

Abstract
We used population- and individual-based genetic approaches to assess barriers to movement in black-backed woodpeckers (Picoides arcticus), a fire-specialist that mainly occupies the boreal forest in North America. We tested if male and female woodpeckers exhibited the same movement patterns using both spatially implicit and explicit genetic analyses to define population structure and movement patterns of both sexes among populations. Three genetic groups were identified, a large, genetically continuous population that spans from the Rocky Mountains to Quebec, a small isolated population in South Dakota and a separate population in the western portion of their distribution (Oregon). Patterns of genetic diversity suggest extensive gene flow mediated by both males and females within the continuous boreal forest. However, male-mediated gene flow is the main form of connectivity between the continuously distributed group and the smaller populations of South Dakota and Oregon that are separated by large areas of unforested habitat, which apparently serves as a barrier to movement of female woodpeckers.
Keywords: microsatellite, movement barriers, mtDNA, Picoides arcticus, population genetic structure, sex-biased movement
Introduction
Dispersal is a central process of interest in evolution and ecology, yet many aspects of dispersal are poorly understood. The movement of individuals and their genes has a long-lasting influence on the evolutionary trajectory of a population, as well as on current demographic population dynamics (Clobert et al. 2001). Barriers to dispersal can be characterized as physical or behavioral. Physical barriers are usually large landscape features such as rivers, mountain ranges, or any landscape feature that an organism is incapable of traversing (Gascon et al. 2000). Behavioral barriers to movement are characterized by changes in habitat features that an organism is physically capable of crossing yet does not successfully cross for various reasons (Harris and Reed 2002). Individual organisms may be reluctant to enter a certain habitat due to perceived increase in predation risk (Rodríguez et al. 2001), or simply due to a lack of resources (e.g., foraging) to use during the dispersal event (Bélisle and Desrochers 2002).
A great deal of research has documented the reluctance of many forest-associated species to move short distances across relatively small gaps in forested habitat (Desrochers and Hannon 1997; St. Clair et al. 1998; Bélisle and St. Clair 2001; Bélisle and Desrochers 2002; Gobeil and Villard 2002; Bakker and Van Vuren 2004). Many of these studies are based on translocation experiments where organisms are taken from their territory and forced to make decisions on what habitat to travel through to return to their home territory (Desrochers et al. 1997; Gobeil and Villard 2002; Bakker and Van Vuren 2004). Ecological models have shown that these behavioral decisions about movement through habitat gaps can affect metapopulation dynamics (Russell et al. 2003; Zollner and Lima 2005).
Short-distance movements are different from long-distance dispersal events in which an individual may move a long distance before establishing a new territory. However, few studies have been able to examine patterns of long-distance dispersal events despite the fundamental role it plays in population connectivity (but see Dale et al. 2006). Although studies have documented differential patterns of movement through habitat types at small scales (Desrochers and Hannon 1997; St. Clair et al. 1998; Bélisle and St. Clair 2001; Bélisle and Desrochers 2002; Gobeil and Villard 2002; Bakker and Vuren 2004); patterns of movement documented at one scale may not be the same at a different scale (Morales and Ellner 2002).
Birds are commonly thought to have fewer behavioral limitations to long-distance dispersal given their high vagility and migratory nature (With et al. 1997). However, Harris and Reed (2002) found ecotones, habitat gaps and large water bodies are common behavioral barriers to nonmigratory movements of birds. They predict that birds that are habitat specialists, forest understory species, tropical species, solitary species, and nonmigratory species can be sensitive to habitat gaps (Harris and Reed 2002).
Woodpeckers are an excellent family to test predictions regarding behavioral barriers to movement because many are nonmigratory, habitat specialists, and they are often solitary. Woodpeckers are typically sedentary species that disperse short distances due to their nonmigratory nature (Paradis et al. 1998), high level of monogamy and territorial fidelity (Mikusiński 2006). Very little is known about the genetic population structure and dispersal patterns of woodpeckers in general (Pasinelli 2006), including patterns of sex-biased dispersal. Sex-biased dispersal, where one sex is philopatric or one sex typically disperses more often and/or farther than the other, is common among a variety of organisms (Lambin et al. 2001). In birds, female-biased dispersal is the most common pattern observed (Greenwood 1980; Clarke et al. 1997). The hypothesis for this pattern is that male birds tend to play a greater role in territory and resource defense and benefit more from being familiar with their natal area and therefore, are the philopatric sex (Greenwood 1980; Perrin and Goudet 2001). Female-biased dispersal can be seen in both red-cockaded (Picoides borealis; Daniels and Walters 2000) and acorn woodpeckers (Melanerpes formicivorus; Hannon et al. 1985), which are cooperative breeders with males that tend to stay in the natal territory as helpers.
Dispersal is difficult to measure by directly tracking individuals because most birds are too small to take advantage of advances in GPS technologies and resightings of banded birds in new locations is typically quite low (Dale et al. 2006). Genetic techniques can be used to estimate successful movement that results in reproduction and thus gene flow. Few studies have used genetic techniques to assess movement patterns in woodpeckers (Ellegren et al. 1999), a taxon with 214 recognized species in the family Picidae (Winkler et al. 1995).
Our study focused on black-backed woodpeckers (Picoides arcticus), a fire-dependent species. This species colonizes burned areas within 1 year after a fire, occupies burned areas for 3–5 years, with peak densities 3 years after fire (Caton 1996; Saab et al. 2007). Black-backed woodpeckers are a monogamous, resident species that maintains territories year-round (Dixon and Saab 2000). Individuals likely change habitat patches more than once in their lifetime because their life span (∼6–8 years; Dixon and Saab 2000) is longer than the length of time a habitat patch is optimal. To date, researchers have been unable to study the dispersal or movement patterns of black-backed woodpeckers due to their natural rarity and unpredictable movement patterns once a burned area is no longer optimal habitat.
Black-backed woodpeckers are continuously distributed across the boreal forest, into Alaska and range down into the northern USA (Fig. 1). They also occupy isolated patches in the Black Hills of South Dakota and regions of Oregon and California, mainly on the east side of the Cascades and Sierra Nevadas. They have been documented making long-distance movements during irruptions outside their normal breeding ranges (Yunick 1985), indicating long-distance movements are physiologically possible. Given black-backed woodpeckers occupy ephemeral habitats (Dixon and Saab 2000; Saab et al. 2009), both sexes regularly disperse during the course of their lifetime (Huot and Ibarzabal 2006). The objective of our study was to test if large gaps in forested areas are behavioral barriers to movement for black-backed woodpeckers and if males and females respond to these potential barriers in the same manner.
Figure 1.
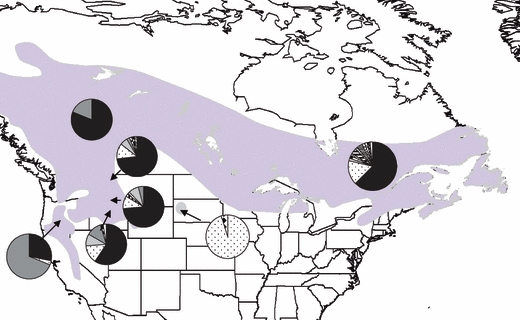
The distribution of black-backed woodpeckers (Natureserve) with the seven sampling locations: Oregon, Idaho, Missoula, Glacier, Alberta and Quebec. The frequency of observed mtDNA cytochrome b haplotypes at each sampling location is represented by pie charts at each location.
Methods
Sampling and DNA extraction
Blood or feather samples were collected in seven sampling locations: Alberta, Idaho, Oregon, Missoula, Glacier, South Dakota, and Quebec (Fig. 1). Blood samples were collected from adults caught at the nest site with either a hoop net or mist net during the 2004–2007 breeding seasons. Blood samples were stored at room temperature in a lysis buffer (Longmire et al. 1988). Individuals were color banded to avoid resampling in concurrent years and to record any dispersal events. We did not sample offspring in the nests to reduce sampling related individuals. A portion of the Idaho samples (n = 29) were feathers collected as part of a radio telemetry study conducted in 1998–2000 (Dudley and Saab 2007); Quebec samples were collected in 2000–2001. The latitude and longitude of individual sample locations was recorded. DNA was extracted from both blood and feather tissues using a DNeasy Tissue Extraction Kit (QIAGEN Inc., Valencia, CA). Blood was incubated for 2–24 h with a final elution of 200 μL and feathers were kept on a rocker for 48 h with a final elution of 100 μL to increase final DNA concentration.
