

Abstract
This short review tells the story of how Reversed Chloroquine drugs (RCQs) were developed. These are hybrid molecules, made by combining the quinoline nucleus from chloroquine (CQ) with moieties which are designed to inhibit efflux via known transporters in the membrane of the digestive vacuole of the malaria parasite. The resulting RCQ drugs can have potencies exceeding that of CQ, while at the same time having physical chemical characteristics that may make them favorable as partner drugs in combination therapies. The need for such novel antimalarial drugs will continue for the foreseeable future.
Keywords: Antimalarial, chloroquine, Plasmodium falciparum, drug development, drug design, structure activity relationship, malaria, drug resistance.
INTRODUCTION
Chloroquine (CQ) was a truly remarkable drug, and the standard of care for malaria over about half a century, beginning in the late 1940s. Speaking about the discovery of CQ in a Review Lecture published in 1963, G. Robert Coatney stated [1], “Results since 1946 have made it the drug of choice for malaria the world over. It was the only antimalarial used by our Armed Forces and most of the Allied Forces in Korea. It has since been employed by most of the Free World Forces when deployed in malarious areas and is the drug of choice in the world-wide malaria eradication program under the World Health Organization.”
Of course, that worldwide malaria eradication program did not succeed, and upon its abandonment the situation regarding malaria grew worse. The emergence and subsequent spread of drug resistance in malaria had made the (otherwise favored) malaria drug CQ ineffective throughout most of the world [2]. Combination therapies [3, 4] have now become the standard of care for malaria chemotherapy, though currently available combinations lack CQ’s simplicity & low cost. Because malaria will doubtless continue to evolve resistance even to the newest cures [5, 6], there is, and will be an ongoing need for a development pipeline of effective drugs until eradication of the disease might become a reality.
In response to this challenging situation, we embarked upon a hybrid-drug approach to overcoming malaria’s drug resistance. Chemosensitizers, or reversal agents (RAs), are molecules that reverse resistance to a drug. In the context of this review, the RA is generally an efflux mechanism inhibitor. RAs for CQ turn the susceptibility of CQ-resistant strains back toward that of CQ-sensitive strains [7-11], mainly by inhibiting the transporter of CQ from the digestive vacuole (DV) – P. falciparum chloroquine-resistance transporter (PfCRT) [12]. We originally hypothesized and showed that CQ-resistance could be reversed by covalently bonding CQ to a RA, producing a single molecule that we term a ‘reversed-CQ’ (RCQ) compound [13]; this is illustrated in Fig. (1). An RCQ compound has a significant advantage over a cocktail of CQ and RA because CQ accumulates strongly in the parasite’s DV, so the linked RA entity accumulates to a much higher concentration than an unlinked RA. The drug:RA ratio is, by definition, 1:1. We have demonstrated the feasibility of this approach by making several dozens of these compounds, and shown that the design is robust and flexible [13-15]. We have generated a large number of potent and novel RCQ compounds that are very effective against both CQ-sensitive and CQ-resistant strains at concentrations far lower than CQ or other similar drugs. In vitro cytotoxicity results for these drugs are favorable [14, 15]. In fact, with the better RCQ drugs we have not observed any adverse effects in mice when the drugs are administered orally with an amount well over an order of magnitude above the in vivo ED50 values, and well above the fatal CQ dose.
Fig. (1).

The original concept leading to RCQ drugs. Note the chiral center in CQ that we eliminate in a RCQ drug.
What follows is a short story about how the RCQ molecules have developed in our laboratory, and a brief description of a few molecule classes that are closely related to the RCQ design. The reader can refer to other publications for reviews covering fundamentals of malaria’s CQ-resistance [16], the discovery and development of resistance reversal drugs [10, 17], hybrid drugs for malaria [18], as well as hybrid drugs generally [19, 20], in addition to a recent review covering the need for new malaria drugs [21].
THE “TARGET”: PfCRT
A protein localized to the membrane defining the digestive vacuole (DV) of P. falciparum was discovered by the Wellems group at NIH to have mutations that are correlated to CQ-resistance [12]. Since the function of this protein was not known, it was designated as the, “P. falciparum Chloroquine Resistance Transporter,” or PfCRT. Although there were multiple mutations found within the locus of the pfcrt gene, a key mutation for CQ-resistance was K76T. Most importantly, incorporation of this mutation into a CQ-sensitive strain induced the CQ-resistance trait. This, and several quickly following papers, e.g., [22-25] established the importance of this point mutation in P. falciparum CQ-resistance, although other mutations and probably other transporters are known to modulate the trait [16, 22, 25-27].
