

Abstract
To clarify the role of mitochondrial electron transport chain (mtETC) in heavy-metal-induced neurotoxicity, we studied action of Cd2+, Hg2+, and Cu2+ on cell viability, intracellular reactive oxygen species formation, respiratory function, and mitochondrial membrane potential of rat cell line PC12. As found, the metals produced, although in a different way, dose- and time-dependent changes of all these parameters. Importantly, Cd2+ beginning from 10 [mu]M and already at short incubation time (3 h) significantly inhibited the FCCP-uncoupled cell respiration; besides, practically the complete inhibition of the respiration was reached after 3 h incubation with 50 [mu]M Hg2+ or 500 [mu]M Cd2+, whereas even after 48 h exposure with 500 [mu]M Cu2+, only a 50% inhibition of the respiration occurred. Against the Cd2+-induced cell injury, not only different antioxidants and mitochondrial permeability transition pore inhibitors were protective but also such mtETC effectors as FCCP and stigmatellin (complex III inhibitor). However, all mtETC effectors used did not protect against the Hg2+- or Cu2+-induced cell damage. Notably, stigmatellin was shown to be one of the strongest protectors against the Cd2+-induced cell damage, producing a 15–20% increase in the cell viability. The mechanisms of the mtETC involvement in the heavy-metal-induced mitochondrial membrane permeabilization and cell death are discussed.
1. Introduction
The highly toxic heavy metals, such as cadmium (Cd2+), mercury (Hg2+), and copper (Cu2+), are environmentally and occupationally widespread pollutants with mutagenic, carcinogenic, and teratogenic effects [1–4]. Being either nonessential (Cd, Hg) or biogenic (Cu) elements, these metals belong to the most harmful factors due to their tendency to accumulate in tissues and organs and to transfer along food chains, high reactivity (in particular, high affinity to thiol groups), and the ability to stimulate reactive oxygen species (ROS) formation and to produce injury in cell functions. In addition, a characteristic feature of Cd2+ is its ability to act not only as a dithiol reagent but also as a Ca2+ agonist most probably due to the extreme closeness of crystal ionic radii of the ions (Cd2+—0.097 nm; Ca2+—0.099 nm) [5].
Although it is well known now that mitochondria are important targets for heavy metals; nevertheless, mechanism(s) of the disturbance of mitochondrial function by heavy metals are not well understood. Not long ago, on rat hepatoma AS-30D cells we have demonstrated that the intracellular ROS changes and mitochondrial dysfunction are involved in cytotoxicity mechanism(s) of such heavy metals as Cd2+, Hg2+, and Cu2+ [6, 7]. Moreover, we have found [6] that Cd2+-induced AS-30D cell death is accompanied by stimulation of ROS production at the mitochondrial respiratory chain complex III level and opening of the mitochondrial permeability transition (MPT) pore (for reviews see [8–11]).
The MPT pore can be defined as a voltage-dependent, nonselective high-conductance inner mitochondrial membrane channel of unknown molecular structure, which allows solutes up to 1500 Da to pass freely in and out of mitochondria. The MPT pore opens under conditions of calcium overload; the opening is greatly enhanced by adenine nucleotide depletion, elevated phosphate, and oxidative stress. The opening of the MPT pore in vivo produces ATP pool exhaustion, disturbance of Ca2+ homeostasis, and efflux of various apoptotic factors from mitochondria [8, 9]. As accepted by the most, the MPT pore represents by itself a transmembrane multiprotein complex located at contact sites between two mitochondrial membranes; the minimum MPT pore configuration likely consists of the matrix protein cyclophilin-D (CyP-D), the potent inhibitor of which is cyclosporin A (CsA), of the adenine nucleotide translocase (ANT) and/or phosphate carrier (PiC) (inner membrane proteins), and the voltage-dependent anion channel or porin (outer membrane protein); however, the involvement of the latter is under doubts at the moment [10, 11]. As considered previously, the ANT represents a crucial core element of the MPT pore; however, the latest evidence indicates that the ANT usually plays a regulatory role rather than provides the transmembrane pore component and point to the PiC as the protein, fulfilling the latter role. The data obtained by Halestrap's group during the last years are consistent with a model of the MPT pore, in which a calcium-triggered conformational change of the PiC, facilitated by CyP-D, induces the pore opening that is enhanced by an association of the PiC with the “c” conformation of the ANT. An interaction of the PiC with the ANT may enable agents that bind to either transporter to modulate the pore opening [10]. The MPT pore opening is inhibited by CsA and different ANT inhibitors, in particular, bongkrekic acid (BKA). The pore opening is strongly promoted by the oxidized state of pyridine nucleotides (PNs) and of critical dithiols, at least, at two discrete redox-sensitive sites, P- and S-site(s), respectively; their localization is still unknown; however, the existence of critical internal and external dithiols is suspected [9, 11, 12]. The MPT pore is characterized by pH- and voltage-dependence, namely, it is closed at acidic pH (intracellular pH below 7.0) and at high transmembrane potential values. The MPT pore function is modulated by electron flux via respiratory complex I, by different ubiquinone analogs, and various regulatory proteins [9]. As known, Ca2+ is a critical factor for opening of the “classical” CsA-sensitive MPT pores. The most of other bivalent metal ions behave as the pore inhibitors, and this is true irrespective of whether cations are accumulated (like Sr2+, Mn2+, and Ba2+) or not (like Mg2+). The experimental data point to the existence of two separate Me2+-binding sites on the MPT pore complex-external (inhibitory) and internal (activating); however, their molecular localization is uncertain up-to-date. H+ is considered to compete with Ca2+ on the Ca2+-trigger site as well, and histidine nature of this site is suspected [8–12]. There are some interesting suppositions in literature in the field about the Me2+-binding sites localization. In particular, it was proposed that respiratory complex I [9], a cardiolipin core of the ANT [13], or its matrix loops [14], could be the hypothetical places of their localization. We also suggested that the respiratory complex III may contain Me2+(Cd2+)-binding loci, crucial for the MPT pore modulation [15]. Further, we hypothesized about possible involvement of the mitochondrial respiratory chain supercomplex, formed by complex I (P-site) and complex III (S-site), in the mitochondrial membrane permeabilization mediated by the MPT pore ([16–18], for details, see Section 4).
The present study was conducted to underscore the role of mitochondria and mitochondrial electron transport chain (mtETC) in heavy metal-induced neurotoxicity, using neuron-like rat PC12 cells and the divalent heavy metal ions as a model system. Portions of this investigation were presented before in an abstract form and as a part of a published lecture [19–21].
2. Materials and Methods
2.1. Chemicals
The most of reagents was purchased from Sigma Aldrich Company (St. Luis, MO, USA). Cyclosporin A was from Novartis (Basel, Switzerland). The rest of the chemicals was of the highest purity, commercially available. The DMEM incubation medium (Dulbecco/Vogt modified Eagle's minimal essential medium) with L-glutamine, horse blood serum, fetal calf serum, and trypsin-EDTA were purchased in Biolot Company (Russia).
2.2. Cell Culture
The experiments were made on cultures of PC12 cells in CO2 incubator in the atmosphere containing 5% of CO2 at 37°C as before [22]. As the incubation medium DMEM with L-glutamine was used, containing 25 μg/mL of penicillin and 25 U/mL of streptomycin, 5% of horse blood serum and 10% of fetal calf serum. The incubation medium was changed every 2-3 days. In some experiments, the incubation medium was DMEM (with L-glutamine and antibiotics) not containing serum. PC12 cells were seeded to 24 well plates in concentration 2.5 × 105 cells in each well to make assays of lactate dehydrogenase (LDH) release or TMRM fluorescence (see below). The experiments started 24 h after the transfer of the cells to the plates or Petri dishes. The preincubation of PC12 cells with different effectors or without them was performed for 30 min at 37°C prior to the exposure of the cells to the heavy metals. Then, to each well or Petri dish, the corresponding concentration of Cd2+, Hg2+ or Cu2+ was added, in particular 10, 50, 100, or 500 μM. CdCl2, HgCl2, or CuCl2 was dissolved in water (10 mM stock solution).
2.3. Cell Viability Assay
The viability of the cells and protective action of different antioxidants, inhibitors of the mitochondrial permeability transition, and the respiratory chain against the heavy-metal-induced cytotoxicity was estimated by spectrophotometric monitoring of the LDH cellular release [22]. The samples were centrifuged for 5 min at 200–300 g to pellet down cells and supernatants collected. The lysis of the cells was performed by using 1% Triton X-100 at room temperature, and then the total activity of LDH was determined in the samples. In particular, the LDH activity was determined measuring NADH level for this purpose. The decrease in optical density of the samples at 340 nm was registered during 5-6 min as previously described [23], using the spectrophotometer M40 (Karl-Zeisse, Germany). The reaction was performed in the medium composed of (mM): 80 Tris-HCl (pH 7.2), 200 NaCl, 1.6 pyruvate, 0.2 NADH. The percent of LDH activity released was determined as the percent of enzyme activity in the incubation medium to the total LDH activity in the sample. The absence of the viable cells corresponds to 100% of LDH activity in the incubation medium. The cell viability was expressed in % of the untreated control.
2.4. Cell Respiration Assay
The cell respiration was determined polarographically with the help of Clark oxygen electrode in a thermostatic water-jacketed vessel with magnetic stirring at 37°C. PC12 cells (107 cells) were incubated in 10 mL of the complete DMEM medium (with serum) in Petri dishes for 3, 5, 24, and 48 h with various concentrations of the corresponding heavy metal, then collected by centrifugation and transferred to the DMEM medium without serum. Under these conditions, oxygen uptake by the cells was used to sustain a steady-state level of mitochondrial membrane potential (ΔΨmito) by compensating for the proton leak and ATP synthesis by mitochondrial ATP synthase (F0F1-ATPase). This was designated as the “steady-state” or basal respiration. Addition of the inhibitor of F0F1-ATPase, oligomycin, typically decreased cell respiration, leaving only that portion of oxygen uptake, which compensated for the proton leak (“resting respiration”, state 4o). A subsequent addition of the chemical protonophore FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) resulted in the maximal rate of respiration, limited only by capacity of the respiratory chain (“uncoupled respiration”, state 3u) [24]. Such protocol enables us to follow the effect of poisoning by heavy metal ions on distinct steps of the mitochondrial energy-coupling process and to estimate cell respiratory control ratios (RCRs) and some additional parameters (coupling efficiency, spare respiratory capacity) that are rich in information about mitochondrial function and dysfunction in intact cell [25].