Genotyping and sequencing
Mitochondrial DNA (mtDNA) was amplified using the polymerase chain reaction (PCR) and primers (L14841 and H15149) for the cytochrome b region (Kocher et al.1989). The reaction volume (50 μL) contained 50–100 ng DNA, 1x reaction buffer (Perkin-Elmer, Waltham, MA), 2.5 mm MgCl2, 200 μm each dNTP, 1 μm each primer, 1 U Taq polymerase (Titanium Taq; Clontech, Mountain View, CA). The PCR program was 94°C/5 min, (94°C/1 min, 55°C/1 min, 72°C/1 min 30 s) × 34 cycles, 72°C/5 min. PCR products (325 bp) were purified using ExoSAP-it (USB, Cleveland, OH) and directly sequenced. Both strands were sequenced using the Thermo Sequenase Cycle Sequencing Kit (USB) and run on either a 4300 DNA Analyzer (Li-Cor Biosciences, Lincoln, NE) or a 3730XL (Applied Biosystems, Foster City, CA, USA). Sequence editing and alignment was completed with sequencher (Genecodes Corp., Ann Arbor, MI)
Samples were genotyped at 11 microsatellite loci: C111, C115, D118, (Vila et al. 2008); RCW4 (added tail), RCW5, RCW17 (added tail) (T. Mullins and S. Haig, personal communication); DIU1, DIU3, DIU4 (Ellegren et al. 1999); HrU2 (Ellegren et al. 1999); Lox4 (Piertney et al. 1998). We added ‘GTTTCTT’ to the 5′ end of the reverse primer of RCW4 and RCW17 to promote the addition of adenine (Brownstein et al. 1996). All PCR amplifications were performed in 10 μL reactions. Three loci (DIU1, DIU3, Lox 4) were analyzed in single PCR reactions containing 2.5 mm MgCl2, 0.2 mm of each dNTP's, 2 μm dye-labeled forward primer and 2 μm reverse primer, 1 U Taq polymerase (Titanium Taq; Clontech), 1x reaction buffer (Perkin-Elmer), and ∼15 ng genomic DNA in 10 μL final reaction volume. Samples were amplified with the following profile: initial denaturation at 94°C for 10 m, followed by 45 cycles of (94°C for 60 s, 58°C for 60 s, 72°C for 60 s). Amplification products were analyzed on 6.5% polyacrylamide gels and visualized on a Li-Cor DNA Analyser 4300 (Li-Cor Biotechnology). Eight loci were analyzed in three multiplex reactions (Table S1) using the QIAGEN Multiplex PCR Kit (QIAGEN Inc.). Samples were amplified with the following profile: initial denaturation at 94°C for 10 m, followed by 45 cycles of (94°C for 60 s, 58°C for 60 s, 72°C for 60 s). Fragment analysis was performed on an ABI 3130xl Genetic Analyzer (Applied Biosystems Inc., Foster City, CA), ABI GS600LIZ ladder was used to determine allele sizes and (Applied Biosystems Inc.) chromatogram output was viewed and analyzed using GeneMapper version 3.7 (Applied Biosystems Inc.). Genotypes were manually checked by two individuals and if there was disagreement on how to score the sample, we reran the genetic analyses. All feather samples were run in a minimum of three separate PCR tubes, a heterozygote genotype was accepted if confirmed a minimum of two times and a homozygote genotype was accepted if confirmed a minimum of three times. We reanalyzed any samples with discrepancies in scoring of alleles to confirm the correct genotype.
Genetic variation
Microsatellite markers were tested for departure from Hardy–Weinberg (H–W) proportions and gametic disequilibrium in genepop (version 1.2; Raymond and Rousset 1995). We calculated observed and expected heterozygosity and average number of alleles/locus in GDA (version 1.1; Lewis and Zaykin 2001). Allelic richness, where the number of alleles is standardized to the smallest sample size, and FIS were calculated in fstat. The presence of null alleles, dropout of large alleles and errors due to stuttering were tested using micro-checker (Van Oosterhout et al. 2004). For mtDNA, haplotype diversity (h) and nucleotide diversity (π) were calculated using DnaSP (version 4.50; Rozas et al. 2003). Haplotype richness was calculated by taking the mean number of haplotypes observed when sampling 21 (minimum number) haplotypes with replacement from the frequency distribution of haplotypes created by sampling 10 000 times.
Population-based analyses
We calculated pairwise FST (Weir and Cockerham 1984) among all sampling locations and tested for isolation by distance based on FST/(1−FST) versus linear geographic distance among sample sites using Mantel tests (Mantel 1967) in the ade4 (Dray et al. 2007) package in the r software environment (http://www.r-project.org/).
Because our study was conducted at such a large spatial scale, we began by assessing hierarchical population structure where individuals at a sampling location (Fig. 1) were considered one group. We conducted an analysis of molecular variance (AMOVA; arlequin 3.11; Excoffier et al. 2005) and a spatial analysis of molecular variance (SAMOVA; Dupanloup et al. 2002) for both marker types. We tested five different hierarchical groupings (Table 1) and tested for significance of the variance components using 1000 permutations. Populations were identified by maximizing the among group percent of variation (FCT) as recommended by Dupanloup et al. (2002). We used principal component analysis (PCA) to visualize how sample sites clustered using pcagen (http://www2.unil.ch/popgen/softwares/pcagen.htm).
Table 1.
Analysis of molecular variance (AMOVA) results of five different groupings of black-backed woodpecker sampling sites for mtDNA and microsatellite loci. Significance values are based on 1000 permutations using arlequin 3.11. Results from spatial analysis of molecular variance (SAMOVA) are nearly identical and therefore, are not shown
| Group | No. groups | Variance component | mtDNA % of variance | Microsatellites % of variance |
|---|---|---|---|---|
| (Rocky Mountains + Quebec + Oregon); (South Dakota) | 2 | Among groups | 35.26 | 3.18 |
| Among sites | 15.24** | 2.22** | ||
| Within sites | 49.5** | 94.61** | ||
| (Rocky Mountains + Quebec); (Oregon); (South Dakota) | 3 | Among groups | 49.99* | 3.54* |
| Among sites | 1.07* | 1.38** | ||
| Within sites | 48.95** | 95.08** | ||
| (Rocky Mountains); (Quebec); (Oregon); (South Dakota) | 4 | Among groups | 37.51 | 2.33 |
| Among sites | 1.4 | 1.27** | ||
| Within sites | 61.08** | 96.4** | ||
| (Missoula + Glacier + Idaho + Quebec) (Alberta); (Oregon); (South Dakota) | 4 | Among groups | 45.01* | 2.71* |
| Among sites | 0.12 | 1.41** | ||
| Within sites | 54.87** | 95.88** | ||
| (Missoula + Glacier); (Idaho); (Alberta); (Quebec); (Oregon); (South Dakota) | 6 | Among groups | 34.13 | 1.9 |
| Among sites | −1.03 | 1.18** | ||
| Within sites | 66.90** | 96.93** |
Rocky Mountains = Idaho, Missoula, Glacier, Alberta.
P, 0.05
P < 0.0001.
Individual-based analyses
We assessed population structure using individual-based approaches. Specifically, we used a Bayesian clustering approach to determine the number of clusters based on gametic disequilibrium and deviations from H–W proportions. We used the program structure (version 2.2; Pritchard et al. 2000; Falush et al. 2003), a widely used approach that does not consider spatial information in the clustering algorithm. Next, we used the program geneland (version 3.1.4; Guillot et al. 2005b), a spatially explicit approach which can infer spatial discontinuities in genetic data when incorporating the spatial location of individual samples as well as a user-defined uncertainty around sampling locations.
In structure, we used the admixture model, with correlated allele frequencies and no prior information regarding where individuals were sampled. We used a burn-in period of 300 000 followed by 1 000 000 iterations for K = 1 through K = 10. We repeated each run four times and averaged log Pr(X|K) across all runs to determine which value of K maximized Pr(X|K).
Although the algorithm in geneland simultaneously estimates all the parameters, Guillot et al. (2005a), recommend a two-step approach. The first step infers the number of populations (K) and the second step holds K constant to assign individuals to populations. We began the geneland analyses by running 10 replicates with the following parameters: maximum rate of Poisson process of 274 (equal to sample size as recommended by Guillot et al. 2005a), allowed K to vary from 1 to 10, maximum number of nuclei in the Poisson–Voronoi tessellation set to 825 (roughly three times the sample size as recommended by Guillot et al. 2005a), 500 000 MCMC iterations with a burn-in period of 100 000 iterations, the Dirichlet model (which has been shown to perform better than alternate models available in geneland; Guillot et al. 2005a) in which allele frequencies are assumed to be independent, and spatial coordinates with an uncertainty of 5 km.
