

Abstract
Disulfiram has been used extensively for alcohol abuse and may have a role in treatment for cocaine addiction. Recent data suggest that disulfiram may also reactivate latent HIV in reservoirs. Disulfiram has complex pharmacokinetics with rapid metabolism to active metabolites, including S-Methyl-N, N-Diethylthiocarbamate (DET-Me) which is formed from cytochrome P450 (CYP450). Assessing disulfiram in HIV-infected individuals with a CYP450 inducing drug (e.g., efavirenz) or a CYP450 inhibiting drug (e.g., HIV-1 protease inhibitors) requires an assay that can measure a metabolite that is formed directly via CYP450 oxidation. Therefore, an assay to measure concentrations of DET-Me in human plasma was validated. DET-Me and the internal standard, S-ethyldipropylthiocarbamate (EPTC) were separated by isocratic ultra performance liquid chromatography using a Waters Acquity HSS T3 column (2.1×100mm, 1.8μm) and detection via electrospray coupled to a triple quadrupole mass spectrometer. Multiple reaction monitoring in positive mode was used with DET-Me at 148/100 and the internal standard at 190/128 with a linear range of 0.500 to 50.0 ng/mL with a five minute run time. Human plasma (500 μL) was extracted using a solid phase procedure. The interassay variation ranged from 1.86 to 7.74% while the intra assay variation ranged from 3.38 to 5.94% over three days. Representative results are provided from samples collected from subjects receiving daily doses of disulfiram 62.5 mg or 250 mg.
Keywords: Disulfiram, S-Methyl-N, N-Diethylthiocarbamate, Ultra Performance Liquid Chromatography, Mass Spectrometry
1. INTRODUCTION
Disulfiram has been used extensively for alcohol dependence [1]. More recently, data suggest that disulfiram may have a role in cocaine addiction [2, 3] and possibly for latent HIV infection [4–7]. Disulfiram has a complex pharmacokinetic profile with rapidly metabolism to active metabolites including S-Methyl-N, N-Diethylthiocarbamate (DET-Me) by enzymatic interaction with cytochrome P450 (CYP450). The use of disulfiram with a CYP450 inducing drug (e.g., efavirenz) or a CYP450 inhibiting drug (e.g., HIV-1 protease inhibitors) requires an assay that can measure a metabolite formed directly via CYP450 oxidation [1, 8–15] (Figure 1). It is important to determine the impact of these medications together on the cytochrome function in vivo. Although the carbamathione metabolite appears to be the active compound for cocaine addiction, DET-Me, which contributes to ALDH inhibition, is likely to be more directly influenced by CYP450-mediated drug interactions. Our current research includes the pharmacokinetics of DET-Me and possible drug interactions that may occur when antiretrovirals are prescribed. With this in mind a method was developed and validated to detect DET-Me in human heparinized plasma [16, 17]. This validation was based on prior guidelines [18, 19] and the FDA Guidance for Industry. The validation of DET-Me is described and concentration profiles in patients are provided.
Figure 1.
