

Abstract
Amyloid fibers and oligomers are associated with a great variety of human diseases including Alzheimer’s disease and the prion conditions. Here we attempt to connect recent discoveries on the molecular properties of proteins in the amyloid state with observations about pathological tissues and disease states. We summarize studies of structure and nucleation of amyloid and relate these to observations on amyloid polymorphism, prion strains, co-aggregation of pathogenic proteins in tissues, and mechanisms of toxicity and transmissibility. Molecular studies have also led to numerous strategies for biological and chemical interventions against amyloid diseases.
What is the amyloid state?
Many proteins enter the so-called amyloid state, in which they form elongated, fibers, with spines consisting of many-stranded β-sheets. The operational definition of amyloid, which has been adopted by the community of pathologists, is that the fibers are unbranched, usually extracellular, and found in vivo; in addition, the fibers bind the dye Congo Red and then show green birefringence when viewed between crossed polarizers (Sipe et al., 2010). By this definition, >25 amyloid-forming proteins have been identified and associated with serious diseases, including amyloid-β peptide (Aβ) with Alzheimer’s disease (AD), islet amyloid polypeptide (IAPP) with diabetes type 2, and prion protein (PrP) with the spongiform encephalopathies.
Biophysicists prefer a molecular-based definition, and thus, they have abandoned the requirement that the fibers are usually extra-cellular and disease-associated. The reasons for this change is that the same disease-related proteins form similar fibers in vitro and many other proteins form similar fibers when denatured (Fandrich et al., 2003) or during their physiological roles (Chapman et al., 2002; Fowler et al., 2007; Si et al., 2003). Accordingly, biophysicists have adopted a structure-related definition for amyloid fibers, in which amyloid fibers display the cross-β fiber diffraction pattern (Figure 1).
Figure 1. Properties of Amyloid Fibers.
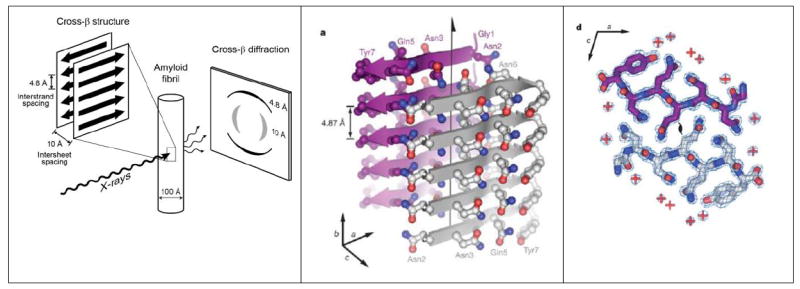
(A) The characteristic cross-β diffraction pattern observed when X-rays are directed on amyloid fibers. The diffuse reflection at 4.8 Å spacing along the meridian (vertical) shows extended protein chains running roughly perpendicular to the fibril and spaced 4.8 Å apart. The even more diffuse reflection at ~10 Å spacing along the equator (horizontal) shows that the extended chains are organized into sheets spaced ~10 Å apart. For less well oriented fibrils, both reflections blur into circular rings. (B) The steric zipper structure of the sequence segment GNNQQNY from the yeast prion Sup35. Five layers of β-strands are shown of the tens of thousands in a typical fibril or microcrystal. The front sheet shows the protein backbones of the strands as gray arrows; the back sheet is in purple. Protruding from each sheet are the sidechains. The arrow marks the fibril axis. (C) The two interdigitating β-sheets are viewed down the axis. Water molecules, shown by red + signs are excluded from the tight interface between the sheets. Red carbonyl groups and blue amine groups form hydrogen bonds up and down between the layers of the sheet (Nelson et al., 2005). Panels B and C reprinted from Nelson et al. 2005.
This pattern was first observed by the pioneering biophysicist William Astbury (Astbury et al., 1935), who stretched a poached egg white into a fiber in the X-ray beam. Astbury reasoned correctly that in such fibers, elongated protein strands must be stacked along the fiber axis, forming protein sheets that are parallel to each other. A decade and a half later, Pauling and Corey built models for these β-sheets, showing that hydrogen bonds hold the strands into sheets.
Proteins may enter the amyloid state when a segment exposes its backbone amide N-H groups and C=O groups, permitting them to couple into hydrogen bonds with other protein chains. Several conditions produce exposed backbone amide groups: denaturation of a normally folded proteins (Chiti et al., 1999); over expression of a protein that overwhelms cellular chaperones and drives it into an inclusion body (Wang et al., 2008); cleavage of a peptide (such as Aβ) from a folded protein; or over production of a natively disordered protein (such as tau or IAPP). Exposure of backbone amide groups is necessary for amyloid formation but not sufficient. In addition, the local concentration of the exposed segment must be sufficiently great to overcome the entropy that opposes formation of ordered fibers. The higher the concentration, the more aggregation is favored. Suppose the concentration of the exposed segment is [P] and that of the aggregated fiber of n units is [nP]. Then (neglecting intermediate states) the free energy change for the process of aggregation is given by:
in which ΔG0 is the standard free energy change for the reaction, and RT is the product of the gas constant and the absolute temperature. Because there are tens of thousands of protein molecules in an amyloid fiber, that is n=thousands, [P]n becomes large when the concentration of P goes up, and the log of the ratio becomes extremely negative, meaning that the free energy of amyloid formation is highly favorable.
In the laboratory, scientists can often produce amyloid fibers from a protein at high concentration by partially denaturing it with a destabilizing solvent, a change in pH, heating, or surface denaturation from agitation. In cells and tissues, amyloid formation occurs with abnormally high expression of a protein. In humans, we do not yet understand all causes of amyloid formation, but increased synthesis or reduced degradation of a given protein, leading to an abnormally high concentration, is a factor (Balch et al., 2008).
For amyloid to form, a nucleus must template the bonding pattern of the fiber spine. As described in Section 2, each fibril spine is built on an intricate pattern of hydrogen bonds and steric interactions. From the atomic structure of one such spine, it was suggested that templating the pattern of the spine requires 3 or 4 protein molecules (Nelson et al., 2005). If so, then 3 or 4 molecules must expose their amyloid-forming segments at the same time and must be at high enough concentration for bonding and consequent templating of the fibril pattern. Thus, nucleation is a rare event, but once the nucleus is formed, single molecules can join the growing fibril one at a time, as they open to expose the proper segment and bond at the ends of fibrils. The result is that amyloid fibril formation is characterized by a slow nucleation phase, followed by a more rapid growth phase (Jarrett and Lansbury, 1993). The pathway for amyloid fiber formation is at present an active area of investigation with evidence that fibers can form by nucleated conformational conversion from an oligomeric state (Lee et al., 2011). Because fibrils grow from their ends, breakage of fibrils affects the kinetics of fibrillar growth (Tanaka et al., 2006), and a full description of fibril formation must include rates of nucleation, growth, and breakage (Knowles et al., 2009).
Concepts of nucleation are important in understanding amyloid and prion diseases. Nucleation is stochastic and the chance for forming a nucleus is lowered as the volume of solution is diminished. For example, a bottle of water can be supercooled to only a few degrees C before ice nucleates. However, as its volume is diminished, water can be increasingly supercooled. Micron-sized drops can be supercooled to -41° C (Kuhns and Mason, 1968). Biological cells are micron sized, and thus, we might expect that amyloid nucleation would be infrequent for intracellular proteins, even when they are at relatively high concentration. Of course if we introduce a nucleus or “seed” from the outside to a supercooled liquid or a supersaturated solution, growth on the nucleus is fast. For instance, try touching an ice chip to the surface of a bottle of supercooled water. Instant crystallization occurs because the ice chip provides the nucleus, or an exact three dimensional pattern, of water molecules in ice. Thus, the concept of seeding is important for understanding propagation of amyloid fibers or prions from cell to cell or organism to organism.
What is the atomic structure of the amyloid spine?
Amyloid fibers share a common “cross-β” spine. In 1959, elongated, unbranched fibrils were reported in electron micrographs of diseased tissues from diverse origins (Cohen and Calkins, 1959), and nine years later X-ray diffraction patterns of such fibrils were identified as Asbury’s cross-β type (Eanes and Glenner, 1968). With the advent of synchrotron X-ray radiation, scientists found that amyloid fibers formed from six different proteins; each one was associated with a different clinical syndrome and showed similar cross-β diffraction (Sunde et al., 1997).
Determining the atomic details of the cross-β spine has been slow because the limited order of fibrils, whether isolated from diseased tissues or from in vitro conversion of native proteins to the amyloid state, presents challenges to crystallographic, NMR, and EM methods. But important features have gradually emerged from studies by solid-state NMR (Benzinger et al., 1998; Tycko, 2011), model-building constrained by X-ray fiber and powder diffraction (Makin et al., 2005; Sunde and Blake, 1998), site-directed spin labeling (Serag et al., 2001; Torok et al., 2002), cryo-electron microscopy (Jimenez et al., 1999; Schmidt et al., 2009), scanning mutagenesis (Williams et al., 2004), and single-crystal X-ray diffraction (Nelson et al., 2005). The most general points to emerge are that: (1) In all amyloid fibers, the strongest repeating feature is a set of β-sheets that are parallel to the fibril axis, with their extended strands near perpendicular to the axis; (2) The β-sheets can be either parallel or anti-parallel—that is, adjacent hydrogen-bonded β-strands within a sheet can run in the same direction or in opposite directions; (3) The sheets are usually “in-register,” meaning that strands align with each other such that identical sidechains are on top of one another along the fibril axis. In parallel sheets, identical sidechains are separated by an inter-strand distance of 4.8 Å (Figure 1), and in antiparallel sheets, they are separated by 2 ×4.8 Å = 9.6 Å.
