

Abstract
The cyclization and planarization of polycyclic aromatic hydrocarbons with concomitant oxygen substitution was achieved through acid catalyzed transetherification and oxygen-radical reactions. The triptycene scaffold enforces proximity of the alcohol and arene reacting partners and confers significant rigidity to the resulting π system, expanding the tool set of iptycenes for materials applications.
Polycyclic aromatic hydrocarbons (PAH) have attracted considerable attention because of the promise these materials hold for molecular electronics.1-5 Inherent to the structure of many PAHs is an overall planar topology, which is generally desired to create the highest π-orbital overlap and electron delocalization.5-7 A common method to access PAHs is by the intramolecular oxidative cyclodehydrogenation of adjacent phenylene vertices through the Scholl reaction8 (1→2). This chemistry has seen broad application because of its ability to form multiple bonds between unfunctionalized C-H bonds under mild conditions with relatively high efficiency. Indeed, two dimensional graphene nanoribbons up to 12 nm in length have been synthesized from preassembled frameworks.9 However, tolerance of heteroatoms—which can dramatically modify the electronic properties of PAHs10—has proven a synthetic challenge.11-16
The rigid three-dimensional structure of iptycene-derived scaffolds has proven to be an exceptionally versatile motif for creating high performance and new material properties.17,18 Recently, our group reported an efficient synthesis of 1,4-dibromotriptycene diols (TD, 3) through a rhodium-catalyzed [2+2+2] cycloaddition.19,20 To expand the diversity of iptycene scaffolds for PAH applications, we envisioned 3 might be elaborated through palladium cross-coupling methods (3→4). The resulting extended π-system (4) bears close proximity to the TD hydroxyl group—enforced by the triptycene scaffold—affording the opportunity for cyclization reactions to create oxygen substituted PAHs (5). We recently demonstrated this principle for cyclization of alkyne π-systems to transition between two classes of conjugated polymer back-bones.19 Herein, we report the extension of this principle for the cyclization and planarization of a variety of arene π-systems—through both oxidative and non-oxidative methods—to give triptycene incorporated PAHs with oxygen heteroatom substituents (4→5). Further, the [2,2,2]-ring system on which the triptycene is based appears to confer considerable rigidity, resulting in extremely sharp photophysical features, introducing a new aspect of iptycene structure for electronic material design.

Compound 3 provided a convenient branch point to explore the proposed cyclization through diversification using palladium catalyzed Suzuki-Miyaura cross-coupling; to this end, 6 was synthesized in excellent yield (Scheme 1). The Scholl reaction is generally thought to proceed through an aromatic cation,21-23 and we envisioned the proximal electron rich oxygen of the hydroxyl group might be coaxed into reaction with the oxidized arene. Unfortunately, under several Scholl-type oxidants (FeCl3,24,25 DDQ,22b,26 CuCl2,27,28), only starting material was isolated. Concurrently, we also tested conditions likely to generate an oxygen-centered radical28 (CeIV,29-32 Pb(OAc)4,33 CuII/S2O8 2-,34,35). Encouragingly, cerium ammonium nitrate produced small amounts of the half-cyclized intermediate, but the CuII/S2O82- protocol gave the fully cyclized 7 in good yield with spot-to-spot conversion by TLC. The structure of 7 was unambiguously confirmed by X-ray crystallography (Figure 1). Reports of the CuII/S2O8 2- conditions have invoked both aromatic radical cations36 and oxygen-centered radical mechanisms,34,35 but we believe the oxygen-radical hypothesis to be the dominant pathway for this system (vide infra).
Figure 1.
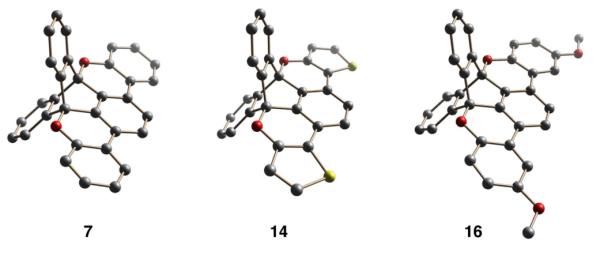
X-ray crystal structures of cyclized compounds.
