

Abstract
Lissencephaly is a severe brain malformation in humans. To study the function of the gene mutated in lissencephaly (LIS1), we deleted the first coding exon from the mouse Lis1 gene. The deletion resulted in a shorter protein (sLIS1) that initiates from the second methionine, a unique situation because most LIS1 mutations result in a null allele. This mutation mimics a mutation described in one lissencephaly patient with a milder phenotype. Homozygotes are early lethal, although heterozygotes are viable and fertile. Most strikingly, the morphology of cortical neurons and radial glia is aberrant in the developing cortex, and the neurons migrate more slowly. This is the first demonstration, to our knowledge, of a cellular abnormality in the migrating neurons after Lis1 mutation. Moreover, cortical plate splitting and thalomocortical innervation are also abnormal. Biochemically, the mutant protein is not capable of dimerization, and enzymatic activity is elevated in the embryos, thus a demonstration of the in vivo role of LIS1 as a subunit of PAF-AH. This mutation allows us to determine a hierarchy of functions that are sensitive to LIS1 dosage, thus promoting our understanding of the role of LIS1 in the developing cortex.
Keywords: brain development, lissencephaly, platelet-activating factor, acetylhydrolase, gene targeting
LIS1 was identified as the gene mutated in a severe human developmental brain malformation known as lissencephaly (“smooth brain”) type I (1). Patients with lissencephaly often are severely retarded, epileptic, and die at a young age. The most striking feature of the brains of affected individuals is that they are smooth and largely devoid of the sulci and gyri that characterize the normal brain. The lissencephalic brain exhibits defects in neuronal migration that result in poor organization of cortical layering. A reduced surface area and lack of cortical folds are also seen, possibly because of an overall reduced number of neurons (2). Mutations in two different genes may result in type I lissencephaly: LIS1, an autosomal gene located on chromosome 17p13.3 (1), and doublecortin, an X-linked gene (3, 4). The pattern of expression of LIS1 in the nervous system suggested that the mouse would be a suitable organism for studying the role of LIS1 during brain development (5). Mouse embryos homozygous for the null Lis1 allele (Lis1−/−) die after implantation, whereas heterozygotes are viable and fertile (6). A half dosage of LIS1 affects neuronal migration only slightly in the developing cortex, whereas adult layer organization appears normal. Further gene dosage reduction severely obstructs cortical and hippocampal organization (6). LIS1 interacts with many proteins and is involved in several basic cellular functions, including mitosis, nuclear positioning, and microtubule regulation (for review, see ref. 7). To better understand the function of LIS1 and its role in neuronal migration, we produced Lis1 mutant mice by using cre recombinase-mediated loxP deletion. Our mutation resulted in a shorter LIS1 protein that initiates from the second methionine (M63), thus lacking two-thirds of the coiled-coil N terminus. The shorter protein enabled us to study biochemical parameters of the mutated protein in addition to the developmental phenotype of mutant embryos. The LIS1 mutants described here show a transient delay in the organization and maturation of the dorsal–caudal portion of the cortex, with abnormal morphology of both cortical neurons and radial glia during corticogenesis.
Materials and Methods
Monoclonal anti-LIS1 antibodies have been described (8). Polyclonal antibody specific for the N-terminal domain of LIS1 was generated by injecting rabbits with a peptide corresponding to amino acids 5–13 of LIS1 (amino acid sequence: QRQRDELNRAIAD), coupled to keyhole limpet hemacyanin (Sigma).
Histological and in Situ Hybridization Analyses.
Embryos were collected at the stages indicated, and brains were either dissected from the head or left in situ. All sections discussed in this paper are coronal. Samples for whole-mount RNA in situ hybridization were fixed in 4% paraformaldehyde at 4°C and then processed essentially as described (9). For DiI (Molecular Probes) labeling, embryos were fixed in 4% paraformaldehyde, and then a DiI crystal (saturated solution in DMSO and air dried) was placed in the cortex or within the thalamus later sectioned by the vibratome in 100-μm-thick sections.
BrdUrd/propidium iodide staining of cortical neurons and FACS analysis were performed according to ref. 10. For analysis of cell cycle kinetics and interkinetic nuclear movements, immunocytochemistry and autoradiography were performed on 4 μm coronal sections, as described previously (11). Gel filtration (12) and GST pulldown (13) were performed as described. Platelet-activating factor acetylhydrolase enzymatic activity was tested as described in ref. 14. Microtubule assembly was performed as described previously (15) with the aid of taxol.
