

Abstract
Several immunocytochemical studies have revealed that Abelson tyrosine kinase (c-Abl) is associated with both neuritic plaques and neurofibrillary tangles in the brains of patients with Alzheimer’s disease (AD). Additionally, c-Abl has been shown to phosphorylate tau on tyrosine 394. The activity of c-Abl is also involved in the control of the cell cycle and apoptosis. To examine the consequences of c-Abl activation in the adult brain, we have constructed two lines of transgenic mice expressing either a constitutively active form of c-Abl (AblPP/tTA mice) or its sister protein, Arg (ArgPP/tTA mice), with a neuron-specific promoter (CamKIIα) regulated by doxycycline (Tet-Off). Expression of active c-Abl in adult mouse forebrain neurons results in severe neurodegeneration, particularly in the CA1 region of the hippocampus. Neuronal loss was preceded and accompanied by substantial microgliosis and astrocyctosis. Despite careful examination, no c-Abl expression is found in glial cells, indicating that neuronal c-Abl expression is responsible for the gliosis. In contrast, ArgPP/tTA mice have no evidence of neuronal loss or gliosis, even though protein expression and kinase activity levels are similar to those in the AblPP/tTA mice. Given the evidence of c-Abl activation in the human AD brain combined with the pathological phenotype of AblPP/tTA mice, it is likely that aberrant c-Abl activity may play a role in neurodegenerative disease.
Keywords: Alzheimer’s disease, Arg, c-Abl, gliosis, neurodegeneration, transgenic
INTRODUCTION
Alzheimer’s disease (AD) is estimated to affect nearly 30 million people worldwide and is characterized pathologically by progressive neurodegeneration with accompanying cognitive impairment, extracellular plaques composed of amyloid-β (Aβ), and neurofibrillary tangles (NFTs) composed of intracellular hyperphosphorylated tau protein [1]. AD cases are also characterized by neuroinflammation surrounding amyloid plaques [2-6].
Studies of transgenic animal models of amyloid and tau pathology support the theory that the formation of plaques or tangles alone is not sufficient to reproduce the severe neurodegenerative phenotype of AD [7-9]. While the mechanism of neuronal loss in AD remains elusive, studies of human AD and transgenic mice have shown that aberrant cell cycle re-entry occurs in neurons and may play a role in neurodegeneration [10-18].
The tyrosine kinase c-Abl (Abl, Abl1) is a cell cycle regulator, and previous studies have suggested a role for c-Abl in neurodegeneration. Abl is upregulated and plays a role in neuronal apoptosis in cell culture and rodent brains exposed to oxidative stress or fibrillar Aβ [19-23]. Inhibition of c-Abl prevented both Purkinje cell death in Niemann-Pick C mice and Aβ42-induced death in Drosophila neuronal cell culture and led to decreases in the levels of phospho-tau in AβPPsw/PSEN1ΔE9 mice [23-26]. Abl co-localizes with plaques, tangles, and granulovacuolar degeneration bodies (GVD) in human AD and phosphorylates tyrosines 197, 310, and 394 of tau [27-29]. Both the amyloid-β protein precursor (AβPP) and one of its adaptor proteins, Fe65, have been shown to be phosphorylated and possibly regulated by c-Abl [30-32]. Together, these findings suggest that c-Abl may be involved in the pathogenesis of neurodegeneration and AD.
In this study, we investigate the effects of overexpression of active c-Abl and its sister protein, Arg (Abl2), in adult neurons. We have developed two lines of transgenic mice expressing either constitutively active c-Abl (AblPP/tTA mice) or Arg (ArgPP/tTA mice) specifically in neurons of the forebrain under the inducible tet-off system. Both c-Abl and Arg phosphorylate tyrosine 394 of tau, and the kinases are highly homologous, sharing approximately 90% of their sequence in their kinase domains [29, 33]. Our models demonstrate that neuronal over-expression of constitutively active c-Abl, but not Arg, induces severe, progressive neurodegeneration in the CA1 region of the hippocampus and early reactive gliosis.
MATERIALS AND METHODS
Mice
Double transgenic AblPP/tTA and ArgPP/tTA mice were generated by breeding two single transgenic lines of mice. Female AblPP or ArgPP mice, generated in our laboratory, were bred with male CamKIIα-tTA (tTA) mice [34] purchased from Jackson Labs. AblPP and ArgPP single transgenic mice were produced by microinjection of a genetic construct encoding a constitutively active form of c-Abl 1 b (AblPP) or Arg (ArgPP) [29, 35] under the tet-responsive PhCMV–1 promoter, pTRE2 (Clontech) into embryos of FVB mice. Each construct was mutated in the conserved proline residues of the SH2-SH3 linker region of the protein, P242E/P249E for AblPP and P269E/P276E for ArgPP. Transgene insertion was verified by PCR of genomic DNA from mouse tail tissue.
Primers for AblPP:
forward, 5′AGCAGAGCTCGTTTAGTGAACCGT3′;
reverse, 5′GGGCTTCGTGTTCCACAAAGACAT3′.
Primers for ArgPP:
forward, 5′AGCAGAGCTCGTTTAGTGAACCGT3′;
reverse, 5′CTCCACACAGCTGGCAAAGTG3′.
Double transgenic AblPP/tTA or ArgPP/tTA mice carrying both transgenes were produced by breeding of female AblPP or ArgPP mice with male tTA mice. Breedings resulted in the expected ratio of genotype, with 25% of pups carrying both transgenes. The presence of both transgenes was verified by PCR of genomic DNA from tail biopsies. Primers, as above for AblPP and ArgPP, primers for tTA:
forward, 5′CGCTGTGGGGCATTTTACTTTAG3′;
reverse, 5′CATGTCCAGATCGAAATCGTC3′.
All mice were kept in a facility with a 12-h light/dark cycle and provided with food and water ad libitum. Breeding mice were kept on a doxycycline (dox) diet with 200 mg/kg doxycycline (Bio-Serv) throughout breeding, pregnancy, and weaning. Both control and experimental mice were weaned at approximately three weeks of age and were taken off doxycycline at approximately four weeks of age. Mice were euthanized with isoflorane, decapitated, and brains were hemisected sagitally. One hemisphere of each brain was homogenized and frozen for biochemical analysis, while the contralateral hemisphere was fixed in 4% paraformaldehyde overnight at 4°C for histological analysis.
Antibodies
Antibodies are listed in Table 1.
Table 1.