We next applied these conditions to higher order π-systems (11). Compound 8—an intermediate in the synthesis of TDs20—was further elaborated to 9 by a [2+2+2] cycloaddition with diphenylacetylene and Wilkinson’s catalyst. Application of the CuII/S2O8 2- conditions led to successful cyclization of the TD hydroxyl groups (10) as anticipated, but failed to convert 9 to a fully cyclized compound. This observation lends support to our oxygen-radical hypothesis for cyclization, as an arene centered cation might have led to a fully cyclized product by a Scholl-type mechanism. Attempts to cyclize the remaining arenes with FeCl3 led to complex oli-gomeric mixtures (11) most likely coupled para to the installed oxygen following established Scholl reactivity for such compounds.23 The proposed structure 11 is based on UV/vis (see Figure 3b and associated text for explanation) and MALDI-TOF mass spectrometry.
Figure 3.

(a-d) UV/Vis absorbance (solid) and fluorescence (dashed) spectra in CHCl3; (e-h) Cyclic voltammograms in CH2Cl2 with nBu4NPF6 as electrolyte (see Supporting Information for further details).
We also investigated the CuII/S2O8 2- conditions with thiophene derivatives (Scheme 2, 12 and 13). Initial progress was hampered due to rapid deboronation of 2-thiophene boronic acid derivatives under cross-coupling conditions. However, application of a recently developed palladium precatalyst in the Buchwald laboratory37 allowed for milder conditions and returned the desired product in excellent yield. Unfortunately, application of the CuII/S2O8 2-conditions produced complex mixtures and insoluble material. These results indicated the conditions to be too harsh for more electron rich aromatics. Further, we also desired a milder alternative as these oxidative conditions have been shown to oxidize the benzylic position of appended alkanes35—substituents relevant for PAH solubility.
We were inspired by the acid catalyzed transetherification of 3-methoxythiophenes38 and synthesized 13 in a similar manner as for 12. In the context of 13, the rigid TD scaffold holds the alcohol in close proximity to the 3-position of the thiophene (Scheme 2). Thus, protonation of the thiophene ring with p-toluenesulfonic acid in refluxing toluene allowed for 6-membered transition state attack—via a C=OMe+ oxonium resonance structure—and substitution of the TD alcohol at the thiophene 3-position to give the cyclized product 13 in high yield (spot to spot conversion by TLC). The cyclization was confirmed by X-ray crystallography (Figure 1).
The acid catalyzed transetherification of 3-methoxythiophenes has largely been applied for the attachment of alkyl side chains. Its application for annulation and planarization of π-systems is rare.16 We envisioned this reaction might hold promise more generally as a strategy for electron rich, oxygen-substituted, phenyl systems. Such systems have been problematic for PAHs because they are prone to quinone formation under the Scholl conditions. Further, the largely hydrocarbon family of PAHs are generally tolerant of protic acid conditions. To this end, we synthesized 15 from 3 and, in the presence of acid in boiling mesitylene, 16 was synthesized in excellent yield.39 Finally, the structure was confirmed by X-ray crystallography (Figure 1).
The successful cyclization of both transetherification substrates (14 and 16) was marked by the removal of rotational disorder in their 1H NMR spectra (Figure 2). For both 13 and 15, steric clash between the methyl ether and TD hydroxyl groups led to broadening of these signals as well as the aryl-H signals on the “wings” of the TD, possibly due to rotomers and intramolecular hydrogen bonding. Upon cyclization and planarization, the rotational isomerism—in addition to the signals for the hydroxyl proton and methyl ether—was removed.
Figure 2.

1H NMR comparison of (a) 13→14 and (b) 15→16. Signal assignments based on 1D coupling constants and 2D NMR experiments (see Supporting Information for further details).
The effects of the cyclization reaction on all of the π systems under study were most distinctly visualized in their UV/vis and florescence signatures relative to their respective starting materials (Figure 3a-d). The absorbance maxima for each compound red-shift by almost 100 nm after cyclization (reducing the Stokes shift, Table 1). This closing of the HOMO-LUMO gap is ascribed to both the increased planarity and the presence of electronrich oxygen donor atoms that raise the HOMO level. Additionally, distinctly sharper absorbance and emission features (vibrational fine structure) for each compound were also observed. Finally, as expected, there is an associated increase in fluorescence quantum yield for the cyclized products relative to their precursors (Table 1). These effects result from the removal of vibrational relaxation through planarization and the enforced rigidity of the [2,2,2] ring system of the triptycene scaffold. An exception to this being 16, which shows slightly broadened vibrational transitions likely due to rotational relaxation from the methoxy substituents. As mentioned above, the proposed structure of 11 is partially based on its absorbance spectrum (Figure 3b), which is indicative of a tribenzo[fg,ij,rst]pentaphene chromophore40 and supports the notion that portions of the oligomer are at least partially cyclized as 11 did show signs of electrochemical cyclization41 (see Supporting Information for further details).