Results and Discussion
Our targeting construct involved insertion of two loxP sites, with a neomycin resistance gene in Lis1 introns flanking the first coding methionine (Fig. 1A). Heterozygotes of the loxP-neo allele were mated with PGK-Cre mice (16). Offspring of these mice exhibited the expected first coding exon deletion (“floxed locus”) in all tissues examined (data not shown); however, no homozygotes were born (Fig. 1B), and lethality was at the implantation stage. Heterozygotes were viable and fertile. Western blot analysis of brain extracts from wild-type and heterozygous mice revealed two LIS1 immunoreactive bands in heterozygotes, but only one band was present in wild-type mice (Fig. 1D); therefore, the mice with the first coding exon deletions will be referred to as Lis1/sLis1 mice. The shorter protein (sLIS1) present in the heterozygotes is likely to result from translation initiation at the second methionine of LIS1 (Fig. 1C). This assumption was confirmed by specific antibodies recognizing only the N-terminal domain (Fig. 1D). Quantitative analysis revealed that heterozygous mice have higher total levels of LIS1 than wild type because of the addition of expression from the mutant allele (sLIS1). Our mutation is therefore different from the null allele of Lis1 reported previously (6).
Figure 1.

(A) Design of the Lis1-targeting construct, the endogenous locus, and the structure of the gene after deletion by Cre recombinase. The first translated exon is marked in black, and positions of the loxP sites (loxP), the first coding methionine (ATG), neomycin resistance (neo), and thymidine kinase (tk) are marked. (B) Genotyping by PCR of loxP-neo (by using two sets of primers); 1.2 kb is the wild-type allele, and 1.8 kb is the loxP-neo allele. Mice 1–4 are heterozygotes, whereas mouse 5 is wild type. (C) Schematic diagram of normal and mutated LIS1 proteins. The N-terminal portion (95 amino acids) is represented by a white box, and the seven WD tryptophane aspartic acid repeats are indicated by using boxes with different patterns. Cre-mediated deletion of the gene resulted in shortening of the 5′ untranslated region (UTR) of the transcript and the N-terminal portion of the protein. The shorter protein (345 instead of the normal 409 amino acids) initiated at the second methionine (2nd ATG) that resides within the N-terminal portion of the protein. (D) Western blot of brain extract reacted with two anti-LIS1 antibodies, 210 and N-ter. Note that only the Lis1/sLis1 animal (genotype −/+) has the second shorter protein (sLIS1). Both heterozygote and normal littermates have the normal LIS1 protein, as revealed by the two antibodies (indicated by an arrow). The anti-N-ter LIS1 polyclonal antibody was raised against a peptide from the deleted region (indicated by an arrow).
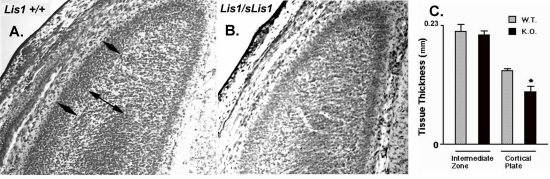
Analysis of the central nervous system in Lis1/sLis1 embryos showed that the parietal and occipital cortex, regions where the phenotype in lissencephaly patients is more pronounced, exhibited apparent alterations. LiS1/sLIS1 embryos at ages embryonic day (E)12.5–14.5 were examined, but only the latter showed irregularities; the emergence of cortical plate (CP) in caudal and medial regions of the cerebral wall of the dorsal telencephalon was abnormal (Fig. 2 A and B). Anticalretinin antibodies, which stain neurons of the primordial plexiform layer (17, 18), show a single layer in the affected cortical domains of LIS1/sLIS1 (Fig. 2D) in contrast to wild type, where the preplate has been split into marginal zone and subplate by the forming CP (Fig. 2C). This result suggests that the CP has not been normally formed. The number of calretinin-positive subplate cells is apparently smaller in the Lis1/sLis1 embryos, and their distribution is irregular. The delay in CP formation was visible in all E14.5 Lis1/sLis1 mice (n = 11) but in none of the wild-type littermates (n = 7). The shape of the affected area was different in the two hemispheres. Seven of the eleven mutant embryos exhibited an extra fold at the level of the dorsomedial cortex, localized in the most affected side (see arrow in Fig. 2 B and D). The extra fold is not present in the wild-type embryos. In both wild-type and Lis1/sLis1 embryos, the splitting of the preplate by the CP occurs gradually from ventrolateral to dorsomedial across the plane of the neopallium. This gradient is paralleled by Lis1 expression, where at E12.5 and E13.5 it is expressed in decreasing gradients from dorsal to ventral and caudal to rostral (see Fig. 6, which is published as supplemental data on the PNAS web site, www.pnas.org). The sites of highest Lis1 expression in the dorsal and caudal cortical regions correspond to the areas of the most pronounced phenotypic defects in Lis1/sLis1 animals. Defects in CP formation are also visible in E15.5 embryos in dorsocaudal regions of the cortex. Measurements of the thickness of the CP and the intermediate zone within the occipital cortex revealed that the width of the CP (but not of the intermediate zone) is reduced significantly in mutant mice (see Fig. 7, supplemental data). Collectively, these data suggest that although the CP does form in the occipital regions of Lis1/sLis1 mice, the process is slowed or delayed.