The structure of PfCRT has been predicted to contain 10 transmembrane helices, with the N- and C- termini extending into the parasite’s cytoplasm, away from the DV [12]. The K76T location is near at the proposed DV-end of the N-terminal (first) transmembrane helix, and so could interact electrostatically with drugs like CQ, which exists as the dication when in the acidic DV; the loss of the positively-charged lysine residue could then form the basis for resistance by reducing this unfavorable interaction such that the drug then can bind and be transported. In any event, the thermodynamics involved in the transport process are not simple, as recently reviewed [27].
THE FIRST RCQ MOLECULE
The fundamental design of a reversed drug molecule is a hybridization of the original drug with a reversal agent (chemosentizer) that can inhibit the resistance mechanism, in this case the export of the drug by an efflux mechanism. For our initial RCQ drug, we chose a reversal agent moiety that was known to produce a buildup of CQ (pKa ~ 8.1 & 10.2 [28]) within the parasite (and so presumably in the DV; pH ~ 5 [28]). Such molecules, including verapamil (VP), are often thought of by their activities as calcium channel blockers. Although VP was the usual reference for a CQ-resistance reversal agent, it is somewhat complex from a chemical perspective. However a simpler choice, imipramine (pKa ~ 9.5 [29]), was also known to be a potent RA [11, 30-32], and is achiral, unlike VP and many other reversal agents known to act against CQ-resistance. The commercial availability of desimipramine led to a simple synthesis for the molecule, 1, also achiral and first published in 2006 [13]. The structure and synthetic route are shown in Fig. (2). This molecule, to our knowledge, was the first reported hybrid drug that intentionally incorporated a chemical-efflux inhibitor coupled to a drug molecule. When tested in vitro against both a CQ-sensitive P. falciparum strain (D6) and against a CQ-resistant strain (Dd2), using the SYBR Green I-based fluorescence assay [33], it was found to be more potent than CQ against either strain, even the CQ-sensitive strain. However, RCQ molecule 1 has a very high estimated clogP of nearly 9, as well as a clogD of 4.2 at pH 7.4 (Marvin 5.5.0.0, 2011, ChemAxon (http://www.chemaxon.com) was used for this calculation), and so was not expected to be a practical drug itself. Nonetheless, we evaluated the in vivo potential of the RCQ drugs in a small mouse trial using P. chabaudi, and found that 1 had >99% suppressive activity at 64 mg/kg/day in a standard 4-day suppressive test. It is important, however, to remember that 1 was a prototype molecule, assembled to test the principle without being optimized. We then returned to the principles of the RA pharmacophore to begin the optimization process.
Fig. (2).

The synthesis and structure of prototype RCQ molecule, 1 [13]. The hydrogen-bond accepting nitrogen is indicated with an arrow, and the reversal agent aromatic groups are circled; the RA moiety will be shown in bold bonds throughout. In the structure of 1, the quinoline and linker are contained in the line-box, and the RA moiety is in the dotted box.
SECOND-GENERATION RCQ MOLECULES
The structural essentials for the CQ-resistance reversing RA molecules identified by Bhattacharjee et al. in 2002 [34] included, “Two aromatic hydrophobic sites and a hydrogen bond acceptor site, preferably at a side chain nitrogen atom. We took this conclusion, based on a modeling / calculation approach based on a training set of 17 compounds, as license to open up the RA structure to modifications in a guided way that would remove the tricyclic nature of the imipramine-based RA end of molecule 1. This might reduce or eliminate any central neural system (CNS) activity of the resultant drugs. In addition, there were the questions of water-solubility and oral availability of the drug to address.
We began to evaluate the structural requirements for an RCQ drug by examining the limits to which one could practically vary the “linker” between the quinoline and the RA’s side-chain nitrogen, as well as between that nitrogen and the RA aromatic rings [14]. We found little differences between RCQ molecules that had between 2 and 4 carbons in the linker between the quinoline and the RA’s side-chain nitrogen, and significant changes could be made to the RA structures themselves. A few examples are shown in Table 1. The length of the chain between the nitrogen and the 2 aromatic groups was varied between 2 and 4 carbons (and changed to a piperazinyl ring) with little change in activity, as well as incorporating a carbonyl (but not converting the central nitrogen into an amide). The aromatic rings were converted from phenyls to benzyls, with little change in activity. A recent paper addresses the ability of this moiety to act specifically as an inhibitor of mutated PfCRT [35]. Two other points that arose from this structure-activity relationship (SAR) work were that having a proton on the quinoline’s 4-amino was beneficial, and that having two RA groups was not a strong advantage. So there was great breadth in scope found for RCQ design, and considerations such as pharmacology and cost could then be addressed with few constraints. In fact, the latitude for RCQ design appeared to be much greater than for developing a ‘simple’ RA.
Table 1.