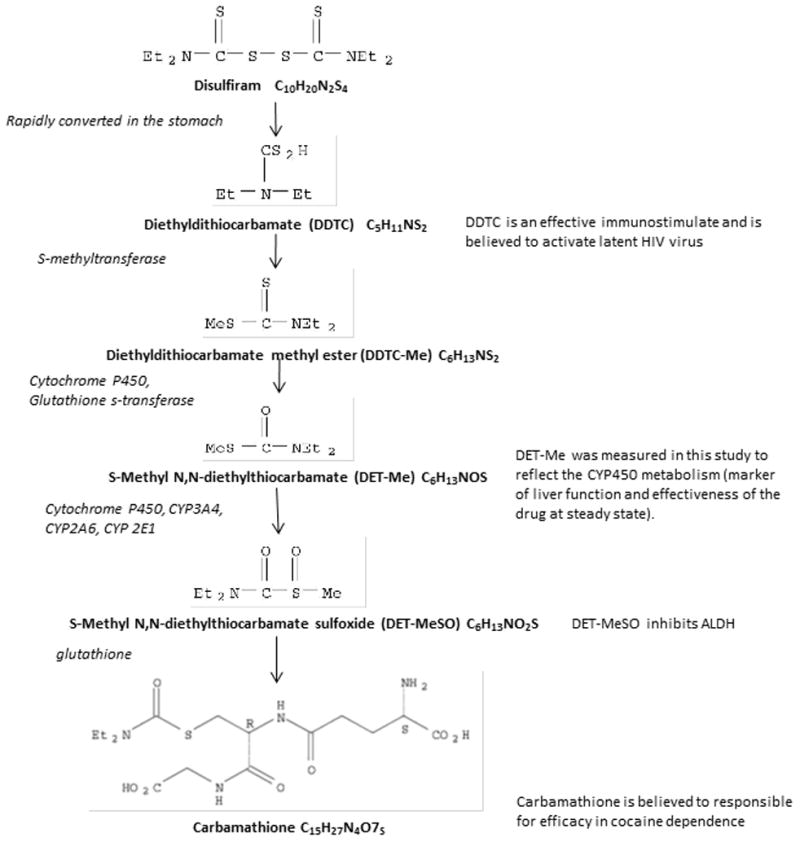
Metabolic pathway for disulfiram in humans
2. MATERIALS AND METHODS
2.1 Chemicals and Reagents
S-Methyl-N, N-Diethylthiocarbamate (DET-Me) was purchased from Toronto Research Chemicals (North York, Ontario) and the internal standard (IS), s-ethyl dipropylthiocarbamate (EPTC) from Fluka (St. Louis, MO). Using SciFinder, similar structural analytes were targeted as an internal standard. Based on commercial availability, safety information, chromatography and mass spectra, s-ethyl dipropylthiocarbamate was chosen. A deuterated source of DET-Me was not available from a commercial source. Liquid chromatography mass spectrometry (LCMS) grade methanol, optima grade formic acid, and LCMS grade water were obtained from Fisher Scientific (Fair Lawn, NJ). Heparinized human plasma was obtained from Biological Specialty Corporation (Colmar, PA).
2.2 Instrumentation
Chromatographic separation was initially developed and validated using HPLC comprised of an Agilent Series 1100 Autosampler and pump and an Applied Biosystems PE/Sciex API 3000 triple quadrupole detector. Separation was achieved using a Waters dC18 Atlantis column with run times of fifteen minutes. To improve on the efficiency and sensitivity, the separation was converted to UPLC. This separation system consisted of Waters (Milford, MA) Acquity UPLC binary solvent manager, UPLC module column manager, UPLC sample manager, and TQD triple quadrupole detector, controlled by Waters Empower 2 software version 6.20.00.00.
2.3 Chromatographic separation
Chromatographic separation was performed using a Waters HSS T3, 2.1 × 100mm, 1.8 μM analytical column preceded with a Waters HSS T3 guard column. The mobile phase consisted of 0.1% formic acid in water: 0.1% formic acid in methanol (22:78, volume/volume). The analysis used an isocratic flow rate at 0.200 mL/min for 5 minutes with a 10 μL sample injection. Detection was performed using electrospray coupled with multiple reaction monitoring (MRM) in positive mode. Mass spectrometry conditions were determined after optimization of DET-Me and the internal standard EPTC with the parameters set as follows: DET-Me monitored at a m/z of148/100 and the internal standard monitored at a m/z of 190/128, cone voltage and collision energy for DET-Me set at 20 and 12 with the cone voltage and collision energy for the internal standard set at 25 and 10;desolvation temperature was set at 350°C; desolvation flow was set a 600 L/hr with the flow path set to waste for the first 1.2 minutes of run and switched to the mass spectrometer to minimize potential contaminates to enter the analyzer. The column temperature was held constant at 30°C.
2.4 Calibration Standards and Quality Controls
Stock solutions were prepared in methanol with a final concentration 5 mg/mL correcting for purity. An intermediate 50 μg/mL solution of DET-Me was prepared in methanol. This 50 μg/mL solution was further diluted down to prepare nine calibration standards that resulted with concentrations when spiked in plasma to be 50, 40, 20, 15, 10, 5, 2.5, 1.25 and 0.5 ng/mL. All stocks and calibration standards were stored at −70°C in glass vials.