Figure 2.

CP formation in E14.5 embryos. (A and B) Nissl stain of Lis1+/+ embryos (A) and heterozygote Lis1/sLis1 littermates (B). The positions of subplate (SP) and CP are indicated. The abnormalities in B are marked by *. The extra fold is indicated by an arrow (B and D). (C and D) Anticalretinin immunostaining of Lis1+/+ embryos (C) and heterozygous Lis1/sLis1 littermates (D). CR, Cajal–Retzius cells; v, ventricle; St, striatum. (E and F) Delay of neocortical innervation by the thalamic fibers in Lis1/sLis1. DiI fluorescence is in red. Although in the wild-type mice the thalamocortical (Th. Cortical) fibers reach the cortex (E), shorter axonal processes are seen in the heterozygotes (F).
It has been hypothesized that the maturation of neurons in the CP plays a crucial role in the invasion of layer 4 by thalamocortical afferent axons (19–22). The retarded development of the CP in Lis1/sLis1 embryos led us to speculate that axonal development might be altered as well. Indeed, injection of DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate) into the thalamus revealed clear labeling of thalamocortical axons in wild-type cortices at E14.5 (Fig. 2E), but little or no labeling was present in Lis1/sLis1 embryos (Fig. 2F). This difference persisted at E15.5; however, by E16.5 and E17.5, the labeling patterns in heterozygotes and their wild-type littermates were similar (data not shown). Additionally, the formation of barrels in the somatosensory cortex was normal (data not shown). The results were verified by immunostaining with calretinin, a calcium-binding protein expressed in the developing dorsal thalamic nuclei and their axons (17, 18) (data not shown). The delay described here may result from intrinsic changes in the properties of thalamocortical fibers and/or from cell autonomous defects in the thalamus. It is also possible that there are delays in the production of guidance cue(s) (or elimination of repellant molecules) by CP neurons (reviewed in ref. 23), or the retarded thalamocortical axon growth may directly affect cortical progenitors as well (23).
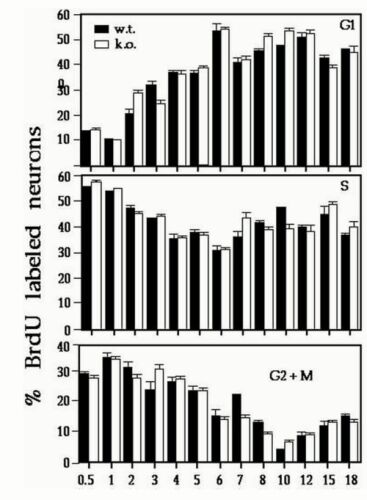
The differences in CP splitting may arise as a consequence of abnormalities in neuronal proliferation or cell cycle and/or neuronal migration. As NudF, the LIS1 homolog in Aspergillus nidulans is known to be involved in nuclear migration (24), hence we have investigated in detail whether the observed phenotype may be caused by an abnormality in cell cycle and/or interkinetic nuclear movement in the ventricular zone. We found that cell cycle kinetics (see Fig. 8, which is published as supplemental data on the PNAS web site, www.pnas.org) and interkinetic nuclear migration (Fig. 3 A and B) within proliferative cells of the ventricular zone were not affected in mutant mice. To visualize interkinetic nuclear migration, we injected [3H]thymidine on E14, then injected BrdUrd 2 h later and killed the mice 30 min later. With this labeling paradigm at the time of death, the BrdUrd-labeled nuclei (Fig. 3 A and B, brown dots) are the cells in S phase, whereas the nuclei labeled only with [3H]thymidine (Fig. 3 A and B, black dots) left S and entered G2/M during the time between the injection of the two labels. To follow the progression of neuronal migration, we injected BrdUrd at E12.5 or E13.5 and killed the mice 48 h after injection. No differences were observed when BrdUrd was injected at E12.5; however, labeling at E13.5 followed by death at E15.5 revealed differences in the pattern and distribution of labeled cells in the occipital cortex (Fig. 3 D and E). In heterozygous embryos, significantly fewer labeled cells were found in the most superficial portion of the CPs, which contains newly arrived postmitotic neurons (Fig. 3E). On average, only 45% of BrdUrd-labeled cells occupied CPs in Lis1/sLis1 animals, compared with 64% in normal littermates (P = 0.0006). This experiment was followed by an experiment where BrdUrd was injected at E13.5, and the mice were killed and sectioned at E17.5 (Fig. 3 F–H). In sections from the dorsal telencephalon (sectioning diagram shown in Fig. 3C), the differences between the wild-type (Fig. 3F) and mutant embryos (Fig. 3 G and H) are clear. More cells are labeled in the Lis1/sLis1 brains, suggesting an alteration in neuron birth date in the mutant embryos. Furthermore, the labeled cells are distributed across the thickness of the CP of the mutant animals, whereas in the wild-type embryos, these cells are confined mainly to the superficial layer of the CP as already observed (Fig. 3D). The sections also show that CP thickness in the mutant embryos is still reduced at E17.5 (note CP arrows in Fig. 3F, in comparison to G and H). These findings may imply that neurons in the mutant migrate more slowly. At birth, in the mutant, the distribution of E13.5 and E14.5 BrdUrd-labeled neurons was not identical to wild type; however, the differences were less striking and did not reach statistical significance (data not shown). In summary, the splitting of the CP is retarded at E14.5 and results in a thinner layer at E15.5 and an abnormal positioning of neurons in the deep (rather than superficial) portions of the CP. These results suggest that CP splitting and neuronal migration are most sensitive to LIS1 function. Interestingly, although we observed no abnormalities in either cell cycle length or interkinetic nuclear migration, we detected more neurons labeled at E13.5 in sections (Fig. 3 G and H). It may be that the time window in which neurons leave the cell cycle is narrower, so more do so within the time frame (25). This observation may be relevant to the given role of LIS1 in chromosome segregation (26). It is also possible that the introduced Lis1 mutation affects neuronal proliferation only within an area limited to the observed phenotype.