Selected RCQ Activities against P. falciparum [14]
| Compound | ClogP a | IC50 b(nM) | IC50 Ratio Dd2/D6 | Cytotoxicity c(nM) | ||
|---|---|---|---|---|---|---|
| D6 (CQS) | Dd2 (CQR) | |||||
| CQ | 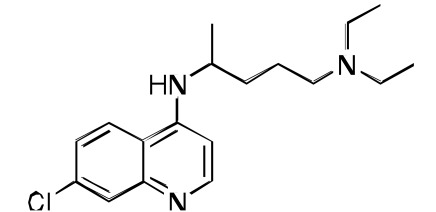 |
5.1 | 7 | 102 | 15 | 12000 |
| 1 | 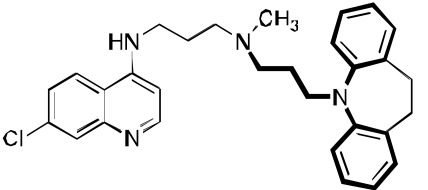 |
8.9 | 3 | 5 | 1.7 | 700 |
| 2 | 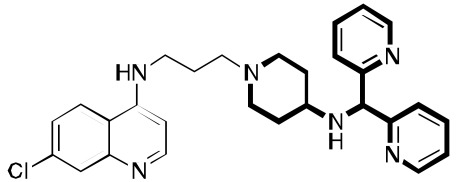 |
3.5 | 1.6 | 1.8 | 1.1 | 6500 |
| 3 | 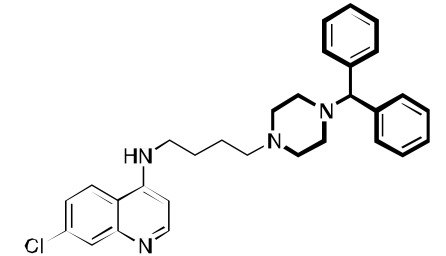 |
7.5 | 1.3 | 1.3 | 1.0 | 1100 |
| 4 | 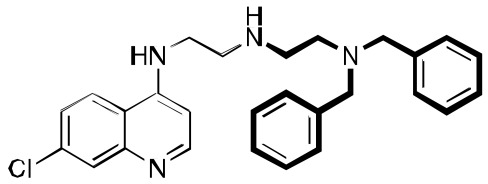 |
7.0 | 10 | 16 | 1.6 | 2200 |
| 5 | 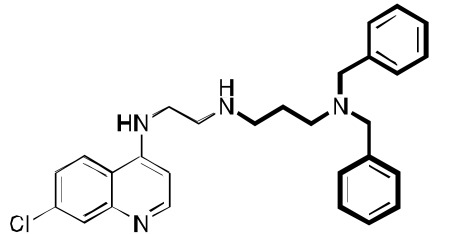 |
7.3 | 7 | 16 | 2.3 | N.D. |
| 6 | 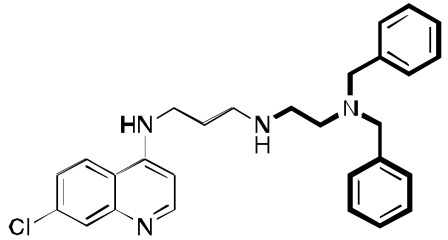 |
7.3 | 2 | 6 | 3.0 | N.D. |
| 7 | 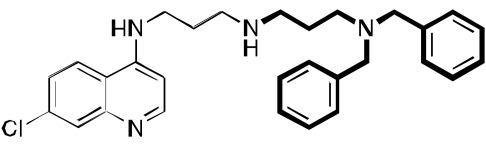 |
7.6 | 23 | 34 | 1.5 | 700 |
| 8 | 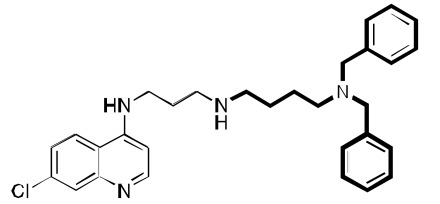 |
7.4 | 8 | 11 | 1.4 | 1300 |
Evaluated using ChemDraw software.
Averages of at least 3 runs (± 15%). The uncertainties are estimated based on weighing uncertainties for the various compounds (which are free-bases and often oils), as well as on variability between determinations that were performed on different weeks.
Cytotoxicities are against mouse spleen lymphocytes. These values are estimated to be ±50%, based on weighing uncertainties for the various compounds (which are free-bases and often oils), as well as on variability between determinations that were performed on different weeks. N.D.: not determined.