2.5. ROS Production Assay
The intracellular ROS production was measured spectrofluorometrically using the fluorescent dye dichlorodihydrofluorescein diacetate (DCFH2-DA) as the ROS-sensitive probe [26, 27]. PC12 cells were seeded to 6 well plates to determine ROS formation and accumulation (1 × 106 of cells in a well). The experiments started 24 h after the transfer of the cells to the plates, they were performed in DMEM containing L-glutamine and this medium will be described as DMEM. The incubation with inhibitors and heavy metals was made as previously described. 20 min before the end of each incubation, DCFH2-DA was added to the incubation medium in final concentration of 10 μM. In order to get rid of the dye excess, the cells were washed by Hanks' balanced salt solution. The fluorescence of the reaction product of ROS with dichlorodihydrofluorescein was estimated at the spectrofluoremeter Schimadzu 1501 (Japan), measuring the emission at λ = 522 nm, the length of the excitation wave being equal to 475 nm [26]. The ROS content was measured in arbitrary units reflecting the intensity of the fluorescence of reaction product. In some cases, DCF fluorescence was measured also by Fluoroscan FL (ThermoFisher) using Em = 538 nm, Ex = 485 nm.
2.6. ΔΨmito Changes Assay
ΔΨmito was determined with the aid of a cationic fluorescent probe, TMRM (tetramethylrhodamine methyl ester) [28]. All incubations were conducted in the DMEM medium as described before (see above). Then, PC12 cells were suspended in Hanks' balanced salt solution (106 of cells in mL) and were loaded with TMRM (200 nM) for 30 min at 37°C in the dark. Afterwards, the cells were centrifuged for 3 min at 60 g and resuspended in PBS (phosphate buffered saline) medium. The samples were analyzed by flow cytometry. A flow cytometer EPIXC-XL (Beckman Coulter, USA) was used equipped with an argon laser emitting at 488 nm. Orange fluorescence caused by TMRM was detected in FL2 575 ± 15 nm. Approximately, 20,000 events per sample were acquired for the analysis performed. The fluorescence intensity of TMRM (reflecting ΔΨmito) was monitored on a log scale and expressed as mean fluorescence intensity, MnI (in arbitrary units). The flow cytometric data were analyzed with WinMDI 2.9 (Joseph Trotter, Scripps Research Institute, La Jolla, CA, USA). The fluorescence of samples in the presence of inhibitors is expressed as the percentage of controls (without inhibitors).
2.7. Statistics
Data are expressed as mean values ± SE for at least three independent experiments, unless otherwise indicated. The statistics were analyzed with ANOVA and Student's t-tests, with P < 0.05 assumed as the significance threshold.
3. Results
To understand molecular mechanism(s) of heavy-metal-induced cell dysfunction and the role of mitochondria in neurotoxic action of such heavy metals, as Cd2+, Hg2+, and Cu2+, we investigated the effects of these divalent metal ions on cell viability, intracellular ROS formation, ΔΨmito, and respiratory function of rat neuron-like PC12 cells. The cell viability was estimated by LDH release from the cells, the respiration rates were measured polarographically, the ROS production—spectrofluorometrically using DCFH2-DA, and the membrane potential—with the help of flow cytometry and a fluorescent probe TMRM.
We found that already after 3 h of incubation of the cells with 50 μM Hg2+, the cell viability decreased on 25%; the same [Cd2+] and its twofold increase were not effective, whereas 500 μM Cd2+ produced close to the 50% decline of the cell viability at that time (Figure 1). 100 μM Cd2+ reduced the cell viability on 25% only after 5 h treatment (Tables 4 and 5). It is important to say that the highest [Cu2+] used (i.e., 500 μM) became effective only after 48 h of incubation of the cells with the metal; however, even in this case, the cell viability reduced not more than 10% (not shown data).
Figure 1.
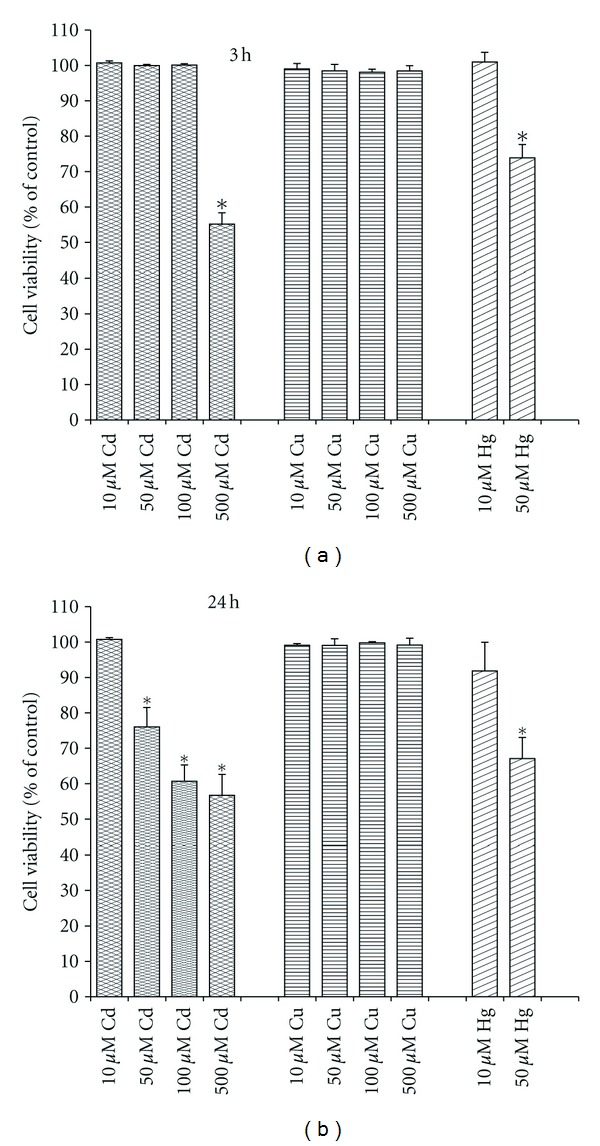
Time- and dose-dependent response to Cd2+, Cu2+, and Hg2+ on PC12 cell viability assessed by LDH release. The results are expressed in % to corresponding control (*P < 0.05 compared to untreated control; n = 4–9 independent experiments).
Table 4.
Action of different respiratory chain effectors on viability of PC12 cell assessed by LDH release after different time of incubation with different concentration of Cd2+.
| Treatment (1 μM) | Control cells (3, 5, 24 h) | 500 μM Cd2+ (3 h) | 100 μM Cd2+ (5 h) | 100 μM Cd2+ (24 h) |
|---|---|---|---|---|
| Control | 100 | 55 ± 3* | 74 ± 4* | 61 ± 5* |
| FCCP | 96 ± 2 | 62 ± 10 | 89 ± 4** | 75 ± 10 |
| Rotenone | 94 ± 1 | 59 ± 10 | 84 ± 8 | 73 ± 7 |
| Myxothiazol | 95 ± 1 | 52 ± 16 | 85 ± 7 | 71 ± 5 |
| Antimycin A | 96 ± 2 | 48 ± 14 | 82 ± 7 | 72 ± 6 |
| Stigmatellin | 99 ± 2 | 78 ± 5** | 90 ± 5** | 76 ± 1** |
The cell viability is expressed in % to untreated control. Mean ± SE; n = 3–11 independent experiments; *P < 0.05 compared to untreated control; **P < 0.05 compared to Cd(II)-treated control.
Table 5.
Action of different antioxidants and MPT pore inhibitors on viability of PC12 cells assessed by LDH release after different time of incubation with different concentration of Cd2+.
| Treatment | Control cells (3, 5, 24 h) | 500 μM Cd (3 h) | 100 μM Cd (5 h) | 100 μM Cd (24 h) |
|---|---|---|---|---|
| Control | 100 | 55 ± 3* | 74 ± 4* | 61 ± 5* |
| Ruthenium Red (10 μM) | 99 ± 3 | 71 ± 6** | 92 ± 3** | 60 ± 4 |
| BKA, 5 μM | 100 ± 2 | 70 ± 5** | 82 ± 6 | 58 ± 2 |
| CsA, 1 μM | 101 ± 2 | 76 ± 4** | 72 ± 6 | 59 ± 18 |
| NAC (10 mM, 2 h) | 104 ± 4 | 94 ± 2** | 96 ± 2** | 92 ± 5** |
| Vit E, 500 μM | 103 ± 1 | 65 ± 6** | 86 ± 7** | 83 ± 2** |
| Mannitol (50 mM) | 100 ± 1 | 92 ± 6** | ||
| BHT, 150 μM | 99 ± 2 | 93 ± 1** | 82 ± 1** | |
| TEMPO (1 mM) | 97 ± 3 | 87 ± 8** |
The cell viability is expressed in % to untreated control. Mean ± SE; n = 3–11 independent experiments; *P < 0.05 compared to untreated control; **P < 0.05 compared to Cd(II)-treated control.
All heavy metal ions under study produced, although in a different way, dose- and time-dependent changes in intracellular ROS generation of PC12 cells. In particular, 100 and 500 μM of Cd2+ induced a significant increase in the ROS formation after 30 min of incubation with the cells, whereas after 3 h already all Cd2+ concentrations under test stimulated strongly the intracellular ROS production (Figure 2). It is seen also that after 24 h of incubation of the cells with the metal, the ROS level in the presence of 10 μM Cd2+ returned to baseline. At the same time, 50–500 μM Cd2+ decreased significantly the ROS production compared to control level. As to Hg2+, already during short incubation time (30 min) all concentrations of the metal studied enhanced the intracellular ROS generation (Figure 2). Moreover, again, like in the case of Cd2+, after 24 h incubation of the cells in the presence of 10 μM of Hg2+, the ROS production level reached the baseline whereas 50–500 μM Hg2+ significantly decreased the ROS production compared with the control. In the case of Cu2+, there were no significant changes in the ROS production after 30 min of incubation while already after 3 h all Cu2+ concentrations under test induced the significant stimulation of the ROS formation (Figure 2). Notably, the ROS generation level observed in the presence of 500 μM Cu2+ was high even after 24 h of incubation of the cells with the metal.