Figure 3.

(A and B) Interkinetic nuclear movement in the ventricular zone. BrdUrd-labeled cells that are in dark brown are in the S phase in the outer half of the ventricular zone, whereas [3H]thymidine cells that have black dots are in G2 and M and are mostly located at the ventricular surface. The position and number of the single- and double-labeled cells are similar in wild-type Lis1+/+ (A) and Lis1/sLis1 (B) embryos, indicating that both interkinetic nuclear movements and cell cycle kinetics are similar. (C) Illustration of the position of the sections (in blue box) in F–H. (D, E, and F) Distribution of cells labeled with BrdUrd at E13.5 in the CP of E15.5 embryos. Sections were cut through the occipital cortex of E15.5 embryos, and the difference in the distribution of BrdUrd-labeled cells between wild type and mutants was analyzed. Although in the Lis1+/+ mice (D), most of the labeled cells (dark) are in the superficial portion of the CP, in the Lis1/sLis1 mice (E), the labeled cells are more equally distributed. The CP was marked, divided in half (the position of the division is marked by an arrow), and labeled cells counted in the superficial (CPs) and interior portions. (F–H) Distribution of cells labeled with BrdUrd at E13.5 in the CP of E17.5 embryos. Sections were cut through the occipital cortex of E17.5 embryos, and the difference in the distribution of BrdUrd-labeled cells between wild type and mutants was analyzed. The width of the CP is marked by an arrow. The position of the subplate (SP) is marked as well. (F) Section in a wild-type embryo. (G and H) Sections in mutant embryos.
To study the initial stages of axonal development and the dendritic morphology of neurons in the cortex, we introduced a crystal of DiI into different levels of the cerebral wall to label neurons and radial glia. The labeled developing pyramidal neurons in wild-type E14.5 embryos are oriented radially (Fig. 4A), with normal apical dendrites directed toward the pial surface (Fig. 4A, white arrow). Each soma is large and polygonal, with a single axon that emerges basally and extends into the primordial white matter (Fig. 4A, yellow arrowhead). Quite strikingly, the labeled neurons in the CP of heterozygous Lis1/sLis1 display abnormal morphology and organization (Fig. 4B). A significant number of neurons had altered multipolar morphology. Interestingly, clusters of labeled cells were often located at the pial surface (marked with white arrowheads, Fig. 4B). In wild-type mice, isolated tangentially oriented single cells were only occasionally observed near the pia (not shown), but such clusters of cells were not detected. In addition, in Lis1/sLis1 embryos, the overall organization of the radial glia is strikingly abnormal: many glial fibers follow a rather curvaceous path in dorsomedial cortical areas, although they remain parallel to each other along this trajectory (Fig. 4D). This finding is in contrast to the normally rather straight pathway followed by the radial glia observed at E15.5 in wild-type embryos (Fig. 4C, arrowhead, and ref. 27). Radial glia are fundamental for normal development of the CP, because they serve as a scaffold for neuronal migration. Furthermore, they also serve to route some ingrowing axons into the CP (reviewed by refs. 28 and 29). Thus, the unusual radial glia in the Lis1/sLis1 mice may offer an explanation for some of the alterations observed. However, it should be noted that in reeler, radial glia differentiate poorly and form a sparse misaligned fiber scaffold, although the primary defect is not in the radial glial cells themselves (30). Thus, it is not clear whether the abnormalities in radial glial cell morphology are an additional primary defect of the mutation in sLis1 or whether they are caused by the neuronal abnormalities. It is undoubtedly important, however, that the most pronounced phenotype during cortical development is in the most caudal part of the brain. No abnormalities were observed in other brain areas or organs; hence, we do not think that general nonspecific growth retardation is a likely explanation for the observed phenotype. Specifically, in this area, we detected a delay in CP formation, possibly slower neuronal migration, delay in thalamocortical axon growth, and abnormal morphology of radial glia and neurons. At E14.5, a unilateral extra fold was observed in the cortex of most mutant mice, possibly as a consequence of abnormal radial glia organization. Importantly, this portion of the telencephalon shows the highest level of Lis1 expression in the mouse cortex (see Fig. 6, www.pnas.org) and is the region where most noticeable brain malformations occur in LIS1-mutated lissencephaly patients (31).