The architecture of at least the simplest cross-β amyloid spines has been clarified by determining short segments of amyloid-forming proteins (Apostol et al., 2010; Ivanova et al., 2009; Nelson et al., 2005; Sawaya et al., 2007; Sievers et al., 2011; Wiltzius et al., 2009). The segments examined are those that seem to be the adhesive parts of amyloid proteins. In isolation from the rest of the protein, they form microcrystals and related fibers with morphological similarity to fibers of the entire parent proteins (Balbirnie et al., 2001). The atomic structures of the microcrystals reveal that the motif of the amyloid protofilament consists of a pair of β-sheets that run the length of the fiber-like crystals (Figure 1B). Each sheet is a standard Pauling-Corey β-sheet, in which each strand is hydrogen-bonded to the strand above and below it through its backbone amide groups.
When the sidechains contain amides (glutamine and asparagine), those amides also form hydrogen bonds to the identical residue in the strands above and below. This creates parallel arrays of hydrogen bonds running along the fiber axis. The electrostatic interactions of all of these aligned hydrogen bonds mutually polarize one another, producing hydrogen bonds even stronger than those in ice (Tsemekhman et al., 2007). The stability of such interdigitated beta sheets explains the persistence of amyloid fibers and prions.
Within the protofilament, the sidechains emanating from the two sheets are tightly interdigitated, as shown in Figure 1C, like the teeth of a zipper. The interface between the sheets is devoid of water, and hence this motif has been termed the “dry steric zipper.” Dozens of atomic structures of dry steric zipper have been determined by X-ray crystallography and share the following properties: (1) Steric zippers form from self-complementary amino acid sequences, in which their sidechains can mutually interdigitate. The sequences can be polar or non-polar, with large sidechains or small, but they fit together in complementary fashion; (2) Steric zippers have dry interfaces between the two sheets. Thus, the hydrophobic effect contributes to amyloid stability, as does the strong hydrogen bonding; (3) The β-strands are most often in-register, maximizing inter-strand hydrogen bonding, and permitting stacking of glutamine (Gln), asparagine (Asn), and tyrosine (Tyr) residues. Although all steric zippers are expected to be formed from complementary sequences, the sequences do not need to be self-complementary. There is strong evidence from solid-state NMR studies (Luhrs et al., 2005; Petkova et al., 2002) that in Aβ, some close interactions are between β-strands that differ in sequence (see Figure 4). Such “hetero-steric zippers” have not yet been observed in X-ray crystal structures.
Figure 4. Models for amyloid fibrils larger than a single steric-zipper spine.
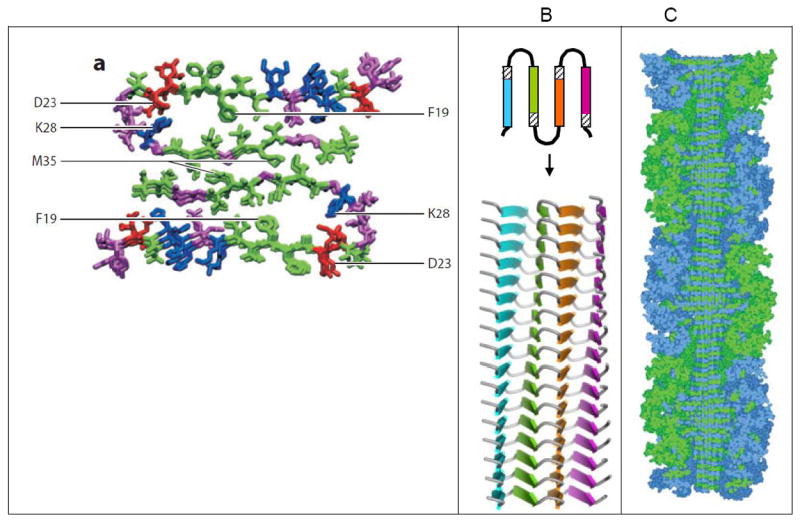
(A) Model for Aβ1-40 based on solid-state NMR data with additional constrains from electron microscopy (Tycko, 2011). The view is down the fibril axis, showing two molecules of Aβ, each with a U-turn or “β-arch”. Where the green segments of the two molecules abut, they appear to form a homo-steric zipper (Class 1 in Figure 2), and a hetero-zipper could exist between the two arms of each U. Both types of steric zipper need to be confirmed by higher resolution structures. (B) A proposed structure for longer amyloid proteins is a “superpleated β-structure” (Kajava et al., 2010), in which the protein chain forms several U-turns/beta-arches. The view of the upper diagram is down the fibril axis; the view of the lower is perpendicular to the fibril axis. In the lower diagram, each protein chain is hydrogen bonded to the ones above and below (not shown). Hetero-zippers may exist between pairs of differently colored β-strands. This type of structure has been proposed for several proteins in the amyloid state including Ure2p, Sup35p, and α-synuclein. (C) A model for a designed amyloid of ribonuclease A with ten glutamine residue inserted between the core and C-terminal domains (Sambashivan et al., 2005) based on X-ray and electron microscopy data and steric constraints. The view is perpendicular to a cut-away of the fibril. The twisting steric zipper can be seen at the center. Globular subunits of ribonuclease A, which are essentially in their native conformation, are at the periphery. The amyloid-like fibrils of this designed amyloid show enzymatic activity, confirming that ribonuclease molecules retain native-like structure.
The most common sheet-to-sheet arrangement for steric zippers is face-to-face (Class 1; Figure 1B), but other arrangements occur (Figure 2). In these other arrangements, the two sheets can be: face-to-back (Classes 2 and 4); pack with opposite edges up rather than both edges up (Class 4); or contain antiparallel strands (Classes 5-8), rather than parallel strands (Classes 1-4) (Sawaya et al. 2007). To date, no examples in Class 3 have been observed.
Figure 2. Steric-zipper protofilaments.
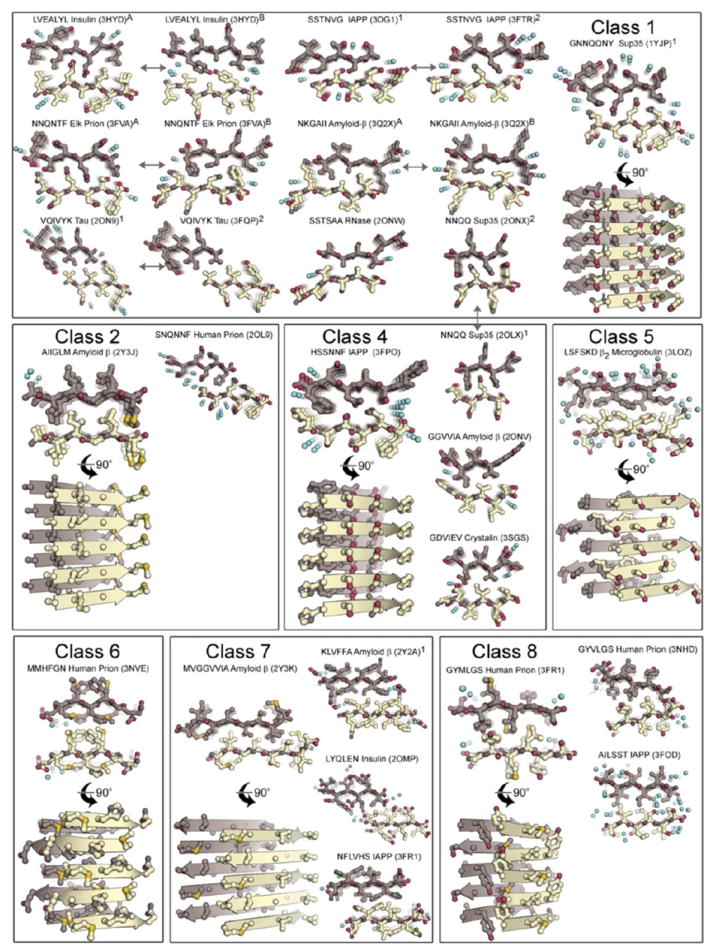
Twenty-eight atomic structures of steric-zipper protofilaments from amyloid-forming proteins, determined by X-ray diffraction. All are viewed projected down the protofilament axis, revealing the two sheets (one gold and one purple) with their interdigitated sidechains. Selected zippers are also viewed perpendicular to the protofilament axis, with five layers of β-strands shown with backbones as arrows. Water molecules are shown as aqua spheres; notice their absence from the interfaces between the paired sheets.
Some amyloid spines are more complex than single steric zippers. For instance, several different steric zippers all formed by the same protein can occur in the spine. In fact some 13 different steric zippers have been found for the 42-residue sequence of Aβ (Colletier et al., 2011). Many proteins, including the PrP and Sup35 prions, Aβ, and IAPP, have several potential steric-zipper forming segments within their sequences. In a recent report, Lewandowski et al. (2011) provide solid-state NMR evidence that fibers of the yeast prion Sup25 contain three distinct steric zippers (one is shown in Figure 1B).
A second source of increased complexity is the likelihood of hetero-zippers formed from cross-complementary sheets. Hetero-zippers have been found by solid-state NMR in the structure of Het-s, a fungal prion (Wasmer et al., 2008) (Figure 3). This structure, termed a solenoid by its discoverers, consists of a stack of two-layer protein loops. Each loop contains two extended strands with their sidechains interdigitating in a similar manner as those in a steric zipper. Each molecule of Het-s contributes two such loops that stack on top of each other. This pair of loops then stacks on top of, and beneath, pairs of loops from its adjacent molecules in the fiber. The entire structure is amyloid-like. The β-sheet interactions in Het-s have been selected by evolution, in contrast to some interactions that are found in the spontaneous aggregation of disease-related proteins. Hetero-zippers probably are also found in spontaneous aggregates of proteins, such as those of Aβ, but they have not yet been fully defined at high resolution.
Figure 3. Structure of a hetero-zipper.