To test the robustness of our geneland results, we varied several input parameters to see if we obtained the same estimate of K. We varied uncertainty on the spatial coordinates from 0 to 50 km. We ran the same analysis as above with the nine locus dataset without using the null allele model to determine if the results would change based on these two different models.
Once K was identified, we ran 100 replicates of the model with the same parameters as above and K held constant. We ranked the models by mean logarithm of posterior probability and conducted postprocessing analyses on the top 10 models runs. We used a burn-in period of 100 000 iterations, a spatial domain of 400 pixels along the X-axis and 200 pixels along the Y-axis and checked the runs visually for consistency.
Phylogeographic analyses
We used several approaches to assess historical versus contemporary processes, which can be difficult to separate when interpreting sequence data (Edwards and Bensch 2009). To visualize the relationship among mtDNA haplotypes, we used network 4.5 (http://www.fluxus-engineering.com) to create a median-joining network. We estimated Tajima's D and Fu's F to test whether patterns of sequence divergenece in mtDNA followed the pattern expected under a model of neutral evolution. We calculated both metrics in DnaSP and significance was tested using coalescent simulations (Rozas et al. 2003). Departures from neutrality in these metrics are often interpreted as evidence for population expansion (Tajima 1989; Fu 1997).
Sex-biased movement patterns
Sex-biased movements can be estimated by using highly variable autosomal markers, such as microsatellites, to compare estimates of population structure between males and females or differences in assignment indices between males and females (Prugnolle and de Meeus 2002). Methods based on autosomal markers require the sampling of recent immigrants or that bias in dispersal is strong (Goudet et al. 2002). When dispersal rates are >10%, the metric that performs best is the comparison of pairwise FST estimates between males and females (Goudet et al. 2002). Sex-biased dispersal should result in pairwise FST estimates that are lower in the dispersing sex (Goudet et al. 2002). We estimated FST among the inferred populations for each sex separately because rates of dispersal in black-backed woodpeckers are likely much >10% given the ephemeral nature of their habitat.
Another method to estimate sex-biased dispersal is to examine different patterns of genetic structure in sex-linked or sex-transmitted markers (i.e., mtDNA) compared to autosomal markers (Prugnolle and de Meeus 2002). Estimates of FST based on microsatellite markers can be biased low due to their highly variable nature (Hedrick 2005a). To account for this potential bias, we calculated standardized estimates of pairwise estimates of FST (GST′) for both marker types (Hedrick 2005a; Meirmans 2006). The maximum FST was calculated by recoding each population to have unique alleles/haplotypes to maximum among population variation, while maintaining observed levels of variation (Hedrick 2005a; Meirmans 2006). We also plotted observed and standardized FST values on plots that show the expected values of FST for both mtDNA and nuclear markers under island model of migration and following isolation (Zink and Barrowclough 2008).
Results
Genetic variation
We found 16 variable sites in the 325 base pairs sequenced in the cytochrome b region of the mitochondrial genome. We identified 18 haplotypes, ranging from two in South Dakota to 12 in Quebec (Table 2). Haplotype diversity (h) and nucleotide diversity (π) were highest in Idaho (h = 0.616, π = 0.0035) and lowest in South Dakota (h = 0.095, π = 0.0006). One haplotype was very common (>60%), a second was relatively common (16%), and eight haplotypes were only detected once (Fig. 1; Table 3).
Table 2.
Genetic diversity and neutrality tests for all sampling locations, including the number of individuals sampled (n), number of haplotypes observed at each location, haplotype diversity (h), nucleotide diversity (π), haplotype richness (HR); FIS, fixation index; AR, allelic richness; HE, expected heterozygosity; Fu's F, test for neutrality and DTaj, Tajima's D-test for neutrality; standard errors are in parentheses
| n | No. of haplotypes | h | π | HR | FIS | HE | AR | Fu's F | DTaj | |
|---|---|---|---|---|---|---|---|---|---|---|
| Idaho | 42 | 6 | 0.616 (0.012) | 0.004 | 4.57 | −0.01 | 0.58 | 5.46 (1.35) | −0.92 | −0.54 |
| Missoula | 49 | 6 | 0.450 (0.012) | 0.002 | 4.00 | 0.12 | 0.58 | 5.52 (1.18) | −1.90 | −1.11 |
| Glacier | 48 | 7 | 0.457 (0.012) | 0.002 | 4.12 | 0.01 | 0.58 | 5.69 (1.15) | −3.25 | −1.45 |
| Alberta | 21 | 2 | 0.324 (0.024) | 0.002 | 1.98 | 0.02 | 0.63 | 6.36 (1.30) | 1.94 | 0.38 |
| Quebec | 56 | 12 | 0.589 (0.010) | 0.002 | 5.58 | 0.05 | 0.60 | 5.76 (1.38) | −10.60** | −1.87* |
| Oregon | 32 | 3 | 0.462 (0.013) | 0.003 | 2.47 | 0.08 | 0.58 | 5.13 (0.90) | 1.54 | 0.64 |
| S. Dakota | 27 | 2 | 0.074 (0.013) | 0.001 | 1.55 | 0.01 | 0.46 | 3.57 (0.52) | −0.07 | −1.51 |
| All locations | 275 | 18 | 0.613 (0.029) | 0.003 | 0.05 | 0.60 | 6.03 (1.33) | −11.46* | −1.50 |
P < 0.05
P < 0.0001.
Table 3.
Haplotype frequencies in each of the seven sampling locations (see Fig. 1)
| Haplotype | Missoula | Glacier | Alberta | Idaho | S. Dakota | Oregon | Quebec |
|---|---|---|---|---|---|---|---|
| 1 | 0.74 | 0.73 | 0.81 | 0.60 | 0.00 | 0.28 | 0.63 |
| 2 | 0.10 | 0.13 | 0.00 | 0.14 | 0.96 | 0.03 | 0.16 |
| 3 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 4 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 5 | 0.08 | 0.06 | 0.00 | 0.10 | 0.00 | 0.00 | 0.02 |
| 6 | 0.04 | 0.02 | 0.19 | 0.12 | 0.00 | 0.69 | 0.00 |
| 7 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 8 | 0.00 | 0.02 | 0.00 | 0.02 | 0.04 | 0.00 | 0.02 |
| 9 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 10 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 |
| 11 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 |
| 12 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.04 |
| 13 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 |
| 14 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.04 |
| 15 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 |
| 16 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 |
| 17 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 |
| 18 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 |
Ten of the 11 microsatellites were polymorphic in all the populations; Locus DIU1 was monomorphic in South Dakota. After correcting for multiple comparisons (Rice 1989), two loci had significant departures from H-W proportions, DIU1 and RCW17; four pairwise comparisons were significant for gametic disequilibrium. The average number of alleles per locus ranged from 3.64 in South Dakota to 6.91 in Quebec. Allelic richness was lowest in South Dakota (3.57) and highest in Alberta (6.36). South Dakota had the lowest levels of heterozygosity (HO = 0.46), other sites ranged from (0.51–0.62; Table 2).
Null alleles were apparently present at three loci: DIU1, RCW17 and C111. Both DIU1 and RCW17 had relatively high estimated frequencies of null alleles (0.20, 0.15) while the estimated frequency of the null allele at C111 occurred at a relatively low frequency (0.06). We conducted most analyses on both a full and reduced data set, with the same general pattern resulting from both datasets; most results presented are from the dataset with nine loci, after removing DIU1 and RCW17 (Chapuis and Estoup 2007). geneland results are from the full dataset because the algorithm implemented can estimate frequencies of null alleles (Guillot et al. 2008).