Quality control samples were prepared using separate stock solutions in human heparinized plasma. Aliquots were made in polypropylene tubes and stored at −70°C. Six replicates of each quality control (low [LQC] at 1.50 ng/mL, medium [MQC] at 17.5 ng/mL and high [HQC] at 35.0 ng/mL) were assayed each day to measure intra-assay variation over three days. Inter-assay variation was determined across the three days of validation.
2.5 Sample Preparation
For clinical samples, voluntary, written, informed consent was obtained. This study was approved by the University of California San Francisco Institutional Review Board to evaluate disulfiram (62.5 mg daily and 250 mg daily) in combination with two major classes of HIV therapeutics. Heparinized blood from healthy volunteers receiving either disulfiram 62.5 mg daily or 250 mg daily for 4 days were obtained at over 24 hours. Whole blood was centrifuged and 800 x g. Plasma was transferred into cryovials and stored at −70°C. Sample shipment was completed overnight on dry ice and stored at −70°C. Plasma calibration standards, quality controls and patient samples were prepared using a solid phase extraction procedure. After the addition of 20 μL of the working internal standard solution (100 ng/mL) to 500 μL of plasma, the sample was vortexed, centrifuged and passed over a preconditioned Waters Oasis 30mg HLB cartridge. The cartridges were washed with LCMS grade water and 1% methanol in LCMS grade water, respectively. Each sample was eluted with 500 μL of LCMS grade methanol. After the eluent was collected, 250 μL of LCMS grade water was added to the solution and 15 μL was injected onto the equilibrated separation system.
Calibration curves were linearly regressed using a weighting of 1/ (concentration)2. For each day ≥75% of the calibration points needed to be within 15% of their nominal value. Patient samples and quality controls were back calculated within the calibration range. Waters Empower2 version 6.20.00 (Milford, MA) was used for these calculations. After validation was completed, the method was used to analyze clinical samples.
2.6 Intra-assay and Inter-assay Variation and Accuracy
Intra-assay and inter-assay variation and accuracy were measured using quality controls prepared at three levels. All three levels were used in replicates of six on three separate days. In the same manner, the lower limit of quantitation (LLOQ) was also tested. Precision (coefficient of variation) and accuracy (% deviation from targeted concentration) were calculated at each quality control level. Intra-assay precision was expressed as relative standard deviation (CV%) between QC of the same value on each of the three days, resulting in a range of results. Inter-assay precision was calculated by defining the CV% over all days at each QC level. Acceptance criteria for precision was ≤ 20% for the LLOQ and ≤15% for the remaining quality controls and the acceptance criteria for accuracy needed to be ≤20% for the LLOQ and ≤15% for all other quality controls.
2.7 Matrix Effect
Matrix effect (ionization enhancement or suppression), process efficiency, and extraction recovery efficiency were tested for DET-Me and the internal standard. Testing was performed using equivalent amounts of DET-Me at concentration of the high, middle and low quality control samples in three different sources of human heparinized plasma obtained from an outside vendor. One set was prepared in an eluent (neat samples-no extraction), the second set was prepared in the plasma extracts after extraction (post-preparative) and the third set was prepared in the plasma spiked prior to extraction (pre-preparative). Precision, accuracy and recovery were calculated comparing the area ratio responses for the analyte to internal standard for all three sets over the different matrices. Specificity was tested by injecting DET-Me and monitoring for the internal standard transitions and injecting the internal standard and monitoring for DET-Me transitions. Specificity was also tested by injecting different lots of independent matrices prepared in plasma without the addition of DET-Me.
2.8 Stability Testing
Stability testing was conducted using quality control samples at the high and low concentrations. Samples were stored at −70°C for approximately two years (1 year and 11 months) prior to analysis and were tested against a freshly prepared calibration curve. Stability was also assessed during validation to include three freeze-thaw cycles and short term stability at room temperature for 5.5 hours.