Antibodies
| Group | Antibody | Epitope | Dilution
|
Source | |||
|---|---|---|---|---|---|---|---|
| IB | IF | IHC | ELISA | ||||
| Abl | 2411 | c-Abl C-terminus | 1 : 1000 | Santa Cruz | |||
| K12 | c-Abl kinase domain | 1 : 2000 | 1 : 4000 | Santa Cruz | |||
| pY412 | c-Abl pY412 | 1 : 1000 | Cell Signaling | ||||
| Arg | AR19 | Arg N-terminus | 1 : 500 | 1 : 1000 | Peter Davies | ||
| AR23 | Abl, Arg N-terminus | 1 : 1000 | Peter Davies | ||||
| P-Tyr | 4 G10 | Phospho-tyrosine | 1 : 1000 | 1 : 1000 | 1 : 5000 (tau) | Millipore | |
| 1 : 1000 (Abl) | |||||||
| Neurons | NeuN | NeuN | 1 : 10000 | Millipore | |||
| Astrocytes | GFAP | GFAP | 1 : 1000 | 1 : 500 | Invitrogen | ||
| Microglia | Iba-1 | Iba-1 | 1 : 1000 | Wako | |||
| Tau | DA31 | Tau, 150–190 | Coating 2 μg/ml | Peter Davies | |||
| PHF-1 | Tau pS396/pS404 | 1 : 5000 | Peter Davies | ||||
| CP13 | Tau pS202 | 1 : 5000 | Peter Davies | ||||
| MC-1 | Tau, 5–15, 312–322 | 1 : 200 | Peter Davies | ||||
| YP4 | Tau pY394, pS396 | 1 : 100 | Peter Davies | ||||
| DA9* | Tau, 102–150 | 1 : 5000 | Peter Davies | ||||
| Amyloid | 4G8 | β-amyloid,17–24 | 1 : 1000 | Covance | |||
Biotinylated.
Immunohistochemistry
For all immunohistochemistry experiments, cohorts of 5–6 mice of the same gender were analyzed at each time point. Single transgenic mice were used as controls. Immunohistochemistry was performed on hemisected brains fixed overnight in 4% paraformaldehyde. Fixed brains were cut sagitally into 50 μm sections using a vibratome. Sections were stained according to standard immunohistochemistry protocols. Briefly, sections were incubated in 3% H2O2 in 0.25% TritonX 100 in TBS for 30 min at room temperature. Sections were blocked in 5% milk /TBS for 1 h at room temperature and incubated in primary in 5% milk/TBS at 4°C overnight. Sections were washed, and biotinylated secondary antibody 1 : 1000 in 20% Superblock (Pierce) in TBS + 0.05% Triton X100 was applied for 2 h at room temperature. Sections were washed and incubated with Streptavidin-HRP 1 : 1000 in 20% Superblock (Pierce) in TBS + 0.05% Triton X100 for 1 h at room temperature. Staining was visualized with diaminobenzidine (DAB).
Immunofluorescence
Vibratome sections of AblPP/tTA, ArgPP/tTA, and control mouse brains were incubated in 3% H2O2 in 0.25% TritonX 100 in TBS for 30 min at room temperature. Sections were blocked in 5% milk for 1 h at room temperature and incubated in primary antibodies (K12, AR19, and AR23) diluted in 5% milk at 4°C overnight. Sections were washed, and biotinylated secondary antibody 1 : 1000 in 20% Superblock (Pierce) in TBS + 0.05% TritonX100 was applied for 2 h at room temperature. Sections were washed and incubated in Alexafluor568 conjugated streptavidin diluted 1 : 1000 in 20% Superblock (Pierce) in TBS + 0.05% TritonX100 for 1 h at room temperature and mounted on microscope slides. Following a 30-min incubation in 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen), sections were washed and then incubated in 0.3% Sudan Black in 70% EtOH for 10 min at room temperature. Finally, slides were cover-slipped using Prolong® Gold anti-fade reagent with DAPI (Invitrogen). A Leica SP2 Scanning Laser Confocal Microscope was used to visualize fluorescence.
Western blotting
Cohorts of 5–6 double transgenic mice (AblPP/tTA or ArgPP/tTA) were analyzed at each time point. Single transgenic mice were used as controls. Brain hemisections were dissected into forebrain and hindbrain. In some cases, hippocampus and cerebral cortex were dissected away from the rest of the forebrain and used for immunoblotting, ELISA, and kinase activity assays. The brains were homogenized in homogenization buffer (TBS with 10 mM NaF, 1 mM Na3VO4, 2 mM EGTA) with complete Mini protease inhibitors (Roche) and kept at −80°C. Prior to use, homogenates were thawed and spun at 14,000 rpm for 10 min at 4°C. Supernatants were analyzed in all experiments by SDS-PAGE. Western blotting was performed according to standard protocols. Protein concentrations were normalized prior to use and all western blots were normalized to β-actin or tubulin loading controls.
Abl kinase assay
Supernatants of single transgenic control, AblPP/tTA, and ArgPP/tTA mouse brain homogenates (12.5 μg protein per reaction) were added to ice cold mastermix consisting of: 5 μl 10X kinase buffer (200 mM HEPES, pH 7.4, 10 mM MnCl2, 10 mM MgCl2, 10 mM DTT, 10 mM Na3VO4, 10 mM NaATP) + 5ug Abltide-Biotin (Millipore) + ddH2O to 50 μl. Recombinant Abl enzyme (1 μg/reaction) was used as a positive control. Reactions were incubated at 30°C for 30 min. Reaction was stopped by addition of 950 μl ice-cold 2% BSA. Cohorts of 4 mice per time point were used.
Abl activity ELISA
Flat-bottomed 96 well plates were coated with neutravidin diluted in coating buffer (20 mM K2HPO4, 20 mM KH2PO4, 0.8% NaCl, 1 mM EDTA, 0.05% NaN3, pH 7.2) overnight at 4°C. Samples (n = 4 per timepoint) were diluted 1 : 40 (1 : 800 final dilution) and added to the top row of the plate in duplicate. Samples were serial-diluted down the plate and allowed to incubate for 1 h at room temperature. A biotinylated peptide derived from tau (biotin-KSGDRSG(pY)SSPGSPG) was used as a phosphopeptide standard. Plates were washed, and 4G10 anti-phosphotyrosine antibody (Millipore) diluted 1 : 1000 in 5% milk/TBS was added to each well, allowed to incubate for 1 h at room temperature on a shaker. Plates were washed, and anti-IgG2b HRP-conjugated secondary (Southern Biotech) diluted 1 : 1000 in 5% milk/TBS was added to each well and incubated for 1 h at room temperature. After washing, plates were developed with Horseradish Peroxidase Substrate Kit (Biorad). Color was allowed to develop for 15 min. Plates were read with an Infinite m200 plate reader (Tecan) at 405 nm.
Phospho-Tau ELISA
Flat-bottomed 96 well plates were coated with purified anti-tau antibody (DA31) (2 μg/mL) [36] in coating buffer overnight at 4°C. Plates were blocked with Starting Block (Pierce) for 90 min at room temperature with shaking. Samples (n = 4 per timepoint) were diluted in 20% Superblock (Pierce) in TBS, added to the plate, and serial diluted then incubated overnight at 4°C with shaking. After washing, either 4G10 anti-phosphotyrosine (Millipore) or biotinylated DA9, diluted in 20% Superblock (Pierce) -TBS was added to the plate and incubated for 1 h at room temperature. After washing, HRP conjugated anti-IgG2b or streptavidin-HRP (Southern Biotech), diluted 1 : 1000 in 5% Milk/TBS was added to the plate and allowed to incubate for 1 h at room temperature. Plates were washed and TMB-Ultra (Pierce) was added to each well for 20 min. TMB reaction was stopped with 4N H2SO4. Plates were read with an Infinite m200 plate reader (Tecan) at 450 nm. Biotinylation of DA9 was peformed using EZ-link NHS-PEO solid phase biotinylation kit (Pierce).