This conclusion underscores the point that there are fundamental differences in the requirements for the design of an effective RA drug, as opposed to RCQ drugs. First, the RCQ molecule’s fundamental activity must be antimalarial activity. The functional requirement of a RA molecule is the ability to inhibit efflux of CQ in a malaria strain; the function of a RA moiety in a RCQ drug is to enhance buildup of the drug in the DV (presumably by decreasing efflux of itself). This latter property does not necessarily require that it be able to inhibit efflux of CQ. In fact, it may be that the best RCQ drugs may not interact with an efflux effector such as PfCRT at all: RCQ drugs could bind other transporters in the DV, or they could simply have lower binding to all such transporters. This is because binding is the first step in transport, so that reduced binding could lead to a higher RCQ drug concentration in the DV, even though it would be a less effective RA for CQ export. Therefore, it may be easier to design an optimized RCQ drug RA than it would be to design the corresponding RA moiety.
AN IMPROVED RCQ MOLECULE
We continued the SAR work, taking advantage of the above-referenced flexibility to find a molecule, 2, that has great water solubility, has good in vitro antimalarial activity against a variety of CQ-resistant and CQ-sensitive P. falciparum strains, and is orally curative in mice. Its IC50 values against D6, Dd2, and 7G8 were about 1, 2, and 2 nM, as compared to CQ values of 7, 102, and 106 nM. Having a clogP value of about 3.5 (CQ has a value of about 5), 2 was anticipated to have good water solubility, especially as a salt formulation (e.g., chloride or phosphate). Low cytotoxicity values (in our studies, to mouse spleen lymphocytes), and the resulting favorable therapeutic index values were found for many RCQ molecules. For example, the therapeutic index values for CQ and 2 were found to be about 120 and 4000, respectively (ratio of IC50 for mouse spleen lymphocytes / P. falciparum strain Dd2). In vivo work in mice found that 2 could cure mice of a P. berghei infection at an equimolar dose to 30 mg/kg/day for CQ. Remarkably, while CQ kills at < 200 mg/kg in mice, we could orally dose 2 to > 400 mg/kg in mice without evident adverse effects for at least 3 days (unpublished).
MECHANISM(S) OF ACTION
While the full story about the mode(s) of action for CQ is not certain, it has been widely assumed to involve interaction with heme, a byproduct of hemoglobin digestion by the Plasmodium parasite. We therefore evaluated the binding of heme by both CQ and 1 using UV-vis spectroscopy. A binding stoichiometry of one drug molecule to one heme μ-oxo dimer was assumed. At pH 5.7, near the DV pH, the measured binding constant (3 × 105 M-1) was found to be the same as measured for CQ (3 × 105 M-1). The measured binding constant at pH 7 (also 3 × 105 M-1) was also found to be comparable to that which we measured for CQ (1 × 106 M-1) under the same conditions. The conclusion is that the RA portion of 1 does not appear to affect the CQ-like binding to heme. This conclusion has been supported by a recent study [36]. Compound 2 Fig. (3) also proved to be an avid molecule for the heme µ-oxo dimer, and was about 3-fold more efficient at inhibiting β-hematin formation as Compound 1. This may indicate that the high potencies of the drugs is a combination of both enhanced accumulation and enhanced interaction with heme, both relative to CQ. We also tested the ability to accumulate in P. falciparum-parasitized red cells. In this test, compound 1 did, in fact, accumulate to a stronger degree than did CQ, as measured by the drop of drug concentration in the surrounding medium. Light microscopic analysis of surviving parasites in high concentrations of 1 showed a strongly swelled DV, as well as very little apparent hemozoin crystals. CQ under the same conditions showed the same qualitative effects, but to a lesser extent. All this is consistent with an inhibition of hemozoin formation mechanism for the RCQ molecules, in analogy with the accepted mechanism of CQ, except that the RCQ molecule is generally more potent across the range of effects.
Fig. (3).

Two additional key RCQ molecules [15]. RCQ drug 2 has good water solubility and oral efficacy, and RCQ 3 has the same linker length between the 4-amino group and the next nitrogen as does CQ. Both have high potencies against CQ-sensitive and CQ-resistant P. falciparum strains.
We found that the RCQ drugs’ effectiveness is not due to varying the length of the linker between the quinoline and the central nitrogen; this result is important because others have found that such changes alone can overcome CQ-resistance. Thus, we synthesized a compound 3 Fig. (3), which was found to have better antimalarial activity than CQ, and in combination with the work cited above, we concluded that the ability of the RCQ molecules to overcome CQ-resistance by the addition of a RA head group to the 4-aminoquinoline ring is, in fact, independent of the chain length between them, at least if the chain length is between 2 and 4 carbons.
The question of whether the RCQ molecules actually inhibit PfCRT’s ability to export CQ in strains which are CQ-resistant may best be evaluated by measurement systems such as implemented by Martin and Kirk [28, 35]. In fact, molecules such as 1 and 2 do inhibit this ability to inhibit CQ transport by PfCRT76T (unpublished data).