Figure 2.
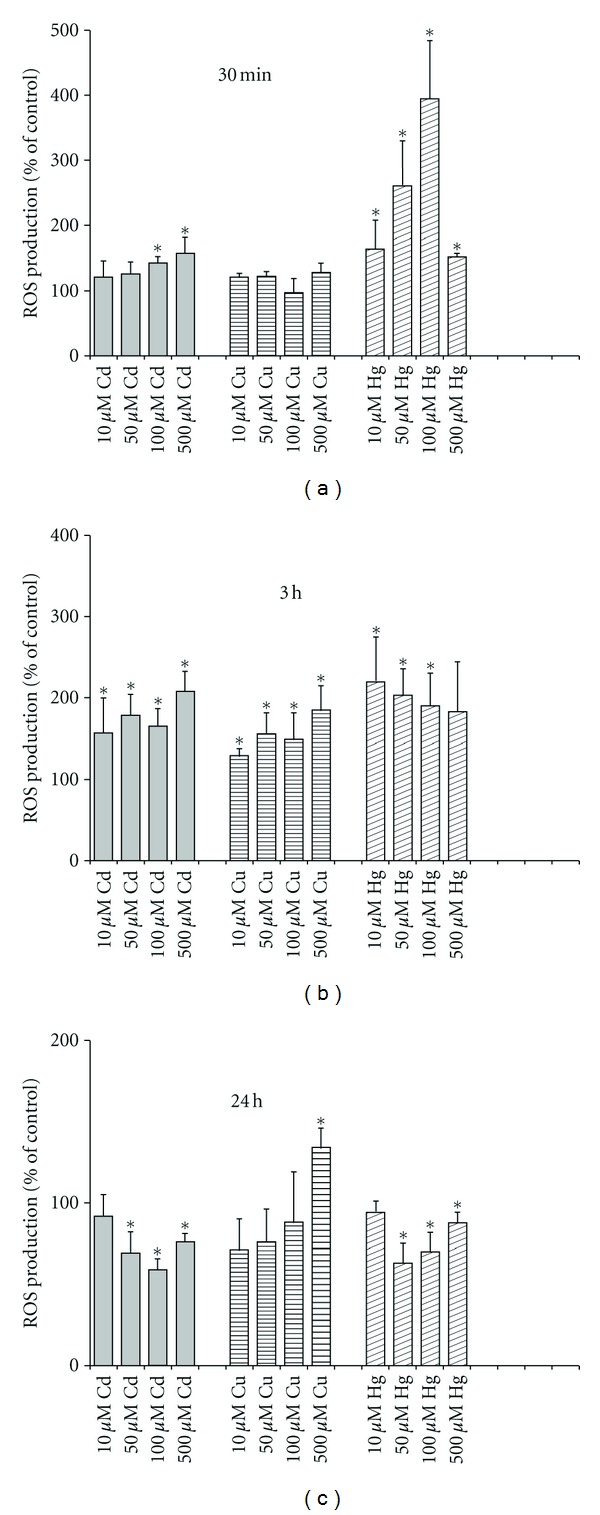
Time- and dose-dependent effects of Cd2+, Cu2+, and Hg2+ on ROS formation by PC12 cells measured spectrofluorometrically using DCFH2-DA as the ROS-sensitive probe. The results are expressed in % to corresponding control (*P < 0.05 compared to untreated control; n = 3–7 independent experiments).
To underscore the involvement of mitochondria in the harmful effects of the metals, we studied their influence on ΔΨmito of PC12 cells. As seen from Figure 3, 50 μM Cd2+ reduced the ΔΨmito more than 25% after 3 h of exposure with the cells, and after 24 h of treatment the ΔΨmito loss reached to 50%. Notably, during 24 h of treatment, 50 μM Cu2+ did not produce any significant action on the ΔΨmito whereas 50 μM Hg2+ evoked practically the complete ΔΨmito dissipation at this time (Figure 3).
Figure 3.
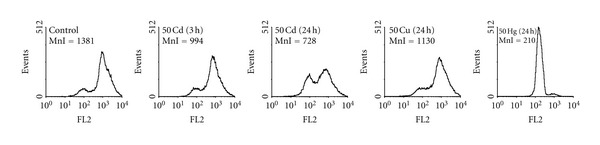
Effects of Cd2+, Cu2+, and Hg2+ on mitochondrial membrane potential of PC12 cells monitored by flow cytometry after staining the cells with the lipophilic cationic probe TMRM. MnI—mean intensity of fluorescence (arbitrary units) is indicated in the upper left corner of each panel. A typical experiment out of at least three independent ones for each metal is shown. For other details, see Section 2.
For assessing mitochondrial dysfunction in cells (see [25]) and the underlying mechanisms, we examined action of the metals on respiration of PC12 cells. With this purpose, we measured the rates of the basal respiration (Table 1), of the resting respiration (Table 2), and of the uncoupled respiration (Table 3) in the absence and in the presence of different concentrations of the heavy metals and after different time of exposure, namely, 3, 5, 24, and 48 h.
Table 1.
Basal respiration rates (%) of PC12 cells after different time of incubation with increasing concentration of Hg2+, Cd2+, or Cu2+.
| Time (h) | Control | [Hg2+], μM | [Cd2+], μM | [Cu2+], μM | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 50 | 10 | 50 | 100 | 500 | 10 | 50 | 100 | 500 | ||
| 3 | 45 ± 4 | 26 ± 5* | 3 ± 3* | 41 ± 7 | 25 ± 3* | 16 ± 4* | 2 ± 2* | 43 ± 7 | 35 ± 7 | 35 ± 5 | 35 ± 5 |
| 5 | 35 ± 6 | 6 ± 3* | |||||||||
| 24 | 39 ± 7 | 30 ± 2 | 2 ± 2* | 32 ± 2 | 13 ± 6* | 7 ± 1* | 0 | 33 ± 5 | 32 ± 2 | 34 ± 5 | 22 ± 8* |
|
| |||||||||||
| 48 | 41 ± 5 | 74 ± 26* | 15 ± 3* | ||||||||
Rate of oxygen uptake is presented in percentage of the maximal (fully uncoupled) respiration of the control cells. Mean ± SE; n = 3 independent experiments; *P < 0.05 compared to untreated control.
Table 2.
Resting respiration rates (%) of PC12 cells after different time of incubation with increasing concentration of Hg2+, Cd2+, or Cu2+.
| Time (h) | Control | [Hg2+], μM | [Cd2+], μM | [Cu2+], μM | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 50 | 10 | 50 | 100 | 500 | 10 | 50 | 100 | 500 | ||
| 3 | 13 ± 3 | 9 ± 1 | 3 ± 3* | 12 ± 3 | 10 ± 1 | 5 ± 1* | 1 ± 1* | 15 ± 3 | 12 ± 2 | 14 ± 3 | 14 ± 1 |
| 5 | 17 ± 2 | 4 ± 3* | |||||||||
| 24 | 14 ± 2 | 12 ± 3 | 2 ± 2* | 10 ± 2 | 5 ± 1* | 4 ± 2* | 0* | 12 ± 1 | 16 ± 2 | 13 ± 1 | 9 ± 3 |
|
| |||||||||||
| 48 | 21 ± 2 | 49 ± 17* | 12 ± 2* | ||||||||
Rate of oxygen uptake is presented in percentage of the maximal (fully uncoupled) respiration of the control cells. Mean ± SE; n = 3 independent experiments; *P < 0.05 compared to untreated control.
Table 3.
Uncoupled respiration rates (%) of PC12 cells after different time of incubation with increasing concentration of Hg2+, Cd2+, or Cu2+.
| Time (h) | [Hg2+], μM | [Cd2+], μM | [Cu2+], μM | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 50 | 10 | 50 | 100 | 500 | 10 | 50 | 100 | ||
| 3 | 66 ± 11* | 15 ± 3* | 77 ± 2* | 48 ± 3* | 35 ± 3* | 4 ± 4* | 85 ± 20 | 81 ± 14 | 83 ± 14 | 77 ± 16 |
| 5 | 21 ± 3* | |||||||||
| 24 | 86 ± 7 | 2 ± 2* | 91 ± 13 | 14 ± 5* | 5 ± 2* | 0 | 94 ± 8 | 82 ± 7 | 72 ± 6* | 58 ± 17* |
|
| ||||||||||
| 48 | 105 ± 5 | 48 ± 5* | ||||||||
Rate of oxygen uptake is presented in percentage of the maximal (fully uncoupled) respiration of the control cells. Mean ± SE; n = 3 independent experiments; *P < 0.05 compared to untreated control.
It should be remind that the participation of the mtETC could be checked by estimation of the uncoupled respiration rate, the inhibition of which is considered a marker of the disturbance of the respiratory chain components. Cd2+, beginning from 10 μM and already at short incubation time (3 h), produced significant inhibition of FCCP-uncoupled respiration of the cells (Table 3). The complete inhibition of the mtETC was reached after 3 h of treatment with 50 μM Hg2+ or 500 μM Cd2+ and after 24 h of treatment with 100 μM Cd2+, whereas even after 48 h of incubation of the cells with 500 μM Cu2+, only 50% inhibition of the maximum respiration rate took place. In opposite, the restoration of the uncoupled respiration rate up to the control level occurred in the presence of 10 μM Cd2+ after 24 h of incubation or in the presence of 10 μM Hg2+ after 48 h of incubation (Table 3). As to Cu2+, only at high concentrations (100 and 500 μM) and starting from 24 h of treatment, the significant changes in the cellular respiration occurred (Tables 1–3). Notably, at 500 μM Cu2+ and after 48 h of incubation, all three values (i.e., the steady-state respiration, the resting respiration, and the uncoupled respiration) were strongly depressed, indicating a potent inhibitory effect on the respiratory chain.