Statistical analysis
One-way ANOVA with Dunnett’s post-test was performed using Graphpad Prism.
RESULTS
Transgene expression is doxycycline dependent and forebrain-specific in AblPP/tTA mice
Forebrain-specific neuronal expression of constitutively active c-Abl (AblPP), consistent with CamKIIα promoter expression, occurred in AblPP/tTA mice (Fig. 1A, B). Abl-PP appeared to be confined to the neuronal cytoplasm, and was not found in nucleus (Fig. 1B). There was dense neuritic staining, in addition to staining of neuronal cell bodies. There was no significant AblPP expression in the cerebellum or brainstem, and no expression of the AblPP transgene was detectable in the absence of the CamKIIα-tTA gene (data not shown). In order to confirm that AblPP expression was tightly regulated, we placed AblPP/tTA mice back on a doxycycline diet after 10 weeks off, for either two or four weeks. AblPP expression was drastically reduced after two weeks back on doxycycline and decreased to control levels after four weeks back on doxycycline, confirming that AblPP expression is tightly regulated (Fig. 1C).
Fig. 1.
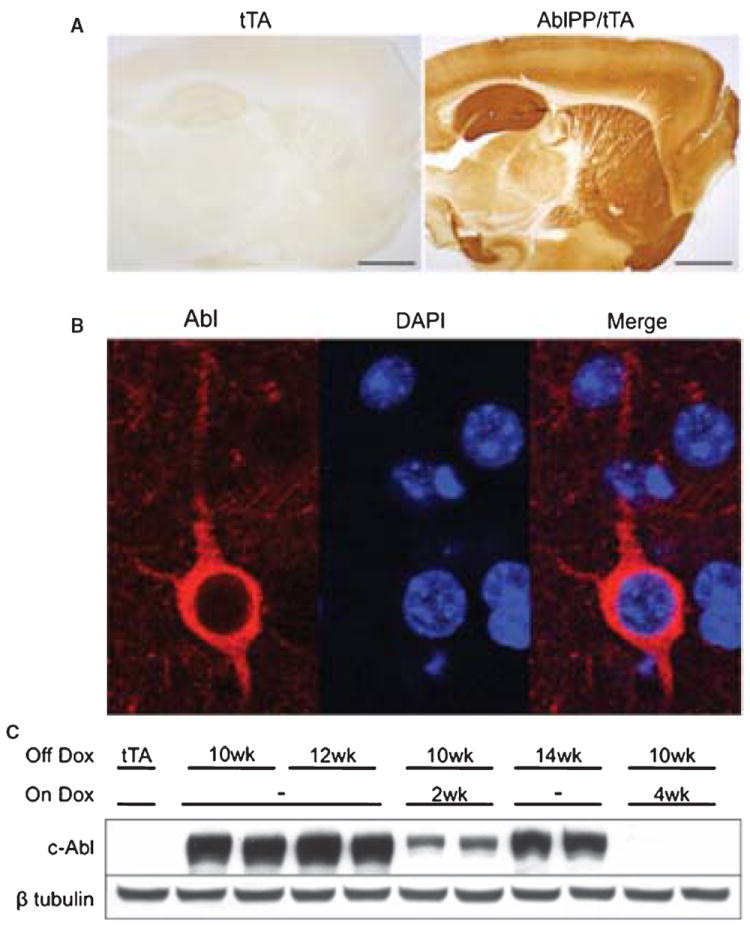
Abl expression in AblPP/tTA mice. A) c-Abl is expressed and active in AblPP/tTA mice. Representative sections of single transgenic control mouse and AblPP/tTA mouse 3 weeks off dox stained for c-Abl (K12). B) c-Abl (K12) is present in cell bodies of neurons in AblPP/tTA mice. Immunofluorescence (K12 – red, DAPI – blue) of cortex of an AblPP/tTA mouse 2 weeks off dox. C) Abl expression is doxycyline dependent. Abl (2411) immunoblot of AblPP/tTA mouse cortex 10 weeks off dox with or without introduction of dox back into diet for 2 or 4 weeks.
AblPP/tTA mice have significantly increased c-Abl kinase activity in the forebrain
Abl activity in mouse forebrain was measured by kinase assay (Fig. 2A, B). Cohorts of 4 AblPP/tTA mice per timepoint exhibited 20–30 fold increases in c-Abl kinase activity over single transgenic control mice, as measured by Abltide peptide phosphorylation, beginning as early as three weeks off doxycycline. No significant differences in Abl activity were found in AblPP/tTA mice at various times off doxycycline. Male AblPP/tTA mice did not survive past 14 weeks off doxycycline, so later time points were not examined. Female AblPP/tTA mice survived to 24 weeks off doxycycline. The reasons for the differences in survival between male and female AblPP/tTA mice remain unclear, as there were no significant differences in Abl activity between male and female AblPP/tTA mice. Western blots showed increased c-Abl phosphorylation at tyrosine 412 (Fig. 2C) in AblPP/tTA mice, a modification associated with increased kinase activity [37, 38].
Fig. 2.
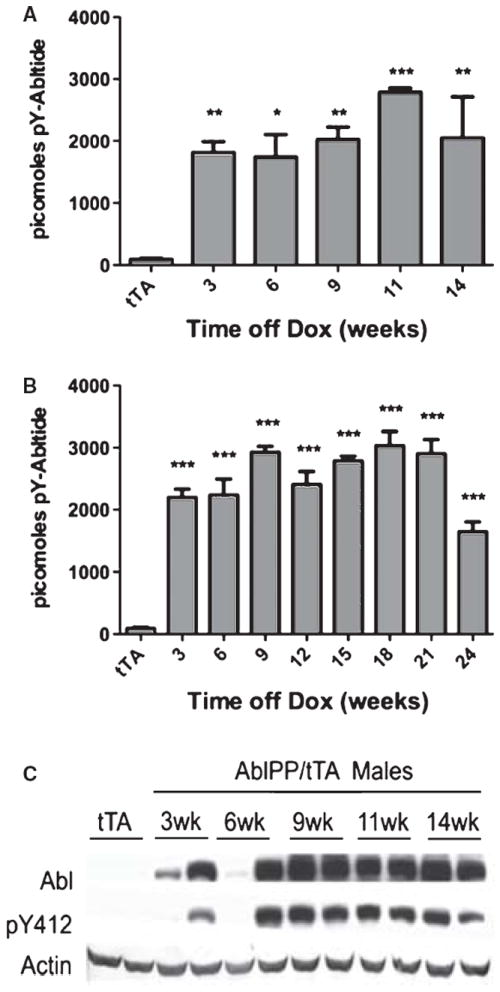
Abl activity in AblPP/tTA mice. A) Male AblPP/tTA mice. B) Female AblPP/tTA mice. Enzyme activity ELISAs of AblPP/tTA mouse cortex. Average picomoles of Abltide phosphorylated per kinase reaction, n = 4 per timepoint. One-way ANOVA with Dunnett’s post-test was used to calculate significance of Abltide phosphorylation in AblPP/tTA mice vs. single transgenic (tTA) controls. *p < 0.05, **p < 0.01, ***p< 0.001. C) Total c-Abl (2411) and phospho-Abl (pY412) immunoblots of male AblPP/tTA mouse cortex 3, 6, 9, 11, and 14 weeks off dox.