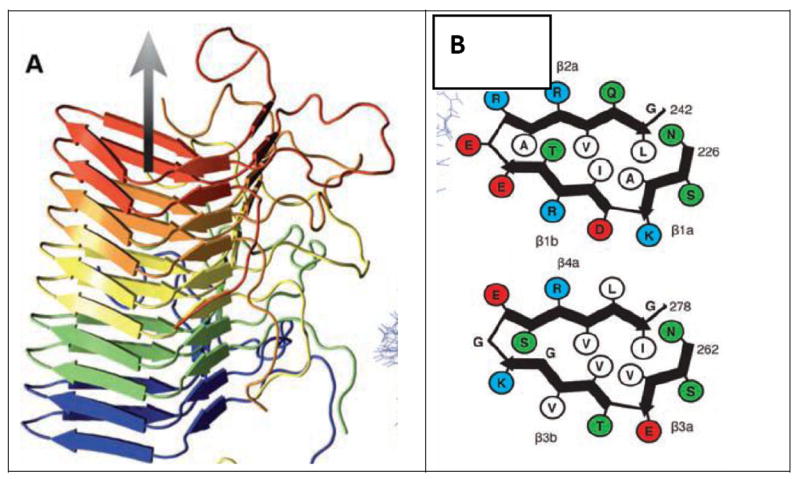
The solid-state NMR-derived structure of Het-s shows hetero-zippers (Wasmer et al., 2008). (A) The protein chain of each molecule (in a single color) contains six β-strands, organized in double loops. The double loops of adjacent molecules sit on top of one another, hydrogen bonded up and down. (B) The two layers are shown schematically with sidechains represented as circles. Each layer may be regarded as a hetero-zipper, in which the sidechains of opposing strands interdigitate. Figure reprinted from Wasmer et al. 2008.
What is the structure of protein segments outside of the amyloid spine?
Although we have atomic structures for some amyloid spines, we do not yet have full atomic structures for whole amyloid fibrils. Full amyloid fibrils are more complicated than the simple spine structures in that some fibrils appear to contain numerous protofilaments and are complex in structure (Lewandowski et al., 2011; White et al., 2009).
The findings about amyloid spines (Section 2) place severe constraints on fibril models. Given that proteins stack in-register with strands spaced 4.8 Å along the fibril axis, the rest of the protein must be flattened out so that each layer is only 4.8Å high or must somehow sit at the periphery of the spine, where it may extend more than 4.8Å along the axis, avoiding overlap with identical domains. A flattened model for Aβ is shown in Figure 4A, based on solid state-NMR measurements (Tycko, 2011). Each Aβ molecule makes a U-turn, called a beta-arch (Kajava et al., 2010). For longer proteins, it has been proposed that U-turns are linked into a serpentine structure, termed a superpleated β-structure (Figure 4B)(Kajava et al., 2010). In a superpleated β-sheet, the entire protein chain is flattened to fit in one 4.8Å layer of the fibril.
Flattening is not necessary for an amyloid-forming protein to retain globular domains. In the model of a designed amyloid of RNase A (Figure 4C), the domains on the periphery of the spine find space to retain their globular structure (Sambashivan et al., 2005). For larger globular domains, a greater circumference of the fibril and a longer protein linker to the steric zipper is required. This means that fibers formed from larger proteins would be expected to have greater diameters. In short, although the spines of amyloid fibers appear similar, fibrils show a great variety of structural complexity.
What is the basis of amyloid cross-seeding?
The observation that amyloid fibrils have spines composed of steric zippers explains why different proteins, when they enter the amyloid state, give fibrils of similar appearance in electron micrographs. The fibrils are all elongated and unbranched, just as their steric zipper spines are elongated and unbranched. The diameters of the fibrils vary because the lengths of the proteins that form them differ and because the number of protofibrils that twist around each other to form the fibril may differ. Thus, we would expect that cross-seeding of amyloid fibril formation is possible in which the seed is formed from another, but similar amyloid fibril. All steric zippers formed from parallel β-strands have one repeat the same: 4.8 Å in the fibril axis direction; similarly, all anti-parallel zippers have one repeat the same: 9.6 Å in the fibril axis (Figure 1). If the seeding steric zipper is complementary in shape to a segment of the seeded protein in solution, we could expect a hetero-steric zipper to form and to serve as a nucleus, as has been shown in vitro for Aβ and IAPP (Andreetto et al., 2010).
In yeast, cross-seeding has been suggested as the mechanism for the observation that one prion can induce the appearance of another (heterologous) prion. However, heterologous prion interactions can also inhibit prion propagation (Bradley et al., 2002). Similar observations have been made with mammalian prions that can either promote or inhibit PrP aggregation (Nilsson et al., 2010). Cross-seeding and cross-inhibition in vivo also has been reported between ApoAII amyloid (AApoAII) and Serum amyloid A (AA) fibrils (Yan et al., 2007). Moreover, AA amyloid can be cross-seeded in susceptible mice with heterologous β-rich proteins (Lundmark et al., 2005). While cross-seeding presumably results from formation of a hetero-zipper, cross-inhibition might result from a capping of fibril ends, a principle used to develop amyloid interfering compounds (Section 7).
In human neurodegenerative diseases, the coexistance of more than one amyloid deposit is a common observation. For example, in Gerstmann-Sträussler-Scheinker Syndrome, colocalisation of APrP and Aβ in the same amyloid plaque has been described (Miyazono et al., 1992). Similarly, in Familial Danish Dementia Aβ and Dan-amyloid (ADan) co-localize in parenchymal and vascular amyloid deposits (Tomidokoro et al., 2005). In Parkinson-related diseases, α-synuclein and tau inclusions can occur in the same cell and form common inclusion bodies (Giasson et al., 2003). Although cross-seeding provides an attractive explanation for these observations (Giasson et al., 2003; Morales et al., 2010), definitive proof is lacking, and other explanations are possible. For example, (i) two amyloid deposits may simply develop independently of each other; (ii) there may be saturable cellular fractions for the removal of misfolded proteins, and thus one aggregated protein may indirectly stimulate aggregation of further proteins by monopolizing clearance mechanisms; and, (iii) co-localization of two amyloids is only apparent at the light-microscopic level and reflects common cellular niches prone to protein aggregation, while at the ultrastructural level, true coaggreagtion of the two amyloids may not occur. Yet other observations indicate that the interaction of amyloidogenic proteins in human brain can impede, rather than promote, aggregation. For example, cystatin C colocalizes with Aβ-plaques in Alzheimer’s disease, but the finding that cystatin C reduces Aβ plaque formation in transgenic mice suggests a mechanism of cross-inhibition rather than cross-seeding (Kaeser et al., 2007).
What are amyloid polymorphisms and amyloid strains?
The term “strain” is used by microbiologists to denote structurally or functionally variant microbes within a given species. Similarly, prion strains in mammals have been assumed to be variants of PrP aggregates that exhibit characteristic biological properties (Aguzzi et al., 2007; Colby and Prusiner, 2011; Collinge and Clarke, 2007). Whereas the genotype of a microbial strain is encoded by its nucleic acid sequence, that of a prion strain is encoded by the “conformation” of PrP (Colby and Prusiner, 2011; Collinge and Clarke, 2007; Prusiner, 1982). The formation of structurally distinct, polymorphic protein fibers is now recognized as a common property of amyloid-forming proteins (Chiti and Dobson, 2006; Goldsbury et al., 2005; Tanaka et al., 2006). For example, synthetic Aβ can aggregate into structurally diverse amyloid fibrils, which retain their conformational properties and cellular toxicities after repeated passage (Petkova et al., 2005).
Hypotheses for the conformational basis of amyloid strains
Three models for the molecular basis of prion strains and amyloid poymorphs have been proposed on the basis of atomic structures of amyloid-like fibers (Figure 2). The models suggest that strains are based in distinct steric zipper spines of the associated amyloid fibers. The first model is termed packing polymorphism, and is illustrated in Figure 2 by the pairs of zippers connected by double-headed arrows. In packing polymorphism, an amyloid segment packs in two or more distinct ways, producing fibrils with different structures and distinctive properties. The simplest form of packing polymorphism is a registration shift in which the two sheets forming the steric zipper in the second polymorph shift their interdigitation from that in the zipper of the first polymorph. Because the nature and position of the sidechains on the outer surface of the fibers differ in the two polymorphs, the properties of the fibers must be different (Greenwald and Riek, 2010; Wiltzius et al., 2009). Thus, in packing polymorphism, one sequence forms two or more “conformations”.
The second structural model for strains is termed segmental polymorphism. In segmental polymorphism, two or more different segments of an amyloid protein are capable of forming steric zipper spines. Figure 2 shows several segments from Aβ which form different steric zippers. Fibrils formed with different steric-zipper spines will each have distinctive properties. Proteins particularly rich in different segments able to form steric zippers include Aβ (Colletier et al., 2011), IAPP (Wiltzius et al., 2009) and PrP (Sawaya et al., 2007).
In a third type of amyloid polymorphism, heterosteric zippers, the zipper is formed from the interdigitation of non-identical beta sheets. Though not yet seen in X-ray structures at the atomic level, heterotypic interactions between sheets are observed in the constrained models derived from solid-state NMR and cyro-EM (Greenwald and Riek, 2010; Schmidt et al., 2009; Tycko, 2011). The existence of such heteroamyloid spines, in addition to self-complementary spines, greatly increases the number of potential amyloid polymorphs and prion strains.
