

Abstract
The development of selectively protected monosaccharide building blocks that can reliably be glycosylated with a wide variety of acceptors is expected to make oligosaccharide synthesis a more routine operation. In particular, there is an urgent need for the development of modular building blocks that can readily be converted into glycosyl donors for glycosylations that give reliably high 1,2-cis-anomeric selectivity. We report here that 1,2-oxathiane ethers are stable under acidic, basic, and reductive conditions making it possible to conduct a wide range of protecting group manipulations and install selectively removable protecting groups such as levulinoyl (Lev) ester, fluorenylmethyloxy- (Fmoc) and allyloxy- (Alloc) carbonates, and 2-methyl naphthyl ethers (Nap). The 1,2-oxathiane ethers could easily be converted into bicyclic anomeric sulfonium ions by oxidization to sulfoxides and arylated with 1,3,5-trimethoxybenzene. The resulting sulfonium ions gave high 1,2-cis anomeric selectivity when glycosylated with a wide variety of glycosyl acceptors including properly protected amino acids, primary and secondary sugar alcohols and partially protected thioglycosides. The selective protected 1,2-oxathianes were successfully employed in the preparation of a branched glucoside derived from a glycogen-like polysaccharide isolated form the fungus Pseudallescheria boydii, which is involved in fungal phagocytosis and activation of innate immune responses. The compound was assembled by a latent-active glycosylation strategy in which an oxathiane was employed as an acceptor in a glycosylation with a sulfoxide donor. The product of such a glycosylation was oxidized to a sulfoxide for a subsequent glycosylation. The use of Nap and Fmoc as temporary protecting groups made it possible to install branching points.
Keywords: stereoselective glycosylations, modular building blocks, auxiliary, sulfonium ions, glucosides
INTRODUCTION
Complex carbohydrates are involved in a wide range of biological processes such as cell-cell recognition, fertilization, embryogenesis, neuronal development, hormone activities, the proliferation of cells and their organization into specific tissues, viral and bacterial infection, and tumor cell metastasis.1 Furthermore, carbohydrates are capable of inducing a protective antibody response, which is a major contributor to the survival of an organism during infection.2 Oligosaccharides have also been found to control the development and defense mechanisms of plants.3 The increased appreciation of the role of carbohydrates in the biological and pharmaceutical sciences has resulted in a revival of interest in carbohydrate chemistry.4
A major obstacle to advances in glycobiology and glycomedicine is the lack of pure and structurally well-defined carbohydrates and glycoconjugates. These compounds are often found in low concentrations and in microheterogeneous forms, greatly complicating their isolation and characterization. In many cases, well-defined oligosaccharides can only be obtained by chemical or enzymatic synthesis.5 Unfortunately, no general method is available for the preparation of complex carbohydrates of biological importance.6 As a result the chemical synthesis of each target compound is a challenging and time-consuming endeavor.
To speed up the process of oligosaccharide synthesis, efforts are underway to identify limited numbers of monosaccharide building blocks that can repeatedly be employed for the synthesis of a wide range of target structures.7 Key features of such building blocks include modification at key positions with selectively removable protecting groups and providing reliably glycoside products in high yield with excellent anomeric control. In respect to the latter, 1,2-transglycosides can reliably be introduced by employing a glycosyl donor modified at C-2 with a protecting group that can perform neighboring group participation during glycosylation. On the other hand, conventional methods to introduce 1,2-cis glycosides require glycosyl donors having a non-assisting functionality at C-2 and in these cases, reaction conditions such as solvent, temperature, and promoter as well as constitution of the glycosyl donor and acceptor (e.g. type of saccharide, leaving group at the anomeric center, protection, and substitution pattern) determine anomeric selectivity.8 1,2-Cis glycosylations give often mixtures of anomers, which requires time-consuming purification protocols resulting in loss of material, as well as limits the use of one-pot multi-step glycosylations9 and automated polymer-supported synthesis.10 Recent advances in anomeric control include the use of protecting groups that sterically shield the β-face of galactosyl donors11 or locking a glycosyl donor in a conformation that allows nucleophilic attack from only one face of an anomeric oxa-carbenium ion.12
We have introduced a stereoselective glycosylation approach based on neighboring group participation by a (S)-phenylthiomethylbenzyl moiety at C-2 of a glycosyl donor (Scheme 1a).13–15 Upon formation of an oxacarbenium ion, the nucleophilic thiophenyl moiety of the C-2 functionality participates leading to the formation of an intermediate sulfonium ion. The formation of the trans-decalin stereoisomer is strongly favored because of the absence of unfavorable gauche interactions. In addition, the alternative cis-decalin system places the phenyl-substituent in an axial position inducing further unfavorable steric interactions. Displacement of the equatorial anomeric sulfonium ion by a sugar alcohol leads to the formation of a 1,2-cisglycoside. We have shown that the (S)-(phenylthiomethyl)benzyl moiety can readily be introduced by reaction of a sugar alcohol with (S)-(phenylthiomethyl)benzyl acetate in the presence of BF3-OEt2 and removed by conversion into an acetyl ester by treatment with BF3-OEt2 in acetic anhydride. The attractiveness of chiral auxiliary mediated glycosylations has been shown by solid phase synthesis of several branched pentasaccharides having only 1,2-cisglycosidic linkages.16 The proposed participation mechanism is supported by low temperature NMR experiments, which unambiguously identified a β-substituted sulfonium ion as a reaction intermediate. Furthermore, the displacement mechanism is supported by the observation that glycosylations with donors having a C-2 auxiliary with incorrect stereochemistry {(R)-(phenylthiomethyl)benzyl} give mixtures of anomers.
Scheme 1.