AblPP transgene expression leads to abundant microgliosis and astrogliosis in the forebrain
Iba1 immunohistochemistry revealed microgliosis in the hippocampi, particularly CA1 regions, of AblPP/tTA mice beginning at approximately 3 weeks off doxycycline. Iba1 staining in the hippocampus continued to be elevated until approximately 18 weeks off doxycycline, after which it began to return to control levels (Fig. 3). Microgliosis appears to be one of the earliest pathological consequences of constituitively active c-Abl expression in the neurons of AblPP/tTA mice.
Fig. 3.
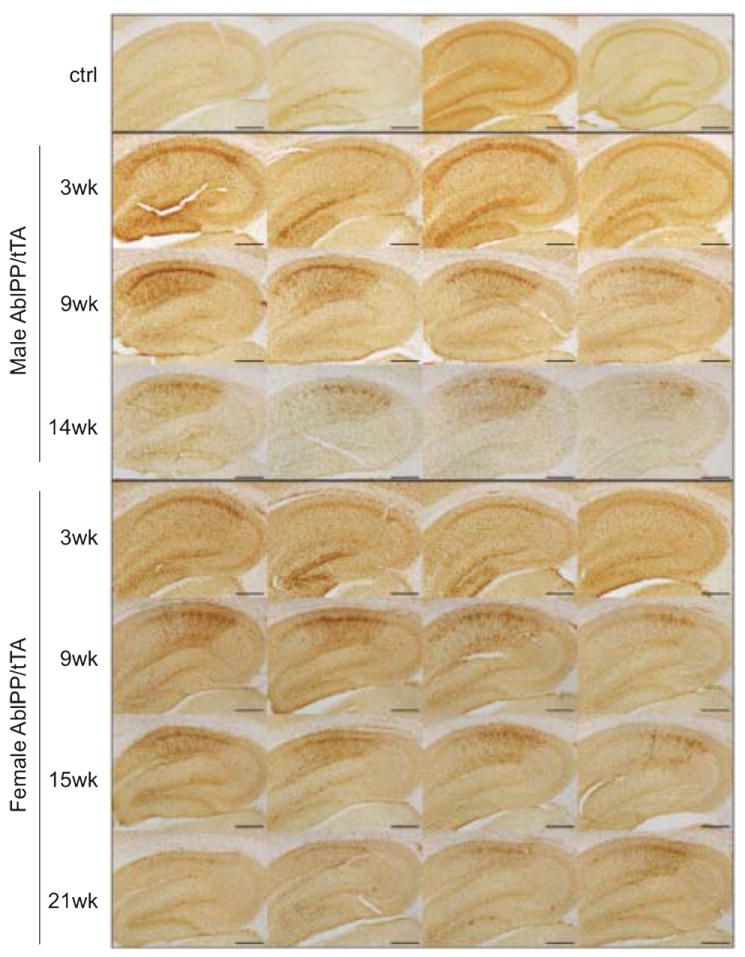
Microgliosis in AblPP/tTA mice. Iba1 immunohistochemistry of AblPP/tTA male and female hippocampi. Top row shows 4 separate single transgenic control mice. Subsequent rows show 4 different mice per timepoint, with #wk indicating weeks off dox. Scale bars = 2 mm.
GFAP immunohistochemistry was also performed on AblPP/tTA mice, and increases in GFAP staining were found in hippocampi of AblPP/tTA mice. GFAP staining was increased in 3 of the AblPP/tTA mice 6 weeks off doxycycline and in all AblPP/tTA mice 9 weeks off doxycycline. It remained high throughout mice at all later time points, with a slight drop in staining in the oldest (24 weeks off dox) mice (Fig. 4). These data suggest that GFAP elevation is another early pathological consequence of neuronal c-Abl expression.
Fig. 4.
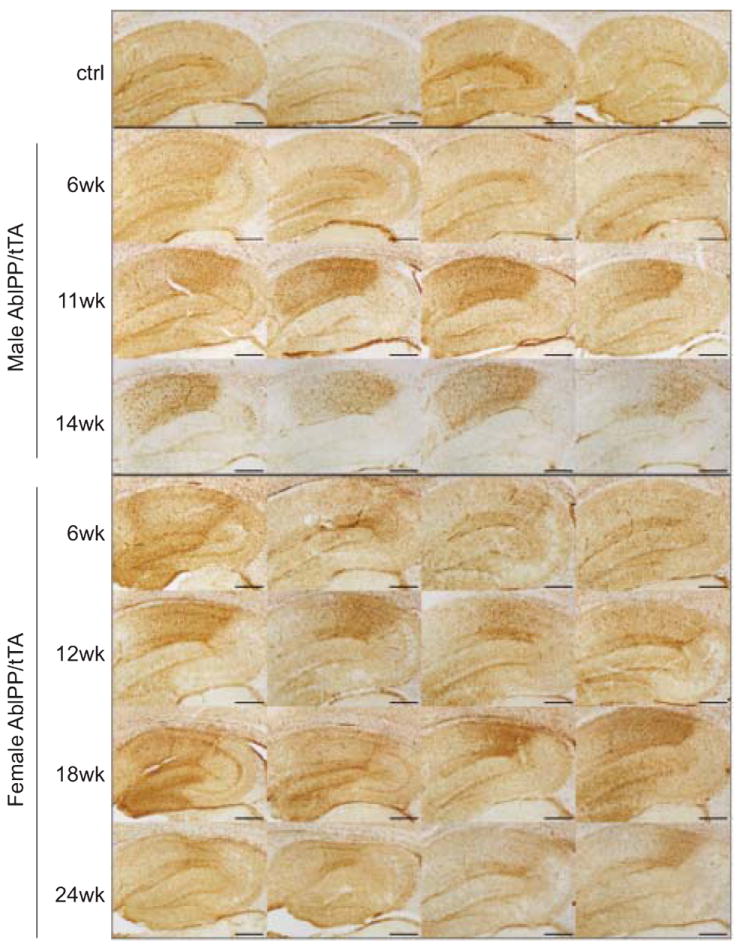
Reactive Astrocytosis in AblPP/tTA mice. GFAP immunohistochemistry of AblPP/tTA males and females throughout the hippocampus. Top row shows 4 separate single transgenic control mice. Subsequent rows show 4 different mice per timepoint, with #wk indicating weeks off dox. Scale bars = 2 mm.