Population-based analyses
Samples collected from sites within the continuously distributed areas (boreal forest: Idaho, Missoula, Glacier, Alberta and Quebec) had lower pairwise FST values for both mtDNA and microsatellite data (Table 4). For mtDNA, pairwise FST values for the continuous sites ranged from 0.00–0.11 while the fragmented sites ranged from 0.36–0.75. Overall, pairwise FST values for microsatellite data were much lower than mtDNA, with values among the continuous sites ranging from 0.006 to 0.022 and from 0.035 to 0.094 among the fragmented sites. The grouping of sites within the continuously distributed locations as one population was supported by AMOVA (Table 1), SAMOVA, and PCA (Fig. 2). Due to similar results between the AMOVA and SAMOVA, we only present AMOVA results. The hierarchical grouping of the Rocky Mountain sites (Glacier, Missoula, Idaho, Alberta) with Quebec, with Oregon and South Dakota as separate groups, explained the most variation among groups for both mtDNA and nuclear DNA (Table 1). PCA reveals that, for both marker types, all sites within the continuously distributed area cluster tightly together and Oregon and South Dakota cluster separately from the continuous sites and each other (Fig. 2).
Table 4.
Pairwise FST values for mtDNA (below diagonal) and microsatellite (above diagonal). Significant values are indicated in bold and with asterisks
| Idaho | Missoula | Glacier | Alberta | Quebec | Oregon | S. Dakota | |
|---|---|---|---|---|---|---|---|
| Idaho | 0.007*** | 0.015** | 0.022*** | 0.019*** | 0.048*** | 0.057*** | |
| Missoula | 0.000 | 0.012*** | 0.014*** | 0.014*** | 0.035*** | 0.044*** | |
| Glacier | 0.001 | 0.000 | 0.012* | 0.017*** | 0.042*** | 0.049*** | |
| Alberta | 0.040 | 0.092** | 0.08*** | 0.006 | 0.050*** | 0.050*** | |
| Quebec | 0.028 | 0.000 | 0.000 | 0.11** | 0.049*** | 0.056*** | |
| Oregon | 0.38*** | 0.51*** | 0.51*** | 0.36*** | 0.54*** | 0.094*** | |
| S. Dakota | 0.43*** | 0.51*** | 0.54*** | 0.73*** | 0.53*** | 0.75*** |
P < 0.05
P < 0.01
P < 0.001.
Figure 2.
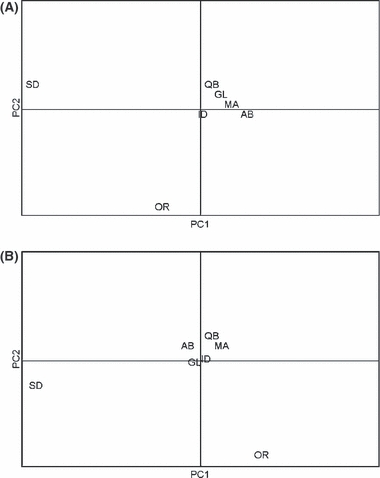
Principal Components Analysis visualizing clustering of sampling locations based on (A) mtDNA; PC1 = 59%, PC2 = 39%; (B) microsatellite data, PC1 = 38%, PC2 = 29%; SD = South Dakota, OR = Oregon, ID = Idaho, MA = Missoula, GL = Glacier, AB = Alberta, QB = Quebec.
Patterns of isolation by distance were more complex. Across all sites there is no pattern of isolation by distance for mtDNA (r = 0.004, P = 0.30) or microsatellites (r = 0.03, P = 0.30). However, the lack of isolation by distance is driven mostly by the absence of a correlation between geographic and genetic distance among sites within the boreal forest (Rocky Mountains and Quebec).
Individual-based analyses
Both structure and a spatially implicit approach in geneland identified one population cluster (K = 1). We focus on results from geneland due to the similarity between the results found from structure and the spatially implicit option available in geneland. When a spatially explicit approach was used, geneland identified three populations (K = 3), with all 10 runs identifying K = 3 with the highest probability (Fig. 3). Individuals assigned to populations with a high probability, with only six individuals ambiguously assigned with probability of assignment ≥0.99 (Fig. 4). Geographic barriers to gene flow were identified with probability of assignment contours (Fig. 4).
Figure 3.
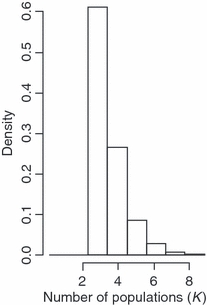
The posterior density distribution of the number of populations (K) estimated using the spatially explicit model in geneland. All 10 replicates of analyses in geneland shared similar distributional plots.
Figure 4.
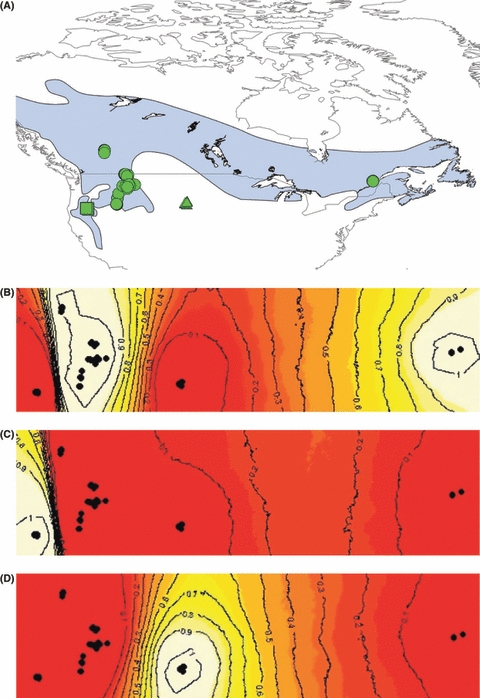
Maps showing the three clusters identified in the spatially explicit analysis conducted in geneland. Figure (A) identifies which cluster each sample was grouped with in assignment tests conducted in geneland (circles = continuous population extending from the Rocky Mountains to Quebec; squares = Oregon; triangles = South Dakota). The three clusters are (B) continuous population extending from the Rocky Mountains to Quebec, (C) western population, (D) South Dakota population. The contours represent probability of assignment to the clusters and display where barriers to gene flow exist. The appearance of a partial barrier to gene flow within (A) is likely an artifact of the lack of samples between Alberta and Quebec given samples at each end of this cluster assign with a high probability to the same population.
Phylogeographic analyses
The haplotype network displayed a star-like pattern, which is often attributed to a recent population expansion (Avise 2004; Fig. 5). However, eight of 13 rare (<0.055%) haplotypes that created this pattern were found in Quebec. Accordingly, only Quebec showed a significant departure from expectations from a neutral model of evolution (Tajima 1989; Fu 1997; Table 2; Fig. 5). Nucleotide diversity among haplotypes was low with one haplotype common to populations within the boreal forest and all other haplotypes differing by few nucleotides (Fig. 5, Table 3). The most common haplotypes in Oregon and South Dakota differed from the most common haplotype within the boreal forest, but were present in the boreal forest population. Locations across the boreal forest population shared several common haplotypes (28%: Hap 1, 2, 5, 6, 8; Fig. 5, Table 3).
Figure 5.
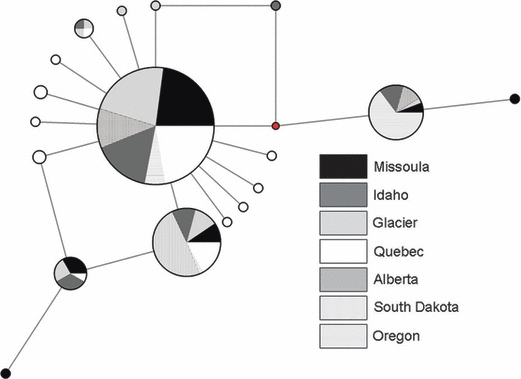
Median-joining network constructed using network 4.5.1.0 (Fluxus Technology Ltd 2005) from 325 bp of cytochrome b region of mtDNA for 275 black-backed woodpeckers sampled in seven different geographic locations across their range. The size of the circles is proportional to the frequency of the haplotype; the number of individuals that share a haplotype in each geographic region is represented by a different color and/or pattern pie slice within each circle.
Sex-biased movement patterns
Pairwise FST estimates between the continuous population found across the boreal forest and the fragmented sites (Oregon and S. Dakota) were consistently lower for males compared to females (Table 5B), a pattern indicating males are dispersing at a higher rate than females. The global FST estimate for females was 0.039 (95% CI: 0.024–0.052), and the global FST estimate for males was 0.023 (95% CI: 0.014–0.031). Pairwise FST estimates for microsatellite data between the continuous and fragmented sites were 4–5 times lower than would be predicted based on island model of migration at mutation–drift equilibrium using the following equation (Brito 2007): FST(msat) = FST(mtDNA)/4−3*FST(mtDNA) (Fig. 6). After standardization, pairwise FST estimates for microsatellite data between the continuous and fragmented sites were >2 times lower than expected based on Wright's island model of migration. For example, pairwise FST(mtDNA) = 0.49 between Oregon and the boreal forest population; under the island model, the expected FST(msat) = 0.19, observed FST(msat) = 0.04. After standardizing, the standardized pairwise FST(mtDNA) = 0.72 between Oregon and the boreal forest population, the expected FST(msat) = 0.39, observed standardized FST(msat) = 0.17 (Table 5A).