Figure 4.

DiI labeling of the cortex. (A and B) E14.5 DiI labeling of cortical cells. (Bar = 1 mm.) The white arrow labels the apical dendrite, and the yellow arrowhead marks the projecting axon. Lis1+/+ (A); Lis1/sLis1, white arrow marks a neuron that has “normal” morphology, and white arrowheads mark clusters of neurons that look abnormal (B). (C and D) E15.5 DiI labeling of cortical cells. (Bar = 25 μm.) Radial glia fibers are marked with arrowheads. Lis1+/+ (E), Lis1/sLis1 (F). V, ventricle; IZ, intermediate zone; CP*, abnormal CP.
To understand the biochemical basis for the phenotypes observed in our mutant mice, we tested the interactions of sLIS1 and microtubules, PAH-AH, and normal LIS1. LIS1 homodimerization was investigated by using a recombinant GST fusion protein containing the first 95 amino acids of LIS1 (referred to as N-LIS1) to pull down the endogenous brain LIS1. This domain is sufficient to confer LIS1 homodimerization (O.R., unpublished data). Although the normal LIS1 protein is pulled down by N-LIS1 from Lis1/sLis1 brain extracts, sLIS1 protein is not (Fig. 5A). This experiment suggests that the mutant protein, which lacks the first 63 amino acids, is not capable of dimerization. We then examined the interaction with the catalytic subunits of PAF-AH. The ability of GST fusion proteins of the bovine α1 and α2 to pull down LIS1 from mouse brain extracts was tested (Fig. 5A). The α1 and α2 catalytic subunits pulled down wild-type LIS1 but not sLIS1 (Fig. 5A), suggesting that LIS1 homodimerization is essential for participation in the enzyme complex. To assess whether the failure of sLIS1 to associate with α1/α2 altered PAF-AH enzymatic activity, acetyl hydrolysis of [3H]PAF was measured in extracts of dorsal telencephalon from E14.5 embryos (in which the histological phenotype was observed) and adult mice (Table 1). PAF-AH activity was significantly higher in E14.5 Lis1/sLis1 embryos compared with wild-type controls. No significant differences were observed in adult mice (Table 1). Next, we examined the interaction of the two LIS1 isoforms with preassembled microtubules. In this assay, the microtubules are assembled in the presence of microtubule-associated proteins (MAPs) and taxol. As demonstrated previously, normal LIS1 protein is found in both the cytoplasmic and microtubule-associated fractions (8). No significant differences were seen in the distribution of LIS1 and sLIS1 (Fig. 5B); the MAP fraction contained 40.7 and 37.3% of total LIS1 and sLIS1 proteins, respectively. This result suggests normal distribution of the mutated protein (sLIS1) between the microtubule cytoskeleton pellet and the cytoplasmic fraction.
Figure 5.

LIS1 and sLIS1 interactions. (A) sLIS1 does not homodimerize and or interact with PAF-AH catalytic subunits. LIS1 homodimerization and PAF-AH catalytic subunits: negative controls include glutathione coated agarose beads, and GST protein show the specificity of the reaction. Western blots (IB) from brain extracts of Lis1+/+ and Lis1/sLis1 (genotype −/+) mice are shown (Left) for comparison. The genotypes are marked under the panels. The interacting proteins were reacted with anti-LIS1 monoclonal antibody (clone 210). (B) sLIS1 assembles with microtubules. Distribution of LIS1 isoforms between the cytoplasmic and microtubule-associated protein fractions. Protein extracts from normal or mutant mouse brains were subjected to microtubule assembly. A representative Western blot by using anti-LIS1 antibodies is shown. The fractions: the two LIS1 isoforms and the genotypes are indicated. (C) Gel filtration analysis of LIS1 and sLIS1. sLIS1 is associated with smaller macromolecular complexes than normal LIS1. Brain extract from heterozygous Lis1/sLis1 mice was size-fractionated by a Sephadex-75 column. Proteins from each fraction were separated by SDS/PAGE and reacted with anti-LIS1 antibody (clone 210). The amount of LIS1 protein was quantified by densitometer (y axis) and plotted against fraction number (x axis). The positions of the void volume and 66 kDa (BSA) markers are shown. The distribution of the normal LIS1 is shown in a thick line, and the distribution of sLIS1is shown by a thin line. (Lower) Representative Western blot.