Sulfonium-ion promoted 1,2-cis glycosylations
a) Neighboring group participation by C-2 (S)-auxiliary leading to 1,2-cis glycosides; b) β-sulfonium ion formation from precyclized oxathiane ketal. In these reactions, hydrolysis of the C-2 auxiliary is a major side product; c) sulfonium ion formation in the presence of a thioether.
Several other studies have shown the usefulness of anomeric sulfonium ions for stereoselective glycosylations. In this respect, a number of donors having an achiral thioether at C-2 have been examined and some of these derivatives gave remarkably good α-anomeric selectivity.13,17 Another interesting approach for forming bicyclic anomeric sulfonium ions involves arylation of 1,2-oxathiane ketals, which can easily be prepared from a thioglycoside (Scheme 1b).18–20 The addition of a thioether to traditional glycosylations or alkylation of thioglycosides can also lead to the formation of anomeric sulfonium ions and enhance alpha-anomeric selectivity (Scheme 1c).21,22
To advance auxiliary mediated glycosylations for building block based oligosaccharide synthesis, it is critical to establish convenient procedures for the preparation of selective protected anomeric sulfonium ions, which can be employed in glycosylations that allow rapid assembly of complex branched compounds. We report here that 1,2-oxathiane ethers are stable under acidic, basic, and reductive conditions making it possible to conduct a wide variety of protecting group manipulations to give panels of selectively protected compounds. The resulting 1,2-oxathiane ethers can easily be converted into bicyclic anomeric sulfonium ions by oxidization to sulfoxides, followed by arylation with 1,3,5-trimethoxybenzene. It is shown that the latter compounds are appropriate glycosyl donors for highly selective 1,2-cis glycosylations provided that they are sufficiently deactivated with electron withdrawing protecting groups. The selective protected 1,2-oxathianes were successfully employed for the preparation of a branched glucoside derived from Pseudallescheria boydii,23 which is involved in fungal phagocytosis and Toll-like receptor activation.
RESULTS AND DISCUSSION
Synthesis and Glycosylations of Oxathiane
We have observed that the (S)-(phenylthiomethyl)benzyl ethers are sensitive to moderately strong acidic conditions complicating a number of important protecting group manipulations. We envisaged that this problem could be addressed by using 1,2-oxathiane ethers as precursors of anomeric sulfonium ions. 1,2-Oxathiane ethers were expected to be stable under acidic conditions because their ring structure prevents the sulfur atom of participating in acid catalyzed cleavage of the benzylic ether linkage, and thus should be compatible with a wide variety of protecting group manipulations. These compounds can, however, be readily converted into an anomeric sulfonium ions by oxidative arylation, for stereoselective 1,2-cis-glycosylations.15,20
To explore the compatibility of 1,2-oxathiane ethers with a range of protecting group manipulations, compounds 9–11 and 13–16 were prepared which differ in the pattern of ether and ester protecting groups (Scheme 2). Furthermore, several of these derivatives are protected with Alloc, Lev or Fmoc protecting groups, which can be selectively removed under mild conditions providing opportunities for further functionalization. The selectively protected 1,2-oxathianes can be oxidized to the corresponding sulfoxides 17–23, which can then be used for the generation of sulfonium ions for subsequent glycosylations.
Scheme 2.

Synthesis of 1,4-oxathiane protected donors
a) TMS2O, TMSOTf, 0 °C, 30 min, then Et3SiH, 3 h (74%); b) PhCH(OMe)2, CSA, reduced pressure, DMF, 50 °C,18 h (87%); c) Ac2O, Py; d) Levulinic acid, DCC, DMAP, DCM (79%); e) AllocCl, TMEDA, DMAP, DCM (87%); f) BnBr, NaH, DMF (79%); g) Et3SiH, TfOH, DCM, −78 °C; h) Et3SiH, PhBCl2, −78 °C; i) Ac2O, Py(9: 85%, g, i 2 steps; 13: 78%, h, i 2 steps; 14: 79%, g, i 2 steps; 15: 92%, g, i 2 steps; 16: 84%, l, i 2 steps); j) Ag2O, BnBr (10: 55%, g, j 2 steps); k) FmocCl, Py/DCM = 1/1 (v/v) (11: 66%, g, k 2 steps); l) EtSH, TsOH, DCM; m) mCPBA, DCM, −78 °C (17: 92%; 18: 96%; 19: 98%; 20: 93%; 21: 98%; 22: 82%; 23: 96%).
Thus, oxathiane 2 was prepared on large scale by a one-pot two-step reaction involving treatment of 1 with TMS2O in the presence of TMSOTf to give an intermediate trimethylsilyl acetal, which was reduced by the addition of Et3SiH. The 4,6-diol of 2 was protected as a benzylidene acetal by treatment with (dimethoxymethyl)benzene in the presence of camphorsulfonic acid (CSA) in DMF under reduced pressure to give 3 in a yield of 87%. The C-3 hydroxyl of compound 3 was protected as an acetyl- and levulinoyl- (Lev) ester,24 fluorenylmethyloxy- (Fmoc)25 and allyloxy- (Alloc)26 carbonate, and benzyl ether using standard conditions to give fully protected 4, 5, 6, and 7, respectively. The benzylidene acetal of 4 was reductively opened using Et3SiH and triflic acid (TfOH) in DCM at −78 °C27 to give compound 8 having a C-4 hydroxyl which was acetylated with Ac2O in pyridine, benzylated under neutral conditions using benzyl bromide and Ag2O in DMF, or treated with FmocCl in a mixture of pyridine and CH2Cl2 to provide selectively protected oxathianes 9, 10, and 11, respectively. Alternatively, the benzylidene acetal of 4 could be opened by treatment with Et3SiH and PhBCl2 at −78 °C27 to give 12 having a C-6 hydroxyl, which was acetylated to give derivative 13. The benzylidene acetals of compounds 5 and 6 could also be selectively opened to give derivatives having a C-4 alcohol, which were acetylated using standard conditions to provide compounds 14 and 15, respectively. Finally, the benzylidene acetal of 7 was removed by treatment with p-toluenesulfonic acid in the presence of ethanethiol to give a diol, which was acetylated to provide derivative 16. The selectively protected oxathianes 9–11 and 13–16 were oxidized by mCPBA at −78 °C to give sulfoxides 17–23 as mixtures of diastereoisomers. The successful preparation of these compounds demonstrates that oxathianes can be subjected to a wide variety of protecting group manipulation and withstand acidic, basic and reductive conditions.
