), a Myc box I deletion mutant (MLP ΔMbI Myc), a Myc box II deletion mutant (MLP ΔMbII Myc), or a Myc box III deletion mutant (MLP ΔMbIII Myc). Scale bars, 50 μm. B, quantification of the proportion of acini with 3 or more cells staining positive for CC3 at days 8, 12, and 16. Error bars, ±1 SEM for the population proportion (χ2 test compared with MLP: *, P = 0.10; **, P < 0.004).
Next, we asked whether Myc function in this setting relies on elements and residues within Myc that are important for induction of proliferation and apoptosis in other settings (5, 29). Initially, we tested the effects of mutations in Myc that disrupt phosphorylation at residue threonine 58 (T58), a process that is important for regulation of Myc by the Fbw7 ubiquitin ligase (30) and for induction of apoptosis in mouse embryo fibroblasts and a lymphoma mouse model (5). Surprisingly, we found that alanine substitutions at T58 (TA), S62 (SA), and T58/S62 (TASA)—all of which disrupt T58 phosphorylation (31)—had little, if any, effect on the ability of Myc to either disrupt acinar morphology (Fig. 1A) or drive apoptosis (Fig. 1A; Supplementary Fig. S1), thus suggesting that signaling pathways that descend on T58 to modulate the apoptotic function of Myc do not operate in MCF10A cells. Although we do not understand the basis for this result, it does show that processes that are essential for stimulation of apoptosis by Myc can be different in different cellular contexts.
We extended these experiments by probing the role of highly conserved Myc boxes I, II, and III in Myc function in 2D and 3D MCF10A cultures (29). The assays were repeated as before on acini chronically expressing Myc variants lacking one of these boxes (Fig. 2; Supplementary Figs. S2 and S3). According to these results, the ΔMbI and ΔMbIII mutants resembled WT Myc in all of the assays, showing that these 2 regions of Myc are dispensable for its behavior in MCF10A cells. The ΔMbII mutant, in contrast, was severely compromised in all Myc functions: Acini expressing ΔMbII Myc were phenotypically normal (Fig. 2A) and resembled those transduced with the MLP control for induction of apoptosis (Fig. 2B; Supplementary Fig. S2), proliferation (Supplementary Fig. S3A and B), and failure to induce expression of Bim (Supplementary Fig. S3C). The requirement of MbII for Myc function in this setting is typical of what is observed in other systems (29, 32) and suggests that interactions of Myc with components of the transcriptional machinery—which are mediated, in large part, via MbII (33, 34)—are required for the ability of Myc to drive ectopic proliferation and apoptosis in developing acini.
Endogenous Myc and retroviral Myc transcripts are reversibly decreased in 3D cell cultures
During the course of the aforementioned experiments, we monitored levels of Myc by Western blotting (Fig. 1A) and were surprised to learn that both endogenous (compare lanes 1 and 6) and retrovirally expressed Myc (compare lanes 2–5 and 7–10) were dramatically downregulated in mature 3D cell cultures. This decrease in Myc levels was not due to trypsinization used to liberate cells from 3D cell cultures, as direct comparison of trypsinization versus harvesting of cultures revealed no differences in detection of Myc by Western blotting (Supplementary Fig. S4), suggesting that decreases in Myc expression occur within the 3D cell culture context. Although Partanen and colleagues (16) reported that MycER protein was expressed in mature acini, they normalized their Western blots to expression of E-cadherin, which we found is similarly reduced as MCF10A cells undergo morphogenesis (Supplementary Fig. S3C), suggesting that relative levels of MycER fusion proteins used in their study could be very different in the 2D and 3D cell culture settings. To test this hypothesis directly, we expressed a MycER fusion protein in MCF10A cells and induced acini to form on Matrigel. This analysis (Fig. 3) showed that—similar to endogenous and constitutively expressed Myc—expression of the MycER protein was significantly lower in the 3D cell culture context (compare lane 2 with lanes 5 and 6). The loss of MycER expression was reversible because MycER levels were restored by harvesting 3D acini and growing the dispersed cells under 2D conditions (Fig. 3A). Importantly, the reduction in Myc levels in maturing acini appeared to be mediated by a reduction in mRNA expression (Fig. 3B) and was observed for both constitutively overexpressed Myc and endogenous Myc (Fig. 3C), showing that transition of MCF10A cells to growth on Matrigel is accompanied by a widespread reduction in Myc expression. Given that levels of MycER, exogenous Myc, and endogenous Myc are all significantly lower under 3D cell culture conditions, we speculate that a significant part of the reported resistance of mature acini to Myc (16) could be due to this global decrease in Myc expression as acini mature.