The hypothesis that distinct steric zipper structures are at the basis of amyloid fiber polymorphism and prion strains is consistent with other observations about steric zippers. Steric zippers can be extremely stable. For example the steric-zipper formed by the segment of Sup35 with amino acid sequence GNNQQNY could not be dissolved by 5% SDS nor 4M urea; dissolution required 100% formic acid, 4M guanidinium hydrocholoride, or 0.5 M NaOH (Balbirnie et al., 2001). Thus, steric zippers share with prion strains robust “conformations” that can conceivably be transmitted from cell to cell or organism to organism. Another similarity between steric zippers and prion strains is that environmental conditions seem to affect the formation of both. For example, incubation of the yeast prion Sup35 at either 4° or 37° produces different prion strains in yeast (Tanaka et al., 2004). Similarly, the differing steric zippers formed from the same protein segment in Figure 2 were created by incubating the segments under different solution conditions.
Amyloid morphotypes
In the brain, Aβ deposits are heterogeneous in histopathological appearance and biochemical composition, both within and among brain regions and patients (Maarouf et al., 2008; Tekirian et al., 1998; Thal et al., 2006). Aβ aggregation can occur in association with the vasculature or in the brain parenchyma as amyloid plaques. Point mutations within the Aβ sequence can either lead to vascular amyloid, amyloid plaques, or both (Herzig et al., 2006). Vascular and parenchymal Aβ deposits differ in the ratio of deposited Aβ ending at amino acid 40 to Aβ ending at amino acid 42 (Herzig et al., 2006). Plus, the Aβ40 : Aβ42 ratio has been linked to different neurotoxicities and clinical Alzheimer’s disease onset (Duering et al., 2005; Kumar-Singh et al., 2006; Kuperstein et al., 2010). In addition, Aβ displays length variations due to truncations at the N terminus (e.g., Aβ starting at residue 3, 11, or 17) and variations in posttranslational modifications (e.g., isomerization, pyroglutamyl formation, phosphorylation, nitration). All these factors can profoundly influence Aβ aggregation and histopathological appearance of the amyloid (De Strooper, 2010; Kumar et al., 2011; Kummer et al., 2011; Miravalle et al., 2005; Tekirian et al., 1998). A predominance of N-truncated and post-translationally modified Aβ distinguishes Aβ-deposits in Alzheimer’s disease compared to normal aging and mouse models (Kuo et al., 2001; Piccini et al., 2005). Although it remains difficult to study the conformational state of Aβ in vivo, indirect measures using luminescent conjugated polythiophene probes that detect particular amyloid conformations suggest the occurrence of conformationally distinct Aβ deposits in brain (Nilsson et al., 2007) (Figure 5). Luminescent conjugated polythiophenes have also been used to discriminate prion strains (Sigurdson et al., 2007).
Figure 5. Rainbow amyloid.
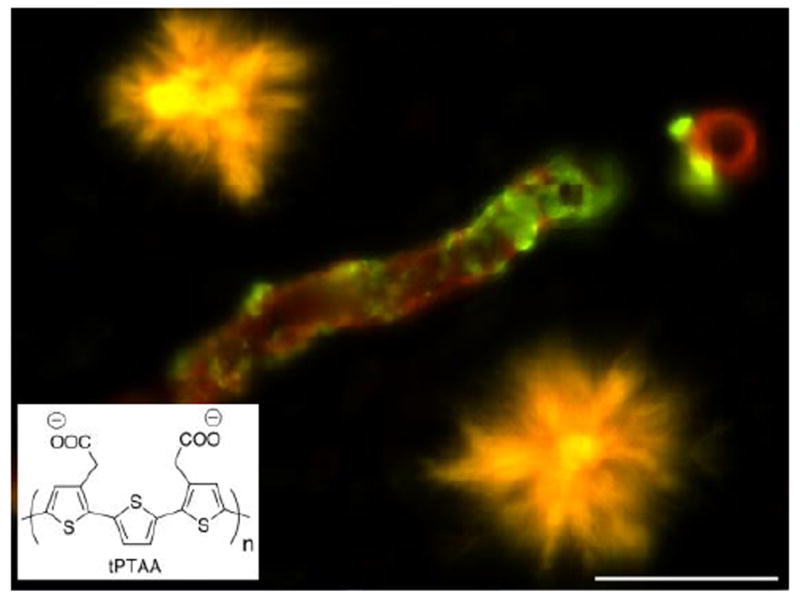
Novel amyloid dyes can be used as surrogate probes of the supramolecular structure of protein aggregates. Shown are Aβ plaques (yellow) and Aβ amyloid angiopathy (green) in an AβPP transgenic mouse (carrying the AβPP Swedish and AβPP Dutch mutation). Note the different spectral signatures upon staining with the luminescent conjugated polythiophene tPTAA (bottom left). The image was recorded using a combination of green and red filters. Scale bar = 20um.
Different Aβ morphotypes in the brain may indicate that local factors influence the Aβ aggregates. They may also represent various stages in the disease process (Thal et al., 2006) or reflect the templated propagation of conformationally distinct seeds (Levine and Walker, 2010). Although these possibilities are not mutually exclusive, the third explanation has gained momentum by the finding that Aβ morphotypes can be transmitted to transgenic mice overexpressing the Aβ precursor protein (AβPP) in vivo (Meyer-Luehmann et al., 2006). These observations are reminiscent of previous transmission studies using transgenic mice that overexpress PrP and deposit PrP amyloid (Peretz et al., 2002), suggesting that the characteristics of prion strains may also apply to multimeric Aβ. However, the link between Aβ conformational variants and distinct clinical subtypes of β-amyloidoses is still lacking.
Hetereogeneous amyloid morphotypes are also observed in other amyloidoses. Transthyretin (TTR) amyloid deposits show variations in fibrils made of full-length versus C-terminal fragments of TTR (Bergstrom et al., 2005), and in familial cases, TTR amyloid deposition varies in the ratio of incorporated wildtype versus mutant TTR (Ihse et al., 2011; Ihse et al., 2008). Strikingly, such amyloid heterogeneity is associated with the organ tropism (i.e., that the amyloid preferentially deposits in particular organs) and clinical manifestation of TTR-amyloidoses (Westermark and Westermark, 2010). Similarly, length variants of the AA protein characterize two different histopathological AA amyloid patterns in the kidney with distinct clinical phenotypes (Westermark et al., 1989). In the brain, tau and α-synuclein inclusions reveal histopathological heterogeneity that is diagnostic of the various tauopathies and α-synucleinopathies, respectively (Goedert et al., 2010). Consistently, α-synuclein and tau fibrils in vitro exhibit conformational diversity (Frost et al., 2009; Heise et al., 2005). Although recent studies have reported the remarkable transmission of disparate proteopathic lesions in transgenic mice (see Section 8), solid evidence for the hypothesis that the heterogeneous disease phenotypes are the result of the (prion-like) templated conversion of conformationally distinct TTR, AA, tau and α-synuclein seeds is still lacking.
Amyloid toxicities and animal models
Not all amyloids are toxic. First described in bacteria, fungi, and yeast, and more recently in mammals, amyloids can function in the formation of biofilms, the binding and storage of peptide hormones, the formation of melanin formation, or the launch of an antiviral innate immune response (Chapman et al., 2002; Fowler et al., 2007; Hou et al., 2011; Maji et al., 2009). The type of amyloid and the controlled growth conditions may account for the lack of toxicity of so-called functional amyloids (Greenwald and Riek, 2010; Watt et al., 2011).
However, most amyloid formation in mammals occurs with aging and is associated with diseases commonly referred to as protein misfolding diseases, aggregation diseases, proteopathies, or more specifically amyloid diseases or amyloidoses (Chiti and Dobson, 2006; Selkoe, 2003). An association of a given amyloid with a disease does not necessarily denote causality. However, a causal relationship between the amyloid formation and amyloid toxicity is suggested from familial cases in which a pathogenic mutation leads to an overproduction of the amyloidogenic protein or enhances the propensity of the protein to aggregate (Table 1). It remains unclear which step of the amyloid formation cascade is toxic, and this step may be different for the various amyloid diseases.
Table 1.
Familial human amyloid diseases in which the mutations promote the formation of amyloid
| Disease | Variant protein | Amyloid |
|---|---|---|
| Alzheimer’s disease | AβPP, PS1/2 | Aβ |
| Hereditary cerebral hemorrhage with amyloidosis, Dutch type | AβPP | Aβ |
| Hereditary cerebral hemorrhage with amyloidosis, Icelandic type | Cystatin C | ACys |
| Familial British Dementia | BriPP | ABri |
| Familial Danish Dementia | BriPP | ADan |
| Parkinson’s disease | α-synuclein | ASyn |
| Frontotemporal lobar degeneration (FTLD)-tau | Tau | ATau |
| Gerstmann-Sträussler-Scheinker | PrP | APrP |
| Greutzfeldt-Jacob Disease | PrP | APrP |
| Amyotrophic lateral sclerosis | SOD1 | ASOD1 |
| Transthyretin familial amyloidosis | TTR | ATTR |
| Hereditary lysozyme amyloidosis | Lysozyme | ALys |
| Hereditary fibrinogen A α-chain amyloidosis | Fibrinogen α-chain | AFib |
| Hereditary ApoAI/II amyloidoses | Apolipoprotein AI/II | AApoAI/II |
| Finnish hereditary amyloidosis | Gelsolin | AGel |
Amyloid toxicity can result from losing the function of a protein or from the sequestration or mislocation of other proteins (Olzscha et al., 2011). This latter mechanism may be the toxicity mechanism for the RNA-binding proteins TDP-43 and FUS (Mackenzie et al., 2010) although their classification as amyloids is uncertain. However, for most amyloid diseases, a gain of toxic function remains a favored hypothesis.