There appears to be an early elevation of microglial markers, beginning at 3 weeks off doxycycline, with an elevation of astrocytic markers beginning between 6 and 9 weeks off doxycycline. While Iba1 levels began to drop after approximately 18 weeks off doxycycline, GFAP levels remained high until approximately 21 weeks off doxycycline. Morphology of microglia and astrocytes in the CA1 region of the hippocampus can be seen in Supplementary Figure 1 (available online: http://www.j-alz.com/issues/25/vol25-1.html#supplementarydata05). Despite careful examination, no c-Abl transgene expression was detected in cells other than neurons (data not shown).
AblPP transgene expression leads to progressive neurodegeneration in the CA1 region of the hippocampus
We observed progressive neuronal loss in the CA1 region of AblPP/tTA mice, culminating in nearly complete ablation of neurons in the CA1 region in the majority of the oldest mice (21 and 24 weeks off dox). We performed NeuN immunohistochemistry on cohorts of 5–6 AblPP/tTA mice at each time point for each gender. NeuN staining in representative sections of the CA1 regions of four different AblPP/tTA mice at the latest time points examined for males (9–14 weeks off dox) and females (18–24 weeks off dox) is shown in Fig. 5A. Neuronal loss was progressive with time off doxycycline. Mice did not begin to display obvious changes in NeuN staining until approximately 9 weeks off doxycycline, and the most striking loss occurred in female mice 18 weeks or more off doxycycline. As stated previously, male AblPP/tTA mice did not survive past 14 weeks off doxycycline, so later time points could not be examined. Neuronal loss was most obvious in CA1, as shown in Fig. 5B.
Fig. 5.
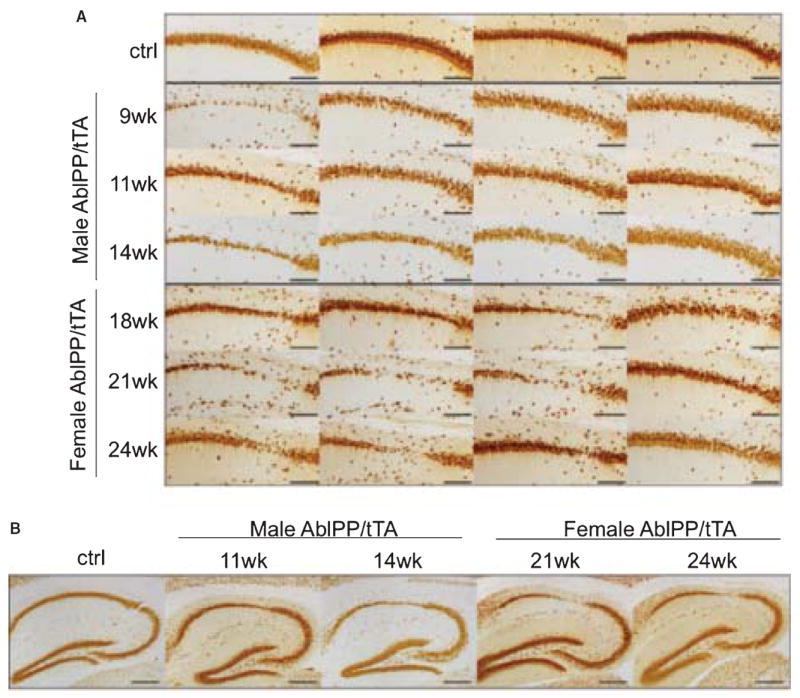
Neurodegeneration in CA1 of AblPP/tTA mice. A) NeuN staining of AblPP/tTA male and female mice CA1. Top row shows 4 separate single transgenic control mice. Subsequent rows show 4 different mice per timepoint, with #wk indicating weeks off dox. Scale bars = 800 μm. B) NeuN immunohistochemistry of control vs. male and female AblPP/tTA mouse hippocampi. Scale bars = 2 mm.
The pathology of degeneration and neuroinflammation in the CA1 region of AblPP/tTA mice was scored based upon three criteria: degree of neuronal loss as observed by NeuN, degree of astrogliosis as observed by GFAP, and degree of microgliosis, as observed by Iba1 (Table 2). All AblPP/tTA animals were given a score of none, + (mild), ++ (moderate), or +++ (severe). A score of none indicates no differences observed in any of the three criteria between the experimental animal and control animals. A score of + indicates increases in one or both inflammatory markers. A score of ++ indicates moderate neuronal loss in the CA1 region as observed by NeuN immunohistochemistry in combination with highly elevated Iba1 and GFAP staining. A score of +++ indicates severe neuronal loss in the CA1 with complete ablation of part of the CA1 in some cases. Out of the 28 mice we examined that had been off doxycycline for 14 or more weeks, 18 exhibited moderate (++) to severe (+++) neuronal loss. Of the mice that were off doxycyline for 18 or more weeks, 10 out of 16 displayed severe neuronal loss, with gaps or holes in CA1, and 13 out of 16 displayed moderate to severe neuronal loss, with moderate to severe thinning of the CA1 pyramidal layer of the hippocampus. Only 3 out of the 49 AblPP/tTA mice that were 9 weeks or more off doxycycline exhibited a similar phenotype to controls, and the majority of the mice more than 14 weeks off doxycycline exhibited a moderate (++) to severe (+++) neurodegenerative phenotype. The mechanism of the c-Abl-induced neuronal loss remains unclear; however, both TUNEL staining and activated caspase 3 Western blotting in AblPP/tTA mice (data not shown) were negative, suggesting that apoptosis is unlikely.
Table 2.
| Time off Dox | Pathology
|
Total | ||||
|---|---|---|---|---|---|---|
| None | + | ++ | +++ | |||
| Males | 3 weeks | 3 | 3 | 0 | 0 | 6 |
| 6 weeks | 4 | 1 | 1 | 0 | 6 | |
| 9 weeks | 0 | 2 | 1 | 2 | 5 | |
| 11 weeks | 0 | 2 | 3 | 1 | 6 | |
| 14 weeks | 1 | 2 | 1 | 2 | 6 | |
| Females | 3 weeks | 4 | 2 | 0 | 0 | 6 |
| 6 weeks | 1 | 4 | 0 | 0 | 5 | |
| 9 weeks | 0 | 2 | 1 | 1 | 4 | |
| 12 weeks | 1 | 4 | 0 | 1 | 6 | |
| 15 weeks | 1 | 3 | 0 | 2 | 6 | |
| 18 weeks | 0 | 1 | 1 | 3 | 5 | |
| 21 weeks | 0 | 0 | 1 | 4 | 5 | |
| 24 weeks | 0 | 2 | 1 | 3 | 6 | |
Numbers indicate the number of mice in each cohort exhibiting a particular score. None indicates no differences observed in Iba1, GFAP, or NeuN staining between the experimental animal and control animals. + Indicates increases in one or both inflammatory markers. ++ Indicates moderate neuronal loss in the CA1 region as observed by NeuN immunohistochemistry in combination with highly elevated Iba1 and GFAP staining. +++ Indicates severe neuronal loss in the CA1 with complete ablation of the CA1 region in some cases.