Figure 3.
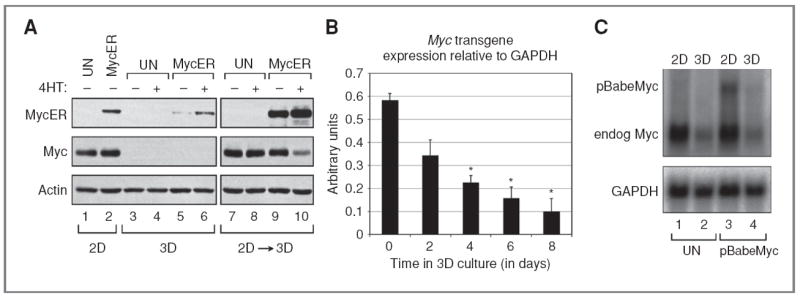
Endogenous and transgenic Myc expression is reversibly reduced in 3D cell cultures. A, Western blot showing Myc ER and Myc expression in 2D, 3D (day 16), and replated (3D → 2D) MCF10A cells, relative to actin, in untransduced (UN) MCF10A cells or in MCF10A cells transduced with pBabeMycER. MCF10A cells were treated with either tamoxifen (4HT; +) or vehicle control (−) while in 3D culture or after replating on 2D. B, time course of transgenic Myc transcripts in 3D culture quantified by qRT-PCR. Exonic Myc transcripts were quantified in MCF10A acini expressing an MSCV-based retroviral Myc transgene. For each sample, the Myc signal from MCF10A acini expressing an empty vector (endogenous transcripts) was subtracted to calculate Myc transgene expression relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Error bars, ±1 SEM (Student t test compared with day 0: *, P < 0.0125; n = 2). C, Northern blot showing expression of transgenic Myc (pBabeMyc LTR) and endogenous (endog) Myc relative to GAPDH in untransduced MCF10A cells or in MCF10A cells transduced with pBabeMyc in 2D or 3D (day 8) culture.
3D cell culture induces widespread changes in gene expression patterns
The fact that mRNA levels for both endogenous and exogenous Myc are diminished in 3D cell culture suggested that widespread transcriptional reprogramming may be induced by interaction of MCF10A cells with the ECM. To address this point, we used microarray analysis to compare the gene expression profiles of MCF10A cells grown as 2D monolayers (proliferating at day 2 and proliferation-arrested at day 5) versus 3D cell cultures (proliferating at day 8 and proliferation-arrested at day 16). Using a conservative FDR of 0.0001, we identified 616 genes that are significantly up- or downregulated in 3D (vs. 2D) cell cultures (Fig. 4A; Supplementary Fig. S5; and Supplementary Tables S1 and S2). Consistent with the loss of endogenous Myc expression reported above, many of the genes that are altered in 3D cell culture are known Myc targets (Supplementary Tables S3 and S4). Importantly, we validated changes in several of the downregulated genes by qPCR (Fig. 4B), suggesting that our microarray data accurately reflected changes in the transcriptional profiles of cells grown under 3D (vs. 2D) cell culture conditions.
Figure 4.