Table 1.
Summary of photophysical data.
| abs λmaxa [nm] |
em λmaxa [nm] |
log εb | Φ F |
τF [ns] |
Eoxc [V] |
|
|---|---|---|---|---|---|---|
| 6 | 242 | 363 | 4.44 | 0.20 | 1.41 | |
| 7 | 358 | 359 | 4.56 | 0.81 | 1.43 | 1.28 |
| 9 | 242 | 4.55 | ||||
| 10 | 360 | 387 | 4.58 | 0.83 | 1.5 | 1.11 |
| 11 | 455b | 465 | 0.70 | 3.1 | ||
| 13 | 275 | 407 | 3.20 | 0.09 | 1.87 | 0.53 |
| 14 | 384 | 386 | 4.58 | 0.30 | 0.97 | 0.83/1.33 |
| 15 | 302 | 375 | 4.00 | 0.03 | 1.39 | 1.08 |
| 16 | 380 | 388 | 4.44 | 0.71 | 2.22 | 0.95/1.24 |
All values measured in CHCl3.
Based on red-most abs λmax.
All values measured in CH2Cl2 with nBu4NPF6 as electrolyte.
Cyclic voltammetry was used to investigate the redox behavior of the cyclized compounds (Figure 3e-h). Planarization and introduction of electron-donating oxygen atoms to the π systems was anticipated to encourage oxidation by raising the HOMO level. This was indeed the case, as both 7 and 10 showed reversible oxidation peaks upon cyclization where 6 and 9 showed no redox behavior over the voltage scanned. Additionally, the more electron rich arenes in 14 and 16 showed two resolved single-electron oxidation peaks at lower potentials relative to 13 and 15 upon cyclization (Table 1).
To summarize, we have developed a strategy that takes advantage of the surrounding molecular architecture for the enforced proximity of reacting partners. The cyclization reactions succeed in the formation of low strain six-membered rings for extended planar PAHs with installed oxygen substituents and incorporated triptycene scaffolds. The trip-tycenes—in addition to fixing the hydroxyl group and arene into a favorable position—contribute significant rigidity to the resulting PAH. This effect on π-systems appears to be a design aspect not yet reported for three-dimensional ip-tycene materials. While this strategy was demonstrated to work under oxidative conditions commonly employed for PAH syntheses, the pre-organized framework also encouraged transetherification reactions with suitably functionalized arenes under Brønsted acid conditions. This acid-catalyzed mode of reactivity has not seen broad use in the field of PAH synthesis, and might provide an avenue towards more heteroatom-substituted platforms. We hope to extend this strategy to other aromatic moieties and side chain functional groups.
Supplementary Material
Scheme 1.
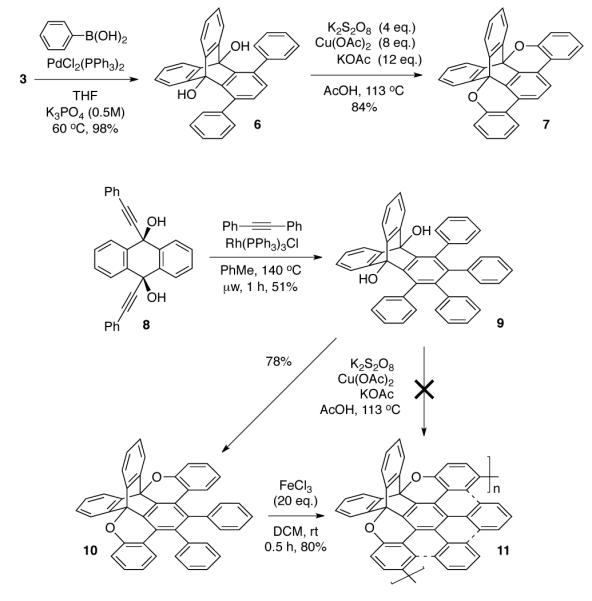
Oxidative cyclization of phenylene substrates.
Scheme 2.