Having the sulfoxides 17–23 at hand, attention was focused on their conversion into anomeric sulfonium ions for subsequent glycosylations. In this respect, activation of the sulfoxides by triflic anhydride in the presence of trimethoxybenzene will result in arylation of sulfur to give an intermediate sulfonium ion.15,18 In the first instance, oxathiane 17 was explored as a glycosyl donor for glycosylations with a variety of different glycosyl acceptors (Table 1). Thus, 17 was activated with trifluoromethanesulfonic anhydride (Tf2O) in the presence of 1,3,5-trimethoxybenzene in DCM at −10 °C. After completion of the electrophilic aromatic substitution and formation of the intermediate sulfonium ion, alcohols 24–28 were added and after a reaction time of 16 h at room temperature, the disaccharides 29–33 were isolated by size exclusion column chromatography and anomeric selectivities determined by careful analysis of 1H NMR spectra. Each glycosylation proceeded with exceptional high alpha-anomeric selectivity, however, in the case of acceptors 24, 25, and 27 a trace amount of beta-amomer was detected. The use of primary and secondary sugar alcohols gave the corresponding disaccharides in good to excellent yields. The glycosylation protocol is also compatible with the use of appropriately protected amino acids and for example glycosylated threonine derivative 31 could be prepared in high yield as only the α-anomer. Furthermore, it was also found that thioglycosides can be employed as glycosyl acceptor and for example the use of 27 and 28 led to clean formation of disaccharides 32 and 33, respectively. The latter type of glycosylation is attractive because it is to be expected that the thioglycosyl products can employed as glycosyl donors in subsequent glycosylations using an appropriate thiophilic reagent thereby offer a rapid strategy for oligosaccharide assembly.
Table 1.
Stereoselective glycosylations between donor 17 and various acceptors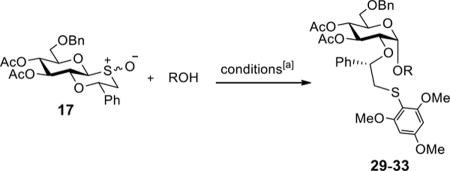
| Entry | ROH | Product | Yield[b] | α:p[c] |
|---|---|---|---|---|
| 1 | 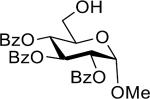 |
29 | 91% | >15:1 |
| 24 | ||||
| 2 | 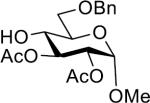 |
30 | 62% | >25:1 |
| 25 | ||||
| 3 | 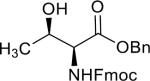 |
31 | 89% | α only |
| 26 | ||||
| 4 | 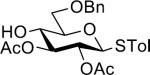 |
32 | 67% | >15:1 |
| 27 | ||||
| 5 | 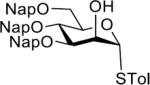 |
33 | 72% | α only |
| 28 |
1,3,5-Trimethoxybenzene, Tf2O, DTBMP, molecular sieves 4 Å, −10 °C, 30 min, then add acceptor, −40 °C to room temperature, 16 h.
Isolated yields of the α/β mixture of disaccharide products.
The α/β ratios were determined by the integration of key signals in the 1H NMR spectra of the disaccharide products after purification by LH-20 size exclusion chromatography.
Next, we explored whether protecting groups commonly employed in oligosaccharide synthesis are compatible with the glycosylation protocol. Thus, glycosyl donors 17, 18, 20–23, 34, and 35 were activated by triflic anhydride in the presence of 1,3,5-trimethoxybenzene to give intermediate sulfonium ions, which were glycosylated with acceptors 36 and 37. As can be seen in Table 2, the expected disaccharides 38–54 were isolated in good to excellent yields. Furthermore, glycosyl donors modified with electron withdrawing protecting groups at C-3, 4, and 6 gave the corresponding disaccharides as only the alpha-anomer (entry 1–4). Glycosyl donors that had an ether function at C-4 or C-6 (entries 5–11) gave exceptional high alpha anomeric selectivities (α:β > 15:1). Highly reactive donors having multiple ether type protecting groups gave in some case disaccharides with modest anomeric selectivities (entries 13 and 17). These observations are in agreement with previous finding15 that highly reactive anomeric sulfonium ions can react through an oxacarbenium ion resulting in loss of α-anomeric selectivity.
Table 2.