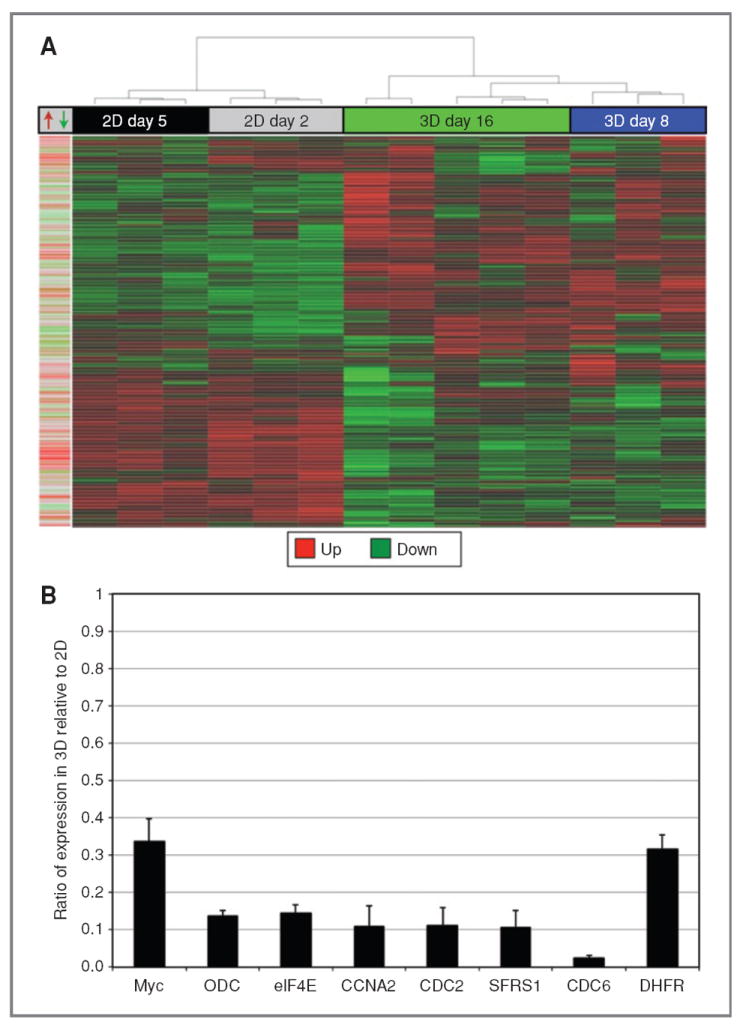
Clustering of Myc target gene expression in 2D and 3D cultures. A, hierarchical clustering analysis of Myc target expression by array and time point (2D MCF10A cells at day 2 or day 5, 3D MCF10A acini at day 8 or day 16). For each array, upregulated genes are represented in red and the downregulated genes are represented in green. Left bar, Myc target gene annotation. From the data set in the Myc Target Gene Database (2), each curated Myc target gene response is labeled in red (upregulated), green (downregulated), or gray (no annotation). B, Myc target gene validation. The 3D:2D ratio of transcripts for each Myc target gene was quantified by qRT-PCR relative to 18S rRNA. Expression of Myc, ornithine decarboxylase (ODC), eukaryotic translation initiation factor 4E (eIF4E), cyclin A2 (CCNA2), cyclin-dependent kinase 1/cell-division cycle 2 (CDK1/CDC2), ASF/SF2 (SFRS1), CDC6, and dihydrofolate reductase (DHFR) is shown. Error bars, ±1 SEM (for each bar, 99.5% confidence interval < 1; n = 3).
Using hierarchical clustering, we identified 2 broad trends in the expression of Myc target genes as cells transitioned from 2D to 3D cell cultures (Fig. 4A): one group of Myc target genes that were expressed at low levels in 2D cell culture and increased in 3D cell culture, and another group that displayed the opposite behavior. The 3D cell culture (days 8 and 16) expression profiles cluster together, in spite of the differences in their proliferative and organizational state, and cluster apart from the highly similar 2D cell culture expression profiles. Moreover, the changes we observed in these 2 groups of genes largely mirror what would be expected on the basis of their known responses to Myc. Many annotated, upregulated (red), and downregulated (green) Myc targets (Fig. 4A, left bar) correlate with the presence of Myc in 2D cell cultures and its absence in 3D cell cultures. Among the Myc target genes that express significantly higher in 3D than 2D cell cultures (Supplementary Table S3), 14 of 21 annotated Myc target genes are identified as downregulated by Myc. Furthermore, of the Myc target genes significantly downregulated in 3D cell culture (Supplementary Table S4), 33 of 37 annotated Myc target genes are identified as upregulated by Myc. The change in expression of these groups of genes occurs early during morphogenesis (it is apparent by day 8; Fig. 4A) and correlates with loss of Myc expression as cells are cultured in 3D culture conditions. These data show that the downregulation of Myc expression that we observe as cells are plated on Matrigel correlates with a reduction in the functional presence of Myc in the cell. The absence of Myc at an early time point in morphogenesis and the effect it has on Myc target genes suggests that controlled morphogenesis could require a reduction in Myc expression to maintain control over proliferation and apoptosis as acini achieve mature organization.