RELATED MOLECULES
October et al. used the chemistry of 3,4-Dihydropyrimidin-2(1H)-ones to construct RCQ molecules [37]; Fig. (4). Although such molecules lack the protonatable nitrogen of the classical pharmacophore, this structure did exhibit excellent potency against CQ-sensitive and CQ-resistant malaria strains. Another set of molecules, linking astemizole to the CQ quinoline end likewise proved hopeful [38]. Substitution of adamantyl groups for the phenyls in the RA pharmacophore has proved somewhat successful for others [39] in overcoming P. falciparum resistance, as well in our laboratory (unpublished). Screening led to related structures, e.g., [40, 41]. The acridine skeleton has been hybridized to CQ to make related RCQ structures [42]. The dibemethin group was appended to quinolines to make related hybrids [43], based on this group’s ability to inhibit PfCRT-mediated efflux of CQ [35], as outlined above. The drug candidate ferroquine [44, 45] may owe much of its potency to the ability to accumulate in the DV and not be exported by mutated PfCRT.
Fig. (4).

Examples of other molecules may be viewed as sharing aspects of RCQ drugs.
Even some older work, eg., bisquinolines; Fig. (4) [46, 47], including piperaquine [48], make up an interesting set of compounds which may be viewed as potential RCQ molecules, especially if nitrogen atoms are used in the segment that links the quinolines. The approved drug, piperaquine makes an interesting case. Fig. (4) shows the essential elements of the RA moiety superimposed on both imipramine and one end of a piperaquine molecule. In fact, one can almost think of CQ itself as a RA pharmacophore, in that the molecule can roughly fit the essential structural elements. Also, binding to PfCRT, a requisite for transport is a trait shared by RA molecules. In fact, PfCRT (mutated or not) may be able to transport some or all of the RA molecules, to various extents [49].
NEXT-GENERATION RCQ MOLECULES
Future efforts in designing RCQ molecules will be aimed in two directions. First, we are attempting to vary the RA end to a greater extent, with the idea that it may not be necessary to retain strong binding to PfCRT. The other direction will be to vary the substituent pattern on the quinoline ring system, as has been done for CQ-like structures by others, e.g., [50]. Both of these directions may help to reduce the hERG binding which gives rise to the potential cardiotoxicity common to quinoline-based malaria drugs [51]. We already have some evidence that the effects of changing the substituents are not the same as found in CQ and its derivatives.
THE ROLE FOR RCQ MOLECULES IN THE WORLD-WIDE MALARIA STRATEGY
One could ask why would we wish to develop new 4-aminoquinoline drugs against malaria, especially in the face of the failure of CQ? The first reason is that the drugs are generally inexpensive to produce, particularly on a large scale. The starting materials are not exotic, and the chemistry required to assemble the molecules is quite safe and simple [13-15]. Second, the RCQ molecules are a fairly strong departure from the side chain of CQ, so that there may be minimal cross-resistance to other strains. In fact, we have evaluated several “field-isolated” strain clones developed from infected patients, and found no significant resistance to any of several of the RCQ drugs. Third, the design is exceedingly flexible. This is important because if resistance does emerge against one drug, then it is possible that a next-generation RCQ could take its place. Forth, the physical-chemical properties can be chosen such that its pharmacokinetic parameters would be a good match for another drug in combination therapy, and such ability could be very useful for optimizing drug combinations. Finally, CQ was really a very good drug for nearly half a century. If we are successful in developing RCQ drugs that are even better in terms of efficacy and safety than CQ, then we could use the advantages of quinoline antimalarial drugs in another attempt at eradicating the disease. Given the RCQ potencies, it is conceivable to aim for single-dose radical cures that are hoped for in an elimination / eradication global strategy [52, 53], especially as a part of combination drugs in which the partners are carefully chosen to complement each other’s abilities and properties.
ACKNOWLEDGEMENTS
The author thanks The Medical Research Foundation of Oregon (Grant 0530) and the National Institutes of Health (Grants AI067837, AI072923, and AI090848) for financial support. There are many people who have contributed to this work, but of special note are collaborators (J.X. Kelly, R. Cooper, M. Smilkstein, R. Brun, S. Wittlin, and M. Riscoe) as well as past and present members of the Peyton research group (S. Burgess, S. Andrews, K. Leibmann, C. Hodson, and others). From DesignMedix, Inc., a start-up company dedicated to overcoming drug resistance in infectious diseases, the author recognizes L. Stevenson, S. Shotwell, W. Morrill, and (again) S. Burgess.
ABBREVIATIONS
- CQ
= Chloroquine
- PfCRT
= Plasmodium falciparum chloroquine resistance transporter
- DV
= Digestive vacuole
- CQ-resistant
= Chloroquine resistant
- CQ-senstive
= Chloroquine sensitive
- RA
= Reversal agent
- RCQ
= Reversed chloroquine compound
- SAR
= Structure-activity relationship
CONFLICT OF INTEREST
The author declares that he has a financial interest in DesignMedix, Inc., which is developing the RCQ drugs.