Table 1.
Acetyl hydrolysis of [3H]PAF
| Dorsal telencephalon | Lis1+/+ | Lis1/sLis1 | Significance |
|---|---|---|---|
| E14.5 | 1,692 ± 57 (n = 27) | 1,870 ± 62 (n = 69) | P < 0.05 |
| Adult | 11,646 ± 448 (n = 24) | 10,736 ± 257 (n = 32) | Not significant |
Acetyl hydrolysis of [3H]PAF was measured in brain cytosolic extracts of E14.5 and adult mice.
It is likely that LIS1 and sLIS1 participate in additional protein complexes in vivo. The fraction distribution of the normal and sLIS1 proteins was compared by using gel filtration (Fig. 5C). Both normal LIS1 protein and sLIS1 reflect high molecular weight complexes, but sLIS1 peaks at about 80 kDa, whereas LIS1 peaks at 150 kDa (12). LIS1 complexes may include LIS1 tubulin, dynein/dynactin complexes, and/or LIS1-PAF-AH complexes, whereas complexes containing sLIS1 are not yet known. However, it is obvious from this experiment that some of the protein interactions in which LIS1 and sLIS1 participate are different. How can these findings be associated with known LIS1 functions? LIS1 is a protein with several known roles: it regulates the microtubule cytoskeleton (discussed below) and controls PAF-AH activity (32, 33). Regulation of the microtubule cytoskeleton may be by a possible direct interaction (8) or indirectly by mediating the activity or localization of cytoplasmic dynein, a microtubule-based motor protein complex (26, 34, 35). Furthermore, LIS1 involvement in microtubule regulation may also stem from its interaction with mammalian homologs of Aspergillus nidulans nudE, mNudE, and Nudel. These proteins were shown to interact with the centrosomes and cytoplasmic dynein (36–40) (reviewed in ref. 7). Within the caudal brain region, we detect abnormal morphology of the neurons. Although the interaction of sLIS1 with preassembled microtubules did not differ significantly from that of LIS1, it is possible that the interactions of sLIS1 with the tubulin subunits alter the dynamic properties of microtubules. We suggest that the abnormal cellular morphology observed is related to LIS1-microtubule regulation function. The mutant protein may also change the interactions with dynein, mNudE, or Nudel in Lis1/sLis1 cells. Thus, it is entirely plausible that changes in the protein composition and posttranslational modifications of protein subunits in specific cell classes will lead to variability in the result of the mutation in different cells. Furthermore, our results showed that the mutant protein can interact with microtubules, perhaps occupying LIS1-binding positions on the microtubules. This interaction may result in a local increase of the normal protein that can enhance specifically the enzymatic activity of PAF-AH α2 dimers (33). The net effect of higher enzymatic activity will be reduced PAF concentration, which may in turn affect the cytoskeleton (41). The observed phenotype could be explained as a developmental and region-specific dominant-negative effect of the truncated protein, with perhaps its greatest effect during periods of proliferation and migration. We find this phenotype very interesting and are presently exploring the molecular mechanism underlying this phenomenon.
Lissencephaly in humans results from a wide range of mutations at the LIS1 locus; the most common disease-causing defects involve large deletions that result in severe lissencephaly (42). The mutation generated in this paper resembles that of a patient with an in-frame N-terminal deletion (43). Indeed, just as our analysis detects partial protein synthesis from the mutated allele, a similar partial protein was detected in this specific patient (43). Compared with classic lissencephaly (42), this patient showed a less severe degree of cortical abnormality, with diffuse pachgyria and small areas of agyria in the posterior convexity. The patient also had mild hypotonia, was able to walk unassisted (at 3 years 4 months), had no epilepsy, was cognitively less impaired, and was able to respond to simple orders. As we have shown here, the phenotype detected in Lis1/sLis1 mice is less severe than that seen in a null allele. Similar to the patient, Lis1/sLis1 mice have no seizures (unpublished data), unlike Lis1−/+ mutant mice, where 5% die from seizures at 3–5 weeks of age (6). In both the null allele and Lis1/sLis1, the adult brain cortices are normal, but abnormalities in the hippocampus were observed only in the null allele (6). Nevertheless, in both mutations, homozygous mutant animals exhibit early lethality, probably relating to LIS1 function in mitosis (26).