In search of the toxic amyloid species
Despite the longstanding knowledge that amyloids are associated with disease (Cohen and Calkins, 1959), we still lack a clear understanding of how amyloids lead to dysfunction, aside from the instances in which amyloids disrupt tissue structure and organ function via simple mass action (Pepys, 2001; Westermark, 2005). This mass action mode of toxicity may well be the most important one for most systemic amyloidoses and for the amyloid associated with the cerebral vessels (cerebral amyloid angiopathy, CAA) (Figure 6). CAA of various types (Aβ, ADan, British amyloid [ABri], Cystatin C amyloid [ACys]) all result in a thickening of the vascular basal lamina, loss of smooth muscle cells, perivascular inflammation, and eventually vessel wall rupture and hemorrhages (Revesz et al., 2009). Similar appearances and toxicities of CAA (independent of the amyloid type) are also seen in Aβ- and ADan-transgenic mouse models (Calhoun et al., 1999; Coomaraswamy et al., 2010). Moreover, correlations between CAA severity and hemorrhage frequency was found in humans and mouse models, suggesting that the mass of amyloid fibrils may be the most important parameter mediating vascular toxicity (Dierksen et al., 2010; Maeda et al., 1993; Winkler et al., 2001).
Figure 6. Histopathology of cerebral β-amyloidosis.
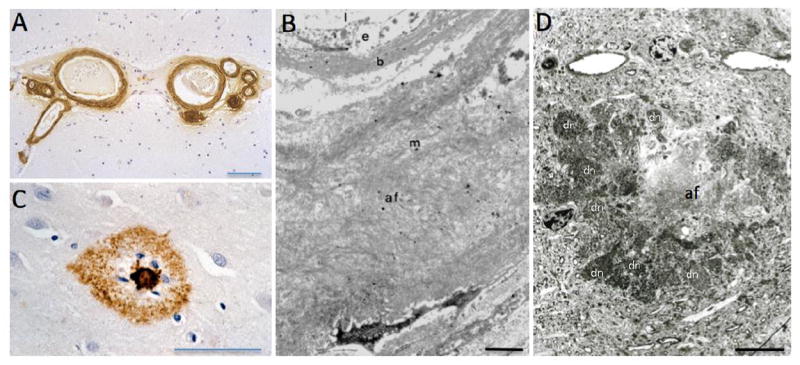
(A) Aβ immunostaining (brown) reveals severe cerebral amyloid angiopathies (CAA) in superficial cortical vessels in a human case. (B) Ultrastructural analysis of Aβ fibrils (af) in the vessel wall of an arteriole with CAA. Note that the amyloid has displaced nearly the entire vascular wall, disrupting normal vessel-neuron communications (b, basal lamina; e, endothelial cells; l, lumen; m, media; (reprinted with permission from Yamada et al., J Neurol 234: 371-6, 1987). (C) Aβ-immunostaining (brown) of an amyloid plaque in a human Alzheimer’s disease case. Note the dense core and glial nuclei (blue) surrounded by a halo of diffuse Aβ immunostaining. (D) Ultrastructure of an Aβ plaque. Note the dense amyloid core with the amyloid fibrils (af) surrounded by numerous dystrophic neuritis (some are labeled with “dn”). The Aβ plaque is from an AβPP transgenic mouse brain due to better tissue preservation compared to postmortem human tissue. Scale bars = 100 um (A), 1 um (B), 50 um (C) 5 um (D).
Other amyloid deposits may not be the predominant toxic entity per se. In Alzheimer’s disease autopsy material, the soluble Aβ species correlate more strongly with the degree of dementia than does the mass of Aβ plaques (Haass and Selkoe, 2007). Indeed, a variety of soluble Aβ multimeric species (e.g., dimers, timers, dodecamers and larger oligomers) have been isolated from the Alzheimer’s disease brain, and they induce synaptic toxicity and dysfunction, both in cell culture and when injected into the rodent brain (Lesne et al., 2006; Shankar et al., 2008). Similarly, synthetic, multimeric Aβ appears to be more toxic than Aβ monomers or fibrils (Haass and Selkoe, 2007; Lambert et al., 1998; Ono et al., 2009), but it is often unclear how the synthetic Aβ species relate to the in vivo counterparts (Meyer-Luehmann et al., 2006; Paravastu et al., 2009). Also, for tau and α-synuclein, soluble oligomeric species appear to be more toxic than the corresponding amyloid fibrils (Haass and Selkoe, 2007; Spires-Jones et al., 2011; Winner et al., 2011).
The physicochemical properties of the toxic oligomeric species are not well understood, and a consistent nomenclature is needed (Glabe, 2008). It is generally assumed that the greater toxicities of oligomers are mediated by their unique structural features (Campioni et al., 2010). The higher relative toxicity of small soluble oligomeric species, however, may also mirror the greater diffusion capability of such small aggregates through the tissue and into various compartments. Along the same lines, the relatively lower toxicity of amyloid fibrils may reflect the fact that many of the toxic structural entities of the fibril are buried in the amyloid mass (Haass and Selkoe, 2007; Keshet et al., 2010).
For Aβ toxicity, both receptor-mediated interactions and non-receptor-mediated membrane interactions have been described (Roychaudhuri et al., 2009; Yankner and Lu, 2009). The most significant toxicity of Aβ is towards the synapse. This is consistent with the profound loss of synapses in the Alzheimer’s disease brain and the observation that oligomeric Aβ species inhibit LTP, an electrophysiological correlate of memory formation (Shankar and Walsh, 2009). Soluble Aβ species bind to post-synaptic structures and interact with various putative ligands, such as PrP, NMDA receptor, EphB2, or downstream signaling events (Cisse et al., 2011; Lacor et al., 2007; Lauren et al., 2009; Snyder et al., 2005; Wei et al., 2010), but their in vivo relevance for Alzheimer’s disease pathogenesis is still unclear. Non-receptor membrane cytotoxicity for Aβ has been suggested through the insertion of Aβ oligomers into membranes, resulting in membrane disruption, possibly with the formation of cation-sensitive ion channels and dysregulation of calcium homeostasis (Glabe and Kayed, 2006; Roychaudhuri et al., 2009). Similar observations have been made with other oligomeric amyloid intermediates, suggesting that membrane disruption may be a more general mechanism in which amyloidogenic proteins exert their toxicity (Glabe and Kayed, 2006; Hebda and Miranker, 2009; Stefani, 2010).
Nevertheless, bona fide amyloid lesions in the brain, such as Aβ plaques, neurofibrillary tangles, and Lewy bodies, are not benign and are probably in equilibrium with their soluble proteinaceous constituents. For example Aβ plaques are responsible for local neuritic dystrophy (Figure 6), gliosis, and eventually can lead to disturbed neural network activity (Hedden et al., 2009; Tsai et al., 2004). Aβ plaques may also serve as a source of the more toxic and soluble Aβ assemblies, consistent with the view that a dynamic continuum of the various amyloid intermediates, not a given protein entity, elicits toxicity (Jan et al., 2011; Martins et al., 2008; Selkoe, 2011; Wogulis et al., 2005)
The value of animal models of human amyloid diseases
Spontaneous amyloidoses do not occur in typical laboratory animals, with the exception of ApoAII amyloidoses in some genetically-defined mouse strains.(Higuchi et al., 1993). Aged non-human primates and some other higher order mammals can develop β-amyloidosis and tau fibrillary inclusions in the brain; however, these animals do not develop the clinical signs of the human diseases (Jucker, 2010). Aged vervet monkeys, but not other non-human primates, can spontaneously develop TTR-amyloidosis. In vervet monkeys, spontaneous amyloidosis has been linked to the TTR lle122 allelic variant, which is also a disease-causing mutation in humans (Ueda et al., 2011). AA amyloidosis caused by inflammation or infection does occur in animals (Gruys, 2004).
The animals models of amyloidoses currently used are largely confined to genetically engineered mouse models. Most models bear transgenes that overexpress the amyloidogenic human protein, or, more often, its mutant counterpart that characterizes the familial forms of the disease (Table 1). This transgene approach has successfully modeled most cerebral amyloidoses (including tauopathies, α-synucleinopathies, prion diseases) and systemic amyloidoses (such as TTR-, gelsolin-, and IAPP-amyloidosis) (Buxbaum, 2009; Jucker, 2010; Page et al., 2009; Wadsworth et al., 2010). In all cases, overexpression of the amyloidogenic human protein appeared necessary because mice with a simple knock-in of the human amyloidogenic proteins do not spontaneously develop amyloid.
These mouse models are extremely valuable for the understanding of the amyloid aspect of the diseases although the models rarely fully recapitulate the clinical phenotype and neurodegeneration observed in humans. Nevertheless, the models have been successfully used to evaluate preclinical amyloid-modifying therapies (Jucker, 2010). Moreover, the incompleteness of most mouse models in recapitulating the entire spectra of the human disease has often been helpful for deciphering and understanding the complexity of the pathomechanisms of the diseases (Jucker, 2010).
Can amyloid formation be inhibited?
Chemical interventions
Discovery of chemical interventions against amyloid conditions have taken at least four different paths. One of the most promising paths is stabilization of the structure of the soluble form of a protein, diminishing the rate at which it is likely to undergo conversion to the amyloid state. The pioneering demonstration of this strategy was on TTR. TTR is a homotetramer that carries serum retinol binding protein and thyroid hormones, such as thyroxine. In several amyloid diseases, one of many mutations can destabilize TTR, leading to fibrous deposits in the heart and peripheral nerves. Using structure-based design, several potent and specific binders to the TTR hormone pocket have been described that inhibit fibril formation (Klabunde et al., 2000). The same strategy could be applied to other amyloid forming proteins that have a stable native structure.
A second approach is to screen for small molecules that disrupt fibril and oligomer formation. This enormous topic is worthy of a review on its own. Writing in 2007, Necula et al. (Necula et al., 2007) list 16 screening studies for molecules that inhibit fibrils of Aβ, and they go on to study molecules that inhibit formation of Aβ oligomers. In a recent study using small-molecule microarrays (Chen et al., 2010), 79 compounds were discovered that rescue cells from cytotoxicity. The authors suggested that a mechanism of rescue is that a compound can accelerate Aβ aggregation past an early-forming toxic oligomer. Screening for compounds that inhibit fibrils of tau is also an active area (Pickhardt et al., 2005). Despite this huge effort, we are unaware of compounds that have yet been found to be efficacious in treating Alzheimer’s disease.