Stability and accuracy was also tested on participant samples collected after disulfiram dosing. Samples were assayed upon receipt and re-analyzed after storage at −70°C for one year. The samples assayed were collected at different times after disulfiram dosing. This analysis was performed by two different analysts and with different valid lots of calibration standards and quality controls. The results from the original analysis and the re-analysis after one year of storage were assessed.
3. RESULTS AND DISCUSSION
3.1 Chromatography
Prior published assays either were performed with gas chromatography or HPLC separation with UV detection with unacceptable limits of quantitation, long run times and larger consumption of organic solvents, extractions of plasma samples with cumbersome liquid/liquid techniques and hazardous chemicals, and need of larger plasma volumes for analysis [1, 20–22]. To improve on these points, we developed an isocratic UPLC method with solid phase extraction. The UPLC HSS T3 column was used to help in retention of the small, polar analyte DET-Me. The run time for each injection was reduced to five minutes. Example chromatograms of the highest calibrator at 50.0 ng/mL with molecular weights and structures of DET-Me and the internal standard EPTC, a matrix blank sample and from a participant sample collected four hours post a 250 mg dose at 7.64 ng/mL are found in Figure 2. The retention times of DET-Me and the internal standard were 2.1 and 4.0 minutes, respectively.
Figure 2.
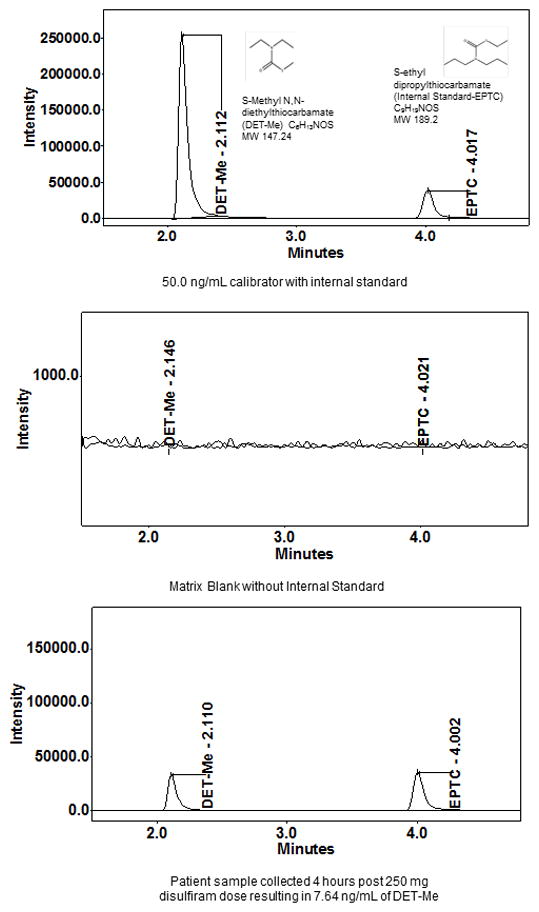
Example of chromatography
3.2 Calibration
The nine calibration standards ranged from 50.0 to 0.500 ng/mL. The lower limit of quantification (0.500 ng/mL) is well above 5 times the signal to noise of the assay. The variation of the calibrators was <4.50%. The regression coefficients (R2) values of the calibration curves were 0.983 or greater. The back calculated calibrator standards concentrations for the method were within ±15% of their target value except for a 2.50 ng/mL calibrator on day three of the validation. This point was ignored and the calibration curve was recalculated. The slopes ranged from 0.0587 to 0.0601 and the y intercepts ranges from 0.00530 to 0.00966 using the weighting of 1/ concentration 2.
3.3 Inter-Assay and Intra-Assay Precision, Accuracy, and Limit of Quantification
Table 1 represents the inter-assay and intra-assay data for each of the DET-Me quality controls across the three days of analysis. A negative bias in the accuracy (% deviation) of all of the quality controls could have been attributed to the preparation of these samples. The reference material, DET-Me purchased from Toronto Research Chemicals, is supplied as an oily material. Special attention in preparing the stock solutions used to prepare either the calibration standards or the quality controls (two separate solutions) is needed. Although there is a negative bias the accuracy (% deviation) the results were within the acceptance criteria of ±15%.
Table 1.