This study provides insight into how different functional alleles of LIS1 may produce the range of phenotypes observed in humans. Our detailed analysis identified that the dorsal telencephalon is most vulnerable to LIS1 dosage, and the developmental brain functions that are most sensitive to normal LIS1 functions include formation of the CP, neuronal migration, neuronal morphology, and radial glia morphology. As full understanding of the molecular complexes formed by LIS1 and its mutant alleles is achieved, the involvement of this gene in cortical development will become clear, and the crucial steps it plays in human cortical development will be elucidated.
Supplementary Material
Acknowledgments
We thank Prof. Peter Lonai (Weizmann Institute, Rehovot, Israel) for Cre mice, Prof. Marion Wassef for help in DiI injections, Dr. Ahuva Kanishinsky, Tanya Burakuva, and Yehudit Hermesh for help with the animals, and Monica Rodenas for technical assistance. This work was supported in part by the Fritz Thyssen Stiftung Foundation, Binational Science Foundation Grant No. 97–00014 (to O.R.), a grant from the Seneca Foundation of Murcia community, 708/CV/99 (to S.M.), a grant from the European Community, EC PL-960146 (to O.R., S.M., and W.W.), Human Frontier Science Program Organization Grant No. RG283199 9 (to O.R. and S.K.M.), and Volkswagen Stiftung (to O.R. and W.W.). O.R. is an Incumbent of the Aser Rothstein Career Development Chair in Genetic Diseases (Weizmann Institute).
Abbreviations
- En
embryonic day n
- CP
cortical plate
References
- 1.Reiner O, Carrozzo R, Shen Y, Whenert M, Faustinella F, Dobyns W B, Caskey C T, Ledbetter D H. Nature (London) 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 2.Reiner O. Mol Neurobiol. 2000;20:143–156. doi: 10.1007/BF02742439. [DOI] [PubMed] [Google Scholar]
- 3.des Portes V, Pinard J M, Billuart P, Vinet M C, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, et al. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 4.Gleeson J G, Allen K M, Fox J W, Lamperti E D, Berkovic S, Scheffer I, Cooper E C, Dobyns W B, Minnerath S R, Ross M E, Walsh C A. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 5.Reiner O, Albrecht U, Gordon M, Chianese K A, Wong C, Sapir T, Siracusa L D, Buchberg A M, Caskey C T, Eichele G. J Neurosci. 1995;15:3730–3738. doi: 10.1523/JNEUROSCI.15-05-03730.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirotsune S, Fleck M W, Gambello M J, Bix G J, Chen A, Clark G D, Ledbetter D H, McBain C J, Wynshaw-Boris A. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 7.Reiner O. Neuron. 2000;28:633–636. doi: 10.1016/s0896-6273(00)00142-2. [DOI] [PubMed] [Google Scholar]
- 8.Sapir T, Elbaum M, Reiner O. EMBO J. 1997;16:6977–6984. doi: 10.1093/emboj/16.23.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Nature (London) 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- 10.McConnell S K, Kaznowski C E. Science. 1991;254:282–285. doi: 10.1126/science.254.5029.282. [DOI] [PubMed] [Google Scholar]
- 11.Hayes N L, Nowakowski R S. Dev Neurosci. 2000;22:44–55. doi: 10.1159/000017426. [DOI] [PubMed] [Google Scholar]
- 12.Sapir T, Eisenstein M, Burgess H A, Horesh D, Cahana A, Aoki J, Hattori M, Arai H, Inoue K, Reiner O. Eur J Biochem. 1999;266:1011–1020. doi: 10.1046/j.1432-1327.1999.00942.x. [DOI] [PubMed] [Google Scholar]
- 13.Caspi M, Atlas R, Kantor A, Sapir T, Reiner O. Hum Mol Genet. 2000;9:2205–2213. doi: 10.1093/oxfordjournals.hmg.a018911. [DOI] [PubMed] [Google Scholar]
- 14.Hattori M, Arai H, Inoue K. J Biol Chem. 1993;268:18748–18753. [PubMed] [Google Scholar]
- 15.Collins C A. Methods in Enzymology. Vol. 196. New York: Academic; 1991. pp. 246–253. [DOI] [PubMed] [Google Scholar]
- 16.Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Transgenic Res. 1998;7:105–112. doi: 10.1023/a:1008868325009. [DOI] [PubMed] [Google Scholar]