It is known also that the cellular respiratory rate in the presence of oligomycin (the resting respiration rate) is a direct measure of the proton leak across the mitochondrial membrane in situ [25]. As seen from Table 2, after 48 h incubation of the cells with 10 μM Hg2+, the resting respiration was stimulated significantly, whereas the uncoupled respiration remained unaffected (Table 3), pointing to some uncoupling effect of that low concentration of the metal. At the same time, the basal respiration in the presence of 10 μM Hg2+ was significantly enhanced as well (Table 1). So, after simple calculations, it becomes evident that low Hg2+ substantially reduced the coupling efficiency of the cells whilst did not affect the spare respiratory capacity of the cells (see Section 2 and [25]).
Further, we studied the action of different effectors of the respiratory chain on the viability of PC12 cells in the presence of the heavy metals which was determined by the LDH release from the cells as before (Table 4). We used two concentrations (0.1 and 1 μM) of FCCP (an artificial uncoupler) and of several selective inhibitors of the mtETC components, namely, of complex I (rotenone) and complex III (myxothiazol, antimycin A, and stigmatellin). Among the mtETC effectors under test, only stigmatellin (1 μM) produced the strong and sustained neuroprotection, increasing the cell viability on 15–20% (Table 4). In the case of Hg2+ (50 μM; 3 h) and Cu2+ (500 μM; 48 h), all mtETC effectors under study did not exhibit significant protective action on the cell survival (data not shown). All this means that the mtETC dysfunction is involved (however, in a different way) in mechanism(s) of neurotoxic action of the heavy metals.
Finally, we investigated action of different antioxidants and inhibitors of the MPT pore on the Cd2+-induced ROS-dependent injury of PC12 cells. We found that all antioxidants under study, namely, N-acetylcysteine (NAC), vitamin E (vit E), butylhydroxytoluene (BHT), 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), and mannitol were effective, however, in a different extent, in protection of the cells against the Cd2+ toxic action, with maximal neuroprotective effect observed after 2 h preincubation of the cells with 10 mM NAC (Table 5). The latter treatment was found to be highly protective against the Hg2+-induced cytotoxicity as well (not shown data). Besides, we revealed that Ruthenium Red, RR, or Ru-360 (selective inhibitors of mitochondrial Ca2+ uniporter, MCU) both taken in concentration of 10 μM were protective only at short times of incubation of the cells with Cd2+ (3 h—for 500 μM Cd2+ and 5 h—for 100 μM Cd2+), increasing the cell viability on 16% and 18%, respectively. By 24 h, both 10 μM and 50 μM of RR did not protect against the 100 μM Cd2+-induced cell death. As seen from Table 5, CsA (a potent pharmacological inhibitor of the MPT pore) also produced only the transient protection against the Cd2+-induced cytotoxicity; in particular, after 3 h, the cell viability in the presence of CsA (1 μM) rose about 20%, whereas at the longer incubations of the cells with Cd2+, it lost the protective action. The same was true for another MPT pore inhibitor—BKA (selective inhibitor of ANT). Both 5 μM and 25 μM of BKA were effective only after 3 h incubation of the cells with Cd2+, increasing the viability approximately on 15%. It is necessary to say that all used concentrations of the effectors under test per se were not cytotoxic under used conditions.
We obtained also that Ru-360 (10 μM), vit E (500 μM), FCCP (1 μM) did not change significantly the ROS formation increase induced by 100 μM Cd2+ after 30 min incubation with the cells, whereas CsA (1 μM) and BKA (7 μM) significantly decreased the ROS production in the presence of Cd2+. However, in the case of 3 h of incubation with Cd2+, BKA lost its protective effect, while vit E, in opposite, became effective. As to FCCP, after 3 h exposure of the cells with 100 μM Cd2+, this protonophoric uncoupler reduced in an equal degree (about 30%) both the ROS production of the Cd2+-treated and the control cells. In turn, stigmatellin (1 μM) did not affect significantly the ROS generation stimulated by 30 min incubation of the cells with 100 μM Cd2+, whereas it reduced on 12% the ROS production enhanced by 3 h incubation of the cells with the same [Cd2+]. This small but significant effect of stigmatellin was observed despite that this concentration of the complex III inhibitor per se stimulated the ROS production of the control cells, upon the average, on 25%.
4. Discussion
The work was made with the aim to elucidate the role of mitochondrial respiratory chain in the heavy-metal-induced neurotoxicity. We studied action of Cd2+, Hg2+, and Cu2+ on cell viability (Figure 1; Tables 4 and 5), respiratory function (Tables 1–3), intracellular ROS generation (Figure 2), and ΔΨmito (Figure 3) of rat neuron-like PC12 cells. As found, the metals produced, however, differently, dose- and time-dependent changes in all these parameters, with Hg2+ being the most neurotoxic.
It is known that the measurement of cell respiratory control is the single most useful general test of mitochondrial function in cell populations [25]. The ratio of the uncoupled rate (state 3u) to the rate with oligomycin present (state 4o) is analogues to the (uncoupled) respiratory control ratio of isolated mitochondria. It is sensitive to changes in substrate oxidation and proton leak (not to ATP turnover) and is a reliable marker of mitochondrial dysfunctions in cells. Using data from Tables 2 and 3, it is possible to estimate the apparent respiratory control ratios of the cells (state 3u/state 4o) in the presence of the heavy metals and to see the dynamics of their changes. As evident, the RCR changed crucially after treatment of the cells with the heavy metals. In addition, under used conditions, the dissipation of ΔΨmito was observed in the presence of Cd2+ and Hg2+ (Figure 3). All this means that the mitochondrial dysfunction participates in mechanisms of the cell dysfunction produced by the heavy metals. Besides, the data on the ROS production (Figure 2) and the cell viability (Figure 1) confirmed our previous suggestion [7] that the increased ROS level alone was not sufficient to induce the cell death by heavy metals and additional factor(s) must have been present that was/were responsible for their cytotoxic action, most likely the blockage of the respiratory chain (see Table 3). The results found in this study are in a good accordance with data shown before on PC12 cells [29–40] and on other types of cells, which obtained by us and other authors ([41–47]; see also [6, 7] and references therein).
As known, the ability of Cd2+ to act not only as SH reagent but also as a Ca2+ agonist makes it an excellent tool to study the MPT, with the aim to solve the problem of the proposed direct participation of the respiratory chain components in this phenomenon and in cell death regulation ([15] and references therein). We discovered herein that the severe mitochondrial dysfunction manifested in the mtETC disturbance and the MPT pore opening was critically involved in mechanism of neurotoxic action of Cd2+. Moreover, on the basis of the obtained in this work evidence, it is possible to conclude that thiol status and the ROS generation changes are important participants of the events which contribute in the cell death induced by Cd2+. In particular, we found that NAC (GSH precursor), RR (MCU inhibitor), CsA and BKA (MPT pore inhibitors), FCCP (protonophoric uncoupler), stigmatellin (complex III inhibitor), and different antioxidants partially prevented the Cd2+-induced necrosis (Tables 4 and 5). Notably, in Cd2+-treated cells, the dose- and time-dependent increase of intracellular ROS production was partially depressed by CsA, BKA, vit E, and stigmatellin. In contrast, FCCP reduced in an equal extent both the ROS production of the Cd2+-treated and of the control cells. All this means that the increase in the ROS generation by Cd2+ is one of the important events in the heavy-metal-produced cytotoxicity as well as underscores the mitochondrial origin of the phenomenon. Moreover, it is likely directly connected with complex III of the mtETC, in particular with its Qo site. Besides, the differences in the effects of stigmatellin and myxothiazol (Table 4) indicate that the increase of ROS formation likely involves the mobility of iron-sulfur protein (ISP) of bc1 complex (i.e., complex III) as a key feature that is in line with previous observations of Armstrong et al. [48] and Muller et al. [50]. It is important that the MPT pore opening is likely also involved in this process that is in a good accordance with findings of He and Lemasters who, using proteomics, revealed that after induction of the MPT, dephosphorylation of the Rieske ISP of complex III took place [51]. In addition, there is interesting evidence in literature in the field that in the binding to complex III of such heavy metal as Zn2+, which partially depressed by Cu2+, ethoxyformic anhydride (i.e., histidyl residues modifier), and N-ethylmaleimide, NEM, the Rieske ISP participates as well ([52], see also [15], and references therein). At last, the absence of significant action of rotenone (complex I inhibitor) confirms the critical role of complex III in the Cd2+-induced neurotoxicity (Table 4).
The results, revealed by us on the intact cells herein and before (PC12 and AS-30D [6, 7]), are reinforced by our previous observations on isolated rat liver mitochondria (RLM) [15–18, 49, 53] where we studied the action of different mtETC and MPT pore inhibitors on the heavy-metal-induced high-conductance mitochondrial swelling in isotonic sucrose medium (a marker for the MPT pore involvement in mitochondrial membrane alterations) [8]. We found that Cd2+-induced high-amplitude swelling in isotonic sucrose medium (highly sensitive to the combined action of CsA, ADP, Mg2+, and dithiothreitol, DTT, [15, 17, 53]) was also sensitive to BKA, rotenone, and stigmatellin [18, 49]. Furthermore, our data agree well with the results obtained by other workers on isolated mitochondria [54, 55]. Not long ago, Wang and coauthors showed that Cd2+ stimulated ROS production in isolated liver, brain, and heart guinea pig mitochondria; moreover, their observations indicated that complex III might be the only site of ROS production induced by Cd2+ [54]. Besides, the results of their kinetic studies and electron turnover experiments confirmed and extended the previous findings of Miccadei and Floridi on isolated RLM [55] and gave them possibility to suggest that Cd2+ might bind between semiubiquinone and cytochrome b566 of the Q0 site of complex III, resulting in accumulation of semiubiquinones at the Q0 site [54]. In addition, on isolated rat hepatocytes, there was shown that just mitochondria were ROS sites for such non-redox or poor redox cycling transition metals as Cd2+, Hg2+, and As3+ [56]. Recently, it was found also that cytotoxic action of Cd2+ was blocked not only by the mitochondrial antioxidant α-lipoic acid but also by the mitochondrial complex III bypass agent 2,6-dichloroindophenol, DCIP (i.e., selective complex III electron acceptor) [45].