Restoring Myc expression to mature acini induces apoptosis
The decrease in Myc (and MycER) expression we observed in MCF10A cells plated on Matrigel suggests that previous reports with regard to the influence of cellular organization on Myc function may be due to decreased Myc burden in the 3D culture setting rather than a fundamental block to the cellular responses to Myc. To directly test this notion, we asked whether we could use adenovirus-mediated gene transfer to efficiently express Myc in mature 3D cell cultures. We, therefore, infected mature (day 18) acini with recombinant adenoviral vectors expressing WT or mutant Myc proteins and measured Myc expression 40 hours after infection by quantitative Western blot (Fig. 5A) and immunofluorescence (Fig. 5B and C). In mature 3D cell cultures, WT Myc and each of the mutants were expressed at levels that were about 10-fold higher than endogenous Myc in 2D cell cultures (Supplementary Fig. S6; Supplementary Table S5) and corresponded to the level of expression we observed from retrovirally expressed Myc in the 2D cell culture setting (data not shown). Thus, by using adenoviral vectors, we were able to express Myc in mature acini at levels similar to those observed in 2D cell cultures, where Myc exhibits a potent effect on cell proliferation and apoptosis.
Figure 5.
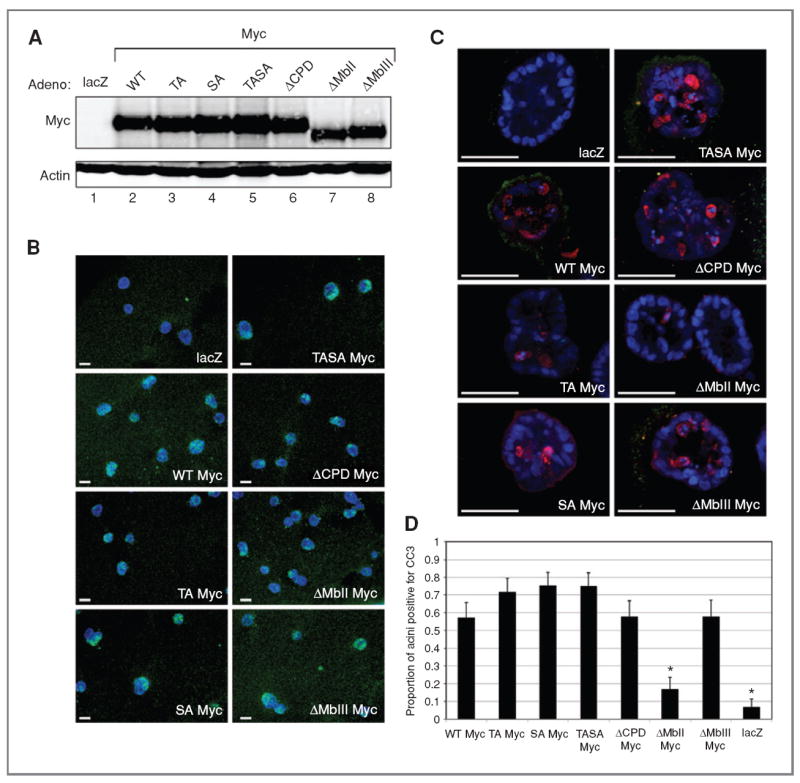
Myc induces apoptosis in mature 3D MCF10A acini. A, quantitative Western blot image showing adenoviral Myc expression relative to actin in mature 3D (day 20) acini (Supplementary Fig. S6A and B). Adenoviral vectors expressing lacZ, Myc (WT Myc), T58A Myc (TA Myc), S62A Myc (SA Myc), T58A S62A Myc (TASA Myc), a CPD-deletion mutant missing L56P57T58P59 (ΔCPD Myc), a Myc box II deletion mutant (ΔMbII Myc), or a Myc box III deletion mutant (ΔMbIII Myc) are shown. B, wide-field images showing adenoviral Myc immunofluorescence (green) in mature 3D (day 20) acini. Nuclei are counterstained with Hoechst 33342 (blue). C, confocal images of CC3 (red) and Ki67 immunofluorescence (green) in MCF10A acini in 3D culture at day 20 with adenoviral vectors from A. Scale bars (B and C), 50 μm. D, quantification of the proportion of acini with 3 or more cells staining positive for CC3. Error bars, ±1 SEM (*, P < 0.007, χ2 test compared with adenoviral WT Myc).