Protecting group pattern effect on stereoselectivity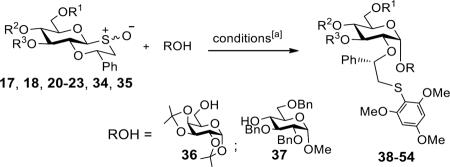
| Entry | Donor | R1 | R2 | R3 | ROH | Product | Yield[a] | α/β ratio[c] |
|---|---|---|---|---|---|---|---|---|
| 1 | 34 | Ac | Ac | Ac | 36 | 38 | 80% | α only |
| 2 | 34 | Ac | Ac | Ac | 24 | 39 | 59% | α only |
| 3 | 34 | Ac | Ac | Ac | 37 | 40 | 48% | α only |
| 4 | 20 | Ac | Bn | Ac | 36 | 41 | 84% | α only |
| 5 | 20 | Ac | Bn | Ac | 37 | 42 | 59% | >15:1 |
| 6 | 17 | Bn | Ac | Ac | 36 | 43 | 94% | α only |
| 7 | 17 | Bn | Ac | Ac | 37 | 44 | 67% | >25:1 |
| 8 | 22 | Bn | Ac | Alloc | 36 | 45 | 67% | α only |
| 9 | 22 | Bn | Ac | Alloc | 24 | 46 | 78% | >15:1 |
| 10 | 21 | Bn | Ac | Lev | 36 | 47 | 85% | >15:1 |
| 11 | 21 | Bn | Ac | Lev | 37 | 48 | 64% | >15:1 |
| 12 | 23 | Ac | Ac | Bn | 36 | 49 | 98% | α only |
| 13 | 23 | Ac | Ac | Bn | 37 | 50 | 46% | 7:1 |
| 14 | 18 | Bn | Bn | Ac | 36 | 51 | 95% | >15:1 |
| 15 | 18 | Bn | Bn | Ac | 37 | 52 | 66% | 10:1 |
| 16 | 35 | Bn | Bn | Bn | 36 | 53 | 98% | >15:1 |
| 17 | 35 | Bn | Bn | Bn | 37 | 54 | 57% | 1.5:1 |
1,3,5-Trimethoxybenzene, Tf2O, DTBMP, molecular sieves 4 Å, −10 °C, 30 min, then add acceptor, −40 °C to r.t., 16 h.
Isolated yields of the α/β mixture of disaccharide products.
The α/β ratios were determined by the integration of key signals in the 1H NMR spectra of the disaccharide products after purification by LH-20 size exclusion chromatography.
Characterization of Sulfonium Ions by NMR
To ascertain that glycosylations of oxathianes proceed through a sulfonium ion intermediates, compound 17, trimethoxybenzene, and DTBMP were dissolved in CDCl3, activated with triflic anhydride and 1H, gCOSY, gHSQC, and HMBC spectra were recorded (Figure 1). As expected, the chemical shift of H-1 of the newly formed product shifted down field (δ = 4.31 and δ = 4.15 to δ = 5.60) and exhibited J1,2 = 9 Hz, which is consistent with a β-anomeric configuration. Full assignment of proton and carbon signals could be made by gCOSY and gHSQC. Careful analysis of proton coupling constants indicated that no conformational distortion of the saccharide ring had occurred. The presence of a carbon-sulfur linkage of the sulfonium ion was established by an HMBC experiment, which allows the determination of three-bond proton-carbon couplings. In particular, a strong correlation was observed between C-1 and H8eq, confirming the presence of a trans-decalin system. Furthermore, the smaller geminal-coupling constant between H8eq and H8ax (12 Hz) indicates that the trimethoxyphenyl substituent of sulfur adopts an equatorial orientation. Comparing chemical shifts of the trimethoxyphenyl (H8eq 3.98, H8ax 4.28 ppm) and phenyl (H8eq 4.32, H8ax 3.66 ppm) substituted sulfonium ion indicates that their aromatic rings adopt different conformations and in particular deshielding of H8ax of the trimethoxylbenzene substituted system indicates that the trimethoxybenzene group is perpendicular to the plane of the sugar ring (see SI for further details).
Figure 1.

NMR study of sulfonium ions. a) thermostability of sulfonium ion 55; b) HMBC spectrum of sulfonium ion 55. c) Comparison of electronic properties of different sulfonium ions by NMR chemical shift (all chemical shifts were referred to TMS).
aStereochemistry at sulfur was assigned by empirical rules using H-8ax,eq geminal coupling.28
bCited data.22
Interestingly, sulfonium 55 was stable at room temperature for 9 h, while subsequent incubation at 45°C for 5 min resulted in complete decomposition. It was also noticed that the excess of trimethoxylbenzene was further consumed after complete formation of the sulfonium ion, and was no longer present after an incubation time of 9 h.
A number of additional sulfonium ions (56–61) were generated and their structure and reactivity studied by NMR. Thus, compounds 55–58 were prepared by arylation of the corresponding sulfides and differ in the pattern of protecting groups at C-3, C-4, and C-6. Compounds 59–61 were prepared by methylation of the corresponding sulfides with methyl triflate and also differ in hydroxyl protection. Finally, acyclic sulfonium ion 62 was formed by methylation of the corresponding ethyl thioglycoside with methyl triflate.
As expected sulfonium ions 55–58 are stable at room temperature but readily react with alcohols to give mainly or exclusively α-glucosides. Interestingly, exposure of sulfonium ion 59 to an excess of methanol did not lead to glucoside formation and this compound was unaltered after a reaction time of 24 h. Benzylated sulfonium ion 61 was slightly more reactive and exposure to an excess of 24 gave the corresponding glucoside in a low yield of 21% as a mixture of anomers. These results clearly demonstrate that the nature of the sulfonium ion is a major determinant of anomeric reactivity. It was also found that the cyclic nature of sulfonium ions influences anomeric reactivity and for example within 15 min acyclic sulfonium ion 62 reacted with excess methanol to give a methyl glycoside whereas the corresponding cyclic sulfonium ion 59 is unreactive under these conditions.22
It is well known that carbon chemical shifts are mainly determined by local electronic environment.29 Interestingly, it was observed that the chemical shift of C-1 of the arylated and alkylation sulfonium ions differ by approximately 2 ppm indicating a lower electronic density at C-1 of alkylated sulfonium ion, which surprisingly did not result a higher reactivity towards alcohols. Probably, the higher reactivity of the arylated sulfonium corresponds to its better leaving group ability. In this respect, a weaker base constitutes in general a better leaving group. Furthermore, it has been established that the relative stabilities of sulfonium ions corresponds with the relative basicities of the corresponding leaving group.30 The fact that thioanisol is a weaker base than dimethyl sulfide30,31 would indicate that arylated sulfonium ion are less stable and provide a better leaving. This is in agreement with the observations described above.