Table 5.
A comparison of estimates of pairwise FST between inferred populations between autosomal and sex-transmitted markers (A) and pairwise FST estimates for males and females based solely on autosomal markers to test for sex-biased movement patterns among populations. (A) Observed and standardized pairwise FST estimates are based on mtDNA (below diagonal) and microsatellite markers (above diagonal). (B) Male and female estimates are based on microsatellite markers for males (below diagonal) and females (above diagonal)
| Observed FST | Standardized FST | ||||||
|---|---|---|---|---|---|---|---|
| Boreal Forest | Oregon | S. Dakota | Boreal Forest | Oregon | S. Dakota | ||
| (A) | |||||||
| Boreal Forest | 0.039 | 0.043 | Boreal Forest | 0.165 | 0.167 | ||
| Oregon | 0.490 | 0.095 | Oregon | 0.716 | 0.200 | ||
| S. Dakota | 0.452 | 0.754 | S. Dakota | 0.669 | 0.885 | ||
| Sex-specific FST | |||||||
|---|---|---|---|---|---|---|---|
| Boreal Forest | Oregon | S. Dakota | |||||
| (B) | |||||||
| Boreal Forest | 0.035 | 0.048 | |||||
| Oregon | 0.027 | 0.088 | |||||
| S. Dakota | 0.024 | 0.042 | |||||
Figure 6.
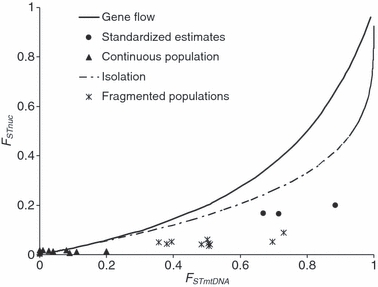
The expected relationship between FSTnuc and FSTmtDNA at mutation–drift equilibrium under Wright's island model of migration (solid black line) and under a model of complete isolation (dashed line). Observed pairwise values of FSTmsat and FSTmtDNA for black-backed woodpeckers are plotted; black triangles are sites within the continuous distribution, asterisks are pairwise values where at least one of the pair are in the fragmented sites and solid black circles are standardized estimates between the three populations inferred from both hierarchical population analyses and individual-level clustering in geneland.
Discussion
We found evidence that large gaps among forested sites apparently act as barriers to the movement of female black-backed woodpeckers and create a higher resistance to movement for male black-backed woodpeckers. Despite the sedentary nature of many woodpeckers, we know black-backed woodpeckers are physiologically capable of long-distance movements based on records of historical irruptions outside their normal distribution into areas south of the boreal forest (Yunick 1985). However, these irruptions occurred almost exclusively outside the breeding season (Yunick 1985) and therefore do not represent natal or breeding dispersal, but are more similar to short-distance migration events.
Shared ancestry and current gene flow can result in similar patterns of genetic structure, especially in species that have recently colonized areas and undergone population expansion. The star-like phylogeny of mtDNA variation (Fig. 5) suggests that black-backed woodpeckers have undergone a fairly recent population expansion. However, private haplotypes in Quebec are a major contributor to this pattern. Samples collected in Quebec have significant departures from a neutral model of evolution (Table 2); since this location is the only one to have significant departures from neutrality, it is unlikely that mtDNA is under selection in this site. A more probable explanation is a recent population expansion in the eastern boreal forest (Fu 1997). The western portion of the boreal forest population shows a pattern of high haplotype diversity in the south (Idaho) that decreases in a northerly direction, indicating a south to north pattern of colonization. High genetic diversity on the east (Quebec, h = 0.589) and west side (Idaho, h = 0.616) of the boreal forest population and the sharing of all common haplotypes (Table 3), as opposed to a single common haplotype, suggest that current gene flow may be responsible for the weak genetic structure observed. Lower genetic diversity within both fragmented populations (Oregon, h = 0.462; S. Dakota, h = 0.074) based on a subset of haplotypes found in the boreal forest suggest shared ancestry without much current gene flow.
Geographical features such as mountain ranges do not appear to create physical barriers to movement given gene flow across the Continental Divide within the Rocky Mountains is the most likely explanation for the weak genetic structure observed. The complete lack of genetic structure for both microsatellite and mtDNA markers across a vast distance (∼3500 km) in the Canadian boreal forest suggest both males and females are dispersing equally when there is continuously distributed habitat.
Population structure and movement
The past few years have seen an explosion of individual-based methods for defining clusters of genetically similar individuals (Manel et al. 2003; Latch et al. 2006; Chen et al. 2007). However, individual-based methods often work best when samples are evenly spaced across the study area. This is because if isolation by distance occurs, these clustering methods can misidentify groups at either end of the spectrum due to a lack of sampling across the distribution of continuously distributed species (Frantz et al. 2009; Schwartz and McKelvey 2009).
In this study, we sampled in a clustered manner, that is, we sampled multiple individuals at several different sites across a large spatial scale. Therefore, we chose to use both traditional population-level based analyses to define groups of individuals (AMOVA, SAMOVA, PCA, FST) and individual-based analyses (geneland). All of the approaches except the spatially implicit individual-based clustering methods defined the same three populations, a large, genetically continuous population (boreal forest) and two fragmented populations (Oregon and South Dakota).
The spatially explicit approach employed in geneland displayed a very low level of uncertainty in estimating the number of populations. All 10 runs estimated K = 3, with subsequent identification of population boundaries and assignment of individuals to the three populations being highly consistent. Two individuals were assigned to more than one population, but they both assigned to the ‘correct’ population with a probability >0.99.
Behavioral barriers to movement
A recent review of patterns of genetic structure in seabirds found that areas between their breeding and nonbreeding distribution indicated potential barriers to dispersal (Friesen et al. 2007), a similar pattern to what we found for female black-backed woodpeckers. However, black-backed woodpeckers’ distribution closely follows the distribution of the boreal forest. There are large areas without contiguous forested habitat between the boreal forest population, the Cascade region, and the Black Hills of South Dakota. These gaps in forested habitat are likely the ultimate cause of the limited gene flow across these geographic regions.
Evidence that large gaps in forested habitat are movement barriers for females can be seen in the population structure we detected and the difference in pairwise FSTmtDNA values between sites that have large gaps without forest between them (fragmented: Oregon and South Dakota) as compared to sites that have forest between them (boreal forest: Idaho, Missoula, Glacier, Alberta and Quebec). Hierarchical population structure is a useful tool to detect barriers to gene flow when you have several subpopulations that may be connected by differing levels of gene flow (Allendorf and Luikart 2007). When we considered three groups: boreal forest, Oregon, and S. Dakota, a large amount of genetic variation was explained among groups and almost no genetic variation was explained among sites within group (Table 1). When we included Oregon with the boreal forest group, the variation among sites increased 15-fold, confirming a barrier likely exists between Oregon and the boreal forest. Additional evidence can be seen in the high pairwise FSTmtDNA values (0.36–0.75) between sites with large gaps in forest between them. These values are similar to those documented among subspecies or separate clades in other birds occupying similar ranges (Gibbs et al. 2000; Mila et al. 2007).
The inclusion of spatial data in geneland identified the general location of barriers to gene flow among the three populations (Fig. 4). Sharp discontinuities in gene flow match the break in large forested areas between the Rocky Mountains and Oregon and the Rocky Mountains and South Dakota. However, the lack of samples in the boreal forest between Alberta and Quebec does not allow geneland to do a good job of assessing connectivity across the boreal forest.