Now that we were capable of expressing Myc efficiently in mature acini, we could ask directly whether these structures are refractory to Myc. Our analysis revealed that this is not the case. Adenovirus-mediated expression of Myc in mature acini potently induced apoptosis, as measured by CC3 staining (Fig. 5C and D). Importantly, induction of CC3 coincided with Myc expression and colocalized with Myc in mature acini (Supplementary Fig. S6) revealing that forced expression of Myc in these structures correlates with high levels of apoptosis. Consistent with the notion that high levels of Myc favor its proapoptotic activities (35), we saw little evidence of enhanced cell proliferation in mature acini overexpressing Myc (as judged by Ki67 staining; Fig. 5C), indicating that the predominant response of mature acini to adenovirus-mediated Myc is apoptosis. As we saw in previous experiments (Figs. 1 and 2), all of the mutant forms of Myc tested were similarly capable of driving apoptosis in mature acini (Fig. 5C and D), with the exception of the ΔMbII mutant, which had little to no activity in this assay, in spite of robust levels of expression. We conclude that, when expressed at comparable levels in mature 3D acini, Myc is capable of potently signaling apoptosis and that it likely requires its transcriptional activities (mediated by MbII) to carry out this function.
Conclusions
Initially, we set out to understand the influence cellular organization has on Myc function. Although mature cellular organization was reported to block Myc function (16, 17), our findings show that a reduction in Myc expression and changes in Myc target gene regulation precede cellular organization. Loss of Myc expression in 3D cell cultures is part of a fundamental change in gene expression that includes modulating the expression of Myc target genes at an early time point in morphogenesis. If Myc does accumulate in both immature and mature acini, such as when expressed via an adenovirus-based vector, it potently induces apoptosis through a mechanism that requires Myc box II. The differences in Myc-induced apoptosis observed between mammary epithelial cells with immature and mature organization are thus due to a sustained loss of Myc expression but not Myc function.
It is critical to understand how Myc function is regulated within organized cells because of the implications for breast cancer progression and therapeutic response. If Myc function were blocked by the organization of a normal TDLU, the hyperplasia induced by Myc and the selective pressure to lose apoptotic effectors of Myc would be delayed until tissue architecture is broken down. Instead, our results show that Myc does function in organized structures, suggesting that, in early breast lesions where cellular architecture is still mostly intact, Myc could be a driving force in cancer progression.
Supplementary Material
Acknowledgments
We thank Gregory Hannon, Scott Lowe, and members of the Muthuswamy and Tansey laboratories for their helpful comments and discussion. We also thank Ross Dickins, Victoria Aranda, Shelley Lorey, Alexandra Lucs, Richard Mulligan, Marissa Nolan, Avi Rosenberg, and Lixing Zhan for technical assistance and reagents.