It should be mentioned, however, that there is some cell type specificity in toxic action of Cd2+, which is known to induce both necrosis and/or apoptosis, and even can produce antiapoptotic effect [57, 58]. For example, in human hepatoma HepG2 cells [59], Cd2+ produced a rapid and transient ROS generation, mitochondrial dysfunction, and apoptosis; moreover, the Cd2+-triggered apoptosis was not inhibited by CsA and BKA while it was blocked by NAC pretreatment. In some other cells [42, 60, 61], the Cd2+-induced apoptosis was mitochondria- and ROS-dependent event that, in opposite, was inhibited by oligomycin [61], CsA [42, 61], and in all cases was depressed by rotenone and antioxidants. In this connection, it is worthy to remind that rotenone decreases ROS production by complex III while enhances ROS production by complex I and the relative contribution of the two complexes to ROS production may vary in different cells [62]. Besides, ROS production by reverse electron transport, RET, in complex I was found to decrease by rotenone as well [63–66].
It seems important to say that findings obtained with Cd2+ as a probe correlate well with data existed in literature when MPT and/or cell death are produced by different other prooxidants and effectors [48, 67–72]. In particular, Armstrong and colleagues showed that ROS produced by complex III were functionally linked to the MPT pore and the MPT pore opening was critical event leading to death of GSH-depleted leukemic CEM and HL60 cells [48, 68] as well as of human B lymphoma (PW) cells [72]. Besides, there is a lot of other evidence in support of mitochondrial respiration and complex III participation in MPT pore opening and cell death regulation, including data obtained with respiration-deficient clones [73–77]. Furthermore, there are enough of observations in support of complex I involvement in MPT and cell death [78–84], including that on PC12 cells [36]. In particular, Toninello and colleagues showed that salicylate interacted with RLM respiratory chain and produced increase of ROS, which in turn oxidized thiol groups and GSH that in the presence of Ca2+ led to the MPT induction; moreover, they found that the reactive group of salicylate for inducing oxidative stress was the hydroxyl group which, by interacting with Fe-S cluster N2 of complex I, produced the ROS increase [84]. It is interesting to note that, as considered by several authors, the major site of superoxide generation of complex I is not flavin, but protein-associated ubisemiquinones which are spin coupled with the Fe-S cluster N2 [85]. The important data were obtained by several groups of investigators under study of mechanism(s) of action of arachidonic acid [86], ceramides [87–90], and tumor necrosis factor-alpha [91, 92] during induction by them of MPT and apoptosis that also indicated the direct participation of complex I and/or complex III in the processes. In addition, data concerning the contribution of the respiratory chain components in ischemia/reperfusion (hypoxia/reoxygenation) injury [93–100] and the modern knowledge about respiratory supercomplexes [101, 102] including findings on the structure of mitochondrial supercomplex formed by complexes I and III [103–107] are also very impressive.
In general, all the above-mentioned data support our hypothesis about the possible involvement of the mtETC components, namely, a supercomplex formed by complex I (P-site) and complex III (S-site) in the mitochondrial membrane permeabilization mediated by the MPT pore (Figure 4, [16–18, 49]). The hypothesis was suggested by us sufficiently long ago [16] when we for the first time speculated that both respiratory complexes I and III might be involved in the mitochondrial membrane permeabilization promoted by Cd2+ and/or Ca2+ plus Pi. On the basis of own findings [15, 53, 108] and data existing in literature in the field at that moment, we postulated a hypothetical model of regulated MPT pore. In particular, a model of conventional Ca2+-activated CsA-sensitive pore originally proposed by Halestrap (for reviews see [10, 14]) in which ANT was considered its crucial core element (in the light of the modern findings it may be also phosphate carrier, PiC) had been integrated with an idea of Fontaine and Bernardi (for review see [9]) concerning mitochondrial respiratory chain complex I involvement in the MPT pore formation and/or regulation. The model had been supplemented by several main postulates, the most important among them were the following. Both complex I and III of the mtETC are places of localization of Ca2+(Me2+)-binding site(s), critical for the MPT induction, and depending on conditions and cell type, either one or both complexes could be involved in triggering of the MPT pore assembly; besides, the complex I of the respiratory chain likely constitutes the P-site while complex III—the S-site of the MPT pore [16]. Later, the model was extended by additional suppositions [17]. In particular, the complex I (P-site) and complex III (S-site) may constitute not only critical Me2+-binding sites but also main loci for ROS generation that was instrumental in oxidation of critical thiol groups and the MPT pore opening [17]. The aforementioned Me2+-binding site(s) are most likely disposed: (i) on the way of reverse electron transfer from succinate to NAD+ (complex I); (ii) on cytochrome b somewhere near heme bL and close to stigmatellin binding site (complex III) [17]. Our recent results [18, 109] extended further this theory and indicated that just complex III could be the critical external dithiol [12]. Moreover, it seems that the supercomplex I-III could be the key component of the regulated (i.e., Ca2+-dependent CsA-sensitive) MPT pore complex, while complex III is likely involved in the “unregulated” (i.e., Ca2+- and/or CsA-insensitive) MPT pore assembly and might concern to the external (inhibitory) Ca2+-binding site. Some of these views become more valid in the light of new interesting experimental data obtained by different investigators during the latest time [109–120]; the comprehensive review of the modern ideas and findings on the issue will be given elsewhere (Belyaeva, in preparation). In summary, it is worthy to note that the question of the participation of mitochondrial respiratory chain components in MPT pore formation and/or regulation as well as localization of the critical Me2+ binding sites and dithiols is so mysterious and intrigues problems, the decision of which, we hope, could bring new understanding of processes of aging and death.
Figure 4.
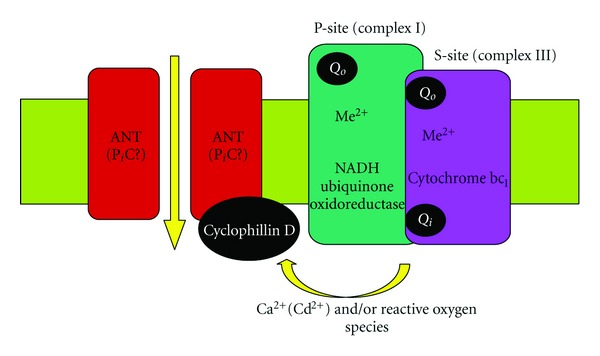
A hypothetical model of mitochondrial permeability transition pore structure: an involvement of a mitochondrial super-complex formed by respiratory chain complexes I and III (modified from [48, 49], see explanation in text).
5. Conclusion
In conclusion, the results obtained in this study give new important information to understand the mechanism(s) underlying the heavy-metal-produced neurotoxic action, pointing to the critical involvement of mitochondrial dysfunction in the heavy-metal-induced cell death. We have shown that against the Cd2+-induced injury, not only well-known antioxidants and the MPT pore and MCU inhibitors are effective but also different mtETC effectors. Among mtETC effectors under test, only stigmatellin produced the significant and sustained protection of PC12 cells. Moreover, stigmatellin was found to be one of the strongest protectors that exhibited its action not only on different types of the cells (PC12 and AS-30D [6]) but on isolated mitochondria as well [18, 49]. All this points to the direct involvement of the mtETC in the Cd2+-induced mitochondrial membrane permeabilization and cell death. In the case of Hg2+ and Cu2+, all mtETC effectors under study did not exhibit significant protective action on PC12 cells. We concluded that the mtETC dysfunction was involved but in a different way in mechanism(s) of neurotoxic action of the heavy metals. Notably, the data obtained herein further support our hypothesis about the direct involvement of the mtETC components in the mitochondrial membrane permeabilization mediated by the MPT pore assembly [16–18, 49].
Acknowledgments
This study was supported partially by a grant from the Russian Foundation for Basic Research to E.A.B. (07-04-00722). The authors thank Dr. Irina Brailovskaya, Irina Zubatkina, Yulia Vlasova, Boris Kormilitsyn, and Elena Dubinina for their fruitful assistance. The authors do not have a direct financial relation with the commercial identity mentioned in the paper that might lead to a conflict of interest for any of the authors.
Abbreviations
- mtETC:
Mitochondrial electron transport chain
- ROS:
Reactive oxygen species
- MPT:
Mitochondrial permeability transition
- CsA:
Cyclosporin A
- CyP-D:
Cyclophilin-D
- DMEM:
Dulbecco/Vogt modified Eagle's minimal essential medium
- LDH:
Lactate dehydrogenase
- ΔΨmito:
Mitochondrial membrane potential
- FCCP:
Carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- RCR:
Respiratory control ratio
- DCFH2-DA:
Dichlorodihydrofluorescein diacetate
- TMRM:
Tetramethylrhodamine methyl ester
- PBS:
Phosphate buffered saline
- NAC:
N-acetylcysteine
- vit E:
Vitamin E
- BHT:
Butylhydroxytoluene
- TEMPO:
Tetramethylpiperidine-1-oxyl
- RR:
Ruthenium Red
- MCU:
Mitochondrial Ca2+ uniporter
- BKA:
Bongkrekic acid
- ANT:
Adenine nucleotide translocase
- PiC:
Phosphate carrier
- PN:
Pyridine nucleotides
- ISP:
Iron-sulfur protein
- NEM:
N-ethylmaleimide
- DTT:
Dithiothreitol
- RLM:
Rat liver mitochondria
- RET:
Reverse electron transport.