Tau and amyloid pathology in AblPP/tTA mice
Neither tau nor Aβ appear to play a significant role in the neurodegenerative phenotype of AblPP/tTA mice. We found no evidence of amyloid pathology by immunohistochemistry with the antibody 4 G8 (data not shown). We performed immunohistochemistry using YP4, CP13, PHF-1, and MC-1 antibodies to tau dually phosphorylated at tyrosine 394 and serine 396, tau phosphorylated at serine 202, tau phosphorylated at serines 396 and 404, and total tau in an abnormal conformation found in tangles in AD, respectively. ELISA analysis showed a significant increase in tyrosine-phosphorylated tau in male AblPP/tTA mice at 6 and 11 weeks off doxycyline, but not at 9 and 14 weeks off doxycycline (Fig. 6A). Female AblPP/tTA mice showed significant increases in tyrosine-phosphorylated tau at all timepoints except 24 weeks off doxycycline (Fig. 6B). While the literature suggests that c-Abl primarily phosphorylates tyrosine 394 of tau, the question of which specific tyrosine residues of tau were being phosphorylated in our model could not be addressed due to a lack of antibodies to specific phospho-tyrosine sites on tau. We observed no difference in staining patterns between control and AblPP/tTA mice using the antibodies YP4, CP13, and MC-1 (data not shown). Sporadic PHF-1 positive cell bodies were observed the hippocampus and cortex of some AblPP/tTA mice between 6 and 12 weeks off doxycycline (Fig. 6C–E). However, PHF-1 staining was not a consistent finding in AblPP/tTA mice.
Fig. 6.
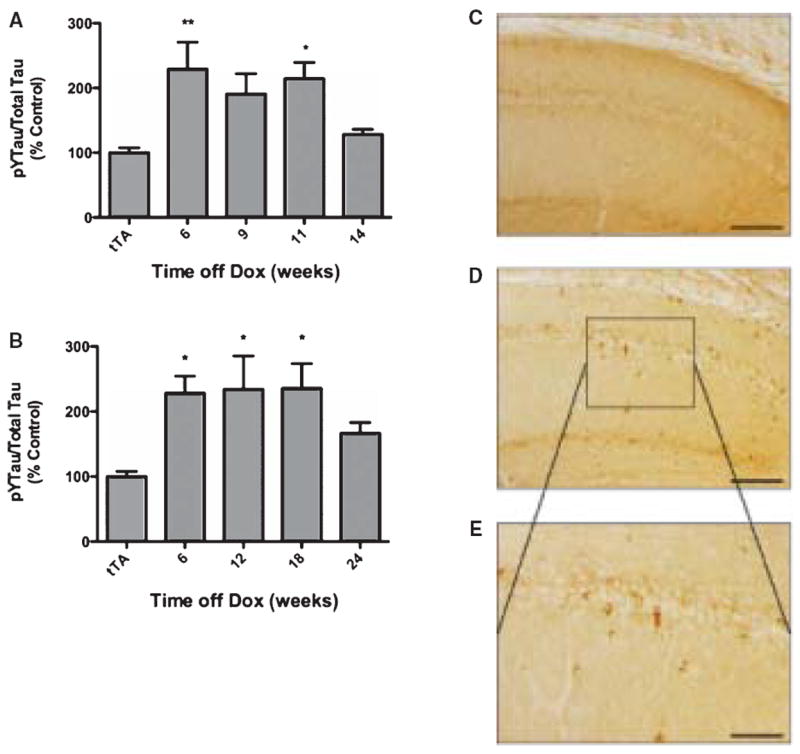
Tau hyperphosphorylation in AblPP/tTA mice. A, B) ELISA analysis of tyrosine phosphorylated (4G10) tau/total (DA9) tau in male (A) and female (B) AblPP/tTA mice. n = 4 for each timepoint. One-way ANOVA with Dunnett’s post-test was used to calculate whether AblPP/tTA mice showed significant increases in tau phosphorylation when compared to control (tTA) mice. *p < 0.05, **p < 0.01, ***p < 0.001. C–E) PHF1 staining of representative sections of AblPP/tTA mouse CA1 (D, E) versus controls (C). Scale bars = 800 μm (C, D), 400 μm (E).
ArgPP/tTA mice do not exhibit the same pathological phenotype as AblPP/tTA mice, despite similar levels of transgene expression and kinase activity
We created a second line of transgenic mice expressing a constitutively active form of the Arg protein (ArgPP), the other member of the Abl family of kinases, under the forebrain specific CamKIIα promoter and the inducible tet-off system. We refer to this line of mice as ArgPP/tTA mice.
Densitometry measurements of Western blots of cortex homogenates of AblPP/tTA and ArgPP/tTA with the AR23 antibody, an antibody with equal affinity for both Abl and Arg proteins, showed that there was no significant difference in protein expression in the cortex of the two lines of mice at both 12 and 24 weeks off doxycycline (Fig. 7A, B). Additionally, Abl kinase assay analysis showed that both lines of transgenic mice had similar levels of kinase activity at 12 and 24 weeks off doxycycline (Fig. 7D).
Fig. 7.