A third approach uses the self-assembling property of amyloid fibers to poison the growth of amyloid fibers with peptides (Sciarretta et al., 2006). A biological system which apparently uses this strategy is Het-S, a native inhibitor of the HET-s prion (Wasner et al. 2008). Adoption of this principle for chemical design is based on our understanding that β-sheets are the fundamental organizing principle of amyloid fibrils and that fibrils grow by addition of new strands to the sheets. The fiber is poisoned or “capped” by adding a peptide that acts as a new strand via hydrogen bonding to the sheet at the fibril’s growing edge but prevents the subsequent addition of another amyloid molecule. Early on it was shown that the segment of Aβ with sequence KLVFF inhibits Aβ aggregation (Tjernberg et al., 1996), but this peptide itself forms steric-zipper fibrils (Colletier et al., 2011). More recent work has emphasized modifications of the blocking peptide, both to inhibit fibrillation of the target protein and to prevent self-fibrillation of the blocker (for review see Sciarretta et al., 2006). Depending on the system it has been found that blocking fiber formation could either increase or diminish the concentration of toxic oligomers (Sciarretta et al., 2006). A protein domain that has been found to inhibit fiber assembly of Aβ is the N-terminal domain of myelin basic protein (Liao et al., 2010).
The fourth approach is a variation of the third: inhibit fiber growth by the structure-based design of peptides targeted to block the ends of fibrils. This approach has become possible by the determination of the atomic structures of steric zippers, and it has been shown to be effective for inhibition in vitro of two different amyloid fibers (Sievers et al., 2011). Based on the structure of the steric zipper segment of the tau protein with sequence VQIVYK, an all D-amino acid inhibitor was designed to cap the ends of VQIVYK fibrils. This 6-residue all D-peptide was found to inhibit fibrillation of both VQIVYK fibers and constructs of tau. The fact that this blocker designed to cap steric zippers also blocks fibrillation of the parent protein of the zipper strengthens the hypothesis that steric zippers form the essential spine of amyloid fibrils.
Biological interventions
Amyloid formation depends on the concentration of the amyloid-forming proteins. Thus, inhibiting the generation of amyloidogenic proteins or of their precursors is a primary therapeutic strategy. For example, suppression of the inflammatory process responsible for serum amyloid A protein (SAA) overproduction is a therapeutic option for AA amyloid and elimination of B cell clones that overproduce immunoglobulin light chains represents a therapeutic option for AL amyloid (Pepys, 2001). Likewise, genetic variability in the expression of amyloidogenic proteins at slightly higher levels than normal may contribute to the risk of amyloidoses (Singleton et al., 2004). However, because of the incomplete mechanistic understanding of such genetic variability, no therapeutic strategies to reduce protein expression at the genetic levels have so far been developed.
Some amyloid-forming proteins are derived from longer precursor proteins that need cleavage to become amyloidogenic. The best-known example is AβPP that is sequentially cleaved by β-secretase and γ-secretase to release the Aβ peptide (De Strooper, 2010). Secretase inhibitors are currently in clinical trials, but current inhibitors may need refinement to avoid unwanted side effects, i.e. blocking cleavage to other substrates (De Strooper, 2010). Other amyloids (e.g. AA, AApoAII, ACys) also consist of protein fragments of larger precursors; however, it is not always clear whether such fragmentation is necessary for the amyloidoses or whether truncation is a secondary event without physiological significance (Westermark, 2005). While the relationship between posttranslational modification of amyloids and disease pathogenesis in general remains ill defined, inhibiting pyroglutamyl formation is pursued as a therapeutic target for Alzheimer’s disease (Schilling et al., 2008).
The finding that vaccination of AβPP transgenic mice can prevent and reduce cerebral β-amyloidosis has stimulated the development of antibody-based immunotherapeutics for Alzheimer’s disease (Brody and Holtzman, 2008). Although mechanistically still unclear, antibodies directed toward Aβ gain access to the brain where they bind to soluble and/or deposited Aβ species and promote their degradation. Phagocytosis of microglia as well as other mechanisms have been proposed for amyloid removal (Brody and Holtzman, 2008). Subsequent human immunotherapy trials showed also a reduction of Aβ deposits in brains of Alzheimer’s disease patients, as predicted from the preclinical mouse work (Jucker, 2010). However, unwanted side effects and lack of cognitive improvements in “immunized” Alzheimer’s disease patients must be overcome in future trials by early preventative, rather than therapeutic, interventions (Golde et al., 2011; Selkoe, 2011). Immunization against other amyloids, such as PrPsc, Tau, and α-synuclein have also been reported in transgenic mouse models (Aguzzi and O’Connor, 2010; Chai et al., 2011; Masliah et al., 2011). Along the same line, immunological depletion (in addition to pharmacological depletion) of serum amyloid P component (SAP) has been developed as a therapeutic strategy. SAP is claimed to stabilize amyloid fibrils and to be associated with most amyloids (Bodin et al., 2010; Pepys et al., 2002).
8. Are amyloid diseases transmissible?
In vitro assembly of amyloid fibrils can be initiated or accelerated by the addition of an amyloid seed (nucleus). Although originally suggested for PrP and Aβ (Jarrett and Lansbury, 1993), nucleated protein aggregation is likely a phenomenon that is common to all amyloids. Thus, from the view of a structural biologist, the features that define the amyloid state, in themselves render amyloids as ‘transmissible’, i.e., an amyloid nucleus can template the aggregation of a homologous protein. Nevertheless, only prions have unequivocally been shown to be infectious at the level of organisms, in which an exogenous, proteinaceous agent (the prion) initiates disease (Aguzzi and Rajendran, 2009). Why is this, and are we sure that infectivity at the organism level is restricted to prions?
A prion-like infectious cycle has been reported for AA amyloid (Westermark and Westermark, 2010). AA amyloid can be transmitted to susceptible hosts through a variety of inoculation routes. Transmission of AA amyloidosis between organisms (horizontal transmission, or ‘infectivity’ in the view of microbiologists) has not been proven unequivocally, but it appears to occur in captive cheetahs through fecal-oral transmission (Zhang et al., 2008). For mouse ApoAII amyloidosis, mother-to-offspring transmission (vertical transmission) has been demonstrated under experimental conditions (Korenaga et al., 2006).
In age-related neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, disease-specific proteopathic deposits occur in predictable region-specific and temporal patterns (Jucker and Walker, 2011). Such observations raise the possibility that the occurrence of the deposits occurs via prion-like propagation and spreading. Indeed, intracerebral inoculation of young, human-sequence AβPP-transgenic mice with brain extract from Alzheimer’s disease patients or aged AβPP-transgenic mice induces β-amyloidosis in a time- and concentration-dependent manner. The induced Aβ deposits first occur in the inoculated brain region, and then spread into neighboring areas often along anatomical and neuronal pathways. After prolonged incubation periods β-amyloid induction spreads throughout most of the entire brain (Hamaguchi et al., 2012; Jucker and Walker, 2011). The amyloid-inducing factor in the brain extract is likely aggregated Aβ in a conformation or polymorph that is most easily generated in the brain, because synthetic Aβ does not show comparable seeding activity in vivo (Langer et al., 2011; Meyer-Luehmann et al., 2006). Similar inoculation experiments have also been performed with α-synuclein- and tau-containing brain extracts; indeed, prion-like propagation spreading of the induced lesions in susceptible mice has also been suggested (Clavaguera et al., 2009; Mougenot et al., 2011). The mechanism by which intracytoplasmic lesions propagate remains puzzling, and its elucidation may provide new insights into basic cell biological processes (Aguzzi and Rajendran, 2009).
More recently, it has been shown that Aβ-containing brain extract can also induce cerebral β-amyloidosis if the extract is applied intraperitoneally (Eisele et al., 2010). These remarkable findings imply that the proteopathic seed can travel from the periphery to the brain. AA amyloid also has been suggested to spread among organs, possibly via the blood (Westermark and Westermark, 2010). Thus, in susceptible hosts and under experimental conditions, transmission, propagation, and spreading of amyloid seeds within and between organs appears possible.
Because of the unusual nature of proteopathic agents and their obligatory relationships to the host, the infectivity of protein-based diseases between individuals is refractory to verification by Koch’s postulates, which were designed to assess the infectivity of microbes (Walker et al., 2006). Hence, it has been suggested that the postulates be modified to account for the physicochemical characteristics of the infectious protein and to recognize the importance of the host in governing susceptibility to the disease (Walker et al., 2006). Naturally, epidemiological evidence also should be brought to bear on this issue. According to these criteria, compelling evidence for the infectivity of prion diseases has been presented (Colby and Prusiner, 2011), but for non-prion amyloidoses such evidence is still largely lacking. Thus, in future studies, there is a need for the epidemiological assessment of the relevance of the experimental amyloid transmission studies. Moreover, there is a need to study host susceptibility traits that allow proteopathic seeds to become “infectious.” It may well turn out that, although mechanistically similar to prionoses at the molecular level, the susceptibility of humans to the horizontal transmission of non-prion amyloid diseases is clinically insignificant under normal conditions. Nevertheless, the remarkable prevalence of this pathogenic principle suggests that common therapeutic strategies might be directed toward a variety of currently untreatable diseases.
9. Outlook
Although the research reviewed here portrays only part of the rapidly advancing knowledge about amyloid diseases, it may be sufficient to define some of the critical questions for the next phase of work.