REFERENCES
- 1. Coatney GR. Pitfalls in a discovery, the chronicle of chloroquine. The American J.Tropic. Med. Hygiene. 1963;12:121–128. doi: 10.4269/ajtmh.1963.12.121. [DOI] [PubMed] [Google Scholar]
- 2. Packard RM. The Making of a Tropical Disease , A Short History of Malaria. Baltimore, Md: Johns Hopkins University Press; 2007. [Google Scholar]
- 3. Kumar A, Katiyar SB, Agarwal A, Chauhan PM. Perspective in antimalarial chemotherapy. Curr. Med. Chem. 2003;10:1137–1150. doi: 10.2174/0929867033457494. [DOI] [PubMed] [Google Scholar]
- 4. Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, von Seidlein L. Artemisinin resistance, current status and scenarios for containment. Nat. Rev. Microbiol. 2010;8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 5. Noedl H, Socheat D, Satimai W. Artemisinin-resistant malaria in Asia. N. Engl. J. Med. 2009;361:540–541. doi: 10.1056/NEJMc0900231. [DOI] [PubMed] [Google Scholar]
- 6. Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 7. Krogstad DJ, Gluzman IY, Kyle DE, Oduola AM, Martin SK, Milhous WK, Schlesinger PH. Efflux of chloroquine from Plasmodium falciparum, mechanism of chloroquine resistance. Science. 1987;238:1283–1285. doi: 10.1126/science.3317830. [DOI] [PubMed] [Google Scholar]
- 8. Martin SK, Oduola AM, Milhous WK. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science. 1987;235:899–901. doi: 10.1126/science.3544220. [DOI] [PubMed] [Google Scholar]
- 9. van Schalkwyk DA, Walden JC, Smith PJ. Reversal of chloroquine resistance in Plasmodium falciparum using combinations of chemosensitizers. Antimicrob. Agents Chemother. . 2001;45:3171–3174. doi: 10.1128/AAC.45.11.3171-3174.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Schalkwyk DA, Egan TJ. Quinoline-resistance reversing agents for the malaria parasite Plasmodium falciparum. Drug Resist. Updat. 2006;9:211–226. doi: 10.1016/j.drup.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 11. Guan J, Kyle DE, Gerena L, Zhang Q, Milhous WK, Lin AJ. Design, synthesis, and evaluation of new chemosensitizers in multi-drug-resistant Plasmodium falciparum. J. Med. Chem. 2002; 45:2741–2748. doi: 10.1021/jm010549o. [DOI] [PubMed] [Google Scholar]
- 12. Fidock AD, Nomura T, Talley KA, Cooper AR, Dzekunov MS, Ferdig TM, Ursos ML, Sidhu AB, Naude B, Deitsch WK, Su XZ, Wootton JC, Roepe PD, Wellems TE. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burgess SJ, Selzer A, Kelly JX, Smilkstein MJ, Riscoe MK, Peyton DH. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 2006;49:5623–5625. doi: 10.1021/jm060399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andrews S, Burgess SJ, Skaalrud D, Kelly JX, Peyton DH. Reversal agent and linker variants of reversed chloroquines, activities against Plasmodium falciparum. J. Med. Chem. 2010; 53:916–919. doi: 10.1021/jm900972u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burgess SJ, Kelly JX, Shomloo S, Wittlin S, Brun R, Liebmann K, Peyton DH. Synthesis, structure-activity relationship, and mode-of-action studies of antimalarial reversed chloroquine compounds. J. Med. Chem. 2010;53:6477–6489. doi: 10.1021/jm1006484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roepe PD. Molecular and physiologic basis of quinoline drug resistance in Plasmodium falciparum malaria. Future Microbiol . 2009;4:441–455. doi: 10.2217/fmb.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henry M, Alibert S, Orlandi-Pradines E, Bogreau H, Fusai T, Rogier C, Barbe J, Pradines B. Chloroquine resistance reversal agents as promising antimalarial drugs. Curr. Drug Targets. 2006;7:935–948. doi: 10.2174/138945006778019372. [DOI] [PubMed] [Google Scholar]
- 18. Walsh JJ, Bell A. Hybrid drugs for malaria. Curr. Pharm. Des. 2009;15:2970–2985. doi: 10.2174/138161209789058183. [DOI] [PubMed] [Google Scholar]