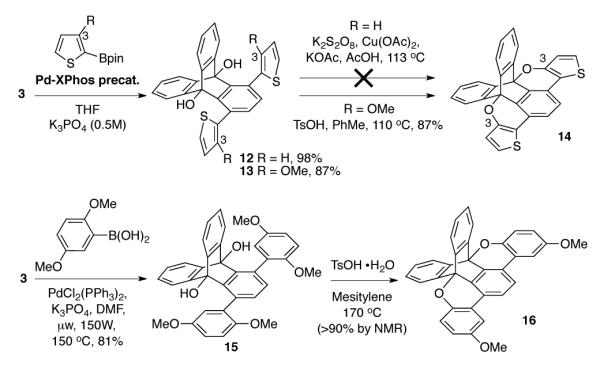
Acid catalyzed transetherification cyclization.a
ACKNOWLEDGMENT
B.V. was supported by the Natural Science and Engineering Council of Canada (NSERC) and in part by an NIH BRP grant (R01 AG026240-01A1. D.J.S. was supported by an NSERC Postdoctoral Fellowship. This work was supported in part by the Air Force Office of Scientific Research (FA9550-10-1-0395). We wish to thank Dr. Yu Lin Zhong for assistance with electrochemical measurements and Dr. Michael Takase for X-ray crystal structures.
Footnotes
Supporting Information. Experimental protocols and analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).O’Neill M, Kelly SM. Adv. Mater. s. 2003;15:1135. [Google Scholar]
- (2).Simpson CD, Wu J, Watson MD, Müllen K. J. Mater. Chem. 2004;14:494. [Google Scholar]
- (3).Grimsdale AC, Müllen K. Angew. Chem. Int. Ed. 2005;44:5592. doi: 10.1002/anie.200500805. [DOI] [PubMed] [Google Scholar]
- (4).Schmidt-Mende L, Fechtenkötter A, Müllen K, Moons E, Friend RH, MacKenzie JD. Science. 2001;293:1119. doi: 10.1126/science.293.5532.1119. [DOI] [PubMed] [Google Scholar]
- (5).Watson MD, Fechtenkötter A, Muüllen K. Chem. Rev. 2001;101:1267. doi: 10.1021/cr990322p. [DOI] [PubMed] [Google Scholar]
- (6).Berresheim AJ, Müller M, Müllen K. Chem. Rev. 1999;99:1747. doi: 10.1021/cr970073+. [DOI] [PubMed] [Google Scholar]
- (7).Wu J, Pisula W, Müllen K. Chem. Rev. 2007;107:718. doi: 10.1021/cr068010r. [DOI] [PubMed] [Google Scholar]
- (8)(a).Scholl R, Mansfeld J. Ber. Dtsch. Chem. Ges. 1910;43:1734. [Google Scholar]; (b) Kovacic P, Jones MB. Chem. Rev. 1987;87:357. [Google Scholar]
- (9).Yang X, Dou X, Rouhanipour A, Zhi L, Räder HJ, Müllen K. J. Am. Chem. Soc. 2008;130:4216. doi: 10.1021/ja710234t. [DOI] [PubMed] [Google Scholar]
- (10).Wu J, Baumgarten M, Debije MG, Warman JM, Müllen K. Angew. Chem. Int. Ed. 2004;43:5331. doi: 10.1002/anie.200460174. [DOI] [PubMed] [Google Scholar]
- (11).Zhang Q, Prins P, Jones SC, Barlow S, Kondo T, An Z, Siebbeles LDA, Marder SR. Org. Lett. 2005;7:5019. doi: 10.1021/ol051972k. [DOI] [PubMed] [Google Scholar]
- (12).Weiss K, Beernink G, Dötz F, Birkner A, Müllen K, Wöll CH. Angew. Chem. Int. Ed. 1999;38:3748. [PubMed] [Google Scholar]
- (13).Wadumethrige SH, Rathore R. Org. Lett. 2008;10:5139. doi: 10.1021/ol8020429. [DOI] [PubMed] [Google Scholar]
- (14).Dou X, Yang X, Bodwell GJ, Wagner M, Enkelmann V, Müllen K. Org. Lett. 2007;9:2485. doi: 10.1021/ol0708018. [DOI] [PubMed] [Google Scholar]
- (15).Feng X, Pisula W, Takase M, Dou X, Enkelmann V, Wagner M, Ding N, Müllen K. Chem. Mater. 2008;20:2872. [Google Scholar]
- (16).Danz M, Tonner R, Hilt G. Chem. Commun. 2012;48:377. doi: 10.1039/c1cc15980a. oxidative subsitution of OMe was observed under Scholl conditions. [DOI] [PubMed] [Google Scholar]
- (17).Swager TM. Acc. Chem. Res. 2008;41:1181. doi: 10.1021/ar800107v. [DOI] [PubMed] [Google Scholar]
- (18).Chong JH, MacLachlan M. J. Chem. Soc. Rev. 2009;38:3301. doi: 10.1039/b900754g. [DOI] [PubMed] [Google Scholar]