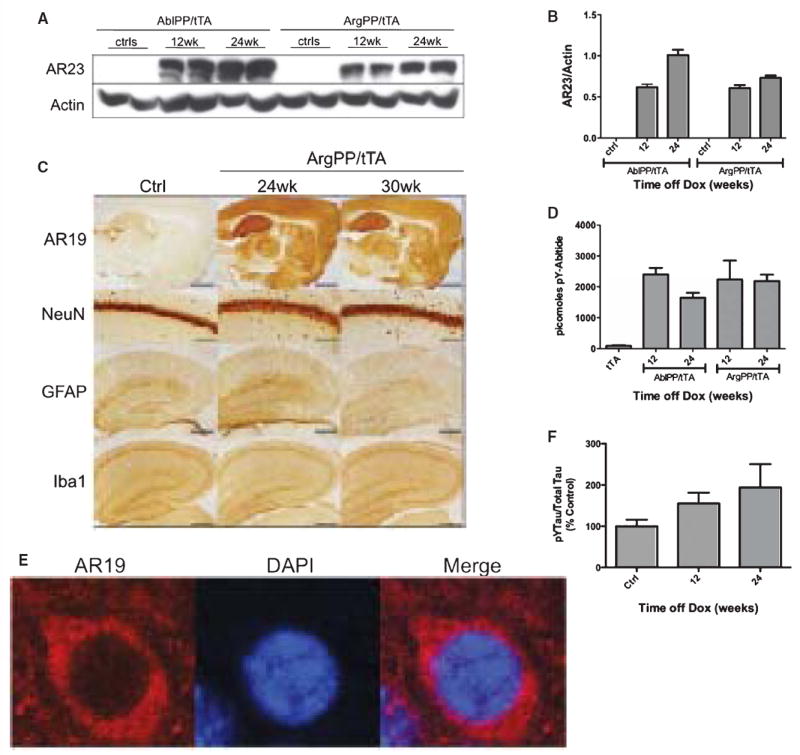
ArgPP/tTA mice do not differ significantly from controls. A) AblPP/tTA and ArgPP/tTA exhibit similar levels of protein expression. Western blotting with AR23 antibody of AblPP/tTA and ArgPP/tTA mouse cortex homogenates 12 and 24 weeks off doxycycline. B) Densitometry analysis of Western blotting in 7A. No significant differences were found between AblPP and ArgPP expression in the two lines of mice. C) Representative sections of control mice and ArgPP/tTA mice 24 and 30 weeks off dox stained for AR19, NeuN, Iba1, and GFAP. Scale bars = 8 mm (AR19), 800 μm (NeuN), 2 mm (GFAP and Iba1). D) Abl activity ELISAs of AblPP/tTA and ArgPP/tTA mouse cortex. Average picomoles of Abltide phosphorylated per kinase reaction, n = 4 per timepoint. E) Arg is expressed in neuronal cell bodies in the ArgPP/tTA mouse. Immunofluorescence of ArgPP/tTA mouse cortex with AR19 (red) and DAPI. F) ELISA analysis of tyrosine phosphorylated (4G10) tau/total (DA9) tau in ArgPP/tTA mice 12 and 24 weeks off dox. n = 4 for each timepoint. One-way ANOVA with Dunnett’s post-test was used to calculate whether ArgPP/tTA mice showed significant increases in tau phosphorlylation when compared to control mice. No significant differences in tau phosphorylation were found.
Despite similar levels of protein expression and kinase activity, ArgPP/tTA mice showed none of the pathological features of AblPP/tTA mice. There was no difference in Iba1, GFAP, or NeuN staining in any of the 36 ArgPP/tTA mice that were examined up to 30 weeks off doxycycline when compared with controls (Fig. 7C). Arg localized to neuronal cell bodies in ArgPP/tTA mice, and did not appear to localize to the nucleus (Fig. 7E). ArgPP/tTA mice exhibited no significant increases in tau tyrosine phosphorylation by ELISA analysis (Fig. 7F).
DISCUSSION
Our model, the AblPP/tTA mouse, develops neurodegeneration and reactive gliosis, both hallmarks of neurodegenerative disease, but no significant plaque or tangle formation, despite the fact that the c-Abl protein is associated with these characteristic lesions of AD in human brain. Together with previous studies showing c-Abl localization in plaques, tangles, and GVD and tyrosine phosphorylated tau in tangles [27-29, 39], our data suggest that c-Abl could be a player in the pathogenesis of neurodegeneration and neuroinflammation associated with AD.
In AD, devastating, forebrain-specific neurodegeneration occurs. Our model, the AblPP/tTA mouse, has severe, progressive neurodegeneration in the CA1 region due to the expression of constitutively active c-Abl in neurons. We focused on the CA1 region in AblPP/tTA mice, as the dense pyramidal cell layer is a well-defined anatomical landmark, and cell loss in this region is clearly seen. The expression of c-Abl throughout the forebrain (Fig. 1) combined with reactive gliosis in the hippocampus (Figs 3, 4) and other areas of the forebrain (data not shown) suggests that neuronal damage or loss may occur in other regions. That activation of c-Abl in neurons leads to neuronal loss in our model suggests that c-Abl may be a potential therapeutic target for neurodegenerative diseases.
Others have shown that in AD, there is an increase in reactive microglia accompanied by large increases in GFAP expression and reactive astrocytosis, especially associated with amyloid plaques [40-42]. In at least one tau transgenic mouse model, microglial activation appears to precede tangle formation [43]. We observe a reactive microgliosis and astrogliosis in our model in the absence of significant tau or amyloid pathology. Early Iba1 and GFAP elevation may be an indicator of early neuronal damage or loss that is not detectable by NeuN staining. The decreases in Iba1 and GFAP staining in the oldest AblPP/tTA mice seem to suggest that the neuroinflammation is due, at least in part, to neuronal damage and that once the affected neurons have died and any remaining debris have been cleared, the glial response may no longer be necessary. However, whether or not the neuroinflammation is solely a response to neuronal damage, or is a critical component leading to neuronal damage, or both, remains unclear. Future studies are necessary to determine the exact nature of the neuroinflammation and what cytokines and signaling molecules may be involved.
Despite careful examination, there was no detectable expression of the AblPP transgene in either microglia or astrocytes in the AblPP/tTA mice. These results imply that gliosis occurs in response to neuronal expression of active c-Abl. While elevation of inflammatory markers can occur in many types of neuronal injury, the duration of the inflammatory pathology in our model is more consistent with a chronic inflammatory phenotype like that observed in neurodegenerative diseases. Although our studies primarily focused on the CA1 pyramidal layer, we also observed changes in Iba1 gliosis throughout the forebrain, and particularly in the striatum. The AblPP/tTA mouse may be a useful model for studying neuroinflammation in a system closely mimicking human neurodegenerative disease in the absence of amyloid plaques and significant NFT.
The AblPP/tTA mouse shows no significant evidence of tau or amyloid pathology. While tyrosine phosphorylation of tau does occur, our model shows no significant evidence of neurofibrillary tangle pathology or tau accumulation. Despite increases in tyrosine-phosphorylated tau, our model has only sporadic staining with the PHF-1 antibody and no NFTs. It has been difficult to reproduce “tangle-like” pathology in mouse models expressing only endogenous mouse tau, as our model does, with only a few known models where this occurs [10, 43-45]. Thus, it is perhaps unsurprising that we observed only sporadic and mild tau pathology in the hippocampus of the AblPP/tTA mouse. It was not possible to determine the specific tyrosine residues that were phosphorylated in our model due to the lack of antibodies to specific phospho-tyrosine sites on tau that are useful for immunohistochemistry. It is possible that the tau tyrosine phosphorylation we observe by ELISA may interfere with the binding of antibodies to other tau epitopes, making phosphorylation at threonine and serine residues and conformational changes difficult to detect.
Studies performed in AD brain and in transgenic mouse and Drosophila models show that neurodegeneration often occurs in neurons devoid of NFT pathology, suggesting that loss of function of tau or disruption of microtubule function may play a larger role in neuronal loss than aggregation of tau [46-48]. We observe a significant increase in tyrosine-phosphorylated tau in the AblPP/tTA mouse at certain time points with minimal evidence of tau aggregation. However, we do not observe significant increases in tyrosine phosphorylation of tau in the ArgPP/tTA mice, suggesting that the tyrosine phosphorylation of tau may be a contributing factor in pathologic phenotype of the AblPP/tTA mice.