Chemical Synthesis of an Immuno-active Glycogen-like Component
Having established a range of selective protected oxathiane building blocks suitable for selective 1,2-cis-glycosylations, attention was focused on the preparation of tetraglucoside 79, which is derived from the cell wall polysaccharide of the fungus Pseudallescheriaboydii.23 This fungus is found in soil and polluted water and can cause infections in immuno-compromised and immuno-competent hosts. Recent structural studies have shown that the cell wall of P. boydii contains a glycogen like component that is composed of a α(1–4)-linked glucopyranoside backbone modified at C-6 with α-Glcp moieties.32 A soluble form of the polysaccharide could inhibit phagocytosis in a dose dependent manner and furthermore enzymatic degradation of the glycogen-like component of P. boydii could reduce the phagocytic index demonstrating that it plays a key role in phagocytosis. It has also been shown that the α-glucan can induce cytokine secretion in a TLR2-dependent manner. The minimal structural motif that can induce these properties is difficult to establish due to structural heterogeneity of the polysaccharide. However, it is to be expected that this problem can be addressed by chemically synthesizing a library of glucans differing in C-6 branching patterns.
α-Glucans represent a considerable synthetic challenge due to the presence of multiple 1,2-cis glycosidic linkages, the low reactivity of C-4 hydroxyls of glucosyl acceptors and the branched nature of the compound. Therefore, this component offered an exciting opportunity to examine the usefulness of oxathianes for the preparation of complex and biological important compounds.
We envisaged that a-glucans such as 79 can be assembled by a latent-active glycosylation strategy33 in which a sulfoxide donor is coupled with an oxathiane acceptor to give a product that can be oxidized to a sulfoxide for further glycosylations (Schemes 3 and 4). To establish the feasibility of such a latent-active glycosylation strategy, sulfoxides 17, 19, and 20 were treated with Tf2O in the presence of 1,3,5-trimethoxybenzene to form an intermediate sulfonium ion, which was glycosylated with oxathiane 63 to give disaccharide 64–66 as mainly alpha-glucosides. These results demonstrate that oxathianes are stable under the condition used for arylation of sulfoxides to give sulfonium ion.
Scheme 3.

Latent-active glycosylation strategy
a The α/β ratios were determined by the integration of key signals in the 1H NMR spectra of the disaccharide products after purification by LH-20 size exclusion chromatography.
Scheme 4.

Synthesis of the glycogen-like glucan isolated from P. boyii
a) i. 10% TFA in DCM, 0 °C, 1 h; ii. Ac2O, Py (67: 89%, 2 steps; 71: 98%, two steps; 74: 76%, 2 steps); b) mCPBA, DCM, −78 °C (98%); c) general glycosylation condition (70: 86%, α only; 73: 65%, α/β>15:1; 76: 70%, α only; 77: 73%, α/β=8:1); d) 20% NMP in DMF, 30 min (91%); e) DDQ, H2O:DCM=1:10 (92%); f) 10% TFA in DCM, 0 °C, 1 h (85%); g) i. NaOMe, MeOH; ii. H2, Pd(OH)2/C, tBuOH-H2O-AcOH=40:1:1 (83%, 2 steps).
It was expected that disaccharide 65 would be a suitable substrate for the preparation of target compound 79. In this respect, the anomeric oxathiane can be oxidized to a sulfoxide and then converted into a sulfonium ion for glycosylation with a properly protected aminopropyl spacer. Furthermore, the Nap34 ether at C-6 and Fmoc carbonate at C-4 are fully orthogonal and will allow glucosylations at these positions to give after deprotection the expected tetrasaccharide 79.
First, the C-2 auxiliary of 65 was converted into an acetyl ester to give 67 using a standard procedure. This additional step was required to avoid oxidation of the (2,4,6-trimethoxyphenyl)sulfane moiety of the non-reducing glucoside during the subsequent oxidation step to convert the oxathiane moiety into a sulfoxides. As expected, compound 67 could be cleanly oxidized to the corresponding sulfoxides, which was activated with Tf2O/1,3,5-trimethoxybenzene to give an anomeric sulfonium ion that was glycosylated with alcohol 69 to provide spacer-containing 70 as only the alpha-anomer. The C-2 auxiliary of 70 was converted into an acetyl ester and the Fmoc group of the resulting compound 71 was cleaved by treatment with N-methyl-2-pyrrolidone (NMP) in DMF to give glucosyl acceptor 72. Next, glycosyl acceptor 72 was coupled with the sulfonium ion derived from sulfoxide 17 using standard conditions to give trisaccharide 73 as mainly the alpha glucoside. The trace amount of unwanted b-anomer could easily be removed by silica gel column chromatography. The C-2 auxilairy of 73 was converted into an acetyl ester by standard procedures to give compound 74, which was converted into glycosyl acceptor 75 by oxidative removal of the Nap ether using 5,6-dicyano-1,4-benzoquinone (DDQ) in a mixture of DCM and water. A Tf2O/1,3,5-trimethoxybenzene mediated glycosylation of 17 with 75 gave tetrasaccharide 77 as an 8/1 mixture of alpha/beta anomers. In order to improve the anomeric selectivity, the glycosylation was repeated with glycosyl donor 20 having an acetyl esters at C-3 and C-6 and a benzyl ether at C-4 and gratifyingly the use of this compound gave tetrasaccharide 76 in a yield of 70% as only the alpha anomer. Deprotection could easily be accomplished by a three-step procedure involving treatment with TFA in DCM to remove the C-2 auxiliary, saponification of the acetyl esters with sodium methoxide in methanol and catalytic hydrogenolysis of the benzyl ether and the reduction of azide using Pd(OH)2/C. The structural identity of α-glucan 77 was confirmed by homo- and heteronuclear two dimensional NMR (gCOSY and gHSQC) experiments and in particular the chemical shifts and coupling constants of the anomeric protons confirmed (1→4) and (1→6)-linked alpha-glucosidic linkages.35