- 17.Fonseca M, del Rio J A, Martinez A, Gomez S, Soriano E. J Comp Neurol. 1995;361:177–192. doi: 10.1002/cne.903610114. [DOI] [PubMed] [Google Scholar]
- 18.Frassoni C, Arcelli P, Selvaggio M, Spreafico R. Neuroscience. 1998;83:1203–1214. doi: 10.1016/s0306-4522(97)00443-0. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh A, Shatz C J. Development (Cambridge, UK) 1993;117:1031–1047. doi: 10.1242/dev.117.3.1031. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh A, Antonini A, McConnell S K, Shatz C J. Nature (London) 1990;347:179–181. doi: 10.1038/347179a0. [DOI] [PubMed] [Google Scholar]
- 21.Katz L C, Shatz C J. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 22.Finney E M, Shatz C J. J Neurosci. 1998;18:8826–8838. doi: 10.1523/JNEUROSCI.18-21-08826.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy H, Dehay C. In: Normal and Abnormal Development of the Cortex. Galaburda A M, Christen Y, editors. Berlin: Springer; 1997. pp. 25–56. [Google Scholar]
- 24.Xiang X, Osmani A H, Osmani S A, Xin M, Morris N R. Mol Biol Cell. 1995;6:297–310. doi: 10.1091/mbc.6.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi T, Nowakowski R S, Caviness V S., Jr J Neurosci. 1996;16:6183–6196. doi: 10.1523/JNEUROSCI.16-19-06183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faulkner N E, Dujardin D L, Tai C Y, Vaughan K T, O'Connell C B, Wang Y, Vallee R B. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 27.Edwards M A, Yamamoto M, Caviness V S J. Neuroscience. 1990;36:121–144. doi: 10.1016/0306-4522(90)90356-9. [DOI] [PubMed] [Google Scholar]
- 28.Super H, Soriano E, Uylings H B. Brain Res Brain Res Rev. 1998;27:40–64. doi: 10.1016/s0165-0173(98)00005-8. [DOI] [PubMed] [Google Scholar]
- 29.Rakic P. Proc Natl Acad Sci USA. 1995;92:11323–11327. doi: 10.1073/pnas.92.25.11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunter-Schaedle K E. J Neurobiol. 1997;33:459–472. doi: 10.1002/(sici)1097-4695(199710)33:4<459::aid-neu9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Pilz D T, Matsumoto N, Minnerath S, Mills P, Gleeson J G, Allen K M, Walsh C A, Barkovich A J, Dobyns W B, Ledbetter D H, Ross M E. Hum Mol Genet. 1998;7:2029–2037. doi: 10.1093/hmg/7.13.2029. [DOI] [PubMed] [Google Scholar]
- 32.Hattori M, Adachi H, Tsujimoto M, Arai N, Inoue K. Nature (London) 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 33.Manya H, Aoki J, Kato H, Ishii J, Hino S, Arai H, Inoue K. J Biol Chem. 1999;274:31827–31832. doi: 10.1074/jbc.274.45.31827. [DOI] [PubMed] [Google Scholar]
- 34.Smith D S, Niethammer M, Ayala R, Zhou Y, Gambello M J, Wynshaw-Boris A, Tsai L H. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Steward R, Luo L. Nat Cell Biol. 2000;2:776–783. doi: 10.1038/35041011. [DOI] [PubMed] [Google Scholar]
- 36.Efimov V P, Morris N R. J Cell Biol. 2000;150:681–688. doi: 10.1083/jcb.150.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Y, Olson E C, Stukenberg P T, Flanagan L A, Kirschner M W, Walsh C A. Neuron. 2000;28:665–679. doi: 10.1016/s0896-6273(00)00145-8. [DOI] [PubMed] [Google Scholar]
- 38.Kitagawa M, Umezu M, Aoki J, Koizumi H, Arai H, Inoue K. FEBS Lett. 2000;479:57–62. doi: 10.1016/s0014-5793(00)01856-1. [DOI] [PubMed] [Google Scholar]
- 39.Niethammer M, Smith D S, Ayala R, Peng J, Ko J, Lee M-S, Morabito M, Tsai L-H. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 40.Sasaki S, Shionoya A, Ishida M, Gambello M, Yingling J, Wynshaw-Boris A, Hirotsune S. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- 41.Clark G D, McNeil R S, Bix G J, Swann J W. NeuroReport. 1995;6:2569–2575. doi: 10.1097/00001756-199512150-00029. [DOI] [PubMed] [Google Scholar]
- 42.Dobyns W B, Reiner O, Carrozzo R, Ledbetter D H. J Am Med Assoc. 1993;270:2838–2842. doi: 10.1001/jama.270.23.2838. [DOI] [PubMed] [Google Scholar]
- 43.Fogli A, Guerrini R, Moro F, Fernandez-Alvarez E, Livet M O, Renieri A, Cioni M, Pilz D T, Veggiotti P, Rossi E, et al. Ann Neurol. 1999;45:154–161. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}