- 19. Morphy R, Kay C, Rankovic Z. From magic bullets to designed multiple ligands. Drug Discov. Today. 2004;9:641–651. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- 20. Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 21. Grimberg BT, Mehlotra RK. Expanding the antimalarial drug arsenal-now, but how? Pharmaceuticals. 2011;4:681–712. doi: 10.3390/ph4050681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol. Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- 23. Mehlotra RK, Fujioka H, Roepe PD, Janneh O, Ursos LM, Jacobs-Lorena V, McNamara DT, Bockarie MJ, Kazura JW, Kyle DE, Fidock DA, Zimmerman PA. Evolution of a unique Plasmodium falciparum chloroquine-resistance phenotype in association with pfcrt polymorphism in Papua New Guinea and South America. Proc. Natl. Acad. Sci. USA. 2001;98:12689–12694. doi: 10.1073/pnas.221440898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mu J, Ferdig MT, Feng X, Joy DA, Duan J, Furuya T, Subramanian G, Aravind L, Cooper RA, Wootton JC, Xiong M, Su XZ. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol. Microbiol. 2003;49:977–989. doi: 10.1046/j.1365-2958.2003.03627.x. [DOI] [PubMed] [Google Scholar]
- 26. Biagini GA, Fidock DA, Bray PG, Ward SA. Mutations conferring drug resistance in malaria parasite drug transporters Pgh1 and PfCRT do not affect steady-state vacuolar Ca2+ Antimicrob. Agents Chemother. 2005;49:4807–4808. doi: 10.1128/AAC.49.11.4807-4808.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roepe PD. PfCRT-mediated drug transport in malarial parasites. Biochemistry. 2010;50:168–171. doi: 10.1021/bi101638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin RE, Marchetti RV, Cowan AI, Howitt SM, Broer S, Kirk K. Chloroquine transport via the malaria parasite's chloroquine resistance transporter. Science. 2009;325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- 29. Martindale W, Reynolds JEF. The extra pharmacopoeia. 30th ed. London: Pharmaceutical Press; 1993. Royal pharmaceutical society of great britain. Dept. Pharmaceutical Sciences. [Google Scholar]
- 30. Adam ME, El Fatih E, Karim AY, Ibrahim KEE, Berger BJ, Wiese M, Babiker HA. Imipramine induced complete reversal of chloroquine resistance in Plasmodium falciparum infections in Sudan. Saudi Pharm. J. 2004;12:130–135. [Google Scholar]
- 31. Bhattacharjee AK, Kyle DE, Vennerstrom JL. Structural analysis of chloroquine resistance reversal by imipramine analogs. Antimicrob. Agents Chemother. 2001;45:2655–2657. doi: 10.1128/AAC.45.9.2655-2657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miki A, Tanabe K, Nakayama T, Kiryon C, Ohsawa K. Plasmodium chabaudi, association of reversal of chloroquine resistance with increased accumulation of chloroquine in resistant parasites. Exp. Parasitol. 1992;74:34–142. doi: 10.1016/0014-4894(92)90040-h. [DOI] [PubMed] [Google Scholar]
- 33. Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhattacharjee AK, Kyle DE, Vennerstrom JL, Milhous WK. A 3D QSAR pharmacophore model and quantum chemical structure--activity analysis of chloroquine(CQ)-resistance reversal. J. Chem. Inf. Comput. Sci. 2002;42:1212–1220. doi: 10.1021/ci0200265. [DOI] [PubMed] [Google Scholar]
- 35. Zishiri VK, Hunter R, Smith PJ, Taylor D, Summers R, Kirk K, Martin RE, Egan TJ. A series of structurally simple chloroquine chemosensitizing dibemethin derivatives that inhibit chloroquine transport by PfCRT. Eur. J. Med. Chem. 2011; 46:1729–1742. doi: 10.1016/j.ejmech.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 36. Otelo VA, Sant'Ana AC, de Faria DL, Menezes CM. Molecular modeling and UV-vis spectroscopic studies on the mechanism of action of reversed chloroquine (RCQ) Bioorg. Med. Chem. Lett. 2011;21:250–254. doi: 10.1016/j.bmcl.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 37. October N, Watermeyer ND, Yardley V, Egan TJ, Ncokazi K, Chibale K. Reversed chloroquines based on the 3,4- dihydropyrimidin-2(1H)-one scaffold, synthesis and evaluation for antimalarial, beta-haematin inhibition, and cytotoxic activity. ChemMedChem. 2008;3:1649–1653. doi: 10.1002/cmdc.200800172. [DOI] [PubMed] [Google Scholar]
- 38. Musonda CC, Whitlock GA, Witty MJ, Brun R, Kaiser M. Chloroquine-astemizole hybrids with potent in vitro and in vivo antiplasmodial activity. Bioorg. Med. Chem. Lett. 2009;19:481–484. doi: 10.1016/j.bmcl.2008.11.047. [DOI] [PubMed] [Google Scholar]