Intra-assay and Inter-assay Variation and Accuracy of S-Methyl-N, N-Diethylthiocarbamate
| Intra-Assay | Inter-Assay | |||||
|---|---|---|---|---|---|---|
| Target Concentration (ng/mL) | LQC | MQC | HQC | LQC | MQC | HQC |
| 1.5 | 17.5 | 35 | 1.5 | 17.5 | 35 | |
| n | 6 | 6 | 6 | 18 | 18 | 18 |
| Mean (ng/mL) | 1.29 to 1.33 | 15.6 to 16.0 | 30.9 to 31.9 | 1.31 | 15.8 | 31.5 |
| Standard Deviation | 0.0488 to 0.0768 | 0.295 to 0.685 | 1.23 to 2.47 | 0.0664 | 0.535 | 1.87 |
| % CV | 3.67 to 5.78 | 1.86 to 4.28 | 3.88 to 7.74 | 5.05 | 3.38 | 5.94 |
| % Deviation | −14.2 to −11.4 | −10.7 to −8.48 | −11.8 to −9.00 | −12.4 | −9.52 | −10.1 |
3.4 Matrix Effect, Selectivity and Specificity
A summary of the results by peak area ratio is shown in Table 2. The mean matrix effect (post preparative versus neat sample) was calculated to be 103% showing no enhancement or suppression. The mean extraction efficiency (pre preparative versus post preparative sample) was 97.2% and the mean process effect (pre-preparative versus neat sample) was 99.8% indicating that the separation system, detector, and the extraction procedure were measuring DET-Me and the internal standard without interference from the matrices and that the method is selective and sensitive.
Table 2.
LC/MS Matrix Effects, Recovery and Efficiency Testing by UPLC Overall Matrix Effects, Recovery and Efficiency
| LQC 1.50 ng/mL | MQC 17.5 ng/mL | HQC 35.0 ng/mL | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Neat | Post | Pre | Neat | Post | Pre | Neat | Post | Pre | |
| n | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| Mean* | 0.125 | 0.118 | 0.113 | 1.42 | 1.55 | 1.48 | 2.95 | 3.06 | 3.09 |
| SD | 0.00506 | 0.00796 | 0.00925 | 0.0425 | 0.0430 | 0.106 | 0.146 | 0.0989 | 0.344 |
| %CV | 4.06 | 6.72 | 8.21 | 3.00 | 2.77 | 7.18 | 4.95 | 3.23 | 11.1 |
| Matrix Effect (post/neat) | Recovery Effect (pre/post) | Process Effect (pre/neat) | |
|---|---|---|---|
| LQC (1.50 ng/mL) | 94.9% | 95.0% | 90.2% |
| MQC (17.5 ng/mL) | 109% | 95.6% | 104% |
| HQC (35.0 ng/mL) | 104% | 101% | 105% |
| Mean | 103% | 97.2% | 99.8% |
| SD | 7.22 | 3.27 | 8.30 |
| %CV | 7.04 | 3.36 | 8.32 |
Mean values = Area of DET-Me/IS response
3.5 Stability
DET-Me was stable in plasma stored at −70°C for approximately two years. The mean values of the long term stability samples (three of the high and low quality controls) were compared to their results from the first day of validation and showed a difference of <−8.2%. Patient samples results from the re-analysis after one year of storage at −70°C were compared to the original analysis (Table 3). These results show that the assay is stable, reliable and reproducible. Samples taken through three freeze/thaw cycles were proven stable with a deviation from the run’s mean quality control results ≤2.5% Short-term stability testing with samples stored at 5.5 hours at room temperature before extracted were stable with a deviation from the mean quality controls of <3.0%.
Table 3.