CONCLUSIONS
Modular building blocks that can readily be converted into glycosyl donors and acceptors have the potential to speedup the process of oligosaccharide assembly. The potential of such a strategy will, however, only be realized when building blocks will be developed that reliably give high 1,2-cis-anomeric selectivities. Previously, we introduced a stereoselective 1,2-cis-glycosylation approach based on neighboring group participation by a (S)-phenylthiomethylbenzyl moiety at C-2 of a glycosyl donor to give a bicyclic b-sulfonium ion intermediate that can readily be displaced by sugar alcohol leading to the formation of 1,2-cis-glycosides. Although the auxiliary mediated approach has many attractive features, the acid sensitivity of the (S)-phenylthiomethylbenzyl moiety complicates certain protecting group manipulation, which in turn makes it difficult to develop robust modular building blocks. We describe here that monosaccharides protected as 1,2-oxathiane ethers are stable to commonly employed protecting groups manipulations making it possible to install a variety of selectively removable protecting groups such as Lev ester, Fmoc and Alloc carbonates, and Nap ether. Furthermore, the 1,2-oxathiane ether could easy be installed by a novel one-pot two-step procedure and it was found that protection of the 1,2-diol facilitated modification of the C-3, C-4 and C-6 alcohols. The resulting 1,2-oxathianes could be employed as glycosyl donors for highly selective 1,2-cisglycosylation by oxidation to sulfoxides followed by arylation with 1,3,5-trimethoxybenzene to give a bicyclic anomeric sulfonium ion. The attractiveness of the new building blocks has been demonstrated by the preparation of a biologically important branched a-glucan, which was assembled by a latent-active glycosylation strategy.
EXPERIMENTAL SECTION
Full experimental is available in Supporting Information. Compounds 1,1824,3625,3726,3827,3934,1535,15 and 3740 were prepared following literature procedures.
General Procedure for the Preparation of Sulfoxide Donors 17 – 23 from their Corresponding Oxathianes 9 – 11 and 13 – 16
m-CPBA (≤ 77%, l.1 eq.) was dissolved in DCM and slowly injected into a cooled (-−78 °C) solution of oxathiane in DCM. The mixture was stirred at −78 °C for 30 min, diluted with DCM (50 mL) and then poured into 10% Na2S2O3 aqueous solution. The organic layer was washed with saturated NaHCO3, dried (MgSO4), filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel chromatography.
General Glycosylation Procedure for Oxathiane Donors with Various Acceptors
A mixture of sulfoxide donor (1 eq.), 1,3,5-trimethoxybenzene (1.5 eq.), 2,6-di-tert-butyl-4-methyl pyridine (2 eq.), and activated molecular sieves (4 Å) in DCM (adjusted donor concentration to 0.15 M) was stirred for 1 h under an atmosphere of argon. After cooling to −10 °C, trifluoromethanesulfonic anhydride (1.1 eq.) was added. After 30 min, the reaction mixture was cooled (−40 °C), and a solution of acceptor (0.8 eq.) in DCM (adjusted donor concentration to 0.1 M) was added slowly. The temperature of the reaction mixture was kept at −40 °C for another 60 min before allowed to warm to room temperature. After 15 h, the reaction mixture was diluted with DCM (10 mL), filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography or sephadex® LH20 size exclusion chromatography (DCM/MeOH = 1/1, 0.2 mL/min).
General Procedure for the Removal of C-2 Auxiliary
Trifluoroacetic acid was added dropwise to a solution of glucoside in DCM at 0 °C adjusting the final concentration to 10% (v/v). The reaction mixture was stirred for 0.5–3 h until TLC indicated complete consumption of starting material. The reaction mixture was diluted with DCM and poured into saturated NaHCO3. The organic layer was dried (MgSO4), filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography.
Procedures for NMR Study of Sulfonium ions
S-(2,3,5-trimethoxylphenyl)-oxathianium triflate: A mixture of R/S sulfoxides (10 µmol), 1,3,5-trimethoxybenzene (16 μmol), 2,6-di-tert- butyl-4-methyl pyridine (21 μmol), and activated molecular sieves (4 Å, pellets) in CDCl3 (1 mL) was shaken for 30 min under an atmosphere of argon. Then 0.5 mL of the solution was transferred to a 5 mm NMR tube and sealed. After cooling to 0 °C, a trifluoromethanesulfonic anhydride stock solution (25 μL, 0.23 M in CDCl3) was added and NMR spectra for 55–58 were recorded at room temperature. S-methyl-oxathianium triflate: Sulfide (10 μmol) was dissolved in dry CDCl3 (1 mL), methyl triflate (0.1 mmol) was added under the atmosphere of argon at room temperature. The reaction mixture was stirred for 2–16 h and monitored by TLC. After the consumption of sulfide, 0.5 mL of the reaction mixture was transferred to a 5 mm NMR tube and sealed. NMR spectra for 59–61 were recorded at room temperature.
Supplementary Material
ACKNOWLEDGEMENT
The authors wish to thank Dr. John Glushka for assisting with NMR experiments and Dr. Paul von Rague Schleyer for helpful discussions. The research was supported by the National Institute of General Medical Sciences (NIGMS) of the U.S. National Institutes of Health (R01GM065248, G.-J.B.).