Black-backed woodpeckers may not be a classic forest species due to their proclivity for burned forests in which most of the standing trees are dead, but it has been well documented that these birds prefer dense stands of dead trees (Saab et al. 2009). Organisms usually avoid dispersing through certain habitat types to avoid predators or a lack of resources during travel (Bélisle and Desrochers 2002). The risk of predation and the amount of resources available for foraging will be more similar in burned and live forests than between any forest type and nonforest type (e.g., grassland, etc.). Nonforested habitat may be somewhat of a physical barrier to movement as it does not provide foraging or roosting resources. Yet, historic records show black-backed woodpeckers traveling far south of their normal distribution in the eastern boreal forest and arriving in areas such as Ohio and Illinois, which would require crossing large expanses of nonforested habitat (Yunick 1985).
Sex-biased movement patterns
Males apparently successfully travel long distances across inhospitable habitat at a much higher rate than females. We propose this because pairwise estimates of FSTmale are lower than FSTfemale and the FSTnuc are lower than FSTmtDNA (Table 5). Dispersal rates between males and females must differ substantially to detect differences using microsatellite markers (Goudet et al. 2002). Pairwise estimates of FSTfemale were 1.3–2.1 times higher than FSTmale (Table 5B), a considerable difference given these methods have a difficult time detecting differential dispersal rates between sexes.
We plotted the expected pairwise values of FSTnuc versus FSTmtDNA under Wright's island model of migration at equilibrium and under a model of complete isolation, along with our observed values (Fig. 6; adapted from Zink and Barrowclough 2008). Generally, points above both lines would indicate female-biased dispersal and points below both lines indicate male-biased dispersal. The pairwise estimates for sites within the continuous population fall within the range of what is expected under a model of gene flow or a model of isolation with extremely large effective population sizes (e.g., Ne > 325 000 after 5000 generations of isolation) which is much larger than the estimates of long term effective population size of other more common warblers (Ne≍ 10 000; Milot et al. 2000) and downy woodpeckers, P. pubescens [Ne(f)≍ 6500; Ball. and Avise 1992). Conversely, the pairwise values of the sites separated by large habitat gaps fall well below expected values under either model (Fig. 6). Finally, the standardized estimates of FST are plotted and fall well below expected values under either model, indicating male-biased movement after correcting for potential bias due to high levels of heterozygosity when using microsatellite markers.
Possible alternative explanations
There are four potential explanations for the observed departure from the expected pairwise values of FSTnuc:FSTmtDNA :lack of equilibrium, high rate of homoplasy in the microsatellite markers compared to mtDNA (Zink and Barrowclough 2008), large amount of heterozygosity due to highly polymorphic microsatellite markers (Hedrick 1999), and differential dispersal rates of males and females. We will discuss the likelihood of these possibilities below.
An ice sheet covered most of the boreal forest until approximately 10 000 years ago (Hewitt 2000). Consequently, most habitat currently occupied by black-backed woodpeckers was probably colonized within the last 10 000 years (Hewitt 2000). If a lack of equilibrium was responsible for the differences between estimates of FSTnuc:FSTmtDNA, then we would expect a pattern opposite of what was observed. The population in South Dakota is likely quite small (low Ne), given black-backed woodpeckers only occupy the Black Hills, a relatively small area (15 500 km2) and they are a rare bird that occupies large territories (50–700 ha; Dixon and Saab 2000; Dudley and Saab 2007). We would expect the pairwise comparison between FSTnuc:FSTmtDNA between South Dakota and other sites to be closest to the predicted pairwise FSTnuc:FSTmtDNA values under an equilibrium scenario. In fact, these sites are the furthest from predicted values (Fig. 6). An alternative reason for the pattern observed in South Dakota could be lack of equilibrium due to a recent bottleneck. While this is plausible, it does not explain why the largest population of black-backed woodpeckers (boreal forest) follows the expected pattern at equilibrium and the two fragmented sites (Oregon and S. Dakota) fall far from expectations.
Homoplasy occurs when two alleles in a population are identical in state but have different origins. Homoplasy is common in microsatellites because they often follow a step-wise mutation model in which each mutation is ‘one step’ or repeat different. Homoplasy can cause two populations to appear more genetically similar than they are because they may share the same alleles from different origins. While homoplasy is certainly common in microsatellite markers and can prevent FSTnuc from going to fixation (FSTnuc = 1.0), it is unlikely to cause as severe an underestimate in FSTnuc as we observed. More importantly, homoplasy would not explain the high FSTmtDNA between sites with large habitat gaps and the low FSTmtDNA between sites with continuous habitat between them, which is a main driver of our pattern.
Because estimates of FST depend on how much variation there is in a population, estimates based on microsatellites are often biased downward due to the large number of alleles per locus and high amount of heterozygosity (Hedrick 2005a). Based on standardized estimates of FSTmtDNA, we would expect estimates of genetic divergence between South Dakota and the boreal forest population to be FSTnuc = 0.335 and our standardized estimate of FSTnuc = 0.167, a value twice as low as expected. Therefore, highly variable loci deflating estimates of genetic divergence does not explain the pattern we observed.
Differential gene flow between males and females is the best explanation for the pattern in our data. Sex-biased dispersal is a common phenomenon in birds, with a majority having female-biased dispersal (Clarke et al. 1997). A review of patterns of avian sex-biased dispersal found 11% (6/53) of bird species to have male-biased dispersal and 15% (8/53) showed equal dispersal between sexes (Clarke et al. 1997; Gibbs et al. 2000). Several studies have documented patterns of sex-biased dispersal based on different patterns of genetic structure observed in mtDNA as compared to nuclear DNA (Gibbs et al. 2000; Helbig et al. 2001; Bouzat and Johnson 2004; Gay et al. 2004; Tiedemann et al. 2004; Jones et al. 2005; Wright et al. 2005; Johnson et al. 2007). Although female-biased dispersal dominates in birds, waterfowl often show a pattern of male-mediated gene flow (Greenwood and Harvey 1982; Lecomte et al. 2009). Red-crested pochards (Netta rufina; Gay et al. 2004), common eiders (Somateria mollissima; Tiedemann et al. 2004), sandhill cranes (Grus canadensis), as well as the yellow warbler (Dendroica petechia; Gibbs et al. 2000) have patterns of genetic structure consistent with male-mediated gene flow (i.e., highly structured mtDNA and weakly structured nuclear DNA). Lekking species tend to have the common pattern of female mediated gene flow with high site fidelity by males that is evidenced by mtDNA structure that is weak compared to nuclear structure (Bouzat and Johnson 2004; Johnson et al. 2007). Most of these species are migratory species with one sex that is philopatric to the natal territory.
The leading hypothesis attributes sex-biased dispersal to advantages to the philopatric sex of obtaining higher quality territories and the dispersing sex leaving to avoid inbreeding (Greenwood 1980). Black-backed woodpeckers’ life history is quite different than most migratory species because they are a resident species that occupy a territory year-round, and then both males and females disperse when their habitat becomes suboptimal (3–5 years postfire). Thus, we would predict equal rates of dispersal for males and females because neither sex can occupy sites near the natal territory long term and the frequent colonization of new habitats should provide a mechanism to avoid inbreeding.
Our findings differ from a simple pattern of male-biased dispersal because females and males both must regularly disperse due to ephemeral nature of their habitat. Additionally, we found evidence that suggests gene flow attributed to both sexes; that is, an absence of genetic structure for both mtDNA and nuclear DNA, over large areas across the boreal forest. Differential gene flow between males and females was confirmed by different estimates of pairwise FST estimates.
Conservation implications
Our data suggest that females do not cross large gaps in habitat and that large gaps in habitat act as a higher resistance landscape to long-distance dispersal for males. Therefore, small isolated populations, such as the one in South Dakota, may deserve higher priority for conservation. For example, land management actions that affect black-backed woodpecker habitat, such as salvage logging, would need to be considered in a spatial and temporal context because fewer habitat patches are available to birds through time. If the population in South Dakota declines due to habitat degradation, recolonization of the areas by females is unlikely. Additionally, it is important to determine if the Oregon population is connected to California or Washington populations when planning management actions that affect populations. Our data suggests there has been gene flow across the boreal forest for a long period of time. Management actions should strive to maintain forested connectivity between burned patches to maintain these levels of gene flow.
We are unaware of any other studies finding evidence for males and females making different decisions regarding crossing large gaps in habitat for long-distance dispersal events. Future studies examining behavioral barriers to movement should consider that patterns of movement of males and females can differ across various types of habitats. This is especially important given the common use of nuclear genetic data to define populations.