References
- 1.Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radical Biology and Medicine. 1995;18(2):321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 2.Valko M, Morris H, Cronin MTD. Metals, toxicity and oxidative stress. Current Medicinal Chemistry. 2005;12(10):1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 3.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Cuypers A, Plusquin M, Remans T, et al. Cadmium stress: an oxidative challenge. BioMetals. 2010;23(5):927–940. doi: 10.1007/s10534-010-9329-x. [DOI] [PubMed] [Google Scholar]
- 5.Nightingale ER. Phenomenological theory of ion solvation. Effective radii of hydrated ions. Journal of Physical Chemistry. 1959;63(9):1381–1387. [Google Scholar]
- 6.Belyaeva EA, Dymkowska D, Wieckowski MR, Wojtczak L. Reactive oxygen species produced by the mitochondrial respiratory chain are involved in Cd2+-induced injury of rat ascites hepatoma AS-30D cells. Biochimica et Biophysica Acta. 2006;1757(12):1568–1574. doi: 10.1016/j.bbabio.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Belyaeva EA, Dymkowska D, Wieckowski MR, Wojtczak L. Mitochondria as an important target in heavy metal toxicity in rat hepatoma AS-30D cells. Toxicology and Applied Pharmacology. 2008;231(1):34–42. doi: 10.1016/j.taap.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochimica et Biophysica Acta. 1995;1241(2):139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 9.Fontaine E, Bernardi P. Progress on the mitochondrial permeability transition pore: regulation by complex I and ubiquinone analogs. Journal of Bioenergetics and Biomembranes. 1999;31(4):335–345. doi: 10.1023/a:1005475802350. [DOI] [PubMed] [Google Scholar]
- 10.Halestrap AP. What is the mitochondrial permeability transition pore? Journal of Molecular and Cellular Cardiology. 2009;46(6):821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 11.Nishida H, Matsumoto A, Tomono N, Hanakai T, Harada S, Nakaya H. Biochemistry and physiology of mitochondrial ion channels involved in cardioprotection. FEBS Letters. 2010;584(10):2161–2166. doi: 10.1016/j.febslet.2009.12.033. [DOI] [PubMed] [Google Scholar]
- 12.Petronilli V, Šileikyte J, Zulian A, et al. Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin-mediated photooxidative stress. Unmasking pore-regulating external thiols. Biochimica et Biophysica Acta. 2009;1787(7):897–904. doi: 10.1016/j.bbabio.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ . Biochemistry. 1996;35(26):8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- 14.Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Current Medicinal Chemistry. 2003;10(16):1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 15.Belyaeva EA, Korotkov SM. Mechanism of primary Cd2+-induced rat liver mitochondria dysfunction: discrete modes of Cd2+ action on calcium and thiol-dependent domains. Toxicology and Applied Pharmacology. 2003;192(1):56–68. doi: 10.1016/s0041-008x(03)00255-2. [DOI] [PubMed] [Google Scholar]
- 16.Belyaeva EA. Regulated mitochondrial permeability transition: a possible involvement of mitochondrial respiratory complexes I and III. Mitochondrion. 2004;4:p. 71. [Google Scholar]
- 17.Belyaeva EA, Glazunov VV, Korotkov SM. Cd2+ versus Ca2+-produced mitochondrial membrane permeabilization: a proposed direct participation of respiratory complexes I and III. Chemico-Biological Interactions. 2004;150(3):253–270. doi: 10.1016/j.cbi.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Belyaeva EA. Mitochondrial respiratory chain inhibitors modulate the metal-induced inner mitochondrial membrane permeabilization. Acta Biochimica Polonica. 2010;57(4):435–441. [PubMed] [Google Scholar]
- 19.Belyaeva EA, Emelyanova LV, Zakharova IO, Sokolova TV. An involvement of mitochondrial respiratory chain dysfunction in mechanism(s) of neurotoxic action of heavy metals: a comparison of effects of Cd2+, Hg2+, and Cu2+ on rat neuron-like cell line PC12. In: Proceedings of the International Conference on Neurochemical Mechanisms of Generation of Adaptive and Pathological Brain Conditions; 2008; St.Petersburg, Russia. Pavlov Institute of RAN; p. 17. [Google Scholar]
- 20.Belyaeva EA. Reactive oxygen species, mitochondrial electron transport chain and permeability transition pore as the key players in heavy metal-induced neurotoxicity. In: Renner-Sattler K, Gnaiger E, editors. Mitochondrial Physiology. The Many Functions of the Organism in Our Cells. 15.06. Mitochondrial Physiology Network; 2010. pp. 65–66. [Google Scholar]
- 21.Belyaeva EA, Sokolova TV, Vlasova YA, Zakharova IO. Mitochondria are critically involved in cadmium(II)-induced neurotoxicity. International Journal of Trends in Medicine. 2011;1(1):14–16. [Google Scholar]
- 22.Sokolova TV, Zakharova IO, Furaev VV, Rychkova MP, Avrova NF. Neuroprotective effect of ganglioside GM1 on the cytotoxic action of hydrogen peroxide and amyloid β-peptide in PC12 cells. Neurochemical Research. 2007;32(8):1302–1313. doi: 10.1007/s11064-007-9304-2. [DOI] [PubMed] [Google Scholar]
- 23.Vassault A. Lactate dehydrogenase: UV-method with pyruvate and NADH. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. Vol. 3. Weinheim, Germany: Chemie; 1983. pp. 118–126. [Google Scholar]
- 24.Nicholls DG, Ferguson SJ. Bioenergetics. 3rd edition. London, UK: Academic Press; 2002. [Google Scholar]
- 25.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochemical Journal. 2011;435(2):297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cafe C, Torri C, Bertorelli L, et al. Oxidative events in neuronal and glial cell-enriched fractions of rat cerebral cortex. Free Radical Biology and Medicine. 1995;19(6):853–857. doi: 10.1016/0891-5849(95)00086-d. [DOI] [PubMed] [Google Scholar]
- 27.Fu W, Luo H, Parthasarathy S, Mattson MP. Catecholamines potentiate amyloid β-peptide neurotoxicity: involvement of oxidative stress, mitochondrial dysfunction, and perturbed calcium homeostasis. Neurobiology of Disease. 1998;5(4):229–243. doi: 10.1006/nbdi.1998.0192. [DOI] [PubMed] [Google Scholar]
- 28.Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophysical Journal. 1988;53(5):785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinkle PM, Osborne ME. Cadmium toxicity in rat pheochromocytoma cells: studies on the mechanism of uptake. Toxicology and Applied Pharmacology. 1994;124(1):91–98. doi: 10.1006/taap.1994.1012. [DOI] [PubMed] [Google Scholar]
- 30.Shafer TJ. Effects of Cd2+, Pb2+ and CH3Hg+ on high voltage-activated calcium currents in pheochromocytoma (PC12) cells: potency, reversibility, interactions with extracellular Ca2+ and mechanisms of block. Toxicology Letters. 1998;99(3):207–221. doi: 10.1016/s0378-4274(98)00225-2. [DOI] [PubMed] [Google Scholar]
- 31.Wätjen W, Cox M, Biagioli M, Beyersmann D. Cadmium-induced apoptosis in C6 glioma cells: mediation by caspase 9-activation. BioMetals. 2002;15(1):15–25. doi: 10.1023/a:1013141926228. [DOI] [PubMed] [Google Scholar]
- 32.Wätjen W, Haase H, Biagioli M, Beyersmann D. Induction of apoptosis in mammalian cells by cadmium and zinc. Environmental Health Perspectives. 2002;110(5):865–867. doi: 10.1289/ehp.110-1241262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Virmani A, Gaetani F, Imam S, Binienda Z, Ali S. Possible mechanism for the neuroprotective effects of L-carnitine on methamphetamine-evoked neurotoxicity. Annals of the New York Academy of Sciences. 2003;993:197–207. doi: 10.1111/j.1749-6632.2003.tb07530.x. [DOI] [PubMed] [Google Scholar]
- 34.Gatti R, Belletti S, Uggeri J, et al. Methylmercury cytotoxicity in PC12 cells is mediated by primary glutathione depletion independent of excess reactive oxygen species generation. Toxicology. 2004;204(2-3):175–185. doi: 10.1016/j.tox.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 35.Wätjen W, Beyersmann D. Cadmium-induced apoptosis in C6 glioma cells: influence of oxidative stress. BioMetals. 2004;17(1):65–78. doi: 10.1023/a:1024405119018. [DOI] [PubMed] [Google Scholar]
- 36.Lee SJ, Youn YC, Han ES, Lee CS. Depressant effect of mitochondrial respiratory complex inhibitors on proteasome inhibitor-induced mitochondrial dysfunction and cell death in PC12 cells. Neurochemical Research. 2005;30(9):1191–1200. doi: 10.1007/s11064-005-8158-8. [DOI] [PubMed] [Google Scholar]
- 37.Lovrić J, Bazzi HS, Cuie Y, Fortin GRA, Winnik FM, Maysinger D. Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots. Journal of Molecular Medicine. 2005;83(5):377–385. doi: 10.1007/s00109-004-0629-x. [DOI] [PubMed] [Google Scholar]
- 38.Marella M, Byoung BS, Matsuno-Yagi A, Yagi T. Mechanism of cell death caused by complex I defects in a rat dopaminergic cell line. Journal of Biological Chemistry. 2007;282(33):24146–24156. doi: 10.1074/jbc.M701819200. [DOI] [PubMed] [Google Scholar]
- 39.Kawakami M, Inagawa R, Hosokawa T, Saito T, Kurasaki M. Mechanism of apoptosis induced by copper in PC12 cells. Food and Chemical Toxicology. 2008;46(6):2157–2164. doi: 10.1016/j.fct.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Fang H, Groom L, et al. Superoxide flashes in single mitochondria. Cell. 2008;134(2):279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toimela T, Tähti H. Mitochondrial viability and apoptosis induced by aluminum, mercuric mercury and methylmercury in cell lines of neural origin. Archives of Toxicology. 2004;78(10):565–574. doi: 10.1007/s00204-004-0575-y. [DOI] [PubMed] [Google Scholar]
- 42.Poliandri AHB, Machiavelli LI, Quinteros AF, Cabilla JP, Duvilanski BH. Nitric oxide protects the mitochondria of anterior pituitary cells and prevents cadmium-induced cell death by reducing oxidative stress. Free Radical Biology and Medicine. 2006;40(4):679–688. doi: 10.1016/j.freeradbiomed.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 43.Reddy PVB, Rama Rao KV, Norenberg MD. The mitochondrial permeability transition, and oxidative and nitrosative stress in the mechanism of copper toxicity in cultured neurons and astrocytes. Laboratory Investigation. 2008;88(8):816–830. doi: 10.1038/labinvest.2008.49. [DOI] [PubMed] [Google Scholar]