- 39. Solaja BA, Opsenica D, Smith KS, Milhous WK, Terzic N, Opsenica I, Burnett JC, Nuss J, Gussio R, Bavari S. Novel 4- aminoquinolines active against chloroquine-resistant and sensitive P. falciparum strains that also inhibit botulinum serotype. A. J. Med. Chem. 2008;51:4388–4391. doi: 10.1021/jm800737y. [DOI] [PubMed] [Google Scholar]
- 40. Ray S, Madrid PB, Catz P, LeValley SE, Furniss MJ, Rausch LL, Guy RK, DeRisi JL, Iyer LV, Green CE, Mirsalis JC. Development of a new generation of 4-aminoquinoline antimalarial compounds using predictive pharmacokinetic and toxicology models. J. Med. Chem. 2010;53:3685–3695. doi: 10.1021/jm100057h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Madrid PB, Wilson NT, DeRisi JL, Guy RK. Parallel synthesis and antimalarial screening of a 4-aminoquinoline library. J. Comb. Chem. 2004;6:437–442. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumar A, Srivastava K, Kumar SR, Puri SK, Chauhan PM. Synthesis of new 4-aminoquinolines and quinoline-acridine hybrids as antimalarial agents. Bioorg. Med. Chem. Lett. 2010;20:7059–7063. doi: 10.1016/j.bmcl.2010.09.107. [DOI] [PubMed] [Google Scholar]
- 43. Zishiri VK, Joshi MC, Hunter R, Chibale K, Smith PJ, Summers RL, Martin RE, Egan TJ. Quinoline antimalarials containing a dibemethin group are active against chloroquinone-resistant Plasmodium falciparum and inhibit chloroquine transport via the P. falciparum chloroquine-resistance transporter (PfCRT) J. Med. Chem. 2011;54:6956–6968. doi: 10.1021/jm2009698. [DOI] [PubMed] [Google Scholar]
- 44. Dive D, Biot C. Ferrocene conjugates of chloroquine and other antimalarials, the development of ferroquine, a new antimalarial. ChemMedChem. 2008;3:383–391. doi: 10.1002/cmdc.200700127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dubar F, Khalife J, Brocard J, Dive D, Biot C. Ferroquine, an ingenious antimalarial drug, thoughts on the mechanism of action. Molecules. 2008;13:2900–2907. doi: 10.3390/molecules13112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vennerstrom JL, Ager AL, Jr, Dorn A, Andersen SL, Gerena L, Ridley RG, Milhous WK. Bisquinolines. 2. Antimalarial N,N-bis(7-chloroquinolin-4-yl)heteroalkanediamines. J. Med. Chem. 1998;41:4360–4364. doi: 10.1021/jm9803828. [DOI] [PubMed] [Google Scholar]
- 47. Vennerstrom JL, Ellis WY, Ager AL, Jr, Andersen SL, Gerena L, Milhous WK. Bisquinolines. 1. N,N-bis(7-chloroquinolin-4-yl)alkanediamines with potential against chloroquine-resistant malaria. J. Med. Chem. 1992;35:2129–2134. doi: 10.1021/jm00089a025. [DOI] [PubMed] [Google Scholar]
- 48. Warhurst DC, Craig JC, Adagu IS, Guy RK, Madrid PB, Fivelman QL. Activity of piperaquine and other 4-aminoquinoline antiplasmodial drugs against chloroquine-sensitive and resistant blood-stages of Plasmodium falciparum. Role of beta-haematin inhibition and drug concentration in vacuolar water- and lipid-phases. Biochem. Pharmacol. 2007;73:1910–1926. doi: 10.1016/j.bcp.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 49. Lehane AM, Kirk K. Efflux of a range of antimalarial drugs and 'chloroquine resistance reversers' from the digestive vacuole in malaria parasites with mutant PfCRT. Mol. Microbiol. 2010 doi: 10.1111/j.1365-2958.2010.07272.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 50. Madrid PB, Sherrill J, Liou AP, Weisman JL, Derisi JL, Guy RK. Synthesis of ring-substituted 4-aminoquinolines and evaluation of their antimalarial activities. Bioorg. Med. Chem. Lett. . 2005;15:1015–1018. doi: 10.1016/j.bmcl.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 51. White NJ. Cardiotoxicity of antimalarial drugs. The Lancet infectious diseases. 2007;7:549–558. doi: 10.1016/S1473-3099(07)70187-1. [DOI] [PubMed] [Google Scholar]
- 52. Alonso P, Brown G, Arevalo-Herrera M, Binka F, Chitnis C, Collins F, Doumbo OK, Greenwood B, Hall BF, Levine MM, Mendis K, Newman RD, Plowe CV, Rodríguez MH, Sinden R, Slutsker L, Tanner M. A research agenda to underpin malaria eradication. PLoS Med. 2011;8(1):e1000406. doi: 10.1371/journal.pmed.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.A research agenda for malaria eradication, drugs. PLoS medicine. 2011;8(1):e1000402. doi: 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]