Re-analysis of Patient Samples after 1 Year of Storage
| DET-Me Results in ng/mL | |||
|---|---|---|---|
| Sample ID | Original Result (ng/mL) | Re-Assayed Result (ng/mL) after 1 year and 3 months | % Difference from Original Result |
| 1 | 0.590 | 0.565 | −4.24 |
| 2 | 4.54 | 4.85 | 6.89 |
| 3 | 5.47 | 5.38 | −1.61 |
| 4 | 7.90 | 8.58 | 8.65 |
| 5 | 2.72 | 2.85 | 4.74 |
| 6 | 1.92 | 1.98 | 3.33 |
| 7 | 1.53 | 1.38 | −9.61 |
| Mean | 1.17 | ||
3.6 Clinical Application
This assay has been utilized to analyze samples collected in clinical pharmacokinetics studies of disulfiram. DET-Me was measured in samples collected at 0, 1, 2, 4, 8, and 10 and in some patients at 12 and 24 hours after the fourth day dose of disulfiram 62.5 mg/d or disulfiram 250 mg/d (Figure 3).
Figure 3.
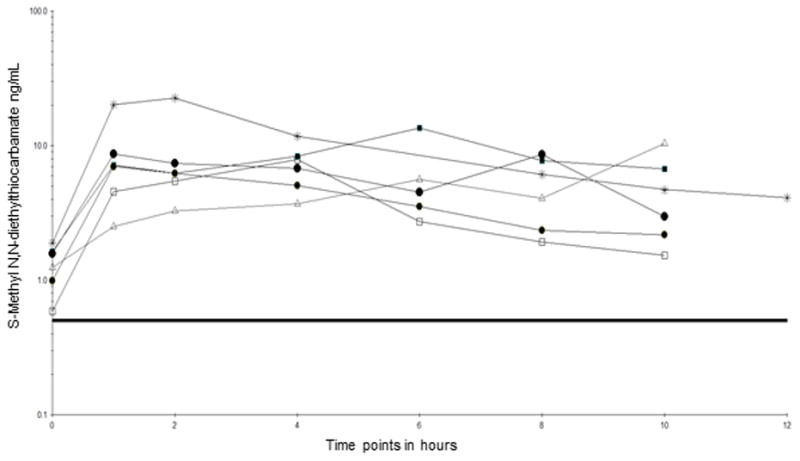
Pharmacokinetic profiles of S-methyl-N,N-diethylthiocarbamate on the fourth day following daily doses of oral 62.5 mg disulfiram
4. CONCLUSIONS
The validated UPLC method is rugged, precise, sensitive and accurate. Although the quality controls show a negative bias in accuracy in the reported inter and intra assay data, the evaluations of the patient samples after storage of one year that were assayed with freshly prepared calibration standards, show both a negative and positive bias. Stability was proven for two years using high and low quality controls that were stored at −70°C, taken through three freeze/thaw cycles and stored at room temperature for five and a half hours. The measurement of DET-Me was not affected by matrix interferences and the sample extraction was proven to be efficient, reproducible and easily performed. The analysis of clinical samples from subjects receiving daily disulfiram indicates that the assay was able to quantify concentrations over the 10–24 hours sampling period.
Highlights.
Disulfiram is rapidly metabolized to active metabolites via CYP450 oxidation.
Metabolites of disulfiram have different therapeutic applications in humans.
Interaction studies have been done with disulfiram and HIV medication.
Measurement of the disulfiram metabolite, S-Methyl-N, N-Diethylthiocarbamate was needed.
A UPLC MS method for this metabolite was developed and validated.