Footnotes
Supporting Information. Full experimental procedures and characterizations of prepared compounds, detailed NMR analysis of sulfonium ion 55, assignment and conformational analysis of diastereomeric sulfonium ions, control glycosylations using donors without C-2 auxiliary, copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Brockhausen I. EMBO Rep. 2006;7:599–604. doi: 10.1038/sj.embor.7400705. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ohtsubo K, Marth JD. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]; Slawson C, Hart GW. Nat. Rev. Cancer. 2011;11:678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Crocker PR, Paulson JC, Varki A. Nat. Rev. Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]; van Vliet SJ, Garcia-Vallejo JJ, van Kooyk Y. Immunol. Cell Biol. 2008;86:580–587. doi: 10.1038/icb.2008.55. [DOI] [PubMed] [Google Scholar]; Astronomo RD, Burton DR. Nat. Rev. Drug Discov. 2010;9:308–324. doi: 10.1038/nrd3012. [DOI] [PMC free article] [PubMed] [Google Scholar]; Morelli L, Poletti L, Lay L. Eur. J. Org. Chem. 2011:5723–5777. [Google Scholar]
- (3).Montesano M, Brader G, Palva ET. Mol. Plant Pathol. 2003;4:73–79. doi: 10.1046/j.1364-3703.2003.00150.x. [DOI] [PubMed] [Google Scholar]; Silipo A, Erbs G, Shinya T, Dow JM, Parrilli M, Lanzetta R, Shibuya N, Newman MA, Molinaro A. Glycobiology. 2010;20:406–419. doi: 10.1093/glycob/cwp201. [DOI] [PubMed] [Google Scholar]
- (4).Brown JR, Crawford BE, Esko JD. Crit. Rev. Biochem. Mol. Biol. 2007;42:481–515. doi: 10.1080/10409230701751611. [DOI] [PubMed] [Google Scholar]; Liang PH, Wu CY, Greenberg WA, Wong CH. Curr. Opin. Chem. Biol. 2008;12:86–92. doi: 10.1016/j.cbpa.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; Buskas T, Thompson P, Boons GJ. Chem. Commun. 2009:5335–5349. doi: 10.1039/b908664c. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ernst B, Magnani JL. Nat. Rev. Drug Discov. 2009;8:661–677. doi: 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]; Laremore TN, Zhang F, Dordick JS, Liu J, Linhardt RJ. Curr. Opin. Chem. Biol. 2009;13:633–640. doi: 10.1016/j.cbpa.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; Stallforth P, Lepenies B, Adibekian A, Seeberger PH. J. Med. Chem. 2009;52:5561–5577. doi: 10.1021/jm900819p. [DOI] [PubMed] [Google Scholar]
- (5).Zhu XM, Schmidt RR. Angew. Chem. Int. Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]; Schmaltz RM, Hanson SR, Wong CH. Chem. Rev. 2011;111:4259–4307. doi: 10.1021/cr200113w. [DOI] [PubMed] [Google Scholar]
- (6).Boltje TJ, Buskas T, Boons GJ. Nat. Chem. 2009;1:611–622. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wong CH, Ye XS, Zhang Z. J. Am. Chem. Soc. 1998;120:7137–7138. [Google Scholar]; Zhu T, Boons GJ. Tetrahedron: Asymmetry. 2000;11:199–205. [Google Scholar]; Werz DB, Ranzinger R, Herget S, Adibekian A, von der Lieth CW, Seeberger PH. ACS Chem. Biol. 2007;2:685–691. doi: 10.1021/cb700178s. [DOI] [PubMed] [Google Scholar]; Arungundram S, Al-Mafraji K, Asong J, Leach FE, 3rd, Amster IJ, Venot A, Turnbull JE, Boons GJ. J. Am. Chem. Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adibekian A, Stallforth P, Hecht ML, Werz DB, Gagneux P, Seeberger PH. Chem. Sci. 2011;2:337–344. [Google Scholar]
- (8).Demchenko AV. Curr. Org. Chem. 2003;7:35–79. [Google Scholar]; Cai F, Wu B, Crich D. Adv. Carbohydr. Chem. Biochem. 2009;62:251–309. doi: 10.1016/S0065-2318(09)00006-7. [DOI] [PubMed] [Google Scholar]; Guo J, Ye XS. Molecules. 2010;15:7235–7265. doi: 10.3390/molecules15107235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Boons GJ. Tetrahedron. 1996;52:1095–1121. [Google Scholar]; Codee JDC, Litjens R, van den Bos LJ, Overkleeft HS, van der Marel GA. Chem. Soc. Rev. 2005;34:769–782. doi: 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]; Tanaka H, Yamada H, Takahashi T. Trends Glycosci. Glycotechnol. 2007;19:183–193. [Google Scholar]
- (10).Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]; Seeberger PH. Chem. Soc. Rev. 2008;37:19–28. doi: 10.1039/b511197h. [DOI] [PubMed] [Google Scholar]
- (11).Imamura A, Ando H, Korogi S, Tanabe G, Muraoka O, Ishida H, Kiso M. Tetrahedron Lett. 2003;44:6725–6728. [Google Scholar]
- (12).Zhu XM, Kawatkar S, Rao Y, Boons GJ. J. Am. Chem. Soc. 2006;128:11948–11957. doi: 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]