- (19).VanVeller B, Robinson D, Swager TM. Angew. Chem. Int. Ed. 2012;51:1182. doi: 10.1002/anie.201106985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Taylor MS, Swager TM. Org. Lett. 2007;9:3695. doi: 10.1021/ol701642j. [DOI] [PubMed] [Google Scholar]
- (21).Rempala P, Kroulík J, King BT. J. Am. Chem. Soc. 2004;126:15002. doi: 10.1021/ja046513d. [DOI] [PubMed] [Google Scholar]
- (22)(a).Rempala P, Kroulik J, King BT. J. Org. Chem. 2006;71:5067. doi: 10.1021/jo0526744. [DOI] [PubMed] [Google Scholar]; (b) Zhai L, Shukla R, Wadumethrige SH, Rathore RJ. Org. Chem. 2010;75:4748. doi: 10.1021/jo100611k. [DOI] [PubMed] [Google Scholar]
- (23).King BT, Kroulík J, Robertson CR, Rempala P, Hilton CL, Korinek JD, Gortari LM. J. Org. Chem. 2007;72:2279. doi: 10.1021/jo061515x. [DOI] [PubMed] [Google Scholar]
- (24).Boden N, Bushby RJ, Headdock G, Lozman OR, Wood A. Liq. Cryst. 2001;28:139. [Google Scholar]
- (25).Boden N, Bushby RJ, Cammidge AN, Duckworth S, Headdock G. J. Mater. Chem. 1997;7:601. [Google Scholar]
- (26).Zhai L, Shukla R, Rathore R. Org. Lett. 2009;11:3474. doi: 10.1021/ol901331p. [DOI] [PubMed] [Google Scholar]
- (27).Simpson CD, Mattersteig G, Martin K, Gherghel L, Bauer RE, Räder HJ, Müllen K. J. Am. Chem. Soc. 2004;126:3139. doi: 10.1021/ja036732j. [DOI] [PubMed] [Google Scholar]
- (28).Kübel C, Eckhardt K, Enkelmann V, Wegner G, Müllen K. J. Mater. Chem. 2000;10:879. [Google Scholar]
- (29).Majetich G, Wheless K. Tetrahedron. 1995;51:7095. [Google Scholar]
- (30).Trahanovsky WS, Macaulay DB. J. Org. Chem. 1973;38:1497. [Google Scholar]
- (31).Trahanov WS, Cramer J. J. Org. Chem. 1971;36:1890. [Google Scholar]
- (32).Trahanov WS, Flash PJ, Smith LM. J. Am. Chem. Soc. 1969;91:5068. [Google Scholar]
- (33).Mihailović ML, Čekovič Ž. Synthesis. 1970:209. [Google Scholar]
- (34).Giordano C, Belli A, Citterio A, Minisci F. J. Org. Chem. 1979;44:2314. [Google Scholar]
- (35).Minisci F, Citterio A, Giordano C. Acc. Chem. Res. 1983;16:27. [Google Scholar]
- (36).Camaioni DM, Alnajjar MS. J. Org. Chem. 1985;50:4456. [Google Scholar]
- (37).Kinzel T, Zhang Y, Buchwald SL. J. Am. Chem. Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38)(a).von Kieseritzky F, Allared F, Dahstedt E, Hellberg J. Tetrahedron Lett. 2004;45:6049. [Google Scholar]; (b) Agarwal N, Mishra SP, Kumar A, Hung C-H, Ravikanth M. Chem. Commun. 2002:2642. doi: 10.1039/b208017f. [DOI] [PubMed] [Google Scholar]; (c) Caras-Quintero D, Bäuerle P. Chem. Commun. 2004:926. doi: 10.1039/b400965g. [DOI] [PubMed] [Google Scholar]
- (39).Liberated methanol reacted with the acid catalyst to from p-toluene sulfonic acid methyl ester under the reaction conditions. This biproduct coeluted with the product during chromatography, diminishing the isolated yield.
- (40).Clar E, McCallum A. Tetrahedron. 1964;20:507. [Google Scholar]
- (41)(a).Rathore R, Kochi JK. J. Org. Chem. 1995;60:7479. [Google Scholar]; (b) Ronlan A, Parker VD. J. Org. Chem. 1974;39:1014. [Google Scholar]; (c) Ronlan A, Hammerich O, Parker VD. J. Am. Chem. Soc. 1973;95:7132. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.