Acknowledgments
The technical support of the Translational Pharmacology Research Core, School of Pharmacy and Pharmaceutical Sciences at the University at Buffalo, is greatly appreciated. The project described was supported by NIDA/NIH RO1 DA 024982 (E. Mccance-Katz: PI) [20–22]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hart BW, Faiman MD. Biochem Pharmacol. 1993;46:2285–2290. doi: 10.1016/0006-2952(93)90619-8. [DOI] [PubMed] [Google Scholar]
- 2.Johansson B. Acta Psychiatr Scand Suppl. 1992;369:15–26. doi: 10.1111/j.1600-0447.1992.tb03310.x. [DOI] [PubMed] [Google Scholar]
- 3.Baker JR, Jatlow P, McCance-Katz EF. Drug and Alcohol Dependence. 2007;87:202–209. doi: 10.1016/j.drugalcdep.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCance-Katz EF, Kosten TR, Jatlow P. Drug and Alcohol Dependence. 1998;52:27–39. doi: 10.1016/s0376-8716(98)00050-7. [DOI] [PubMed] [Google Scholar]
- 5.Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, Bhat S, Zhang H, Margolick JB, Quinn TC, Margolis DM, Siliciano JD, Siliciano RF. J Virol. 2011;85:6060–6064. doi: 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliveto A, Poling J, Mancino MJ, Feldman Z, Cubells JF, Pruzinsky R, Gonsai K, Cargile C, Sofuoglu M, Chopra MP, Gonzalez-Haddad G, Carroll KM, Kosten TR. Drug Alcohol Depend. 2011;113:184–191. doi: 10.1016/j.drugalcdep.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pani PP, Trogu E, Vacca R, Amato L, Vecchi S, Davoli M. Cochrane Database Syst Rev. 2010:CD007024. doi: 10.1002/14651858.CD007024.pub2. [DOI] [PubMed] [Google Scholar]
- 8.Roache JD, Kahn R, Newton TF, Wallace CL, Murff WL, De La Garza R, 2nd, Rivera O, Anderson A, Mojsiak J, Elkashef A. Drug Alcohol Depend. 2011 doi: 10.1016/j.drugalcdep.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faiman MD, Artman L, Haya K. Alcohol Clin Exp Res. 1980;4:412–419. doi: 10.1111/j.1530-0277.1980.tb04841.x. [DOI] [PubMed] [Google Scholar]
- 10.Faiman MD, Dodd DE, Hanzlik RE. Res Commun Chem Pathol Pharmacol. 1978;21:543–567. [PubMed] [Google Scholar]
- 11.Faiman MD, Dodd DE, Minor SS, Hanzlik R. Alcohol Clin Exp Res. 1978;2:366–369. doi: 10.1111/j.1530-0277.1978.tb04745.x. [DOI] [PubMed] [Google Scholar]
- 12.Faiman MD, Dodd DE, Nolan RJ, Artman L, Hanzlik RE. Res Commun Chem Pathol Pharmacol. 1977;17:481–496. [PubMed] [Google Scholar]
- 13.Faiman MD, Jensen JC, Lacoursiere RB. Clin Pharmacol Ther. 1984;36:520–526. doi: 10.1038/clpt.1984.213. [DOI] [PubMed] [Google Scholar]
- 14.Heemskerk AA, van Haandel L, Woods JM, McCance-Katz EF, Williams TD, Stobaugh JF, Faiman MD. J Pharm Biomed Anal. 2011;54:799–806. doi: 10.1016/j.jpba.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaul S, Williams TD, Lunte CE, Faiman MD. J Pharm Biomed Anal. 2010;51:186–191. doi: 10.1016/j.jpba.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viswanathan CT, Bansal S, Booth B, DeStefano AJ, Rose MJ, Sailstad J, Shah VP, Skelly JP, Swann PG, Weiner R. Pharmaceutical Research. 2007;24:1962–1973. doi: 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- 17.DeStefano SBaA. AAPS Journal: Bioanalytical Method Validation and Implementation: Best Practices for Chromatographic and Ligand Binding Assays. 2007;9:6. doi: 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- 18.Shah KKM Vinod P, Findlay John WA, Hill Howard M, Hulse James D, McGilveray Iain J, McKay Gordon, Miller Krys J, Patnaik Rabindra N, Powell Mark L, Tonelli Alfred, Viswanathan CT, Yacobi Avraham. Pharmaceutical Research. 2000;17:7. doi: 10.1023/a:1007669411738. [DOI] [PubMed] [Google Scholar]
- 19.FDA, in, 2001.
- 20.Jensen JC, Faiman MD. J Chromatogr. 1980;181:407–416. doi: 10.1016/s0378-4347(00)81143-3. [DOI] [PubMed] [Google Scholar]
- 21.Johansson B. Clin Chim Acta. 1988;177:55–63. doi: 10.1016/0009-8981(88)90307-5. [DOI] [PubMed] [Google Scholar]
- 22.Hart BW, Faiman MD. Alcohol Clin Exp Res. 1994;18:340–345. doi: 10.1111/j.1530-0277.1994.tb00023.x. [DOI] [PubMed] [Google Scholar]