At the molecular level, we still lack high resolution knowledge of amyloid oligomers in all but the simplest fibers. Recent work has begun to reveal the structural basis of prion strains. Now we need to establish whether amyloid strains play a physiologically significant role in other amyloid diseases and if so, we need a fuller view of amyloid polymorphism. Furthermore, we need a better molecular understanding of the assembly pathways from functional proteins to amyloid oligomers and fibers and their pathways for disassembly. At the level of cellular biology, we need to learn which biological cofactors stabilize and destabilize amyloid structures, and we need to fill in more details of metabolic and signaling pathways that regulate degradation and disposal of amyloids.
An urgent need is the further development of structural and physiochemical techniques that permit the analysis of aggregated proteins in cells and living tissues, as opposed to extracted amyloid or recombinant amyloid. A remaining mystery is the enormously greater potency of seeding by amyloid and prions extracted from tissues compared to recombinant amyloids. Is this greater potency due to undetected biological cofactors in the extracted material, or has the extracted protein been templated into some structure in vivo which the recombinant, apparently identical, material cannot achieve? Can biological factors be discovered which can convert recombinant proteins to forms that are as potent as extracted amyloid?
Another mystery involves the mechanisms and pathways for cellular toxicity of amyloid. Are there common mechanisms of toxicities, or do mechanisms differ between systemic and cerebral amyloid diseases? What are the toxic structures? Are oligomers distinct from small fibers, and what accounts for their toxicity? Why can toxicity of PrP be recapitulated in animal models whereas the toxicity of Aβ in animal models is comparatively modest? What is different about functional amyloids that render them non-toxic?
Finally, the implications for disease of the recently reported experimental transmission of non-prion amyloids need to be established. Are similar or different structures responsible for toxicity and transmission? Can amyloid in the environment seed human diseases, and if so, what protective measures are necessary?
As answers to these questions emerge, a class of diseases that afflict and kill millions will be understood and perhaps controlled by preventative and therapeutic interventions.
Acknowledgments
We thank Lary Walker (Atlanta, GA), Per Westermark (Uppsala, Sweden), and the members of our laboratories for experimental support and comments on this manuscript. The contributions of Michael Sawaya (Los Angeles, CA) and Rebecca Nelson (Los Angeles, CA), M. Tolnay (Basel, Switzerland), M. Yamada (Kanazawa, Japan), P. Nilsson (Linkoping, Sweden), and S. Eberle (Tübingen, Germany) to figures and text are greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nature reviews Molecular cell biology. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature reviews Drug discovery. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A. Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angewandte Chemie. 2010;49:3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]
- Apostol MI, Sawaya MR, Cascio D, Eisenberg D. Crystallographic studies of prion protein (PrP) segments suggest how structural changes encoded by polymorphism at residue 129 modulate susceptibility to human prion disease. J Biol Chem. 2010;285:29671–29675. doi: 10.1074/jbc.C110.158303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astbury WT, Dickinson S, Bailey K. The X-ray interpretation of denaturation and the structure of the seed globulins. Biochem J. 1935;29:2351–2360. doi: 10.1042/bj0292351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Benzinger TL, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer’s beta-amyloid(10-35) is parallel beta-sheet with residues in exact register. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom J, Gustavsson A, Hellman U, Sletten K, Murphy CL, Weiss DT, Solomon A, Olofsson BO, Westermark P. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. The Journal of pathology. 2005;206:224–232. doi: 10.1002/path.1759. [DOI] [PubMed] [Google Scholar]
- Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, Mangione PP, Gallimore JR, Millar DJ, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468:93–97. doi: 10.1038/nature09494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(Suppl 4):16392–16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN. Animal models of human amyloidoses: are transgenic mice worth the time and trouble? FEBS letters. 2009;583:2663–2673. doi: 10.1016/j.febslet.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Armstrong AH, Koehler AN, Hecht MH. Small molecule microarrays enable the discovery of compounds that bind the Alzheimer’s Abeta peptide and reduce its cytotoxicity. Journal of the American Chemical Society. 2010;132:17015–17022. doi: 10.1021/ja107552s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annual review of biochemistry. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–1203. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harbor perspectives in biology. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colletier JP, Laganowsky A, Landau M, Zhao M, Soriaga AB, Goldschmidt L, Flot D, Cascio D, Sawaya MR, Eisenberg D. Molecular basis for amyloid-{beta} polymorphism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Coomaraswamy J, Kilger E, Wolfing H, Schafer C, Kaeser SA, Wegenast-Braun BM, Hefendehl JK, Wolburg H, Mazzella M, Ghiso J, et al. Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7969–7974. doi: 10.1073/pnas.1001056107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiological reviews. 2010;90:465–494. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA, Kumar A, Neal KL, Betensky RA, Frosch MP, et al. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Annals of neurology. 2010;68:545–548. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duering M, Grimm MO, Grimm HS, Schroder J, Hartmann T. Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiology of aging. 2005;26:785–788. doi: 10.1016/j.neurobiolaging.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Eanes ED, Glenner GG. X-ray diffraction studies on amyloid filaments. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1968;16:673–677. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandrich M, Forge V, Buder K, Kittler M, Dobson CM, Diekmann S. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15463–15468. doi: 10.1073/pnas.0303758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends in biochemical sciences. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem. 2009;284:3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends in neurosciences. 2010;33:317–325. doi: 10.1016/j.tins.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsbury C, Frey P, Olivieri V, Aebi U, Muller SA. Multiple assembly pathways underlie amyloid-beta fibril polymorphisms. Journal of molecular biology. 2005;352:282–298. doi: 10.1016/j.jmb.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Gruys E. Protein folding pathology in domestic animals. Journal of Zhejiang University Science. 2004;5:1226–1238. doi: 10.1631/jzus.2004.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature reviews Molecular cell biology. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hamaguchi T, Eisele YS, Varvel NH, Lamb BT, Walker LC, Jucker M. The presence of Abeta seeds, and not age per se, is critical to the initiation of Abeta deposition in the brain. Acta Neuropathol. 2012;123:31–37. doi: 10.1007/s00401-011-0912-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebda JA, Miranker AD. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annual review of biophysics. 2009;38:125–152. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, Buckner RL. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. The Journal of neuroscience. 2009;29:12686–12694. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol. 2006;16:40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi K, Kitado H, Kitagawa K, Kogishi K, Naiki H, Takeda T. Development of congenic strains of mice carrying amyloidogenic apolipoprotein A-II (Apoa2c). Apoa2c reduces the plasma level and the size of high density lipoprotein. FEBS letters. 1993;317:207–210. doi: 10.1016/0014-5793(93)81277-7. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihse E, Suhr OB, Hellman U, Westermark P. Variation in amount of wild-type transthyretin in different fibril and tissue types in ATTR amyloidosis. Journal of molecular medicine. 2011;89:171–180. doi: 10.1007/s00109-010-0695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihse E, Ybo A, Suhr O, Lindqvist P, Backman C, Westermark P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. The Journal of pathology. 2008;216:253–261. doi: 10.1002/path.2411. [DOI] [PubMed] [Google Scholar]
- Ivanova MI, Sievers SA, Sawaya MR, Wall JS, Eisenberg D. Molecular basis for insulin fibril assembly. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18990–18995. doi: 10.1073/pnas.0910080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, Muhs A, Lashuel HA. Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J Biol Chem. 2011;286:8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Jimenez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. The EMBO journal. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nature Medicine. 2010;16:1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in alzheimer disease and other neurodegenerative disorders. Annals of neurology. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser SA, Herzig MC, Coomaraswamy J, Kilger E, Selenica ML, Winkler DT, Staufenbiel M, Levy E, Grubb A, Jucker M. Cystatin C modulates cerebral beta-amyloidosis. Nature genetics. 2007;39:1437–1439. doi: 10.1038/ng.2007.23. [DOI] [PubMed] [Google Scholar]
- Kajava AV, Baxa U, Steven AC. Beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. The FASEB journal. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet B, Yang IH, Good TA. Can size alone explain some of the differences in toxicity between beta-amyloid oligomers and fibrils? Biotechnology and bioengineering. 2010;106:333–337. doi: 10.1002/bit.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nature structural biology. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- Korenaga T, Yan J, Sawashita J, Matsushita T, Naiki H, Hosokawa M, Mori M, Higuchi K, Fu X. Transmission of amyloidosis in offspring of mice with AApoAII amyloidosis. The American journal of pathology. 2006;168:898–906. doi: 10.2353/ajpath.2006.050350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns IE, Mason BJ. The supercooling and freezing of small water droplets falling in air and other gases. Proc R Soc A. 1968;302:437–452. [Google Scholar]