Acknowledgments
We would like to thank J. Brininstool, M. Byall, M. Burriss, D. Cerasale, M. Edwards, E. Johnson, T. Loya, L. McFarland, C. Forristal, S. Meloy, K. Nittinger, L. Reynolds, E. Rindal, T. Swearingen, S. Story, C. Street, and N. Walker for assistance collecting field samples. J. Dudley provided several samples from Idaho, S. Pelech provided the Alberta samples, T. Juntti and M. Rumble provided several South Dakota samples. Laboratory assistance was provided by S. Amish, C. Engjker, K. Pilgrim, and S. Painter. We would like to thank Susan Haig, Tom Mullins, and Marta Vila for providing unpublished primers. Funding for this project was provided by Montana Fish, Wildlife and Parks, Bureau of Land Management, Glacier National Park, The Glacier Fund, U.S.D.A. Forest Service, Y2Y Science Grants and Wilburforce Foundation, McIntire-Stennis Cooperative Research Program, National Center for Landscape Fire Analysis, Northwest Scientific Association and Five Valleys Audubon. Support for J. Pierson was provided by the American Association of University Women, P.E.O. Scholar Award, National Science Foundation and The University of Montana.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. The primers, number of alleles,length range, annealing temperature and primer concentrations used in either multiplexed or single PCR reactions. Primer sequences canbe found in the reference listed; ‘GTTTCTT’ was addedto the 5′ end of the reverse primer of RCW4 and RCW17 to promote the addition of adenine (Brownstein et al. 1996).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Literature cited
- Allendorf FW, Luikart G. Conservation and the genetics of populations. Malden: Blackwell Publishing; 2007. [Google Scholar]
- Avise JC. Molecular markers, natural history and evolution. 2nd edn. Sunderland: Sinauer Associates, Inc; 2004. [Google Scholar]
- Bakker VJ, Van Vuren DH. Gap-crossing decisions by the red squirrel, a forest-dependent small mammal. Conservation Biology. 2004;18:689–697. [Google Scholar]
- Ball J, Avise JC. Mitochondrial DNA phylogeographic differentiation among avian populations and the evolutionary significance of subspecies. The Auk. 1992;109:626–636. [Google Scholar]
- Bélisle M, St. Clair CC. Cumulative effects of barriers on the movement of forest birds. Conservation Ecology. 2001;5:9. [Google Scholar]
- Bélisle M, Desrochers A. Gap-crossing decisions by forest birds: an empirical basis for parameterizing spatially-explicit, individual-based models. Landscape Ecology. 2002;17:219–231. [Google Scholar]
- Bouzat JL, Johnson K. Genetic structure among closely spaced leks in a peripheral population of lesser prairie-chickens. Molecular Ecology. 2004;13:499–505. doi: 10.1046/j.1365-294x.2003.02068.x. [DOI] [PubMed] [Google Scholar]
- Brito PH. Contrasting patterns of mitochondrial and microsatellite genetic structure among Western European populations of tawny owls (Strix aluco. Molecular Ecology. 2007;16:3423–3437. doi: 10.1111/j.1365-294X.2007.03401.x. [DOI] [PubMed] [Google Scholar]
- Brownstein M, Carpten D, Smith JR. Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitates genotyping. BioTechniques. 1996;20:1004–1010. doi: 10.2144/96206st01. [DOI] [PubMed] [Google Scholar]
- Caton EL. Effects of fire and salvage logging on the cavity nesting bird community in Northwestern Montana. Missoula: University of Montana; 1996. Ph.D. Thesis. [Google Scholar]
- Chapuis MP, Estoup A. Microsatellite null alleles and estimation of population differentiation. Molecular Biology and Evolution. 2007;24:621–631. doi: 10.1093/molbev/msl191. [DOI] [PubMed] [Google Scholar]
- Chen C, Durand E, Forbes F, François O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Molecular Ecology Notes. 2007;7:747–756. [Google Scholar]
- Clarke AL, Sæther BE, Røskaft E. Sex biases in avian dispersal: a reappraisal. Oikos. 1997;79:429–438. [Google Scholar]
- Clobert J, Danchin E, Dhondt A, Nichols JD. Dispersal. New York: Oxford University Press; 2001. [Google Scholar]
- Dale S, Steifetten O, Osiejuk TS, Losak K, Cygan JP. How do birds search for breeding areas at the landscape level? Interpatch movements of male ortolan buntings. Ecography. 2006;29:886–898. [Google Scholar]
- Daniels SJ, Walters JR. Inbreeding depression and its effects on natal dispersal in red-cockaded woodpeckers. The Condor. 2000;102:482–491. [Google Scholar]
- Desrochers A, Hannon SJ. Gap crossing decisions by forest songbirds during the post-fledging period. Conservation Biology. 1997;11:1204–1210. [Google Scholar]
- Dixon R, Saab V. Black-backed woodpecker (Picoides arcticus. In: Poole A, Gill F, editors. The Birds of North America. Philadelphia, PA: The Birds of North America, Inc; 2000. pp. 1–20. No. 509. [Google Scholar]
- Dray S, Dufour AB, Chessel D. The ade4 package-II: two-table and K-table methods. R News. 2007;7:47–52. [Google Scholar]
- Dudley JG, Saab VA. Home range size of black-backed woodpeckers in burned forests of southwestern Idaho. Western North American Naturalist. 2007;67:593–600. [Google Scholar]
- Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Molecular Ecology. 2002;11:2571–2581. doi: 10.1046/j.1365-294x.2002.01650.x. [DOI] [PubMed] [Google Scholar]
- Edwards S, Bensch S. Looking forwards or looking backwards in avian phylogeography? A comment on Zink and Barrowclough 2008. Molecular Ecology. 2009;18:2930–2933. doi: 10.1111/j.1365-294X.2009.04270.x. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Carlson A, Stenberg I. Genetic structure and variability of whitebacked woodpecker (Dendrocopos leucotos) populations in northern Europe. Hereditas. 1999;130:291–299. [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin version 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz AC, Cellina S, Krier A, Schley L, Burke T. Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? Journal of Applied Ecology. 2009;46:493–505. [Google Scholar]
- Friesen VL, Burg TM, McCoy KD. Mechanisms of population differentiation in seabirds. Molecular Ecology. 2007;16:1765–1785. doi: 10.1111/j.1365-294X.2006.03197.x. [DOI] [PubMed] [Google Scholar]
- Fu YX. Statistical test of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon C, Malcolm JR, Patton JL, Silva MNFD, Bogart JP, Lougheed SC, Peres CA, et al. Riverine barriers and the geographic distribution of Amazonian species. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13672–13677. doi: 10.1073/pnas.230136397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay L, Defos DRP, Mondain-Monval J-Y, Crochet PA. Phylogeography of a game species: the red-crested pochard (Netta rufina) and consequences for its management. Molecular Ecology. 2004;13:1035–1045. doi: 10.1111/j.1365-294X.2004.02117.x. [DOI] [PubMed] [Google Scholar]
- Gibbs HL, Dawson RJG, Hobson KA. Limited differentiation in microsatellite DNA variation among northern populations of the yellow warbler: evidence for male-biased gene flow? Molecular Ecology. 2000;9:2137–2147. doi: 10.1046/j.1365-294x.2000.01136.x. [DOI] [PubMed] [Google Scholar]
- Gobeil JF, Villard MA. Permeability of three boreal forest landscape types to bird movements as determined from experimental translocations. Oikos. 2002;98:447–458. [Google Scholar]
- Goudet J, Perrin N, Waser P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Molecular Ecology. 2002;11:1103–1114. doi: 10.1046/j.1365-294x.2002.01496.x. [DOI] [PubMed] [Google Scholar]
- Greenwood P. Mating systems, philopatry, and dispersal in birds and mammals. Animal Behaviour. 1980;28:1140–1162. [Google Scholar]
- Greenwood P, Harvey PH. The natal and breeding dispersal of birds. Annual Review of Ecology and Systematics. 1982;13:1–21. [Google Scholar]
- Guillot G, Estoup A, Mortier F, Cosson JF. A spatial statistical model for landscape genetics. Genetics. 2005a;170:1261–1280. doi: 10.1534/genetics.104.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot G, Mortier F, Estoup A. Geneland: a program for landscape genetics. Molecular Ecology Notes. 2005b;5:712–715. [Google Scholar]
- Guillot G, Santos F, Estoup A. Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics. 2008;24:1406–1407. doi: 10.1093/bioinformatics/btn136. [DOI] [PubMed] [Google Scholar]