- 44.Roy DN, Mandal S, Sen G, Biswas T. Superoxide anion mediated mitochondrial dysfunction leads to hepatocyte apoptosis preferentially in the periportal region during copper toxicity in rats. Chemico-Biological Interactions. 2009;182(2-3):136–147. doi: 10.1016/j.cbi.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 45.Monroe RK, Halvorsen SW. Environmental toxicants inhibit neuronal Jak tyrosine kinase by mitochondrial disruption. NeuroToxicology. 2009;30(4):589–598. doi: 10.1016/j.neuro.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 46.Chen YW, Huang CF, Yang CY, Yen CC, Tsai KS, Liu SH. Inorganic mercury causes pancreatic β-cell death via the oxidative stress-induced apoptotic and necrotic pathways. Toxicology and Applied Pharmacology. 2010;243(3):323–331. doi: 10.1016/j.taap.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 47.Polunas M, Halladay A, Tjalkens RB, Philbert MA, Lowndes H, Reuhl K. Role of oxidative stress and the mitochondrial permeability transition in methylmercury cytotoxicity. NeuroToxicology. 2011;32(5):526–534. doi: 10.1016/j.neuro.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armstrong JS, Yang H, Duan W, Whiteman M. Cytochrome bc1 regulates the mitochondrial permeability transition by two distinct pathways. Journal of Biological Chemistry. 2004;279(48):50420–50428. doi: 10.1074/jbc.M408882200. [DOI] [PubMed] [Google Scholar]
- 49.Belyaeva EA, Saris N-E. Cd2+-produced disturbance of mitochondrial function: the cause/consequence relationships. In: Collery P, Maymard I, Theophanides T, Khassanova L, Collery T, editors. Metal Ions in Biology and Medicine. Vol. 10. Paris, France: John Libbey Eurotext; 2008. pp. 328–333. [Google Scholar]
- 50.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. Journal of Biological Chemistry. 2004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 51.He L, Lemasters JJ. Dephosphorylation of the Rieske iron-sulfur protein after induction of the mitochondrial permeability transition. Biochemical and Biophysical Research Communications. 2005;334(3):829–837. doi: 10.1016/j.bbrc.2005.06.170. [DOI] [PubMed] [Google Scholar]
- 52.Lorusso M, Cocco T, Sardanelli AM, Minuto M, Bonomi F, Papa S. Interaction of Zn2+ with the bovine-heart mitochondrial bc1 complex. European Journal of Biochemistry. 1991;197(2):555–561. doi: 10.1111/j.1432-1033.1991.tb15944.x. [DOI] [PubMed] [Google Scholar]
- 53.Belyaeva EA, Glazunov VV, Korotkov SM. Cyclosporin A-sensitive permeability transition pore is involved in Cd2+-induced dysfunction of isolated rat liver mitochondria: doubts no more. Archives of Biochemistry and Biophysics. 2002;405(2):252–264. doi: 10.1016/s0003-9861(02)00400-9. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Fang J, Leonard SS, Rao KMK. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radical Biology and Medicine. 2004;36(11):1434–1443. doi: 10.1016/j.freeradbiomed.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Miccadei S, Floridi A. Sites of inhibition of mitochondrial electron transport by cadmium. Chemico-Biological Interactions. 1993;89(2-3):159–167. doi: 10.1016/0009-2797(93)90006-k. [DOI] [PubMed] [Google Scholar]
- 56.Pourahmad J, O’Brien PJ, Jokar F, Daraei B. Carcinogenic metal induced sites of reactive oxygen species formation in hepatocytes. Toxicology in Vitro. 2003;17(5-6):803–810. doi: 10.1016/s0887-2333(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 57.Yuan C, Kadiiska M, Achanzar WE, Mason RP, Waalkes MP. Possible role of caspase-3 inhibition in cadmium-induced blockage of apoptosis. Toxicology and Applied Pharmacology. 2000;164(3):321–329. doi: 10.1006/taap.2000.8921. [DOI] [PubMed] [Google Scholar]
- 58.Gunawardana CG, Martinez RE, Xiao W, Templeton DM. Cadmium inhibits both intrinsic and extrinsic apoptotic pathways in renal mesangial cells. American Journal of Physiology. 2006;290(5):F1074–F1082. doi: 10.1152/ajprenal.00067.2005. [DOI] [PubMed] [Google Scholar]
- 59.Oh SH, Lim SC. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicology and Applied Pharmacology. 2006;212(3):212–223. doi: 10.1016/j.taap.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 60.Lemarié A, Lagadic-Gossmann D, Morzadec C, Allain N, Fardel O, Vernhet L. Cadmium induces caspase-independent apoptosis in liver Hep3B cells: role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease G and apoptosis-inducing factor. Free Radical Biology and Medicine. 2004;36(12):1517–1531. doi: 10.1016/j.freeradbiomed.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 61.Shih CM, Ko WC, Wu JS, et al. Mediating of caspase-independent apoptosis by cadmium through the mitochondria-ROS pathway in MRC-5 fibroblasts. Journal of Cellular Biochemistry. 2004;91(2):384–397. doi: 10.1002/jcb.10761. [DOI] [PubMed] [Google Scholar]
- 62.Bernacchia A, Biondi A, Genova ML, Lenaz G, Falasca A. The various sources of mitochondrial oxygen radicals: a minireview. Toxicology Mechanisms and Methods. 2004;14(1-2):25–30. doi: 10.1080/15376520490257383. [DOI] [PubMed] [Google Scholar]
- 63.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 64.Korshunov SS, Korkina OV, Ruuge EK, Skulachev VP, Starkov AA. Fatty acids as natural uncouplers preventing generation of O2 · − and H2O2 by mitochondria in the resting state. FEBS Letters. 1998;435(2-3):215–218. doi: 10.1016/s0014-5793(98)01073-4. [DOI] [PubMed] [Google Scholar]
- 65.Korshunov SS, Krasnikov BF, Pereverzev MO, Skulachev VP. The antioxidant functions of cytochrome c. FEBS Letters. 1999;462(1-2):192–198. doi: 10.1016/s0014-5793(99)01525-2. [DOI] [PubMed] [Google Scholar]
- 66.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochemical Journal. 2002;368(2):545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salvi M, Brunati AM, Clari G, Toninello A. Interaction of genistein with the mitochondrial electron transport chain results in opening of the membrane transition pore. Biochimica et Biophysica Acta. 2002;1556(2-3):187–196. doi: 10.1016/s0005-2728(02)00361-4. [DOI] [PubMed] [Google Scholar]
- 68.Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. The FASEB Journal. 2002;16(10):1263–1265. doi: 10.1096/fj.02-0097fje. [DOI] [PubMed] [Google Scholar]
- 69.Armstrong JS, Whiteman M, Rose P, Jones DP. The Coenzyme Q10 analog decylubiquinone inhibits the redox-activated mitochondrial permeability transition: role of mitochondrial complex III. The Journal of biological chemistry. 2003;278(49):49079–49084. doi: 10.1074/jbc.M307841200. [DOI] [PubMed] [Google Scholar]
- 70.Toninello A, Clari G, Mancon M, Tognon G, Zatta P. Aluminum as an inducer of the mitochondrial permeability transition. Journal of Biological Inorganic Chemistry. 2000;5(5):612–623. doi: 10.1007/s007750000144. [DOI] [PubMed] [Google Scholar]
- 71.Braguini WL, Cadena SMSC, Carnieri EGS, Rocha MEM, De Oliveira MBM. Effects of deltamethrin on functions of rat liver mitochondria and on native and synthetic model membranes. Toxicology Letters. 2004;152(3):191–202. doi: 10.1016/j.toxlet.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 72.Armstrong JS, Steinauer KK, Hornung B, et al. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death and Differentiation. 2002;9(3):252–263. doi: 10.1038/sj.cdd.4400959. [DOI] [PubMed] [Google Scholar]
- 73.Hail N, Lotan R. Mitochondrial respiration is uniquely associated with the prooxidant and apoptotic effects of N-(4-Hydroxyphenyl)retinamide. Journal of Biological Chemistry. 2001;276(49):45614–45621. doi: 10.1074/jbc.M106559200. [DOI] [PubMed] [Google Scholar]
- 74.Hail N, Youssef EM, Lotan R. Evidence supporting a role for mitochondrial respiration in apoptosis induction by the synthetic retinoid CD437. Cancer Research. 2001;61(18):6698–6702. [PubMed] [Google Scholar]
- 75.Hail N, Lotan R. Apoptosis induction by the natural product cancer chemopreventive agent deguelin is mediated through the inhibition of mitochondrial bioenergetics. Apoptosis. 2004;9(4):437–447. doi: 10.1023/B:APPT.0000031449.57551.e1. [DOI] [PubMed] [Google Scholar]
- 76.Hail N, Jr., Lotan R. Cancer chemoprevention and mitochondria: targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Molecular Nutrition and Food Research. 2009;53(1):49–67. doi: 10.1002/mnfr.200700527. [DOI] [PubMed] [Google Scholar]
- 77.Lee JY, Jung GY, Heo HJ, et al. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicology Letters. 2006;166(3):212–221. doi: 10.1016/j.toxlet.2006.07.305. [DOI] [PubMed] [Google Scholar]
- 78.Vinogradov AD. Catalytic properties of the mitochondrial NADH-ubiquinone oxidoreductase (Complex I) and the pseudo-reversible active/inactive enzyme transition. Biochimica et Biophysica Acta. 1998;1364(2):169–185. doi: 10.1016/s0005-2728(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 79.Grivennikova VG, Kapustin AN, Vinogradov AD. Catalytic activity of NADH-ubiquinone oxidoreductase (Complex I) in intact mitochondria: evidence for the slow active/inactive transition. Journal of Biological Chemistry. 2001;276(12):9038–9044. doi: 10.1074/jbc.M009661200. [DOI] [PubMed] [Google Scholar]
- 80.Maklashina E, Sher Y, Zhou HZ, Gray MO, Karliner JS, Cecchini G. Effect of anoxia/reperfusion on the reversible active/de-active transition of NADH-ubiquinone oxidoreductase (complex I) in rat heart. Biochimica et Biophysica Acta. 2002;1556(1):6–12. doi: 10.1016/s0005-2728(02)00280-3. [DOI] [PubMed] [Google Scholar]