- Kumar S, Rezaei-Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, Mc Donald JM, Wullner U, Glebov K, Heneka MT, et al. Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. The EMBO journal. 2011;30:2255–2265. doi: 10.1038/emboj.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Human mutation. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, Konig S, Roeber S, et al. Nitration of Tyrosine 10 Critically Enhances Amyloid beta Aggregation and Plaque Formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, et al. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem. 2001;276:12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, et al. Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. The EMBO journal. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. The Journal of Neuroscience. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. The Journal of Neuroscience. 2011;31:14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol. 2011;7:602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Levine H, 3rd, Walker LC. Molecular polymorphism of Abeta in Alzheimer’s disease. Neurobiol Aging. 2010;31:542–548. doi: 10.1016/j.neurobiolaging.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski JR, van der Wel PC, Rigney M, Grigorieff N, Griffin RG. Structural complexity of a composite amyloid fibril. Journal of the American Chemical Society. 2011;133:14686–14698. doi: 10.1021/ja203736z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao MC, Hoos MD, Aucoin D, Ahmed M, Davis J, Smith SO, Van Nostrand WE. N-terminal domain of myelin basic protein inhibits amyloid beta-protein fibril assembly. J Biol Chem. 2010;285:35590–35598. doi: 10.1074/jbc.M110.169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark K, Westermark GT, Olsen A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6098–6102. doi: 10.1073/pnas.0501814102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarouf CL, Daugs ID, Spina S, Vidal R, Kokjohn TA, Patton RL, Kalback WM, Luehrs DC, Walker DG, Castano EM, et al. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Molecular neurodegeneration. 2008;3:20. doi: 10.1186/1750-1326-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Maeda A, Yamada M, Itoh Y, Otomo E, Hayakawa M, Miyatake T. Computer-assisted three-dimensional image analysis of cerebral amyloid angiopathy. Stroke. 1993;24:1857–1864. doi: 10.1161/01.str.24.12.1857. [DOI] [PubMed] [Google Scholar]
- Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D’Hooge R, De Strooper B, et al. Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. The EMBO journal. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PloS one. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry. 2005;44:10810–10821. doi: 10.1021/bi0508237. [DOI] [PubMed] [Google Scholar]
- Miyazono M, Kitamoto T, Iwaki T, Tateishi J. Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Straussler syndrome. Acta Neuropathol. 1992;83:333–339. doi: 10.1007/BF00713522. [DOI] [PubMed] [Google Scholar]
- Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, Soto C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. The Journal of Neuroscience. 2010;30:4528–4535. doi: 10.1523/JNEUROSCI.5924-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchere J, Lakhdar L, Legastelois S, Baron T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiology of aging. 2011 doi: 10.1016/j.neurobiolaging.2011.06.022. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson KP, Aslund A, Berg I, Nystrom S, Konradsson P, Herland A, Inganas O, Stabo-Eeg F, Lindgren M, Westermark GT, et al. Imaging distinct conformational states of amyloid-beta fibrils in Alzheimer’s disease using novel luminescent probes. ACS chemical biology. 2007;2:553–560. doi: 10.1021/cb700116u. [DOI] [PubMed] [Google Scholar]
- Nilsson KP, Joshi-Barr S, Winson O, Sigurdson CJ. Prion strain interactions are highly selective. The Journal of neuroscience. 2010;30:12094–12102. doi: 10.1523/JNEUROSCI.2417-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page LJ, Suk JY, Bazhenova L, Fleming SM, Wood M, Jiang Y, Guo LT, Mizisin AP, Kisilevsky R, Shelton GD, et al. Secretion of amyloidogenic gelsolin progressively compromises protein homeostasis leading to the intracellular aggregation of proteins. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11125–11130. doi: 10.1073/pnas.0811753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of beta-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys MB. Pathogenesis, diagnosis and treatment of systemic amyloidosis. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2001;356:203–210. doi: 10.1098/rstb.2000.0766. discussion 210-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, Lovat LB, Bartfai T, Alanine A, Hertel C, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417:254–259. doi: 10.1038/417254a. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, DeArmond SJ, Prusiner SB, Scott MR. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron. 2002;34:921–932. doi: 10.1016/s0896-6273(02)00726-2. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, Giliberto L, Armirotti A, D’Arrigo C, Bachi A, et al. beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280:34186–34192. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- Pickhardt M, von Bergen M, Gazova Z, Hascher A, Biernat J, Mandelkow EM, Mandelkow E. Screening for inhibitors of tau polymerization. Current Alzheimer research. 2005;2:219–226. doi: 10.2174/1567205053585891. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118:115–130. doi: 10.1007/s00401-009-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambashivan S, Liu Y, Sawaya MR, Gingery M, Eisenberg D. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature. 2005;437:266–269. doi: 10.1038/nature03916. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nature Medicine. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Sachse C, Richter W, Xu C, Fandrich M, Grigorieff N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) amyloid fibrils reveals similar protofilament structures. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta KL, Gordon DJ, Meredith SC. Peptide-based inhibitors of amyloid assembly. Methods in enzymology. 2006;413:273–312. doi: 10.1016/S0076-6879(06)13015-3. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nature Medicine. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- Serag AA, Altenbach C, Gingery M, Hubbell WL, Yeates TO. Identification of a subunit interface in transthyretin amyloid fibrils: evidence for self-assembly from oligomeric building blocks. Biochemistry. 2001;40:9089–9096. doi: 10.1021/bi010655s. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Molecular neurodegeneration. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. 2003;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Polymenidou M, Schwarz P, Leclerc M, Hammarstrom P, Wuthrich K, Aguzzi A. Prion strain discrimination using luminescent conjugated polymers. Nature methods. 2007;4:1023–1030. doi: 10.1038/nmeth1131. [DOI] [PubMed] [Google Scholar]
- Singleton A, Myers A, Hardy J. The law of mass action applied to neurodegenerative disease: a hypothesis concerning the etiology and pathogenesis of complex diseases. Human molecular genetics. 2004;13(Spec No 1):R123–126. doi: 10.1093/hmg/ddh093. [DOI] [PubMed] [Google Scholar]
- Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17:101–104. doi: 10.3109/13506129.2010.526812. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nature neuroscience. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT. Are tangles as toxic as they look? J Mol Neurosci. 2011;45:438–444. doi: 10.1007/s12031-011-9566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. The FEBS journal. 2010;277:4602–4613. doi: 10.1111/j.1742-4658.2010.07889.x. [DOI] [PubMed] [Google Scholar]
- Sunde M, Blake CC. From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Quarterly reviews of biophysics. 1998;31:1–39. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. Journal of molecular biology. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- Tekirian TL, Saido TC, Markesbery WR, Russell MJ, Wekstein DR, Patel E, Geddes JW. N-terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. Journal of neuropathology and experimental neurology. 1998;57:76–94. doi: 10.1097/00005072-199801000-00009. [DOI] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Science of aging knowledge environment : SAGE KE. 2006;2006:re1. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- Tomidokoro Y, Lashley T, Rostagno A, Neubert TA, Bojsen-Moller M, Braendgaard H, Plant G, Holton J, Frangione B, Revesz T, et al. Familial Danish dementia: co-existence of Danish and Alzheimer amyloid subunits (ADan AND A{beta}) in the absence of compact plaques. J Biol Chem. 2005;280:36883–36894. doi: 10.1074/jbc.M504038200. [DOI] [PubMed] [Google Scholar]
- Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and dynamic features of Alzheimer’s Abeta peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nature neuroscience. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Tsemekhman K, Goldschmidt L, Eisenberg D, Baker D. Cooperative hydrogen bonding in amyloid formation. Protein science. 2007;16:761–764. doi: 10.1110/ps.062609607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. Solid-state NMR studies of amyloid fibril structure. Annual review of physical chemistry. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Ageyama N, Nakamura S, Nakamura M, Chambers JK, Misumi Y, Mizuguchi M, Shinriki S, Kawahara S, Tasaki M, et al. Aged vervet monkeys developing transthyretin amyloidosis with the human disease-causing Ile122 allele: a valid pathological model of the human disease. Lab Investigation. 2011 doi: 10.1038/labinvest.2011.195. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Wadsworth JD, Asante EA, Collinge J. Review: contribution of transgenic models to understanding human prion disease. Neuropathology and applied neurobiology. 2010;36:576–597. doi: 10.1111/j.1365-2990.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L, Levine H, Jucker M. Koch’s postulates and infectious proteins. Acta Neuropathol. 2006;112:1–4. doi: 10.1007/s00401-006-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Maji SK, Sawaya MR, Eisenberg D, Riek R. Bacterial inclusion bodies contain amyloid-like structure. PLoS biology. 2008;6:e195. doi: 10.1371/journal.pbio.0060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- Watt B, Tenza D, Lemmon MA, Kerje S, Raposo G, Andersson L, Marks MS. Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS genetics. 2011;7:e1002286. doi: 10.1371/journal.pgen.1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nature neuroscience. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark GT, Sletten K, Westermark P. Massive vascular AA-amyloidosis: a histologically and biochemically distinctive subtype of reactive systemic amyloidosis. Scandinavian journal of immunology. 1989;30:605–613. doi: 10.1111/j.1365-3083.1989.tb02468.x. [DOI] [PubMed] [Google Scholar]
- Westermark GT, Westermark P. Prion-like aggregates: infectious agents in human disease. Trends in molecular medicine. 2010;16:501–507. doi: 10.1016/j.molmed.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Westermark P. Aspects on human amyloid forms and their fibril polypeptides. The FEBS journal. 2005;272:5942–5949. doi: 10.1111/j.1742-4658.2005.05024.x. [DOI] [PubMed] [Google Scholar]
- White HE, Hodgkinson JL, Jahn TR, Cohen-Krausz S, Gosal WS, Muller S, Orlova EV, Radford SE, Saibil HR. Globular tetramers of beta(2)-microglobulin assemble into elaborate amyloid fibrils. Journal of molecular biology. 2009;389:48–57. doi: 10.1016/j.jmb.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. Journal of molecular biology. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Wiltzius JJ, Landau M, Nelson R, Sawaya MR, Apostol MI, Goldschmidt L, Soriaga AB, Cascio D, Rajashankar K, Eisenberg D. Molecular mechanisms for protein-encoded inheritance. Nature structural & molecular biology. 2009;16:973–978. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. The Journal of Neuroscience. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wogulis M, Wright S, Cunningham D, Chilcote T, Powell K, Rydel RE. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. The Journal of Neuroscience. 2005;25:1071–1080. doi: 10.1523/JNEUROSCI.2381-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Fu X, Ge F, Zhang B, Yao J, Zhang H, Qian J, Tomozawa H, Naiki H, Sawashita J, et al. Cross-seeding and cross-competition in mouse apolipoprotein A-II amyloid fibrils and protein A amyloid fibrils. The American journal of pathology. 2007;171:172–180. doi: 10.2353/ajpath.2007.060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Lu T. Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284:4755–4759. doi: 10.1074/jbc.R800018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Une Y, Fu X, Yan J, Ge F, Yao J, Sawashita J, Mori M, Tomozawa H, Kametani F, et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7263–7268. doi: 10.1073/pnas.0800367105. [DOI] [PMC free article] [PubMed] [Google Scholar]