- Hannon SJ, Mumme RL, Koenig WD, Pitelka FA. Replacement of breeders and within-group conflict in the cooperatively breeding acorn woodpecker. Behavioral Ecology and Sociobiology. 1985;17:303–312. [Google Scholar]
- Harris RJ, Reed JM. Behavioral barriers to non-migratory movements of birds. Annales Zoologici Fennici. 2002;39:275–290. [Google Scholar]
- Hedrick PW. Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution. 1999;53:313–318. doi: 10.1111/j.1558-5646.1999.tb03767.x. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. A standardized genetic differentiation measure. Evolution. 2005;59:1633–1638. [PubMed] [Google Scholar]
- Helbig AJ, Salomon M, Bensch S, Seibold I. Male-biased gene flow across an avian hybrid zone: evidence from mitochondrial and microsatellite DNA. Journal of Evolutionary Biology. 2001;14:277–287. [Google Scholar]
- Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- Huot M, Ibarzabal J. A comparison of the age-class structure of black-backed woodpeckers found in recently burned and unburned boreal coniferous forests in eastern Canada. Annales Zoologici Fennici. 2006;43:131–136. [Google Scholar]
- Johnson JA, Burnham KK, Burnham WA, Mindell DP. Genetic structure among continental and island populations of gyrfalcons. Molecular Ecology. 2007;16:3145–3160. doi: 10.1111/j.1365-294X.2007.03373.x. [DOI] [PubMed] [Google Scholar]
- Jones KL, Krapu GL, Brandt DA, Ashley MV. Population genetic structure in migratory sandhill cranes and the role of Pleistocene glaciations. Molecular Ecology. 2005;14:2645–2657. doi: 10.1111/j.1365-294X.2005.02622.x. [DOI] [PubMed] [Google Scholar]
- Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, Wilson AC. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:6196–6200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambin X, Aars J, Piertney SB. Dispersal, intraspecific competition, kin competition, and kin facilitation: a review of the empirical evidence. In: Clobert J, Danchin E, Dhondt A, Nichols J, editors. Dispersal. New York: Oxford University Press; 2001. [Google Scholar]
- Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OEJ. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conservation Genetics. 2006;7:295–302. [Google Scholar]
- Lecomte N, Gauthier G, Giroux J, Milot E, Bernatchez L. Tug of war between continental gene flow and rearing site philopatry in a migratory bird: the sex-biased dispersal paradigm reconsidered. Molecular Ecology. 2009;18:593–602. doi: 10.1111/j.1365-294X.2008.04067.x. [DOI] [PubMed] [Google Scholar]
- Lewis PO, Zaykin D. 2001. Genetic data analysis: computer program for the analysis of allelic data, Version 1.0. http://lewis.eeb.uconn.edu/lewishome/software.html (accessed June 2008)
- Longmire JL, Lewis AK, Brown NC, Buckingham JM, Clark LM, Jones MD, Meincke LJ, et al. Isolation and molecular characterization of a highly polymorphic centromeric tandem repeat in the family Falconidae. Genomics. 1988;2:14–24. doi: 10.1016/0888-7543(88)90104-8. [DOI] [PubMed] [Google Scholar]
- Manel S, Schwartz MK, Luikart G, Taberlet P. Landscape genetics: combining landscape ecology and population genetics. Trends in Ecology & Evolution. 2003;18:189–197. [Google Scholar]
- Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Research. 1967;27:209–220. [PubMed] [Google Scholar]
- Meirmans PG. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution. 2006;60:2399–2402. [PubMed] [Google Scholar]
- Mikusiński G. Woodpeckers: distribution, conservation, and research in a global perspective. Annales Zoologici Fennici. 2006;43:86–95. [Google Scholar]
- Mila B, Smith TB, Wayne RK. Speciation and rapid phenotypic differentiation in the yellow-rumped warbler (Dendroica coronata) complex. Molecular Ecology. 2007;16:159–173. doi: 10.1111/j.1365-294X.2006.03119.x. [DOI] [PubMed] [Google Scholar]
- Milot E, Gibbs HL, Hobson KA. Phylogeography and genetic structure of northern populations of the yellow warbler (Dendroica petechia. Molecular Ecology. 2000;9:667–681. doi: 10.1046/j.1365-294x.2000.00897.x. [DOI] [PubMed] [Google Scholar]
- Morales JM, Ellner SP. Scaling up animal movements in heterogeneous landscapes: the importance of behavior. Ecology. 2002;83:2240–2247. [Google Scholar]
- Paradis E, Baillie SR, Sutherland WJ, Gregory RD. Patterns of natal and breeding dispersal in birds. Journal of Animal Ecology. 1998;67:518–536. [Google Scholar]
- Pasinelli G. Population biology of European woodpecker species: a review. Annales Zoologici Fennici. 2006;43:96–111. [Google Scholar]
- Perrin N, Goudet J. In: Inbreeding, kinship, and the evolution of natal dispersal. In: Clobert J, Danchin E, Dhondt A, Nichols J, editors. Dispersal. New York: Oxford University Press; 2001. [Google Scholar]
- Piertney SB, Marquiss M, Summers R. Characterization of tetranucleotide microsatellite markers in the Scottish crossbill (Loxia scotica. Molecular Ecology. 1998;7:1247–1263. [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, De Meeus T. Inferring sex-biased dispersal from population genetic tools: a review. Heredity. 2002;88:161–165. doi: 10.1038/sj.hdy.6800060. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Rodríguez A, Andrén H, Jansson G. Habitat-mediated predation risk and decision making of small birds at forest edges. Oikos. 2001;95:383–396. [Google Scholar]
- Rozas J, Sanchez-Delbarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Russell RE, Swihart RK, Feng Z. Population consequences of movement decisions in a patchy landscape. Oikos. 2003;103:142–152. [Google Scholar]
- Saab VA, Russell RE, Dudley JG. Nest densities of cavity-nesting birds in relation to postfire salvage logging and time since wildfire. Condor. 2007;109:97–108. [Google Scholar]
- Saab VA, Russell RE, Dudley JG. Nest-site selection by cavity-nesting birds in relation to postfire salvage logging. Forest Ecology and Management. 2009;257:151–159. [Google Scholar]
- Schwartz MK, McKelvey K. Why sampling scheme matters: the effect of sampling scheme on landscape genetic results. Conservation Genetics. 2009;10:441–452. [Google Scholar]
- St. Clair CC, Bélisle M, Desrochers A, Hannon S. Winter responses of forest birds to habitat corridors and gaps. Conservation Ecology. 1998;2:13. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann R, Paulus KB, Scheer M, Von Kistowski KG, Skírnisson K, Bloch D, Dam M. Mitochondrial DNA and microsatellite variation in the eider duck (Somateria mollissima) indicate stepwise postglacial colonization of Europe and limited current long-distance dispersal. Molecular Ecology. 2004;13:1481–1494. doi: 10.1111/j.1365-294X.2004.02168.x. [DOI] [PubMed] [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes. 2004;4:535–538. [Google Scholar]
- Vila M, Robles H, Ciudad C, Olea PP, Baglione V. Isolation and characterization of 12 microsatellite markers in the middle-spotted woodpecker (Dendrocopos medius. Molecular Ecology Resources. 2008;8:415–417. doi: 10.1111/j.1471-8286.2007.01976.x. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Winkler H, Christie DA, Nurney D. A guide to the woodpeckers of the world. New York: Houghton Mifflin Company; 1995. [Google Scholar]
- With KA, Gardner RH, Turner MG. Landscape connectivity and population distributions in heterogeneous environments. Oikos. 1997;78:151–169. [Google Scholar]
- Wright TF, Rodriquez AM, Fleischer RC. Vocal dialects, sex-biased dispersal, and microsatellite population structure in the parrot Amazona auropalliata. Molecular Ecology. 2005;13:1197–1205. doi: 10.1111/j.1365-294X.2005.02466.x. [DOI] [PubMed] [Google Scholar]
- Yunick RP. A review of recent irruptions of the black-backed woodpecker and the three-toed woodpecker in eastern North America. Journal of Field Ornithology. 1985;56:138–152. [Google Scholar]
- Zink RM, Barrowclough GF. Mitochondrial DNA under siege in avian phylogeography. Molecular Ecology. 2008;17:2107–2121. doi: 10.1111/j.1365-294X.2008.03737.x. [DOI] [PubMed] [Google Scholar]
- Zollner PA, Lima SL. Behavioral tradeoffs when dispersing across a patchy landscape. Oikos. 2005;108:219–230. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.