Grant Support
This study was supported by Department of Defense Traineeship Award W81XWH-06-1-0731 (D.R. Simpson), Vanderbilt Ingram Cancer Center support grant CA68465 (Z. Zhao and W.P. Tansey), Cold Spring Harbor Laboratory Cancer Center support grant CA45508 (S.K. Muthuswamy and W.P. Tansey), and Department of Defense grant BC075024 (S.K. Muthuswamy). U.S. Public Health Service grants CA098830 (S.K. Muthuswamy) and CA13106 (W.P. Tansey).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–45. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 2.Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of apoptosis in fibroblasts by c-Myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 4.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–9. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–11. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dow LE, Humbert PO. Polarity regulators and the control of epithelial architecture, cell migration, and tumorigenesis. Int Rev Cytol. 2007;262:253–302. doi: 10.1016/S0074-7696(07)62006-3. [DOI] [PubMed] [Google Scholar]
- 7.Blancato J, Singh B, Liu A, Liao DJ, Dickson RB. Correlation of amplification and overexpression of the c-Myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br J Cancer. 2004;90:1612–9. doi: 10.1038/sj.bjc.6601703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naidu R, Wahab NA, Yadav M, Kutty MK. Protein expression and molecular analysis of c-myc gene in primary breast carcinomas using immunohistochemistry and differential polymerase chain reaction. Int J Mol Med. 2002;9:189–96. [PubMed] [Google Scholar]
- 9.Scorilas A, Trangas T, Yotis J, Pateras C, Talieri M. Determination of c-myc amplification and overexpression in breast cancer patients: evaluation of its prognostic value against c-erbB-2, cathepsin-D and clinicopathological characteristics using univariate and multivariate analysis. Br J Cancer. 1999;81:1385–91. doi: 10.1038/sj.bjc.6693404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efstratiadis A, Szabolcs M, Klinakis A. Notch, Myc and breast cancer. Cell Cycle. 2007;6:418–29. doi: 10.4161/cc.6.4.3838. [DOI] [PubMed] [Google Scholar]
- 11.Allred DC, Mohsin SK, Fuqua SA. Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001;8:47–61. doi: 10.1677/erc.0.0080047. [DOI] [PubMed] [Google Scholar]
- 12.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 14.Mailleux AA, Overholtzer M, Brugge JS. Lumen formation during mammary epithelial morphogenesis: insights from in vitro and in vivo models. Cell Cycle. 2008;7:57–62. doi: 10.4161/cc.7.1.5150. [DOI] [PubMed] [Google Scholar]
- 15.Reginato MJ, Mills KR, Becker EB, Lynch DK, Bonni A, Muthuswamy SK, et al. Bim regulation of lumen formation in cultured mammary epithelial acini is targeted by oncogenes. Mol Cell Biol. 2005;25:4591–601. doi: 10.1128/MCB.25.11.4591-4601.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Partanen JI, Nieminen AI, Makela TP, Klefstrom J. Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci U S A. 2007;104:14694–9. doi: 10.1073/pnas.0704677104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Partanen JI, Nieminen AI, Klefstrom J. 3D view to tumor suppression: Lkb1, polarity and the arrest of oncogenic c-Myc. Cell Cycle. 2009;8:716–24. doi: 10.4161/cc.8.5.7786. [DOI] [PubMed] [Google Scholar]
- 18.Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–6. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- 19.Martin SG, St Johnston D. A role for Drosophila LKB1 in anterior–posterior axis formation and epithelial polarity. Nature. 2003;421:379–84. doi: 10.1038/nature01296. [DOI] [PubMed] [Google Scholar]
- 20.Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865–78. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717–26. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–95. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- 23.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–90. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook J, Russell DW. Molecular Cloning: a Laboratory Manual. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 25.Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci U S A. 1996;93:11400–6. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 27.Xiang B, Muthuswamy SK. Using three-dimensional acinar structures for molecular and cell biological assays. Methods Enzymol. 2006;406:692–701. doi: 10.1016/S0076-6879(06)06054-X. [DOI] [PubMed] [Google Scholar]
- 28.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Rep. 2005;6:177–83. doi: 10.1038/sj.embor.7400333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol. 1994;14:5510–22. doi: 10.1128/mcb.14.8.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang XY, DeSalle LM, McMahon SB. Identification of novel targets of MYC whose transcription requires the essential MbII domain. Cell Cycle. 2006;5:238–41. doi: 10.4161/cc.5.3.2409. [DOI] [PubMed] [Google Scholar]
- 33.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–74. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 34.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–62. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–57. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.