- 81.Kanno T, Sato EF, Muranaka S, et al. Oxidative stress underlies the mechanism for Ca2+-induced permeability transition of mitochondria. Free Radical Research. 2004;38(1):27–35. doi: 10.1080/10715760310001626266. [DOI] [PubMed] [Google Scholar]
- 82.Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain Complex I. Journal of Biological Chemistry. 2004;279(17):17197–17204. doi: 10.1074/jbc.M310329200. [DOI] [PubMed] [Google Scholar]
- 83.García N, Correa F, Chávez E. On the role of the respiratory complex I on membrane permeability transition. Journal of Bioenergetics and Biomembranes. 2005;37(1):17–23. doi: 10.1007/s10863-005-4119-9. [DOI] [PubMed] [Google Scholar]
- 84.Battaglia V, Salvi M, Toninello A. Oxidative stress is responsible for mitochondrial permeability transition induction by salicylate in liver mitochondria. Journal of Biological Chemistry. 2005;280(40):33864–33872. doi: 10.1074/jbc.M502391200. [DOI] [PubMed] [Google Scholar]
- 85.Ohnishi ST, Ohnishi T, Muranaka S, et al. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. Journal of Bioenergetics and Biomembranes. 2005;37(1):1–15. doi: 10.1007/s10863-005-4117-y. [DOI] [PubMed] [Google Scholar]
- 86.Cocco T, Di Paola M, Papa S, Lorusso M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radical Biology and Medicine. 1999;27(1-2):51–59. doi: 10.1016/s0891-5849(99)00034-9. [DOI] [PubMed] [Google Scholar]
- 87.García-Ruiz C, Colell A, Marí M, Morales A, Fernández-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species: role of mitochondrial glutathione. Journal of Biological Chemistry. 1997;272(17):11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 88.Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. Journal of Biological Chemistry. 1997;272(39):24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- 89.Poppe M, Reimertz C, Münstermann G, Kögel D, Prehn JHM. Ceramide-induced apoptosis of D283 medulloblastoma cells requires mitochondrial respiratory chain activity but occurs independently of caspases and is not sensitive to Bcl-xL overexpression. Journal of Neurochemistry. 2002;82(3):482–494. doi: 10.1046/j.1471-4159.2002.01007.x. [DOI] [PubMed] [Google Scholar]
- 90.Therade-Matharan S, Laemmel E, Carpentier S, et al. Reactive oxygen species production by mitochondria in endothelial cells exposed to reoxygenation after hypoxia and glucose depletion is mediated by ceramide. American Journal of Physiology. 2005;289(6):R1756–R1762. doi: 10.1152/ajpregu.00480.2004. [DOI] [PubMed] [Google Scholar]
- 91.Sánchez-Alcázar JA, Schneider E, Martínez MA, et al. Tumor necrosis factor-α increases the steady-state reduction of cytochrome b of the mitochondrial respiratory chain in metabolically inhibited L929 cells. Journal of Biological Chemistry. 2000;275(18):13353–13361. doi: 10.1074/jbc.275.18.13353. [DOI] [PubMed] [Google Scholar]
- 92.Higuchi M, Proske RJ, Yeh ETH. Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene. 1998;17(19):2515–2524. doi: 10.1038/sj.onc.1202485. [DOI] [PubMed] [Google Scholar]
- 93.Lesnefsky EJ, Gudz TI, Migita CT, et al. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Archives of Biochemistry and Biophysics. 2001;385(1):117–128. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- 94.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. Journal of Biological Chemistry. 2003;278(38):36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 95.Feldkamp T, Kribben A, Roeser NF, et al. Preservation of complex I function during hypoxia-reoxygenation-induced mitochondrial injury in proximal tubules. American Journal of Physiology. 2004;286(4):F749–F759. doi: 10.1152/ajprenal.00276.2003. [DOI] [PubMed] [Google Scholar]
- 96.Solaini G, Harris DA. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochemical Journal. 2005;390(2):377–394. doi: 10.1042/BJ20042006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Experimental Physiology. 2006;91(5):807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 98.Bell EL, Klimova TA, Eisenbart J, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. Journal of Cell Biology. 2007;177(6):1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feldkamp T, Park JS, Pasupulati R, et al. Regulation of the mitochondrial permeability transition in kidney proximal tubules and its alteration during hypoxia-reoxygenation. American Journal of Physiology. 2009;297(6):F1632–F1646. doi: 10.1152/ajprenal.00422.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen Q, Lesnefsky EJ. Blockade of electron transport during ischemia preserves bcl-2 and inhibits opening of the mitochondrial permeability transition pore. FEBS Letters. 2011;585(6):921–926. doi: 10.1016/j.febslet.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schägger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochimica et Biophysica Acta. 2002;1555(1–3):154–159. doi: 10.1016/s0005-2728(02)00271-2. [DOI] [PubMed] [Google Scholar]
- 102.Bianchi C, Fato R, Genova ML, Castelli GP, Lenaz G. Structural and functional organization of Complex I in the mitochondrial respiratory chain. BioFactors. 2003;18(1–4):3–9. doi: 10.1002/biof.5520180202. [DOI] [PubMed] [Google Scholar]
- 103.Blanchi C, Genova ML, Castelli GP, Lenaz G. The mitochondrial respiratory chain is partially organized in a supercomplex assembly: kinetic evidence using flux control analysis. Journal of Biological Chemistry. 2004;279(35):36562–36569. doi: 10.1074/jbc.M405135200. [DOI] [PubMed] [Google Scholar]
- 104.Acín-Pérez R, Bayona-Bafaluy MP, Fernández-Silva P, et al. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Molecular Cell. 2004;13(6):805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dudkina NV, Eubel H, Keegstra W, Boekema EJ, Braun HP. Structure of a mitochondrial supercomplex formed by respiratory-chain complexes I and III. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(9):3225–3229. doi: 10.1073/pnas.0408870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dudkina NV, Kouřil R, Peters K, Braun H-P, Boekema EJ. Structure and function of mitochondrial supercomplexes. Biochimica et Biophysica Acta. 2010;1797(6-7):664–670. doi: 10.1016/j.bbabio.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 107.Lenaz G, Baracca A, Barbero G, et al. Mitochondrial respiratory chain super-complex I-III in physiology and pathology. Biochimica et Biophysica Acta. 2010;1797(6-7):633–640. doi: 10.1016/j.bbabio.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 108.Belyaeva EA, Glazunov VV, Nikitina ER, Korotkov SM. Bivalent metal ions modulate Cd2+ effects on isolated rat liver mitochondria. Journal of Bioenergetics and Biomembranes. 2001;33(4):303–318. doi: 10.1023/a:1010607307099. [DOI] [PubMed] [Google Scholar]
- 109.Belyaeva EA, Korotkov SM, Saris N-E. In vitro modulation of heavy metal-induced rat liver mitochondria dysfunction: a comparison of copper and mercury with cadmium. Journal of Trace Elements in Medicine and Biology. 2011;25(1, supplement):S63–S73. doi: 10.1016/j.jtemb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 110.Guidarelli A, Cerioni L, Cantoni O. Inhibition of complex III promotes loss of Ca2+ dependence for mitochondrial superoxide formation and permeability transition evoked by peroxynitrite. Journal of Cell Science. 2007;120(11):1908–1914. doi: 10.1242/jcs.003228. [DOI] [PubMed] [Google Scholar]
- 111.Dröse S, Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 Complex. Journal of Biological Chemistry. 2008;283(31):21649–21654. doi: 10.1074/jbc.M803236200. [DOI] [PubMed] [Google Scholar]
- 112.Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. Journal of Biological Chemistry. 2008;283(6):3476–3486. doi: 10.1074/jbc.M707528200. [DOI] [PubMed] [Google Scholar]
- 113.Porcelli AM, Angelin A, Ghelli A, et al. Respiratory complex I dysfunction due to mitochondrial DNA mutations shifts the voltage threshold for opening of the permeability transition pore toward resting levels. Journal of Biological Chemistry. 2009;284(4):2045–2052. doi: 10.1074/jbc.M807321200. [DOI] [PubMed] [Google Scholar]
- 114.Galkin A, Abramov AY, Frakich N, Duchen MR, Moncada S. Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury? Journal of Biological Chemistry. 2009;284(52):36055–36061. doi: 10.1074/jbc.M109.054346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fato R, Bergamini C, Bortolus M, et al. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochimica et Biophysica Acta. 2009;1787(5):384–392. doi: 10.1016/j.bbabio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Deng N, Zhang J, Zong C, et al. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Molecular and Cellular Proteomics. 2011;10(2) doi: 10.1074/mcp.M110.000117. Article ID M110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Berry EA, Huang L-S. Conformationally linked interaction in the cytochrome bc1 complex between inhibitors of the Q0 site and the Rieske iron-sulfur protein. Biochimica et Biophysica Acta. 2011;1807(10):1349–1363. doi: 10.1016/j.bbabio.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 118.Wen Y, Li W, Poteet EC, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. Journal of Biological Chemistry. 2011;286(18):16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rodrigues FP, Pestana CR, Dos Santos GA, et al. Characterization of the stimulus for reactive oxygen species generation in calcium-overloaded mitochondria. Redox Report. 2011;16(3):108–113. doi: 10.1179/1351000211Y.0000000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang Y, Li JH, Liu XR, Jiang FL, Tian FF, Liu Y. Spectroscopic and microscopic studies on the mechanisms of mitochondrial toxicity induced by different concentrations of cadmium. Journal of Membrane Biology. 2011;241(1):39–49. doi: 10.1007/s00232-011-9361-y. [DOI] [PubMed] [Google Scholar]