- (13).Kim JH, Yang H, Park J, Boons GJ. J. Am. Chem. Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- (14).Park J, Boltje TJ, Boons GJ. Org. Lett. 2008;10:4367–4370. doi: 10.1021/ol801833n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Boltje TJ, Kim JH, Park J, Boons GJ. Org. Lett. 2011;13:284–287. doi: 10.1021/ol1027267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Boltje TJ, Kim JH, Park J, Boons GJ. Nat. Chem. 2010;2:552–557. doi: 10.1038/nchem.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cox DJ, Fairbanks AJ. Tetrahedron: Asymmetry. 2009;20:773–780. [Google Scholar]
- (18).Fascione MA, Adshead SJ, Stalford SA, Kilner CA, Leach AG, Turnbull WB. Chem. Commun. 2009:5841–5843. doi: 10.1039/b913308a. [DOI] [PubMed] [Google Scholar]
- (19).Fascione MA, Turnbull WB. Beilstein J. Org. Chem. 2010;6:19. doi: 10.3762/bjoc.6.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fascione MA, Kilner CA, Leach AG, Turnbull WB. Chem. Eur. J. 2012;18:321–333. doi: 10.1002/chem.201101889. [DOI] [PubMed] [Google Scholar]
- (21).West AC, Schuerch C. J. Am. Chem. Soc. 1973;95:1333–1335. [Google Scholar]; Lundt I, Skelbæk-Pedersen B. Act. Chem. Scand. 1981;B35:637–642. [Google Scholar]; Park J, Kawatkar S, Kim JH, Boons GJ. Org. Lett. 2007;9:1959–1962. doi: 10.1021/ol070513b. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nokami T, Shibuya A, Mgnabe S, Ito Y, Yoshida J. Chem. Eur. J. 2009;15:2252–2255. doi: 10.1002/chem.200802293. [DOI] [PubMed] [Google Scholar]; Nokami T, Nozaki Y, Saigusa Y, Shibuya A, Manabe S, Ito Y, Yoshida J. Org. Lett. 2011;13:1544–1547. doi: 10.1021/ol200242u. [DOI] [PubMed] [Google Scholar]
- (22).Mydock LK, Kamat MN, Demchenko AV. Org. Lett. 2011;13:2928–2931. doi: 10.1021/ol2009818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lopes LC, da Silva MI, Bittencourt VC, Figueiredo RT, Rollin-Pinheiro R, Sassaki GL, Bozza MT, Gorin PA, Barreto-Bergter E. Mycoses. 2011;54:28–36. doi: 10.1111/j.1439-0507.2011.02105.x. [DOI] [PubMed] [Google Scholar]
- (24).van Boom JH, Burgers PMJ. Tetrahedron Lett. 1976:4875–4878. [Google Scholar]
- (25).Gioeli C, Chattopadhyaya JB. Chem. Commun. 1982:672–674. [Google Scholar]
- (26).Adinolfi M, Barone G, Guariniello L, Iadonisi A. Tetrahedron Lett. 2000;41:9305–9309. [Google Scholar]
- (27).Sakagami M, Hamana H. Tetrahedron Lett. 2000;41:5547–5551. [Google Scholar]
- (28).Lambert JB, Featherman SI. Chem. Rev. 1975;75:611–626. [Google Scholar]
- (29).Martin GJ, Martin ML, Odiot S. Organic Magnetic Resonance. 1975;7:2–17. [Google Scholar]
- (30).Jurić S, Denegri B, Kronja O. J. Phys. Org. Chem. 2012;25:147–152. [Google Scholar]
- (31).Laurence C, Berthelot M, Evain K, Illien B. Can. J. Chem. 2005;83:138–145. [Google Scholar]
- (32).Bittencourt VC, Figueiredo RT, da Silva RB, Mourao-Sa DS, Fernandez PL, Sassaki GL, Mulloy B, Bozza MT, Barreto-Bergter E. J. Biol. Chem. 2006;281:22614–22623. doi: 10.1074/jbc.M511417200. [DOI] [PubMed] [Google Scholar]; Figueiredo RT, Fernandez PL, Dutra FF, Gonzalez Y, Lopes LC, Bittencourt VC, Sassaki GL, Barreto-Bergter E, Bozza MT. J. Biol. Chem. 2010;285:40714–40723. doi: 10.1074/jbc.M110.181255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Roy R, Andersson FO, Letellier M. Tetrahedron Lett. 1992;33:6053–6056. [Google Scholar]; Boons GJ, Isles S. Tetrahedron Lett. 1994;35:3593–3596. [Google Scholar]; Hasty SJ, Kleine MA, Demchenko AV. Angew. Chem. Int. Ed. 2011;50:4197–4201. doi: 10.1002/anie.201007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fuwa H, Fujikawa S, Tachibana K, Takakura H, Sasaki M. Tetrahedron Lett. 2004;45:4795–4799. [Google Scholar]; Szabo ZB, Borbas A, Bajza I, Liptak A. Tetrahedron: Asymmetry. 2005;16:83–95. [Google Scholar]
- (35).Zang LH, Howseman AM, Shulman RG. Carbohydr. Res. 1991;220:1–9. doi: 10.1016/0008-6215(91)80001-4. [DOI] [PubMed] [Google Scholar]; Dinadayala P, Lemassu A, Granovski P, Cerantola S, Winter N, Daffe M. J. Biol. Chem. 2004;279:12369–12378. doi: 10.1074/jbc.M308908200. [DOI] [PubMed] [Google Scholar]
- (36).Ziegler T, Kovac P, Glaudemans CP. Carbohydr. Res. 1989;194:185–198. doi: 10.1016/0008-6215(89)85018-9. [DOI] [PubMed] [Google Scholar]
- (37).Deninno MP, Etienne JB, Duplantier KC. Tetrahedron Lett. 1995;36:669–672. [Google Scholar]
- (38).Mo KF, Fang T, Stalnaker SH, Kirby PS, Liu M, Wells L, Pierce M, Live DH, Boons GJ. J. Am. Chem. Soc. 2011;133:14418–14430. doi: 10.1021/ja205473q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ellervik U, Grundberg H, Magnusson G. J. Org. Chem. 1998;63:9323–9338. [Google Scholar]
- (40).Garegg PJ, Hultberg H. Carbohydr. Res. 1981;93:C10–C11. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.