The transgenic lines described in this work are the first mouse models of c-Abl and Arg activation in adult neurons. The ArgPP/tTA mice exhibited a phenotype indistinguishable from single transgenic control mice, even up to 30 weeks off doxycycline, despite the fact that both AblPP/tTA and ArgPP/tTA mice had similar levels of kinase activity, protein expression, and protein localization in forebrain. While this result is surprising, given the 90% homology between kinase domains of c-Abl and Arg [33] and the fact that both proteins have been shown to phosphorylate tau at tyrosine 394 [27, 29], it does provide strong evidence that the neurodegenerative and neuroinflammatory pathology observed in the AblPP/tTA mice is specifically due to the effects c-Abl activation in neurons rather than transgene integration or protein overexpression. The pathological phenotype of the AblPP/tTA mouse provides strong evidence that c-Abl activation in neurons can lead to progressive neuronal loss and inflammation like that observed in AD and that c-Abl may be a potential therapeutic target in AD and other neurodegenerative diseases.
Supplementary Material
Acknowledgments
The AblPP plasmid used to subclone the AblPP ORF into the TRE2 vector and generate AblPP mice was a kind gift from Dr. Guilio Superti-Furga. This work was supported by NIMH38623 and AG022102, and NIH Medical Scientist Training Program Grant T32GM007288.
Footnotes
Supplementary data available online: http://www.j-alz.com/issues/25/vol25-1.html#supplementarydata05
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=752) The AblPP mice have been deposited in Jackson Labs under Stock number 014544.
References
- 1.Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2010;6:158–194. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Rojo LE, Fernandez JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Sheng JG, Griffin WS, Royston MC, Mrak RE. Distribution of interleukin-1-immunoreactive microglia in cerebral cortical layers: implications for neuritic plaque formation in Alzheimer’s disease. Neuropathol Appl Neurobiol. 1998;24:278–283. doi: 10.1046/j.1365-2990.1998.00122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- 5.Shaftel SS, Griffin WS, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNaull BB, Todd S, McGuinness B, Passmore AP. Inflammation and anti-inflammatory strategies for Alzheimer’s disease – a mini-review. Gerontology. 2010;56:3–14. doi: 10.1159/000237873. [DOI] [PubMed] [Google Scholar]
- 7.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 10.Park KH, Hallows JL, Chakrabarty P, Davies P, Vincent I. Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice. J Neurosci. 2007;27:2969–2978. doi: 10.1523/JNEUROSCI.0186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Husseman JW, Nochlin D, Vincent I. Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000;21:815–828. doi: 10.1016/s0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 13.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P. Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease. Neurobiol Aging. 1998;19:287–296. doi: 10.1016/s0197-4580(98)00071-2. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–784. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, Vincent I. The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer’s disease. Am J Pathol. 2000;157:1983–1990. doi: 10.1016/S0002-9440(10)64837-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao C, Leng Y, Huang W, Liu X, Kufe D. Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J Biol Chem. 2003;278:39609–39614. doi: 10.1074/jbc.M305770200. [DOI] [PubMed] [Google Scholar]
- 20.Cao C, Leng Y, Kufe D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J Biol Chem. 2003;278:29667–29675. doi: 10.1074/jbc.M301292200. [DOI] [PubMed] [Google Scholar]
- 21.Cao C, Leng Y, Li C, Kufe D. Functional interaction between the c-Abl and Arg protein-tyrosine kinases in the oxidative stress response. J Biol Chem. 2003;278:12961–12967. doi: 10.1074/jbc.M300058200. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS. Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species. Neurobiol Dis. 2004;17:326–336. doi: 10.1016/j.nbd.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Cancino GI, Toledo EM, Leal NR, Hernandez DE, Yevenes LF, Inestrosa NC, Alvarez AR. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits. Brain. 2008;131:2425–2442. doi: 10.1093/brain/awn125. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez AR, Klein A, Castro J, Cancino GI, Amigo J, Mosqueira M, Vargas LM, Yevenes LF, Bronfman FC, Zanlungo S. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 2008;22:3617–3627. doi: 10.1096/fj.07-102715. [DOI] [PubMed] [Google Scholar]
- 25.Cancino GI, Perez de Arce K, Castro PU, Toledo EM, von Bernhardi R, Alvarez AR. c-Abl tyrosinekinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Lin H, Lin TY, Juang JL. Abl deregulates Cdk5 kinase activity and subcellular localization in Drosophila neurodegeneration. Cell Death Differ. 2007;14:607–615. doi: 10.1038/sj.cdd.4402033. [DOI] [PubMed] [Google Scholar]
- 27.Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, Lenz C, Price C, Bird IN, Perera T, Kellie S, Williamson R, Noble W, Van Etten RA, Leroy K, Brion JP, Reynolds CH, Anderton BH. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci. 2005;25:6584–6593. doi: 10.1523/JNEUROSCI.1487-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jing Z, Caltagarone J, Bowser R. Altered subcellular distribution of c-Abl in Alzheimer’s disease. J Alzheimers Dis. 2009;17:409–422. doi: 10.3233/JAD-2009-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tremblay MA, Acker CM, Davies P. Tau phosphorylated at tyrosine 394 is found in Alzheimer’s disease tangles and can be a product of the Abl-related kinase, Arg. J Alzheimers Dis. 2010;19:721–733. doi: 10.3233/JAD-2010-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkinton MS, Standen CL, Lau KF, Kesavapany S, Byers HL, Ward M, McLoughlin DM, Miller CC. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J Biol Chem. 2004;279:22084–22091. doi: 10.1074/jbc.M311479200. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez MC, Vargas LM, Inestrosa NC, Alvarez AR. c-Abl modulates AICD dependent cellular responses: transcriptional induction and apoptosis. J Cell Physiol. 2009;220:136–143. doi: 10.1002/jcp.21743. [DOI] [PubMed] [Google Scholar]
- 32.Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T. The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J Biol Chem. 2001;276:19787–19792. doi: 10.1074/jbc.M100792200. [DOI] [PubMed] [Google Scholar]
- 33.Kruh GD, Perego R, Miki T, Aaronson SA. The complete coding sequence of arg defines the Abelson subfamily of cytoplasmic tyrosine kinases. Proc Natl Acad Sci U S A. 1990;87:5802–5806. doi: 10.1073/pnas.87.15.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 35.Barila D, Superti-Furga G. An intramolecular SH3-domain interaction regulates c-Abl activity. Nat Genet. 1998;18:280–282. doi: 10.1038/ng0398-280. [DOI] [PubMed] [Google Scholar]
- 36.Davies P. Characterization and use of monoclonal antibodies to tau and paired helical filament tau. Methods Mol Med. 2000;32:361–373. doi: 10.1385/1-59259-195-7:361. [DOI] [PubMed] [Google Scholar]
- 37.Brasher BB, Van Etten RA. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 2000;275:35631–35637. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- 38.Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- 39.Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol. 1998;149:329–340. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- 41.Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209. doi: 10.1016/s0006-8993(03)02361-8. [DOI] [PubMed] [Google Scholar]
- 42.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging. 2004;25:663–674. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 44.Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:12867–12872. doi: 10.1073/pnas.0601075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 47.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 48.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.